

Abstract
Burkholderia cenocepacia is an opportunistic human pathogen that encodes two LuxI-type acylhomoserine lactone (AHL) synthases and three LuxR-type AHL receptors. Of these, cepI and cepR form a cognate synthase/receptor pair, as do cciI and cciR, while cepR2 lacks a genetically linked AHL synthase gene. Another group showed that a cepR2 mutant overexpressed a cluster of linked genes that appear to direct the production of a secondary metabolite (Malott et al., 2009). We found that these same genes were upregulated by octanoylhomoserine lactone (OHL), which is synthesized by CepI. These data suggest that several cepR2-linked promoters are repressed by CepR2 and that CepR2 is antagonized by OHL. Fusions of two divergent promoters to lacZ were used to confirm these hypotheses, and promoter resections and DNase I footprinting assays revealed a single CepR2 binding site between the two promoters. This binding site lies well upstream of both promoters, suggesting an unusual mode of repression. Adjacent to the cepR2 gene is a gene that we designate cepS, which encodes an AraC-type transcription factor. CepS is essential for expression of both promoters, regardless of the CepR2 status or OHL concentration. CepS therefore acts downstream of CepR2, and CepR2 appears to function as a CepS antiactivator.
Introduction
The genus Burkholderia encompasses over 50 species that occupy extremely diverse ecological niches (Vanlaere et al., 2009). Some species are of interest in the bioremediation of xenobiotic contaminants (Chen et al., 2003). Other members are capable of forming nitrogen-fixing root nodules with legumes (Bontemps et al., 2010). Some members protect host plants against fungal pathogens (Parke & Gurian-Sherman, 2001), while other species are pathogenic against plants, animals, and humans. B. mallei causes glanders in equines, while B. pseudomallei causes melioidosis in a variety of animals. Both can also be transmitted to humans, and are select agents of concern as possible bioweapons (Godoy et al., 2003, Wheelis, 1998).
Seventeen pathogenic species, including B. cenocepacia, B. cepacia, B. vietnamiensis, and B. multivorans are members of the Burkholderia cepacia complex, or BCC (Vandamme et al., 1997, Vanlaere et al., 2009, Vanlaere et al., 2008). Among these, B. cenocepacia is recognized as an opportunistic pathogen of humans and is a particular threat to cystic fibrosis (CF) patients (Mahenthiralingam et al., 2005, Vandamme et al., 1997). Colonization of the CF lung by B. cenocepacia (Vandamme et al., 2003) tends to occur in patients already infected with Pseudomonas aeruginosa (Jones & Webb, 2003, Vandamme et al., 1997). B. cenocepacia strains are resistant to most antibiotics, making them virtually impossible to eradicate (Nzula et al., 2002).
Many or possibly all Burkholderia spp. encode at least one regulatory system that resembles the LuxR and LuxI proteins of Vibrio fischeri, where LuxI synthesizes an acylhomoserine lactone (AHL)-type pheromone, also called an autoinducer, and LuxR is an AHL-dependent transcriptional regulator (Ng & Bassler, 2009). Regulatory systems of this family are found in countless proteobacteria, where they are thought to allow bacteria to estimate their population size and for individual bacteria to coordinate their physiology with their siblings (Stevens et al., 2012, Galloway et al., 2011, Churchill & Chen, 2011). In general, target genes are transcribed preferentially at population densities high enough to favor AHL accumulation, a phenomenon sometimes referred to as quorum sensing.
A few members of this family are antagonized by their cognate autoinducers, and bind DNA only in their absence (Tsai & Winans, 2010). Most of these are closely related to each other and include EsaR of Pantoea stewartii, ExpR of Pectobacterium caratovorum (formerly Erwinia caratovora), and YenR of Yersinia enterocolitica (Castang et al., 2006, Cui et al., 2005, Fineran et al., 2005, Minogue et al., 2005, Sjoblom et al., 2006, Tsai & Winans, 2011). At least one LuxR-type protein that is not closely related to EsaR, ExpR, or YenR is also antagonized by its cognate (Delrue et al., 2005). We will demonstrate that B. cenocepacia encodes a protein with similar properties.
B. cenocepacia J2315 encodes three LuxR homologs and two LuxI homologs (Lewenza et al., 1999, Malott et al., 2005, Malott et al., 2009). Among these, CepR and CepI are well conserved within the BCC (Venturi et al., 2004). CepI synthesizes primarily octanoylhomoserine lactone (OHL), and lower levels of hexanoylhomoserine lactone (HHL) (Aguilar et al., 2003, Gotschlich et al., 2001, Huber et al., 2001, Lewenza et al., 1999). Null mutations in cepI or cepR increase the production of the siderophore ornibactin, and decrease the production of secreted lipases and metalloproteases ZmpA and ZmpB (Kooi et al., 2006, Lewenza et al., 1999, Lewenza & Sokol, 2001, Sokol et al., 2003). CepI and CepR are also required for swarming motility and biofilm formation (Huber et al., 2001) and for pathogenicity in several animal models (Kothe et al., 2003, Sokol et al., 2003). B. cenocepacia J2315 also encodes CciI and CciR, which are found on a genomic island called cci (cenocepacia island), that is found only in a subset of B. cenocepacia strains (Malott et al., 2005). The CepIR and CciIR systems extensively interact, in that CciR negatively regulates cepI, while CepR is required for expression of the cciIR operon (Malott et al., 2005). Transcriptional profiling studies indicate that CepR and CciR regulate many of the same genes, but do so in opposite ways (O'Grady et al., 2009).
B. cenocepacia also encodes a third LuxR-type transcription factor, CepR2, whose gene is not linked to any apparent AHL synthase gene. In an elegant study, the cepR2 gene was reported to be autorepressed and repressed by CciR (Malott et al., 2009). A cepR2 mutation increased the expression of 64 genes and decreased the expression of 127 others (Malott et al., 2009). These included genes involved in virulence, chemotaxis, heat shock, and signal transduction, and pyochelin production. Differential expression was strongest in a group of genes that are closely linked to cepR2, including cepR2 itself, an adjacent gene bcam0189, which encodes an AraC type protein (that we designate CepS), a two gene operon (bcam0191-0190), a divergent five-gene operon (bcam0192-0196), and a nearby four gene operon (bcam0199-0202). Bcam0190-0196 are predicted to direct the synthesis of a secondary metabolite, while Bcam0199-0202 are predicted to direct the efflux of a small molecule. All of these genes were expressed more strongly in the mutant than in wild type, indicating that CepR2 inhibits their expression. CepR2 was fully functional in the absence of any AHL. In a heterologous system, the ability of CepR2 to activate a lux operon was not affected by the addition of any AHL. It was concluded that CepR2 functions independently of AHLs and does not detect them (Malott et al., 2009).
Members of our laboratory are interested in the genetic and biochemical properties of several LuxR-type proteins, including CepR. To further those studies, we used oligonucleotide microarrays to identify genes that are differentially expressed by exogenous OHL, and were surprised to find that several genes that are induced by OHL were previously found to be repressed by CepR2 (Malott et al., 2009). Taking the two findings together, this would suggest that OHL antagonizes CepR2 activity, though this model was difficult to reconcile with the report that CepR2 was unaffected by any AHL (Malott et al., 2009). This puzzle prompted the current set of experiments, which include determining the roles of CepR2, OHL, and CepS on promoter activity in whole cells as well as biochemical assays of the ability of CepR2 to bind OHL and its ability to bind to specific DNA sequences near target promoters.
Results
In previous studies, we identified a set of genes that are directly regulated by CepR (Wei et al., 2011). In an effort to identify additional members of this regulon, we cultured the cepI mutant strain CLW101 in the presence and absence of 1 μM OHL, and screened for differential gene expression using oligonucleotide microarrays. This strain contains a PcepI-lacZ fusion that was created by an insertion of Tn5lac in cepI (Weingart et al., 2005), which allows us to do two parallel tests for induction, one by assaying for β-galactosidase activity, and the other assaying for lacZ mRNA, as our microarrays include probes for this transcript. Cultures containing OHL expressed 100 to 200 times moreβ-galactosidase than identical cultures lacking OHL (Table 1). OHL caused a 3.2–3.5 fold increase in lacZ mRNA abundance as measured by the microarrays (Table 1). These data indicate that the microarrays reflected expression of this gene but show a compressed induction ratio, perhaps due to lacZ mRNA being less stable than β-galactosidase protein. Another CepR-regulated operon composed of aidA and aidB, was also strongly induced by OHL (Table 1).
Table 1.
Transcriptional profiles of cells cultured in the presence or absence of 1 μM OHL.a
| OHL Induction Ratio (S.D.) | |||||
|---|---|---|---|---|---|
| Gene | Trial 1 | Trial 2 | Average | cepR2 vs. WT (Malott et al., 2009) | Alternate Name, Comments, References |
| lacZ | 3.51 (0.81) | 3.24 (0.32) | 3.38 | n.a.b | cepI-lacZ reporter |
| bcal0510 | 7.61 (1.58) | 6.41 (0.46) | 7.01 | n.d.c | CepR-regulated (Wei et al., 2011) |
| bcal0831 | 1.66 (0.20) | 2.88 (0.81) | 2.27 | n.d. | |
| bcal0833 | 0.87 (0.15) | 1.29 (0.28) | 1.08 | n.d. | phbB |
| bcal2118 | 1.55 (0.24) | 3.56 (0.93) | 2.55 | n.d. | |
| bcal3178 | 1.49 (0.41) | 3.09 (0.54) | 2.29 | n.d. | |
| bcam0030 | 2.63 (1.41) | 5.36 (2.27) | 3.99 | n.d. | |
| bcam0031 | 2.57 (1.21) | 4.48 (3.18) | 3.52 | n.d. | |
| bcam0184 | 1.34 (0.28) | 4.06 (3.23) | 2.70 | n.d. | Lectin |
| bcam0185 | 1.43 (0.45) | 11.8 (1.96) | 6.59 | n.d. | Lectin |
| bcam0186 | 3.12 (0.40) | 1.81 (0.52) | 2.46 | 2.9 (0.8) | bclA (lectin) |
| bcam0187 | 0.99 (0.22) | 3.24 (0.32) | 2.12 | n.d. | |
| bcam0188 | 2.45 (0.99) | 2.62 (0.69) | 2.53 | 21.1 (3.4) | cepR2 |
| bcam0189 | 1.03 (0.26) | 2.15 (0.46) | 1.59 | 188 (46) | cepS |
| bcam0190 | 2.00 (0.81) | 3.25 (0.54) | 2.62 | 11.6 (0.9) | Aminotransferase Class III |
| bcam0191 | 1.96 (0.37) | 4.11 (1.24) | 3.04 | 9.5 (3.0) | Non-ribosomal peptide synthase |
| bcam0192 | 4.92 (1.24) | 4.63 (2.79) | 4.78 | 113 (27) | Conserved hypothetical |
| bcam0193 | 3.08 (1.28) | 6.15 (1.73) | 4.62 | 171 (74) | Conserved hypothetical |
| bcam0194 | 7.46 (1.69) | 5.18 (1.70) | 6.32 | 151 (48) | Conserved hypothetical |
| bcam0195 | 3.41 (1.76) | 6.60 (1.33) | 5.00 | 58.3 (62) | Non-ribosomal peptide synthase |
| bcam0196 | 7.08 (3.02) | 3.50 (3.19) | 5.29 | 80.5 (31) | Conserved hypothetical |
| bcam0393 | 2.45 (0.89) | 3.34 (1.43) | 2.90 | n.d. | |
| bcam0634 | 3.59 (0.08) | 2.64 (0.15) | 3.12 | n.d. | |
| bcam1413a | 3.58 (1.60) | 3.50 (3.19) | 3.54 | n.d. | aidC (Wei et al., 2011) |
| bcam1742 | 1.86 (0.65) | 2.60 (1.10) | 2.23 | n.d. | |
| bcam1869 | 1.86 (0.65) | 3.26 (1.23) | 2.56 | n.d. | CepR-regulated (Wei et al., 2011) |
| bcam2307 | 2.96 (0.60) | 7.09 (3.23) | 5.02 | −1.7 ± (0.7) | zmpB |
| bcam2308 | 2.00 (0.57) | 2.31 (0.85) | 2.15 | n.d. | |
| bcas0153 | 2.34 (0.42) | 1.78 (0.21) | 2.06 | n.d. | |
| bcas0292 | 14.9 (11.8) | 39.4 (41.2) | 27.2 | n.d. | aidB, CepR-regulated (Wei et al., 2011) |
| bcas0293 | 293 (299) | 198 (156) | 245 | 1.7 ± (0.2) | aidA, CepR-regulated (Wei et al., 2011) |
| bcas0409 | 2.31 (0.60) | 2.64 (0.67) | 2.47 | 1.6 ± (0.3) | zmpA |
| All Genes | 1.00 (1.26) | 1.00 (1.37) | 1.00 | ||
Strain CLW101 contains a chromosomal PcepI-lacZ fusion (Weingart et al., 2005). In Trial 1 the culture lacking OHL expressed 2.7 Miller units of β-galactosidase, while the culture containing OHL expressed 290 units. In Trial 2, the culture lacking OHL expressed 1.5 units, while the culture containing OHL expressed 195 units.
Not applicable
Not determined
In this transcriptional profiling experiment, we also detected OHL-inducible expression of a number of additional genes (Table 1), including several that are closely linked to cepR2. Interestingly, all of the OHL-inducible genes linked to cepR2 were previously found to be expressed more strongly in a cepR2 mutant than in a wild type strain (Malott et al., 2009). The two studies taken together could suggest that apo-CepR2 represses these genes, and that its ability to repress them is somehow antagonized by OHL. These genes are expressed in six apparent operons (Fig. 1), including cepR2 and another possible regulatory gene cepS (both of which are monocistronic). The operon containing bcam0184-0186 and the divergent bcam0187 were induced rather weakly compared to the others and were not pursued in the present study. We focus first on the promoters of the bcam0191-0190 operon and of the divergent the bcam0192-0196 operon. Later, we will describe the regulation of cepR2 and cepS.
Fig. 1. Resections of the bcam0191 and bcam0192 promoters.
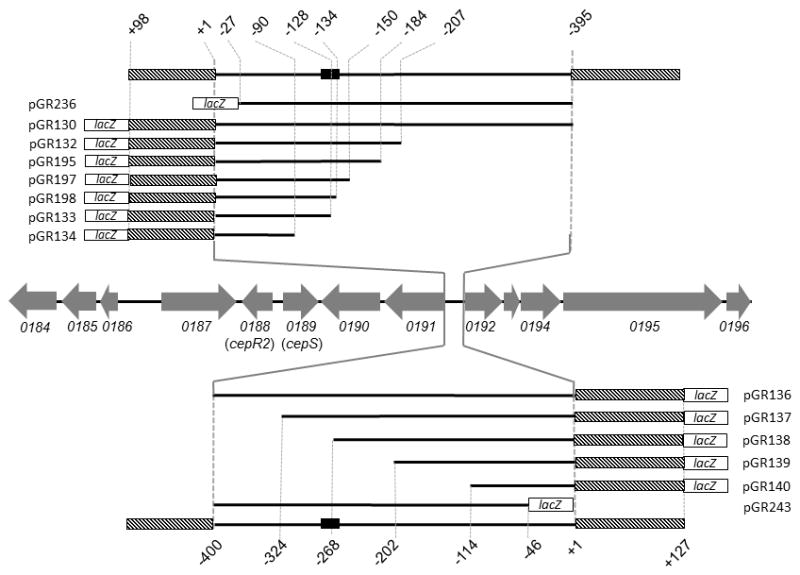
Chromosomal organization of OHL-inducible genes is indicated using gray arrows. Genes are named in accordance with the genome sequence of strain J2315 (Holden et al., 2009). Promoter-proximal portions of the bcam0191 and bcam0192 genes are indicated using hatched boxes. Endpoints of each resection are calculated with respect to the translation start site of the regulated gene. The solid black box represents the CepR2 binding site.
Regulation of bcam0191 and bcam0192 by CepR2 and CepS
In order to study the bcam0191 and bcam0192 promoters more closely, we fused each to lacZ on a low copy plasmid. Plasmid pGR130 contains the bcam0191 promoter on a 493 nucleotide fragment (Fig. 1), while plasmid pGR136 contains the bcam0192 promoter on a 527 nucleotide fragment. Expression of these fusions was tested in strains containing or lacking cepR2 or cepS, and in the presence or absence of exogenous OHL. All strains lacked cepI, and so they did not synthesize OHL.
A cepR2+ strain expressing the bcam0191-lacZ fusion (pGR130) expressed 35 units of β-galactosidase in the absence of OHL (Table 2). This fusion was induced approximately 11-fold by OHL, in reasonable agreement with the transcriptional profiling experiments described above. This fusion was also expressed 11-fold more strongly in a strain lacking CepR2 than in a CepR2+ strain, in agreement with the data of the Malott study (Malott et al., 2009). Addition of OHL did not stimulate expression in the strain lacking CepR2 (Table 2). These data are consistent with the hypothesis that the bcam0191 promoter is repressed by CepR2 and that repression is somehow antagonized by OHL.
Table 2.
Regulation of the promoters of bcam0191 and bcam0192 by CepR2, CepS, and OHL.
| Fusiona | Chromosomal genotypeb | Plasmid genotype | OHL (uM) | β-Galactosidase | Normalized Valuesc |
|---|---|---|---|---|---|
| bcam0191 | WT | none | 0 | 35 ± 8 | (1) |
| WT | none | 1 | 377 ± 30 | 10.8 | |
| GR141 (cepR2-) | pSRKKm | 0 | 383 ± 24 | 10.9 | |
| GR141 (cepR2-) | pSRKKm | 1 | 397 ± 32 | 11.3 | |
| GR141 (cepR2-) | pGR192 (cepR2) | 0 | 33 ± 4 | 0.95 | |
| GR141 (cepR2-) | pGR192 (cepR2) | 1 | 387 ± 21 | 11.1 | |
| GR145 (cepS-) | pSRKGm | 0 | 5 ± 3 | 0.14 | |
| GR145 (cepS-) | pSRKGm | 1 | 3 ± 2 | 0.1 | |
| GR145 (cepS-) | pGR193 (cepS) | 0 | 41 ± 8 | 1.2 | |
| GR145 (cepS-) | pGR193 (cepS) | 1 | 412 ± 32 | 11.8 | |
| bcam0192 | WT | none | 0 | 32 ± 3 | (1) |
| WT | none | 1 | 362 ± 15 | 11.3 | |
| GR141 (cepR2-) | pSRKKm | 0 | 376 ± 31 | 11.8 | |
| GR141 (cepR2-) | pSRKKm | 1 | 368 ± 25 | 11.5 | |
| GR141 (cepR2-) | pGR192 (cepR2) | 0 | 36 ± 1 | 1.1 | |
| GR141 (cepR2-) | pGR192 (cepR2) | 1 | 373 ± 24 | 11.7 | |
| GR145 (cepS-) | pSRKGm | 0 | 3 ± 1 | 0.1 | |
| GR145 (cepS-) | pSRKGm | 1 | 4 ± 2 | 0.13 | |
| GR145 (cepS-) | pGR193 (cepS) | 0 | 43 ± 1 | 1.3 | |
| GR145 (cepS-) | pGR193 (cepS) | 1 | 396 ± 36 | 12.4 |
A bcam0191-lacZ transcriptional fusion was provided by pGR130, while a bcam0192-lacZ fusion was provided using pGR136. The vector for both plasmids, pYW302, expressed only 1–2 units of β-galactosidase activity.
All strains are derived from K56-I2, which carries an insertion mutation in cepI. Strains were cultured at 37°C in LB supplemented with 0.5 mM IPTG, appropriate antibiotics, and containing or lacking OHL as indicated, to an OD600 of 0.4, and assayed for β-galactosidase activity. Data were obtained from a single representative experiment using three independent isolates of each strain, each assayed once. Mean value and standard deviations are indicated.
β-galactosidase activity is normalized to that of the wild type strain carrying the indicated plasmid and cultured in the absence of OHL.
As described above, the cepS gene is adjacent to cepR2, and encodes a possible transcription factor of the AraC family. The genetic linkage of cepS to cepR2 and to bcam0191 suggested a possible role in their regulation. We therefore deleted cepS and tested for the expression of the bcam0191-lacZ fusion in this mutant. Loss of cepS caused a severe decrease in expression of this promoter, both in the presence and absence of OHL (Table 2). The lack of stimulation by OHL in a cepS mutant indicates that when CepR2 is inactive and CepS is absent, expression is very low. In other words CepS is epistatic to CepR2.
Similar results were obtained from the divergent bcam0192 promoter (Table 2). In a strain expressing CepR2, the fusion in pGR136 was expressed 11-fold more strongly in the presence of OHL than in its absence. In a strain lacking CepR2, the fusion was expressed 12-fold more strongly than in the presence of apo-CepR2 (Table 2) and was unaffected by OHL. The cepS mutant expressed this promoter at low levels that were unaffected by OHL. Evidently, the bcam0192 promoter is repressed by apo-CepR2 and activated by CepS, similar to the bcam0191 promoter.
Reconstitution of regulated expression in a heterologous host
We sought to determine whether CepR2 and CepS regulate the bcam0191 and bcam0192 promoters directly, and therefore attempted to reconstitute regulated expression in E. coli MC4100. Plasmid pGR130 was introduced into derivatives of MC4100 containing plasmids that express CepR2 and/or CepS. In a strain expressing neither CepR2 nor CepS, the bcam0191-lacZ fusion expressed approximately 120 units of β-galactosidase and was not significantly affected by OHL (Table 3). The fusion was repressed approximately 4-fold by CepR2. Surprisingly, OHL had little or no effect on CepR2-mediated repression. CepS enhanced expression of the fusion about 2.5 fold in the presence or absence of OHL. When both proteins were provided in the absence of OHL, expression fell to the same levels as with CepR2 alone (Table 3). However, when OHL was provided, expression increased to the same levels as with CepS alone. Very similar data were obtained using E. coli strains expressing the bcam0192-lacZ fusion (Table 3). The ability of CepR2 and CepS to regulate expression of these promoters in E. coli indicates that they both are likely to act directly upon them.
Table 3.
Regulated expression of the bcam0191 and bcam0192 promoters in E. colia.
| Fusionb | Plasmids expressing B. cenocepacia genes | OHL (uM) | β-Galactosidase Activity | Normalized Valuec |
|---|---|---|---|---|
| bcam0191 | none | 0 | 121 ± 11 | (1) |
| None | 1 | 137 ± 15 | 1.13 | |
| pGR192 (cepR2) | 0 | 29 ± 8 | 0.24 | |
| pGR192 (cepR2) | 1 | 35 ± 8 | 0.29 | |
| pGR276 (cepS) | 0 | 310 ± 21 | 2.6 | |
| pGR276 (cepS) | 1 | 305 ± 17 | 2.5 | |
| pGR192 (cepR2), pGR276 (cepS) | 0 | 35 ± 8 | 0.29 | |
| pGR192 (cepR2), pGR276 (cepS) | 1 | 307 ± 30 | 2.5 | |
| bcam0192 | None | 0 | 101 ± 11 | (1) |
| None | 1 | 93 | 0.92 | |
| pGR192 (cepR2) | 0 | 23 ± 4 | 0.23 | |
| pGR192 (cepR2) | 1 | 32 ± 3 | 0.32 | |
| pGR276 (cepS) | 0 | 300 ± 11 | 3.0 | |
| pGR276 (cepS) | 1 | 344 ± 23 | 3.4 | |
| pGR192 (cepR2), pGR276 (cepS) | 0 | 32 ± 3 | 0.32 | |
| pGR192 (cepR2), pGR276 (cepS) | 1 | 293 ± 30 | 2.9 |
All strains were derived from MC4100. Strains were cultured at 37°C in LB supplemented with 0.5 mM IPTG, appropriate antibiotics, and containing or lacking OHL as indicated to an OD600 of 0.4, and assayed for β-galactosidase activity. Data were obtained from a single representative experiment using three independent isolates of each strain, each assayed once. Mean value and standard deviations are indicated.
A bcam0191-lacZ transcriptional fusion was provided by pGR130, while a bcam0192-lacZ fusion was provided using pGR136.
β-galactosidase activity is normalized to that of the wild type strain carrying the indicated plasmid and cultured in the absence of OHL.
Localization of DNA sequences required for regulated gene expression of bcam0191 and bcam0192
The intergenic region between bcam0191 and bcam0192 start codons is 396 nucleotides in length, and contains a strongly AT-rich region characteristic of many bacterial promoters (Fig. S1). In order to identify the essential sequences required for regulated expression of bcam0191, we made several resections of this promoter from its 5’ end (Fig. 1) and fused the remaining sequences to lacZ. Plasmids pGR132, pGR195, pGR133, and pGR134 resemble pGR130, but contain 207, 184, 128, and 90 nucleotides upstream of the bcam0191 translation start site, respectively. Plasmid pGR236 contains sequences from nucleotides −395 to −27 (Fig. 1). These plasmids were introduced into B. cenocepacia strain K56-I2, and the resulting strains were assayed for β-galactosidase activity. The fusion in pGR132 was expressed at 3-fold higher levels than that of pGR130 in the absence of OHL, while the two fusions were expressed at similar levels in the presence of OHL (Table 4). Both fusions were expressed at equally high levels in the absence of CepR2 and at equally low levels in a cepS mutant. Similar data were obtained using pGR195 and pGR236. Together, sequences required for OHL-responsive expression are limited to nucleotides −184 to −27.
Table 4.
Regulation of resected bcam0191 promoters by CepR2, CepS, and OHLa.
| Plasmid | Fragment | Genotype | OHL (uM) | β-Galacto- sidase | Normalized Valueb |
|---|---|---|---|---|---|
| pGR130 | −395 – +98 | WT | 0 | 35 ± 8 | (1) |
| WT | 1 | 377 ± 30 | 10.8 | ||
| GR141 (cepR2-) | 0 | 381 ± 30 | 10.9 | ||
| GR141 (cepR2-) | 1 | 365 ± 17 | 10.4 | ||
| GR145 (cepS-) | 0 | 7 ± 1 | 0.2 | ||
| GR145 (cepS-) | 1 | 2 ± 0 | 0.01 | ||
| pGR132 | −207 – +98 | WT | 0 | 94 ± 27 | (1) |
| WT | 1 | 361 ± 34 | 3.8 | ||
| GR141 (cepR2-) | 0 | 327 ± 28 | 3.5 | ||
| GR141 (cepR2-) | 1 | 349 ± 31 | 3.7 | ||
| GR145 (cepS-) | 0 | 9 ± 1 | 0.1 | ||
| GR145 (cepS-) | 1 | 7 ± 1 | 0.07 | ||
| pGR195 | −184 – +98 | WT | 0 | 74 ± 13 | (1) |
| WT | 1 | 361 ± 35 | 4.9 | ||
| GR141 (cepR2-) | 0 | 326 ± 28 | 4.4 | ||
| GR141 (cepR2-) | 1 | 389 ± 31 | 5.3 | ||
| GR145 (cepS-) | 0 | n.d. | n.d. | ||
| GR145 (cepS-) | 1 | n.d. | n.d. | ||
| pGR133 | −128 – +98 | WT | 0 | 339 ± 9 | (1) |
| WT | 1 | 361 ± 20 | 1.1 | ||
| GR141 (cepR2-) | 0 | 339 ± 29 | 1.0 | ||
| GR141 (cepR2-) | 1 | 360 ± 11 | 1.1 | ||
| GR145 (cepS-) | 0 | 1 ± 1 | 0.003 | ||
| GR145 (cepS-) | 1 | 1 ± 1 | 0.002 | ||
| pGR134 | −90 – +98 | WT | 0 | 10 ± 2 | (1) |
| WT | 1 | 6 ± 4 | 1.0 | ||
| GR141 (cepR2-) | 0 | 9 ± 4 | 1.0 | ||
| GR141 (cepR2-) | 1 | 10 ± 3 | 1.0 | ||
| GR145 (cepS-) | 0 | 2 ± 1 | 0.2 | ||
| GR145 (cepS-) | 1 | 2 ± 1 | 0.2 | ||
| pGR236 | −395 – 27 | WT | 0 | 45 ± 6 | (1) |
| WT | 1 | 248 ± 26 | 5.5 | ||
| GR141 (cepR2-) | 0 | 248 ± 19 | 5.5 | ||
| GR141 (cepR2-) | 1 | 226 ± 20 | 5.0 | ||
| GR145 (cepS-) | 0 | 3.3 ± 1 | 0.07 | ||
| GR145 (cepS-) | 1 | 2.5 ± 1 | 0.06 |
Derivatives of strain K56-I2 containing the indicated cepR2 or cepS mutations and the indicated plasmids were cultured at 37° C in LB supplemented with 0.5 mM IPTG and 300 μg ml-1 tetracycline, in the presence or absence of 1 μM OHL to an optical density of approximately 0.4 and assayed for β-galactosidase activity. Data were obtained from a single representative experiment using three independent isolates of each strain, each assayed once. Mean value and standard deviations are indicated.
β-galactosidase activity is normalized to that of the wild type strain carrying the indicated plasmid and cultured in the absence of OHL.
The fusion of pGR133 was expressed at equally high levels in the presence or absence of OHL (Table 4), and was not affected by a CepR2 mutation (Table 4). It was expressed at very low levels in a cepS mutant. These data indicate that pGR133 lacks some sequence required for repression by CepR2. Plasmid pGR195 contains all such sequences and is 56 nucleotides longer than pGR133 at the 5’ end.
The fusion of pGR134 was expressed at low levels in all backgrounds and was not responsive to OHL. This plasmid therefore lacks sequences required for promoter expression, either the promoter itself or the CepS binding site. Plasmid pGR133 contains all sequences required of CepS-dependent expression and is 38 nucleotides longer (Fig. 1).
We noticed an imperfect dyad symmetrical DNA sequence (GACAGCCCGATTTGCGGATGTC, symmetrical bases are underlined) present in all CepR2-repressed plasmids and absent or partially absent in all CepR2-nonresponsive ones. To determine whether this sequence plays a role in regulation, we constructed two additional plasmids, pGR197 and pGR198 (Fig. 1 and Fig. 2). The first plasmid contains this sequence plus 13 additional promoter-distal bases, while the second plasmid lacks five bases of this sequence. Plasmid pGR197 was induced by OHL in an E. coli strain expressing CepR2, while pGR198 was not affected (Fig. 2). We used site-directed mutagenesis to alter small groups of nucleotides within this dyad symmetry. Plasmids pGR259, pGR260, and pGR261 have 3- or 4-nucleotide mutations in the upstream half of this sequence. All three mutations significantly reduced induction by OHL (Fig. 2), providing additional evidence that this dyad is essential for CepR2 activity. We will demonstrate that this site is bound by CepR2 in vitro (see below).
Fig. 2. Resections and alterations of the CepR2 binding site.

The dyad symmetrical CepR2 binding site is indicated using inverted arrows. All sequences shown were part of bcam0191-lacZ fusions. B. cenocepacia sequences are capitalized while vector sequences are shown in lower case. Vector sequences that fortuitously match the original DNA sequence are capitalized. Site-directed mutations of the CepR2 binding site are underlined.β-galactosidase specific activities were determined for cells cultured for 12 hours in the presence or absence of OHL. The values shown are the means and standard deviations (error bars) of three independent experiments.
Similar experiments were carried out to identify cis-acting sites necessary for regulated expression of the divergent gene bcam0192. Four plasmids, pGR137, pGR138, pGR139, and pGR140 were constructed that resemble pGR136 but have 324, 268, 202, or 114 nucleotides of upstream DNA, respectively (Fig. 1). Plasmid pGR243 also resembles pGR136 but contains sequences from nucleotides -400 to -46 (Fig. 1). Significantly, pGR137 contains all of the dyad symmetry described above and 21 additional nucleotides, while pGR138 lacks half of the dyad, and pGR139 and pGR140 lack all of it. Plasmid pGR137 resembled pGR136 in that it was derepressed by OHL and by a cepR2 mutation, and was expressed at very low levels in a cepS mutant (Table 5). In contrast, the fusions in pGR138 and pGR139 were expressed at high levels and not significantly affected by CepR2 or OHL. They were expressed at low levels in a cepS mutant. Plasmid pGR140 expressed its fusion at very low levels under all conditions. Expression of the fusion of pGR243 was similar to wild type, indicating that all sequences required for regulation lie upstream of nucleotide −46. These data suggest that the dyad symmetry is required for regulation of the bcam0192 promoter, just as it was for the divergent bcam0191 promoter. In both cases, the repressor binding site appears to lie well upstream of the regulated promoters.
Table 5.
Regulation of the promoter of bcam0192 by CepR2, CepS, and OHLa.
| Plasmid | Fragment | Genotype | OHL (uM) | β-Galacto- sidase | Normalized Valueb |
|---|---|---|---|---|---|
| pGR136 | −400 – +127 | WT | 0 | 32 ± 3 | (1) |
| WT | 1 | 367± 30 | 11.5 | ||
| GR141 (cepR2-) | 0 | 376 ± 31 | 11.8 | ||
| GR141 (cepR2-) | 1 | 368 ± 25 | 1.0 | ||
| GR145 (cepS-) | 0 | 3 ± 1 | 0.1 | ||
| GR145 (cepS-) | 1 | 4 ± 2 | 0.01 | ||
| pGR137 | −324 – +127 | WT | 0 | 78 ± 10 | (1) |
| WT | 1 | 357 ± 15 | 4.8 | ||
| GR141 (cepR2-) | 0 | 356 ± 23 | 4.6 | ||
| GR141 (cepR2-) | 1 | 374 ± 34 | 1.0 | ||
| GR145 (cepS-) | 0 | 7 ± 3 | 0.9 | ||
| GR145 (cepS-) | 1 | 5 ± 2 | 0.01 | ||
| pGR138 | −268 – +127 | WT | 0 | 217 ± 23 | (1) |
| WT | 1 | 263 ± 15 | 1.2 | ||
| GR141 (cepR2-) | 0 | 374 ± 18 | 1.7 | ||
| GR141 (cepR2-) | 1 | 382 ± 23 | 1.7 | ||
| GR145 (cepS-) | 0 | 4 ± 2 | 0.02 | ||
| GR145 (cepS-) | 1 | 7 ± 3 | 0.03 | ||
| pGR139 | −202 – +127 | WT | 0 | 370 ± 21 | (1) |
| WT | 1 | 375 ± 23 | 1.0 | ||
| GR141 (cepR2-) | 0 | 364 ± 26 | 1.0 | ||
| GR141 (cepR2-) | 1 | 384 ± 43 | 1.0 | ||
| GR145 (cepS-) | 0 | 12.5 ± 4 | 0.03 | ||
| GR145 (cepS-) | 1 | 18.7 ± 9 | 0.05 | ||
| pGR140 | −114 – +127 | WT | 0 | 5 ± 7 | (1) |
| WT | 1 | 4 ± 9 | 0.8 | ||
| GR141 (cepR2-) | 0 | 8 ± 6 | 1.6 | ||
| GR141 (cepR2-) | 1 | 5 ± 3 | 1.0 | ||
| GR145 (cepS-) | 0 | 3 ± 2 | 0.7 | ||
| GR145 (cepS-) | 1 | 3 ± 1 | 0.7 | ||
| pGR243 | −400 – -46 | WT | 0 | 75 ± 14 | (1) |
| WT | 1 | 332 ± 32 | 4.4 | ||
| GR141 (cepR2-) | 0 | 392 ± 32 | 5.2 | ||
| GR141 (cepR2-) | 1 | 421 ± 28 | 5.6 | ||
| GR145 (cepS-) | 0 | 2.4 ± 1.2 | 0.03 | ||
| GR145 (cepS-) | 1 | 5.6 ± 1.2 | 0.07 |
Derivatives of strain K56-I2 containing the indicated cepR2 or cepS mutations and the indicated plasmids were cultured at 37°C in LB supplemented with 300 μg ml−1 tetracycline to an optical density of approximately 0.4 in the presence or absence of 1 μM OHL, and assayed for β-galactosidase activity. Data were obtained from a single representative experiment using three independent isolates of each strain, each assayed once. Mean value and standard deviations are indicated.
β-galactosidase activity is normalized to that of the wild type strain carrying the indicated plasmid and cultured in the absence of OHL.
Regulation of the cepR2 and cepS promoters
The microarray data described above shows that OHL may cause induction of cepR2 and cepS, though the effect is very slight. In contrast, microarray data of Malott and colleagues indicate that both these genes are expressed far more strongly in a cepR2 mutant than in a cepR2+ strain (Malott et al., 2009). Although these data do not directly contradict ours, the two datasets are nonetheless somewhat difficult to reconcile.
In order to study the expression of the cepR2 and cepS genes further, we constructed plasmids containins PcepR2-lacZ or PcepS-lacZ fusions. These plasmids were introduced into strains lacking one or the other of these genes, and cultured in the presence or absence of OHL. Both fusions gave similar results. In the strain containing cepR2 and cepS, expression was increased about 2-fold by OHL (Table S1). Perhaps surprisingly, this slight increase also was detected in a cepR2 mutant, indicating that CepR2 is not required. The cepS mutation caused a mild decrease in expression of both promoters, but did not affect the very slight stimulation by OHL. These data tend to support our microarray data. In the Discussion, we will present a possible explanation for the data of the Malott study.
Specificity of CepR2 for AHL-type pheromones
Throughout this study, we have used strains that have null mutations in cepI, and have been providing exogenous OHL where indicated. These strains still have cciI, and therefore presumably synthesize hexanoyl-HSL (HHL), and smaller amounts of similar pheromones. The fact that OHL influences CepR2 indicates that CciI-synthesized AHLs do not activate this fusion, at least not fully. However, they could in principle play some role in CepR2 function. To address this question, we assayed the expression of a bcam0191-lacZ fusion in the presence of different AHL-type pheromones, with acyl groups that vary in length and substitution. Among these, OHL was the most effective at derepressing the fusion (Fig. 3). The only other pheromone that showed significant activity was 3-oxooctanoyl-HSL (OOHL). Decanoyl-HSL (DHL) showed a trace of activity when provided at high concentrations, while five other AHL pheromones (hexanoyl-HSL, 3-oxo-hexanoyl-HSL, 3-oxo-decanoyl, dodecanoyl-HSL, and 3-oxo-dodecanoyl-HSL) were inactive (data not shown). We conclude that endogenous levels of pheromones synthesized by CciI did not detectably impact CepR2 activity.
Fig. 3. Detection of cognate and heterologous AHLs by CepR2.

Strains K56-I2(pGR130) and K56-I2(pGR136) were used to test the induction of the bcam0191 (A) and bcam0192 promoters (B), respectively. Strains were cultured with AHLs in the indicated amounts for 12 hours, and assayed forβ-galactosidase specific activity; OHL (triangles), 3-oxooctanoyl-HLS (OOHL, squares), and decanoyl-HSL (DHL, circles). The values shown are the mean standard deviation (error bars) from triplicate experiments. Five other AHLs (hexanoyl-HSL, 3-oxo-hexanoyl-HSL, 3-oxo-decanoyl-HSL, dodecanoyl-HSL, and 3-oxo-dodecanoyl-HSL) did not detectably induce expression of the fusion (data not shown). The values shown are the means and standard deviations (error bars) of three independent experiments. RFU: relative fluorescence units.
Ability of cells expressing CepR2 to sequester AHLs
The hypothesis that CepR2 is antagonized by OHL and OOHL predicts that it should be able to bind these AHLs stably and preferentially. To test this, we overexpressed CepR2 using the T7 promoter in E. coli in the presence of each of eight different AHLs, then washed the cells of each culture to remove unbound or weakly bound AHLs, and bioassayed for CepR2-bound AHLs. Of the eight AHLs tested, OHL was detected at the highest levels, followed by OOHL and ODHL (3-oxodecanoyl-HSL) (Fig. 4). Trace amounts of HHL and OHHL (3-oxohexanoyl-HSL) were bound, while DHL, dDHL and OdDHL (dodecanoyl-HSL and 3-oxododecanoyl-HSL) were not detectably sequestered. These data agree fairly well with the preference for OHL in vivo as described above, except that DHL was more active then ODHL in the former assay, while ODHL was sequestered more effectively than DHL. It appears that ODHL can bind CepR2 without altering its DNA binding properties as profoundly as other AHLs.
Fig. 4. Ability of CepR2 overproduced in E. coli to sequester eight different AHLs.
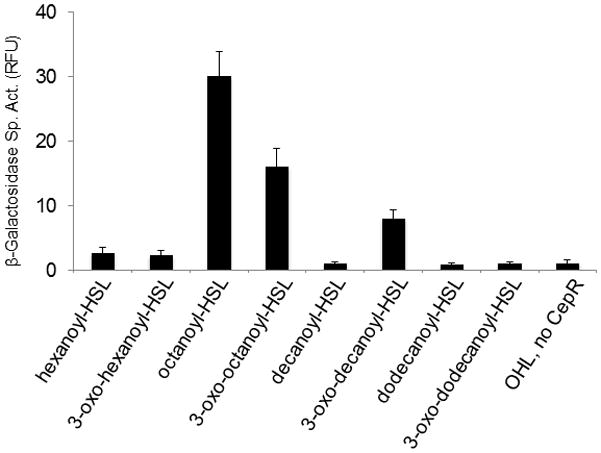
E. coli strain BL21(DE3)(pGR107) was incubated in medium containing 10 nM of the indicated AHL. Bound AHLs were extracted and bioassayed (Zhu et al., 1998). The bioassay strain was calibrated using each AHL. The values shown are the means and standard deviations (error bars) of three independent experiments. RFU: relative fluorescence units.
AHL-independent folding of CepR2
Several LuxR-type transcription factors that require AHLs for activity fail to fold into a soluble, protease resistant form in the absence of AHLs (Zhu & Winans, 1999, Zhu & Winans, 2001, Urbanowski et al., 2004, Schuster et al., 2004, Weingart et al., 2005). In contrast, several LuxR-type proteins that are antagonized by cognate AHLs fold into soluble, protease-resistant forms in the absence of their cognate pheromones (Tsai & Winans, 2011, Minogue et al., 2002, Castang et al., 2006). Solubility of some LuxR-type proteins is also enhanced by artificial overexpression of the chaperone GroESL (Chai & Winans, 2009, Choi & Greenberg, 1992). We assayed the accumulation of soluble CepR2 in the presence and absence of OHL, and in strains that express normal or elevated levels of GroESL. CepR2 was detected in a soluble form only when GroESL was overproduced (Fig. 5). The yield of soluble CepR2 may have been enhanced somewhat by OHL, but it was significantly soluble in the absence of OHL. CepR2 therefore resembles at least three other LuxR-type proteins that function as apo-proteins in that none requires its ligand for folding into a soluble form.
Fig. 5. Ability of CepR2 to fold into a soluble form requires GroESL but does not require OHL.
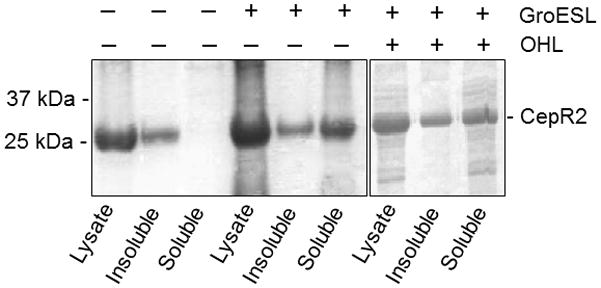
E. coli strain BL21(DE3)(pGR107) containing or lacking pT7-GroESL was cultured in medium containing or lacking 1 μM OHL, lysed, and clarified extracts were size- fractionated by SDS-PAGE and stained using Coomassie Brilliant Blue. Results are representative of three experiments with similar results.
Electrophoretic mobility shift assays with CepR2
Data described above suggested that CepR2 binds to a dyad symmetrical DNA sequence in the intergenic region between bcam0191 and bcam0192. We sought to obtain biochemical support for this hypothesis by carrying out electrophoretic mobility shift assays (EMSA) using radiolabelled DNA fragments containing this sequence. Several attempts to purify CepR2 failed to yield soluble and active protein. However clarified supernatants from an E. coli strain that overexpresses CepR2 were found to be active and were used for all binding experiments.
Clarified supernatants containing apo-CepR2 shifted a DNA fragment containing 83 nucleotides of DNA that contains this sequence (Fig. 6, Fragment 2) under conditions including a 10,000-fold excess non-specific competitor DNA. The extract containing apo-CepR2 did not shift two fragments containing nearby sequences (Fragments 1 and 3). We also tested two fragments identical to Fragment 2 (denoted Fragments 4 and 5) that contained either a 3-nucleotide or 4-nucleotide alterations in the dyad sequence (Fig. 6, bottom panel). Binding was virtually abolished with these mutant DNA fragments.
Fig. 6. Electrophoretic mobility shift assays of fragments containing the CepR2 binding site.
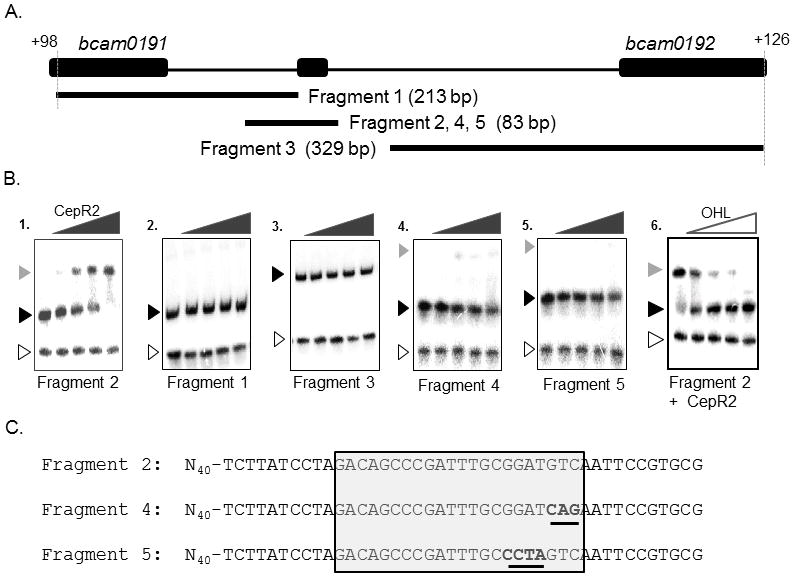
A: location and size of DNA fragments used in Part B. B. Clarified supernatants containing CepR2 were used for all binding reactions. A 65-bp PCR amplified lacZ DNA fragment was used as a negative control (open arrowhead). Free DNA is indicated using a black arrowhead, while CepR2-DNA complexes are indicated using a grey arrowhead. CepR2 supernatants were diluted serially in 3.16-fold increments in reactions with DNA fragments in the absence of OHL (gels 1–5). In gel 6, binding reactions containing CepR2 and Fragment 2 were amended with OHL to final concentrations of 0 μM, 0.032 μM, 0.1 μM, 0.315 μM, and 1.0 μM. C. Sequence of fragments containing the wild type CepR2 binding site (Fragment 2) or near-identical fragments having the indicated sequence alterations (Fragments 4 and 5). The dyad symmetrical CepR2 binding site is boxed, and altered sequences are underlined. Results are representative of at least two experiments with similar results.
The data described above using fusions indicates that CepR2 is antagonized by OHL and suggests that its ability to bind DNA might be inhibited by this pheromone. To test this, we set up binding reactions using Fragment 2, apo-CepR2, and a range of OHL concentrations. As predicted, OHL inhibited DNA binding by CepR2 (Fig. 6, right panel).
Earlier in this study we provided evidence that CepR2 does not autoregulate, nor does it regulate cepS. Supporting these conclusions, CepR2 did not detectably shift a DNA fragment containing the cepR2-cepS intergenic region (Fig. S2).
DNase I footprinting of the CepR2 binding site
In order to further localize the CepR2 binding site, we carried out DNase I footprinting experiments using fluorescently end-labeled DNA fragments containing this sequence. Clarified supernatants containing apo-CepR2 protected a region of approximately 20 nucleotides that contains this dyad symmetry (Fig. 7). On the basis of promoter resections, point mutations, EMSA, and DNase I footprinting, we conclude that CepR2 binds specifically to this dyad DNA sequence.
Fig. 7. DNase I protection of the CepR2 binding site by CepR2.
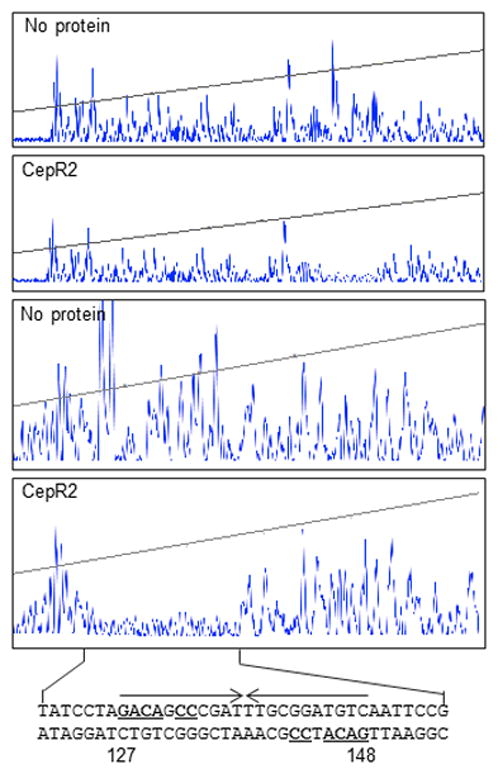
A fluorescently end-labeled DNA fragment was combined with a clarified extract containing CepR2 (second and fourth panel) or an extract lacking CepR2 (first and third panel), partially digested with DNase I, and size fractionated by automated capillary electrophoresis. The bottom two panels are enlargements of the right third of the top two panels. The DNA sequence of the protected region is shown at the bottom. The CepR2 binding site is indicated using inverted arrows, and symmetrical nucleotides are underlined. Nucleotides are numbered with respect to the 5’-end of the fluorescently-labeled amplicon. Results are representative of three experiments with similar results.
Identification of the transcription start sites of bcam0191 and bcam0192
In an effort to identify possible transcription start sites for the two promoters, we isolated total mRNA from strain K56-I2 cultured in the presence or absence of OHL and hybridized it with a 5’ fluorescently labeled oligonucleotide complementary to bcam0191 mRNA, and in a separate reaction, did the same experiment using an oligonucleotide complementary to bcam0192 mRNA. These oligonucleotides were used as primers for DNA synthesis by reverse transcriptase, and resulting cDNA transcripts were size-fractionated by automated capillary electrophoresis.
Using the former primer, the major reverse transcripts were 61, 62, and 63 nucleotides in length, corresponding to apparent start sites lying 54, 55, and 56 nucleotides upstream of the bcam0191 translation start site (Fig. 8). Upstream of these sites are sequences that resemble the -10 and -35 motifs of proteobacterial vegetative promoters. The promoter motif and apparent starts sites are
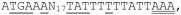 where single underlined sequences resemble consensus promoters, and the double underlines indicate the three apparent transcription start sites. Plasmid pGR134 contains this putative promoter with no additional upstream sequences. It expresses this promoter at very low levels, suggesting that it may lack a binding site for CepS. The CepR2 binding site is centered 75 nucleotides upstream of this putative transcription start site.
where single underlined sequences resemble consensus promoters, and the double underlines indicate the three apparent transcription start sites. Plasmid pGR134 contains this putative promoter with no additional upstream sequences. It expresses this promoter at very low levels, suggesting that it may lack a binding site for CepS. The CepR2 binding site is centered 75 nucleotides upstream of this putative transcription start site.
Fig. 8. Localization of the bcam0191 and bcam0192 promoters.
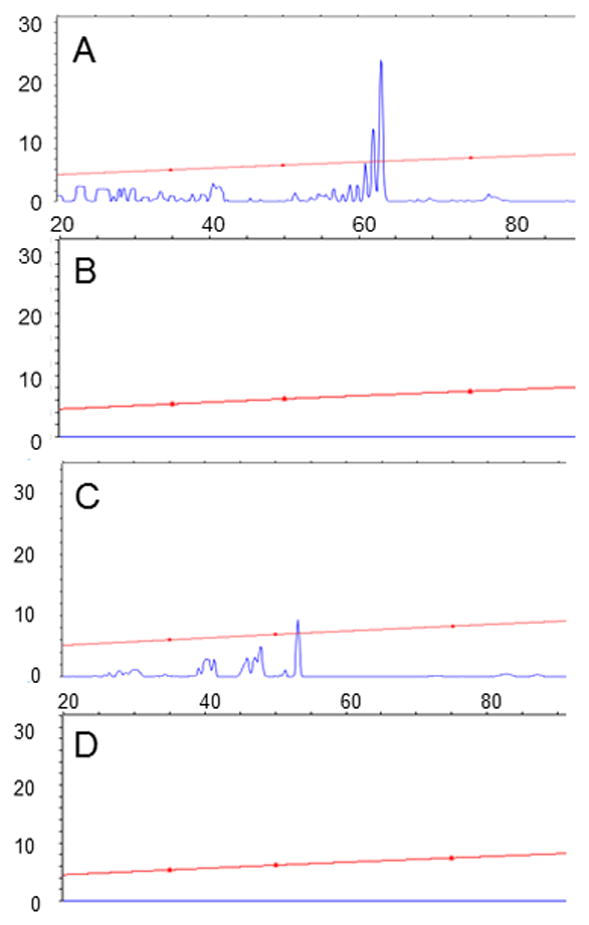
Total RNA was purified from strain K56-I2 cultured in the absence (B and D) or presence (A and C) of 1 μM OHL. Oligonucleotides GR458 and GR459 were used to prime reverse transcription of bcam0191 (A and B) and bcam0192 (C and D) mRNA, respectively, and the resulting cDNA fragments were size-fractionated by automated capillary electrophoresis. Sizes are relative to the 5’ ends of the two fluorescently labeled primers. Results are representative of two experiments with similar results.
Using the fluorescent primer that hybridizes to bcam0192 mRNA, we detected several reverse transcripts ranging in size from 40 to 53 nucleotides (Fig. 8). These correspond to apparent transcription start sites between and 112 and 126 nucleotides upstream of the bcam0192 translation start site. Upstream of these apparent start sites is the sequence
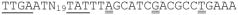 , where single underlined sequences resemble consensus promoters, and the double underlines indicate the apparent transcription start sites. The positioning of a promoter motif with respect to the three candidate start sites suggests that the middle candidate, a G residue, may represent the true transcription start site. The CepR2 binding site is centered 150 nucleotides upstream from this putative transcription start site.
, where single underlined sequences resemble consensus promoters, and the double underlines indicate the apparent transcription start sites. The positioning of a promoter motif with respect to the three candidate start sites suggests that the middle candidate, a G residue, may represent the true transcription start site. The CepR2 binding site is centered 150 nucleotides upstream from this putative transcription start site.
Discussion
CepR2 is active only as an apo-protein
This study was initiated while trying to reconcile transcriptional profiling data of our lab with that of another group. Malott and colleagues showed that a strain lacking CepR2 overexpressed a number of genes tightly linked to cepR2 (Malott et al., 2009), while we had found that OHL stimulated the expression of an overlapping set of genes. The hypothesis that CepR2 was a repressor whose activity was blocked by a cognate pheromone seemed worth exploring, as most LuxR-type proteins require a cognate AHL for activity. Our data confirm that CepR2 is antagonized by OHL, making it functionally similar to VjbR and to members of the EsaR clade, all of which function only as apo-proteins. EsaR-type proteins, CepR2, and VjbR are only distantly related to each other (28–35% identical), suggesting that the ability of these proteins to function only as apo-proteins may have evolved at least three times independently. It seems quite plausible that additional LuxR-type proteins will turn out to be AHL-inhibited rather than AHL-stimulated.
The study of Malott and colleagues provided data that CepR2, when expressed in E. coli, activated the luxI promoter in the absence of any AHL (Malott et al., 2009). Activation was not affected by addition of ten different AHLs, including OHL. It was concluded that CepR2 does not detect AHLs. The ability of CepR2 to function in the absence of AHLs agrees well with our findings. The lack of inhibition by AHLs is also reminiscent of data in the current study. CepR2, when expressed in E. coli, repressed both the bcam0191 and bcam0192 promoters whether or not OHL was provided (Table 3). OHL-responsiveness was restored only when CepS was co-expressed. In both studies, CepR2 was expressed by fusing the cepR2 gene to the Plac promoter. We believe that in both studies, CepR2 may inadvertently have been overexpressed. If so, perhaps this overexpression may overcome the inhibitory activity of OHL. One could imagine that CepR2 binds DNA only as a dimer, that OHL weakens dimerization, and the overexpression of CepR2 may shift the equilibrium toward dimers, such that enough dimers exist to populate the binding site and repress transcription (in our study) or activate transcription (in the Malott study).
CepR2 acts as a repressor
Although EsaR-type members of the LuxR family are sometimes referred to as repressors, at least some of them can act as both repressors and activators, depending largely on the position of their binding sites relative to the target promoter (Schu et al., 2011, Tsai & Winans, 2011). In the present study, CepR2 was demonstrated to act as a repressor. However, it is plausible that it could also activate one or more other promoters in this organism. The fact that CepR2 can activate the luxI promoter of V. fischeri in a system reconstituted in E. coli provides further evidence that it could act as an activator in B. cenocepacia. Malott and colleagues reported that the cepR2 mutation caused decreased expression of 127 genes, though the effects were generally modest (Malott et al., 2009). CepR2 also enhanced expression pchR, a regulator of a pyochelin biosynthesis operon, and as expected, did so in the absence of pheromone.
CepR2 inhibited expression of two target promoters in E. coli, and CepS activated both, just as they did in B. cenocepacia, strongly suggesting that these proteins act directly. The expression of both promoters in the absence of these proteins was far higher in E. coli than in B. cenocepacia, probably due at least in part to a ColE1 replication origin in the reporter plasmid that replicates at high copy number in E. coli but which is inactive in B. cenocepacia. It was initially surprising that OHL did not seem to block CepR2 repression, though these results were rationalized as due to CepR2 overproduction. The fact that OHL-responsiveness was restored by CepS could be due to synergistic effects of OHL and CepS.
Regulation of cepR2 and cepS
In the present study, we found that the divergent cepR2 and cepS genes were very slightly up-regulated by OHL in transcriptional profiling experiments. Fusions between these promoters and lacZ confirmed these results, and showed curiously, that the effect was CepR2-independent. In another study, a mutation in cepR2 was described as causing a large increase in the expression of cepR2 and of cepS (Malott et al., 2009) . We believe that the apparent discrepancy between those data and ours could be due to cis-acting effects of the cepR2 mutation used in the Malott study. In that study, a cepR2 null mutation was constructed using a trimethoprim resistance cassette inserted near the 5’ end of the gene (Fig. S3). Significantly, this cassette has two divergent promoters (DeShazer & Woods, 1996). We believe that transcription from one promoter may have continued into cepR2 while transcription from the other promoter continued into cepS. If so, the mutant would express both genes at higher levels than the wild type, exactly as reported. However, the implication that this enhanced expression originated at the native promoters of the two genes would have to be re-evaluated. If we are right that the cepR2 mutation caused increased expression of cepS, the increased accumulation of CepS protein could increase the expression of all CepS-dependent promoters described in the Malott study. In other words, the high level expression of these genes could be due both to the lack of CepR2 and to CepS overexpression.
Identity of a secondary metabolite
The functions of the regulated genes remain a matter for speculation. Analysis of these protein sequences suggests a role in synthesizing a secondary metabolite. The N-terminal half of Bcam0195 is predicted to bind ATP and leucine, while the C-terminal half contains a phosphopantatheine binding site and a reductase domain. bcam0191 is a condensation domain while Bcam0190 is an aminotransferase. Based on these homologies, one could hypothesize that this pathway could convert a yet unknown ketone into an amine, condense it to leucine, and then reduce the dipeptide into a terminal aldehyde. Further chemistry probably could occur on the reactive aldehyde (Michael Burkart, personal communication).
Opposing roles for CepR2 and CepS
The two CepR2-repressed promoters that we examined are unusual in that the repressor binding site appears to lie upstream of the regulated promoters. In the case of bcam0191, the binding site is centered 75 nucleotides upstream of the transcription start site, while in the case of bcam0192, the binding site is centered 150 nucleotides upstream. These positions are unusual, as repressor binding sites generally lie within the target promoter or directly downstream (Perez-Rueda et al., 1998). We believe that this unusual promoter geometry can be explained only in the context of CepS, a positive regulator of both promoters. Data obtained from promoter resections can be used to predict the region of the CepS binding site. We have several 5’ resections that are blind to CepR2 yet are still CepS-dependent, indicating that CepS must bind downstream of CepR2. Data presented in this paper strongly suggest that CepS binds DNA between the promoter and the CepR2 binding site.
A cepS mutant expressed both promoters at very low levels irrespective of OHL status. This indicates that when CepS is absent, CepR2 has no effect on expression of these promoters. In other words, CepS appears to work downstream of CepR2, and CepR2 appears to act by inhibiting CepS activity. One possibility is that CepR2 binding sterically blocks CepS binding, and that OHL, by blocking CepR2 activity, allows CepS to bind and activate the two promoters. If so, there must be two CepS binding sites, as plasmid pGR133 and pGR138, which share no B. cenocepacia DNA, have two different CepS-dependent promoters and therefore two different CepS binding sites. When CepR2 and CepS function were reconstituted in E. coli, CepR2 was able to decrease expression even in the absence of CepS, while this was not true in B. cenocepacia. It seems possible therefore that CepR2 may regulate these promoters in two ways, one dependent on CepS, and one that is independent.
The interactions of the CepR2 repressor and the CepS activator are somewhat reminiscent of the CytR repressor and CAP activator of E. coli, which function antagonistically at several promoters (Shin et al., 2001, Tretyachenko-Ladokhina et al., 2006, Valentin-Hansen et al., 1996). CytR binds to a site centered 70 nucleotides upstream of the deoP2 promoter, flanked by two binding sites for CAP, one centered at −40.5 and the other at −93.5. Binding of CytR does not dislodge CAP, but may block the proper positioning of the C-terminal domain of the alpha subunit of RNA polymerase (RNAP). By analogy, apo-CepR2 could act by blocking the interactions between CepS and RNAP (Fig. 9) or might block the binding of CepS to a site near these promoters.
Fig. 9. A model of proposed activities of CepR2 and CepS.
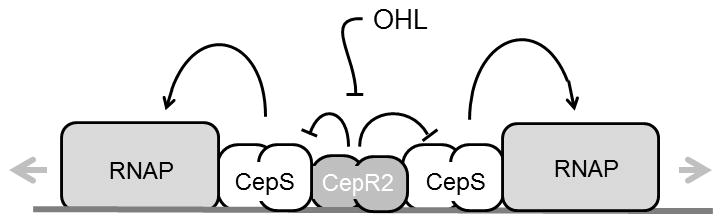
In this model, apo-CepR2 binds to a single site between the bcam0191 and bcam0192 promoters. Bound CepR2 inhibits the stimulatory activity of CepS, which binds between CepR2 and the two target promoters. At high-cell density, OHL accumulates and releases CepR2 from the DNA, permitting CepS to activate both promoters. CepR2 thus functions as an antiactivator of CepS.
Experimental Procedures
Strains, oligonucleotides, and growth conditions
Bacterial strains and plasmids used in this study are listed in Tables S2 and S3. Oligonucleotide primers (IDT, Coralville, Iowa) used for PCR amplification and DNA-mutagenesis are listed in Table S4. Burkholderia cenocepacia and Escherichia coli were cultured at 37°C in Luria-Bertani (LB) medium and Agrobacterium tumefaciens was cultured at 28°C in AT minimal medium. Antibiotics were added where described at the following concentrations: 100 μg ml−1 ampicillin, 100 μg ml−1 kanamycin, 35 μg ml−1 chloramphenicol, and 12 μg ml−1 tetracycline for E. coli; 300 μg ml−1 tetracycline, 700μg ml−1 kanamycin, 400 μg ml−1 gentamicin for B. cenocepacia; 100 μg ml−1 spectinomycin and 15μg ml−1 tetracycline for A. tumefaciens. Media was supplemented with 500 μM isopropyl β-d-galactopyranoside (IPTG) where indicated.
Transcriptional activity of bcam0191 and bcam0192 promoters
Recombinant DNA techniques were performed using standard methods (Sambrook & Russell, 2001). The intergenic region containing promoter and regulatory elements for each divergent promoter was resected by PCR amplification. For each resection, the amplicon was cloned into the promoterless transcriptional lacZ reporter plasmid pYWN302 at KpnI and XbaI sites creating a transcriptional reporter fusion. Reporter fusion plasmids were transformed into B. cenocepacia or E. coli strain MC4100 by electroporation (Cangelosi et al., 1991). To assay promoter activity, overnight cultures were diluted to 1:100 into LB medium and grown at 37°C to an OD600 of 0.35 with the appropriate antibiotics and 1μM OHL. E. coli strains containing cloned Plac-cepR2 or Plac-cepS fusions were also supplemented with IPTG to a final concentration of 0.5 mM. Cultures aliquots (150 μL) were transferred to the wells of opaque microtitre plates containing 4 μl of a 1.5 mg/ml solution of 4-methylumbelliferyl-β-D-galactopyranoside (MUG) dissolved in DMSO. β-galactosidase specific activities were measured using a Biotek Synergy HT microplate fluorescence reader. The data are averages of three independent experiments, each experiment performed with three independent isolates of each strain. Standard deviations are indicated in parentheses.
To construct a plasmid expressing a regulated Plac-cepR2 fusion, the cepR2 gene was cloned into the pSRKKm broad-host range vector (Khan et al., 2008) to create pGR192. This promoter is regulated by LacIq encoded on the plasmid and is induced with IPTG. pSRKGm was used to construct plasmid pGR193, which expresses an IPTG-inducible cepS gene. Expression from both pSRK vectors were induced using 0.5 mM IPTG. Constitutive expression of CepS was obtained by cloning the cepS gene into plasmid pSW208 to create pGR276.
Construction of deletion mutations in cepR2 and cepS
To create an internal deletion cepR2 mutant, oligonucleotides GR329 and GR330 were used to PCR amplify a 741-nucleotide fragment upstream of cepR2, while oligonucleotides GR331 and GR332 were used to PCR amplify a 737-nucleotide fragment downstream of cepR2. These fragments were digested with EcoRI, ligated, and PCR amplified using oligonucleotides GR329 and GR332, creating a 1.5 kb fragment with a 633 nucleotide deletion of cepR2 (nucleotides 21–652 of the cepR2 reading frame). This fragment was digested using HindIII and XbaI and ligated into pEX18Tet-pheS (Barrett et al., 2008), and introduced into strain SM10(λpir) by transformation, creating pGR178. This plasmid was introduced into B. cenocepacia K56-I2 by conjugation. Tetracycline-resistant single-crossover recombinant mutants were screened by PCR for correct integration of the plasmid and double crossover recombinants were selected using M9 agar supplemented with 0.1% p-chlorophenylalanine (Sigma-Aldrich) (Barrett et al., 2008). The resulting colonies were screened by PCR amplification for the 633 nucleotide cepR2 deletion and verified by DNA sequencing (Cornell Biotechnology Resource Center). The resulting cepR2 deletion was designated GR141.
A similar strategy was used to delete cepS. Oligonucleotides GR345 and GR346 were used to amplify a 474-nucleotide fragment upstream of cepS, while GR347 and GR348 were used to PCR amplify a 492-nucleotide fragment downstream of cepS. These fragments were digested with SpeI, and PCR amplified using oligonucleotides GR345 and GR348, yielding a 0.95 kb fragment that contains a 0.9 kb deletion of cepS. This fragment was digested using BamHI and EcoRI and ligated into pEX18Tet-pheS, to create pGR182. The cepS deletion was crossed into the genomic DNA of strain K56-I2 as described above, creating strain GR145.
AHL detection by CepR2
To measure CepR2 AHL ligand specificity, strains K56-I2(pGR130) and K56- I2(pGR136), was cultured at 37°C to mid-log phase (OD600 0.4) in 2 ml LB medium supplemented with tetracycline and AHLs at concentrations ranging from 1 pM to 1 μM. Promoter activity was determined by measuring β-galactosidase activity of three isolates from each strain as described above in three independent experiments.
Overexpression of CepR2
To overexpress CepR2 in E. coli, the cepR2 gene was PCR amplified using oligonucleotides GR295 and GR288 and inserted into pRSETa (Invitrogen) after digesting both with NdeI and XhoI, creating pGR107. E. coli strain BL21 (DE3) (Novagen) harboring plasmids pGR107 and pT7-groESL (which expresses the chaperone GroESL) were grown in LB medium supplemented with 0.4% glucose, 400 μg ml−1 ampicillin and 35 μg ml−1 chloramphenicol at 37°C. At an OD600 of 0.4, cultures were cooled to 28°C and 10 μM OHL was added as indicated. Protein expression was induced using 0.5 mM IPTG and growth was continued for three additional hours at 28°C. Cells were harvested and resuspended in TEDG buffer (50 mM Tris-HCl pH 8.0, 1 mM EDTA, 1 mM DTT, 20% glycerol) supplemented with 200 mM NaCl. Cells were disrupted using a French press (three passages, 10,000 psi) and the lysate was clarified by ultracentrifugation (106,000 × g, 30 min, 4°C). Protein fractions from lysates obtained from three cultures grown independently were analyzed on SDS-PAGE gels stained with Coomassie blue.
AHL sequestration assays
E. coli strain BL21(DE3)(pGR107) was used to test for the sequestration of AHLs. Cells were cultured at 18°C in 10 ml LB medium supplemented with 100 μg ml−1 ampicillin. When the OD600 reached 0.4, IPTG was added to a final concentration of 0.5 mM, and AHLs were added at a final concentration of 10 μM. When the cultures reached an OD600 of 0.7 (approximately 4 hours), they were harvested, washed twice with LB, then washed three times with TE buffer (10 mM Tris (pH 8), 0.5 mM EDTA) and resuspended in lysis buffer (200 mM Tris (pH 8), 400 mM EDTA, 0.7 mM sucrose). Cell-associated autoinducers were extracted twice with ethyl acetate:acetonitrile (99.5:0.5 v/v) (HPLC grade, Fisher). Organic phase extracts were pooled and dried under nitrogen gas. Pellets were resuspended in 10 μl ethyl acetate and added to cultures inoculated with the biosensor strain A. tumefaciens WCF47(pCF218)(pCF372), which detects a wide range of AHLs (Zhu et al., 1998). The detection of each AHL was calibrated using known concentrations of each AHL. Cultures from three isolates were grown for 12 h at 28° and assayed for β-galactosidase specific activity in three independent experiments.
Electrophoretic mobility shift assays
For all EMSA reactions, a clarified supernatant from BL21(DE3)(pGR107)(pT7-groESL) was dialyzed against EMSA buffer (50 mM Tris-HCl pH 7.0, 2 mM EDTA, 2 mM DTT, 60 μM potassium acetate, 39 μM potassium glutamate, 20% glycerol). DNA fragments were PCR amplified using oligonucleotides described in Table S2 and end-labeled with T4 polynucleotide kinase and [γ-32P]-ATP (Perkin Elmer). Binding reactions contained 2.5 pM of DNA and varying concentrations of CepR2 protein in a 15 μl total volume containing EMSA buffer, 20 μg ml-1 of calf thymus DNA, and 20 μg ml-1 of BSA. Reactions were incubated at room temperature for 30 minutes, and complexes were size-fractionated at 4°C using 10% polyacrylamide gels (Dgel Sciences) containing 20 mM Tris-acetate pH 8.5, and 1 mM EDTA (0.5 × TAE). Gels were analyzed using a Storm B840 Phosphorimager (Molecular Dynamics). All binding reactions were performed in at least two experiments with similar results.
DNase I protection assay
A fluorescently labeled 84-bp fragment was PCR amplified using primers GR280 and GR458 (Table S3). Binding reactions contained ~ 200 ng DNA and a clarified supernatant of strain BL21(DE3)(pGR107)(pT7-groESL) (10 mg ml−1 total protein) or BSA (for control) diluted in 20 μl EMSA buffer and incubated at room temperature for 30 minutes. MgCl2 (2.5 mM), CaCl2 (0.5 mM) and 0.1 units of DNase I (Ambion) were added to the reaction and allowed to incubate at room temperature for 2.5 minutes. The reaction was stopped by addition of 0.75 μl stop solution (20 mM EDTA (pH 8.0), 200 mM NaCl, 1% SDS). DNA was purified with the Qiagen PCR kit and eluted in 20 μl water. DNA fragments were analyzed using an Applied BioSystems 3730xl DNA Analyzer (Cornell University Life Sciences Core Laboratories Center).
Primer extension assays
Strain K56-I2 was cultured to mid-log phase in LB with or without 1 μM OHL at 37°C. DNA-free mRNA preparations were isolated from 2 ml cell culture aliquots using Qiagen RNeasy Plus Mini kit. Residual DNA in mRNA extracts was degraded using Turbo DNA-free kit (Applied Biosystems) and mRNA was purified by isopropanol precipitation. cDNA transcripts containing bcam0191 and bcam0192 transcriptional start sites were obtained with the Superscript III RT kit (Invitrogen) using GR458 or GR459 fluorescently labeled primers, respectively. cDNA transcripts were purified (Qiagen PCR purification kit) and DNA fragment analysis was performed as above.
Transcriptional profiling
Whole genome microarray slides containing 3–5 different probes for each gene of the B. cenocepacia genome were purchased from Agilent (AMADID #016249). Bacterial strains were cultured to exponential phase in AT minimal medium and subjected to RNA extraction as described previously (Cho & Winans, 2005). Preparation of fluorescent cDNA was performed following a published procedure (Hegde et al., 2000). Hybridization and washing of slides was performed according to the manufacturer’s protocol. Fluorescence intensity was analyzed using a GenePix 400B scanner (Axon). Induction ratios were calculated after normalization with locally weighted linear regression (lowess) analysis. Experiments were performed in duplicate, with independent bacterial culturing, RNA preparation, cDNA probe synthesis, dye coupling and hybridizations. The Cy3 and Cy5 dyes were swapped in the two trials.
Supplementary Material
Acknowledgments
We acknowledge members of our laboratory for helpful discussions. We also thank Drs. L. Eberl and P. Sokol for openly sharing ideas and unpublished data, and Dr. M. Burkart (U.C.S.D.) for his insights about the possible functions of genes regulated by CepR2. This work was funded by a grant from the NIGMS (RO1-GM042893).
References
- Aguilar C, Friscina A, Devescovi G, Kojic M, Venturi V. Identification of quorum-sensing-regulated genes of Burkholderia cepacia. J Bacteriol. 2003;185:6456–6462. doi: 10.1128/JB.185.21.6456-6462.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett AR, Kang Y, Inamasu KS, Son MS, Vukovich JM, Hoang TT. Genetic tools for allelic replacement in Burkholderia species. Appl Environ Microbiol. 2008;74:4498–4508. doi: 10.1128/AEM.00531-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bontemps C, Elliott GN, Simon MF, Dos Reis FB, Junior, Gross E, Lawton RC, Neto NE, de Fatima Loureiro M, De Faria SM, Sprent JI, James EK, Young JP. Burkholderia species are ancient symbionts of legumes. Mol Ecol. 2010;19:44–52. doi: 10.1111/j.1365-294X.2009.04458.x. [DOI] [PubMed] [Google Scholar]
- Cangelosi GA, Best EA, Martinetti G, Nester EW. Genetic analysis of Agrobacterium. Methods Enzymol. 1991;204:384–397. doi: 10.1016/0076-6879(91)04020-o. [DOI] [PubMed] [Google Scholar]
- Castang S, Reverchon S, Gouet P, Nasser W. Direct evidence for the modulation of the activity of the Erwinia chrysanthemi quorum-sensing regulator ExpR by acylhomoserine lactone pheromone. J Biol Chem. 2006;281:29972–29987. doi: 10.1074/jbc.M601666200. [DOI] [PubMed] [Google Scholar]
- Chai Y, Winans SC. The chaperone GroESL enhances the accumulation of soluble, active TraR protein, a quorum-sensing transcription factor from Agrobacterium tumefaciens. J Bacteriol. 2009;191:3706–3711. doi: 10.1128/JB.01434-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WM, Moulin L, Bontemps C, Vandamme P, Bena G, Boivin-Masson C. Legume symbiotic nitrogen fixation by beta-proteobacteria is widespread in nature. J Bacteriol. 2003;185:7266–7272. doi: 10.1128/JB.185.24.7266-7272.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Winans SC. VirA and VirG activate the Ti plasmid repABC operon, elevating plasmid copy number in response to wound-released chemical signals. Proc Natl Acad Sci U S A. 2005;102:14843–14848. doi: 10.1073/pnas.0503458102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Greenberg EP. Genetic dissection of DNA-binding and luminescence gene activation by the Vibrio fischeri LuxR protein. J Bacteriol. 1992;174:4064–4069. doi: 10.1128/jb.174.12.4064-4069.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill ME, Chen L. Structural basis of acyl-homoserine lactone-dependent signaling. Chem Rev. 2011;111:68–85. doi: 10.1021/cr1000817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Chatterjee A, Hasegawa H, Dixit V, Leigh N, Chatterjee AK. ExpR, a LuxR homolog of Erwinia carotovora subsp. carotovora, activates transcription of rsmA, which specifies a global regulatory RNA-binding protein. J Bacteriol. 2005;187:4792–4803. doi: 10.1128/JB.187.14.4792-4803.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delrue RM, Deschamps C, Leonard S, Nijskens C, Danese I, Schaus JM, Bonnot S, Ferooz J, Tibor A, De Bolle X, Letesson JJ. A quorum-sensing regulator controls expression of both the type IV secretion system and the flagellar apparatus of Brucella melitensis. Cell Microbiol. 2005;7:1151–1161. doi: 10.1111/j.1462-5822.2005.00543.x. [DOI] [PubMed] [Google Scholar]
- DeShazer D, Woods DE. Broad-host-range cloning and cassette vectors based on the R388 trimethoprim resistance gene. Biotechniques. 1996;20:762–764. doi: 10.2144/96205bm05. [DOI] [PubMed] [Google Scholar]
- Fineran PC, Slater H, Everson L, Hughes K, Salmond GP. Biosynthesis of tripyrrole and beta-lactam secondary metabolites in Serratia: integration of quorum sensing with multiple new regulatory components in the control of prodigiosin and carbapenem antibiotic production. Mol Microbiol. 2005;56:1495–1517. doi: 10.1111/j.1365-2958.2005.04660.x. [DOI] [PubMed] [Google Scholar]
- Galloway WR, Hodgkinson JT, Bowden SD, Welch M, Spring DR. Quorum sensing in Gram-negative bacteria: small-molecule modulation of AHL and AI-2 quorum sensing pathways. Chem Rev. 2011;111:28–67. doi: 10.1021/cr100109t. [DOI] [PubMed] [Google Scholar]
- Godoy D, Randle G, Simpson AJ, Aanensen DM, Pitt TL, Kinoshita R, Spratt BG. Multilocus sequence typing and evolutionary relationships among the causative agents of melioidosis and glanders, Burkholderia pseudomallei and Burkholderia mallei. J Clin Microbiol. 2003;41:2068–2079. doi: 10.1128/JCM.41.5.2068-2079.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotschlich A, Huber B, Geisenberger O, Togl A, Steidle A, Riedel K, Hill P, Tummler B, Vandamme P, Middleton B, Camara M, Williams P, Hardman A, Eberl L. Synthesis of multiple N-acylhomoserine lactones is widespread among the members of the Burkholderia cepacia complex. Syst Appl Microbiol. 2001;24:1–14. doi: 10.1078/0723-2020-00013. [DOI] [PubMed] [Google Scholar]
- Hegde P, Qi R, Abernathy K, Gay C, Dharap S, Gaspard R, Hughes JE, Snesrud E, Lee N, Quackenbush J. A concise guide to cDNA microarray analysis. Biotechniques. 2000;29:548–562. doi: 10.2144/00293bi01. [DOI] [PubMed] [Google Scholar]
- Holden MT, Seth-Smith HM, Crossman LC, Sebaihia M, Bentley SD, Cerdeno-Tarraga AM, Thomson NR, Bason N, Quail MA, Sharp S, Cherevach I, Churcher C, Goodhead I, Hauser H, Holroyd N, Mungall K, Scott P, Walker D, White B, Rose H, Iversen P, Mil-Homens D, Rocha EP, Fialho AM, Baldwin A, Dowson C, Barrell BG, Govan JR, Vandamme P, Hart CA, Mahenthiralingam E, Parkhill J. The genome of Burkholderia cenocepacia J2315, an epidemic pathogen of cystic fibrosis patients. J Bacteriol. 2009;191:261–277. doi: 10.1128/JB.01230-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber B, Riedel K, Hentzer M, Heydorn A, Gotschlich A, Givskov M, Molin S, Eberl L. The cep quorum-sensing system of Burkholderia cepacia H111 controls biofilm formation and swarming motility. Microbiology. 2001;147:2517–2528. doi: 10.1099/00221287-147-9-2517. [DOI] [PubMed] [Google Scholar]
- Jones AM, Webb AK. Recent advances in cross-infection in cystic fibrosis: Burkholderia cepacia complex, Pseudomonas aeruginosa, MRSA and Pandoraea spp. J R Soc Med. 2003;96:66–72. [PMC free article] [PubMed] [Google Scholar]
- Khan SR, Gaines J, Roop RM, 2nd, Farrand SK. Broad-host-range expression vectors with tightly regulated promoters and their use to examine the influence of TraR and TraM expression on Ti plasmid quorum sensing. Appl Environ Microbiol. 2008;74:5053–5062. doi: 10.1128/AEM.01098-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooi C, Subsin B, Chen R, Pohorelic B, Sokol PA. Burkholderia cenocepacia ZmpB is a broad-specificity zinc metalloprotease involved in virulence. Infect Immun. 2006;74:4083–4093. doi: 10.1128/IAI.00297-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothe M, Antl M, Huber B, Stoecker K, Ebrecht D, Steinmetz I, Eberl L. Killing of Caenorhabditis elegans by Burkholderia cepacia is controlled by the cep quorum-sensing system. Cell Microbiol. 2003;5:343–351. doi: 10.1046/j.1462-5822.2003.00280.x. [DOI] [PubMed] [Google Scholar]
- Lewenza S, Conway B, Greenberg EP, Sokol PA. Quorum sensing in Burkholderia cepacia: Identification of the LuxRI homologs CepRI. J Bacteriol. 1999;181:748–756. doi: 10.1128/jb.181.3.748-756.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewenza S, Sokol PA. Regulation of ornibactin biosynthesis and N-acyl-L-homoserine lactone production by CepR in Burkholderia cepacia. J Bacteriol. 2001;183:2212–2218. doi: 10.1128/JB.183.7.2212-2218.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahenthiralingam E, Urban TA, Goldberg JB. The multifarious, multireplicon Burkholderia cepacia complex. Nat Rev Microbiol. 2005;3:144–156. doi: 10.1038/nrmicro1085. [DOI] [PubMed] [Google Scholar]
- Malott RJ, Baldwin A, Mahenthiralingam E, Sokol PA. Characterization of the cciIR quorum-sensing system in Burkholderia cenocepacia. Infect Immun. 2005;73:4982–4992. doi: 10.1128/IAI.73.8.4982-4992.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malott RJ, O'Grady EP, Toller J, Inhulsen S, Eberl L, Sokol PA. A Burkholderia cenocepacia orphan LuxR homolog is involved in quorum-sensing regulation. J Bacteriol. 2009;191:2447–2460. doi: 10.1128/JB.01746-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minogue TD, Carlier AL, Koutsoudis MD, von Bodman SB. The cell density-dependent expression of stewartan exopolysaccharide in Pantoea stewartii ssp. stewartii is a function of EsaR-mediated repression of the rcsA gene. Mol Microbiol. 2005;56:189–203. doi: 10.1111/j.1365-2958.2004.04529.x. [DOI] [PubMed] [Google Scholar]
- Minogue TD, Wehland-von Trebra M, Bernhard F, von Bodman SB. The autoregulatory role of EsaR, a quorum-sensing regulator in Pantoea stewartii ssp. stewartii: evidence for a repressor function. Mol Microbiol. 2002;44:1625–1635. doi: 10.1046/j.1365-2958.2002.02987.x. [DOI] [PubMed] [Google Scholar]
- Ng WL, Bassler BL. Bacterial quorum-sensing network architectures. Annu Rev Genet. 2009;43:197–222. doi: 10.1146/annurev-genet-102108-134304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nzula S, Vandamme P, Govan JR. Influence of taxonomic status on the in vitro antimicrobial susceptibility of the Burkholderia cepacia complex. J Antimicrob Chemother. 2002;50:265–269. doi: 10.1093/jac/dkf137. [DOI] [PubMed] [Google Scholar]
- O'Grady EP, Viteri DF, Malott RJ, Sokol PA. Reciprocal regulation by the CepIR and CciIR quorum sensing systems in Burkholderia cenocepacia. BMC Genomics. 2009;10:441. doi: 10.1186/1471-2164-10-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parke JL, Gurian-Sherman D. Diversity of the Burkholderia cepacia complex and implications for risk assessment of biological control strains. Annu Rev Phytopathol. 2001;39:225–258. doi: 10.1146/annurev.phyto.39.1.225. [DOI] [PubMed] [Google Scholar]
- Perez-Rueda E, Gralla JD, Collado-Vides J. Genomic position analyses and the transcription machinery. J Mol Biol. 1998;275:165–170. doi: 10.1006/jmbi.1997.1465. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning, third edition. Cold Spring Harbor Laboratory; Cold Spring Harbor: 2001. [Google Scholar]
- Schu DJ, Ramachandran R, Geissinger JS, Stevens AM. Probing the impact of ligand binding on the acyl-homoserine lactone-hindered transcription factor EsaR of Pantoea stewartii subsp. stewartii. J Bacteriol. 2011;193:6315–6322. doi: 10.1128/JB.05956-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster M, Urbanowski ML, Greenberg EP. Promoter specificity in Pseudomonas aeruginosa quorum sensing revealed by DNA binding of purified LasR. Proc Natl Acad Sci U S A. 2004;101:15833–15839. doi: 10.1073/pnas.0407229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M, Kang S, Hyun SJ, Fujita N, Ishihama A, Valentin-Hansen P, Choy HE. Repression of deoP2 in Escherichia coli by CytR: conversion of a transcription activator into a repressor. Embo J. 2001;20:5392–5399. doi: 10.1093/emboj/20.19.5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoblom S, Brader G, Koch G, Palva ET. Cooperation of two distinct ExpR regulators controls quorum sensing specificity and virulence in the plant pathogen Erwinia carotovora. Mol Microbiol. 2006;60:1474–1489. doi: 10.1111/j.1365-2958.2006.05210.x. [DOI] [PubMed] [Google Scholar]
- Sokol PA, Sajjan U, Visser MB, Gingues S, Forstner J, Kooi C. The CepIR quorum-sensing system contributes to the virulence of Burkholderia cenocepacia respiratory infections. Microbiology. 2003;149:3649–3658. doi: 10.1099/mic.0.26540-0. [DOI] [PubMed] [Google Scholar]
- Stevens AM, Schuster M, Rumbaugh KP. Working together for the common good: cell-cell communication in bacteria. J Bacteriol. 2012;194:2131–2141. doi: 10.1128/JB.00143-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretyachenko-Ladokhina V, Cocco MJ, Senear DF. Flexibility and adaptability in binding of E. coli cytidine repressor to different operators suggests a role in differential gene regulation. J Mol Biol. 2006;362:271–286. doi: 10.1016/j.jmb.2006.06.085. [DOI] [PubMed] [Google Scholar]
- Tsai CS, Winans SC. LuxR-type quorum-sensing regulators that are detached from common scents. Mol Microbiol. 2010;77:1072–1082. doi: 10.1111/j.1365-2958.2010.07279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CS, Winans SC. The quorum-hindered transcription factor YenR of Yersinia enterocolitica inhibits pheromone production and promotes motility via a small non-coding RNA. Mol Microbiol. 2011;80:556–571. doi: 10.1111/j.1365-2958.2011.07595.x. [DOI] [PubMed] [Google Scholar]
- Urbanowski ML, Lostroh CP, Greenberg EP. Reversible acyl-homoserine lactone binding to purified Vibrio fischeri LuxR protein. J Bacteriol. 2004;186:631–637. doi: 10.1128/JB.186.3.631-637.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin-Hansen P, Sogaard-Andersen L, Pedersen H. A flexible partnership: the CytR anti-activator and the cAMP-CRP activator protein, comrades in transcription control. Mol Microbiol. 1996;20:461–466. doi: 10.1046/j.1365-2958.1996.5341056.x. [DOI] [PubMed] [Google Scholar]
- Vandamme P, Holmes B, Coenye T, Goris J, Mahenthiralingam E, LiPuma JJ, Govan JR. Burkholderia cenocepacia sp. nov.--a new twist to an old story. Res Microbiol. 2003;154:91–96. doi: 10.1016/S0923-2508(03)00026-3. [DOI] [PubMed] [Google Scholar]
- Vandamme P, Holmes B, Vancanneyt M, Coenye T, Hoste B, Coopman R, Revets H, Lauwers S, Gillis M, Kersters K, Govan JR. Occurrence of multiple genomovars of Burkholderia cepacia in cystic fibrosis patients and proposal of Burkholderia multivorans sp. nov. Int J Syst Bacteriol. 1997;47:1188–1200. doi: 10.1099/00207713-47-4-1188. [DOI] [PubMed] [Google Scholar]
- Vanlaere E, Baldwin A, Gevers D, Henry D, De Brandt E, LiPuma JJ, Mahenthiralingam E, Speert DP, Dowson C, Vandamme P. Taxon K, a complex within the Burkholderia cepacia complex, comprises at least two novel species, Burkholderia contaminans sp. nov. and Burkholderia lata sp. nov. Int J Syst Evol Microbiol. 2009;59:102–111. doi: 10.1099/ijs.0.001123-0. [DOI] [PubMed] [Google Scholar]
- Vanlaere E, Lipuma JJ, Baldwin A, Henry D, De Brandt E, Mahenthiralingam E, Speert D, Dowson C, Vandamme P. Burkholderia latens sp. nov., Burkholderia diffusa sp. nov., Burkholderia arboris sp. nov., Burkholderia seminalis sp. nov. and Burkholderia metallica sp. nov., novel species within the Burkholderia cepacia complex. Int J Syst Evol Microbiol. 2008;58:1580–1590. doi: 10.1099/ijs.0.65634-0. [DOI] [PubMed] [Google Scholar]
- Venturi V, Friscina A, Bertani I, Devescovi G, Aguilar C. Quorum sensing in the Burkholderia cepacia complex. Res Microbiol. 2004;155:238–244. doi: 10.1016/j.resmic.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Wei Y, Ryan GT, Flores-Mireles AL, Costa ED, Schneider DJ, Winans SC. Saturation mutagenesis of a CepR binding site as a means to identify new quorum-regulated promoters in Burkholderia cenocepacia. Mol Microbiol. 2011;79:616–632. doi: 10.1111/j.1365-2958.2010.07469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingart CL, White CE, Liu S, Chai Y, Cho H, Tsai CS, Wei Y, Delay NR, Gronquist MR, Eberhard A, Winans SC. Direct binding of the quorum sensing regulator CepR of Burkholderia cenocepacia to two target promoters in vitro. Mol Microbiol. 2005;57:452–467. doi: 10.1111/j.1365-2958.2005.04656.x. [DOI] [PubMed] [Google Scholar]
- Wheelis M. First shots fired in biological warfare. Nature. 1998;395:213. doi: 10.1038/26089. [DOI] [PubMed] [Google Scholar]
- Zhu J, Beaber JW, More MI, Fuqua C, Eberhard A, Winans SC. Analogs of the autoinducer 3-oxooctanoyl-homoserine lactone strongly inhibit activity of the TraR protein of Agrobacterium tumefaciens. J Bacteriol. 1998;180:5398–5405. doi: 10.1128/jb.180.20.5398-5405.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Winans SC. Autoinducer binding by the quorum-sensing regulator TraR increases affinity for target promoters in vitro and decreases TraR turnover rates in whole cells. Proc Natl Acad Sci U S A. 1999;96:4832–4837. doi: 10.1073/pnas.96.9.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Winans SC. The quorum-sensing transcriptional regulator TraR requires its cognate signaling ligand for protein folding, protease resistance, and dimerization. Proc Natl Acad Sci U S A. 2001;98:1507–1512. doi: 10.1073/pnas.98.4.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.