

Abstract
Background
TAR DNA-binding protein 43, encoded by the TARDBP gene, has been identified as the major pathological protein of frontotemporal lobar dementia (FTLD) with or without amyotrophic lateral sclerosis (ALS) and sporadic ALS. Subsequently, mutations in the TARDBP gene have been detected in 2% to 3% of patients with ALS (both familial and sporadic ALS). However, to our knowledge, there is only 1 description of 2 patients with FTLD and TARDBP gene mutations who later developed motor neuron disease.
Objective
To describe cognitive abnormalities in 3 Italian families with familial ALS and TARDBP gene mutations.
Design, Setting, and Participants
Genetic, neuropsychological, and neuroimaging analyses in 36 patients with familial non–superoxide dismutase 1 gene (SOD1) ALS and 280 healthy controls.
Main Outcome Measure
We identified 3 index cases of familial ALS carrying the p.Ala382Thr missense mutation of the TARDBP gene and with clinical, neuroimaging, and neuropsychological features of FTLD.
Results
The p.Ala382Thr missense mutation of the TARDBP gene was absent in the 280 controls. It was present in all affected members of the 3 families for whom DNA was available. All affected members of the 3 families developed FTLD after the onset of ALS, confirmed by neuropsychological testing and hypometabolism in frontal associative areas assessed with fludeoxyglucose F 18 positron emission tomography and computed tomography.
Conclusions
Three apparently unrelated families with familial ALS carrying the p.Ala382Thr TARDBP missense mutation developed FTLD. In these families, FTLD co-segregates with ALS. Patients with ALS carrying TARDBP mutations may develop FTLD.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder of adult life, characterized by a progressive impairment of motor function at spinal and bulbar levels. The disorder is sporadic in approximately 95% of cases, while it is inherited in the remaining cases (familial ALS [FALS]), typically with autosomal dominant transmission. To date, mutations in 3 genes have been described as causative for ALS, namely SOD1 (OMIM 147450), TARDBP (OMIM 605078), FUS/TLS (OMIM 137070), and FIG4 (OMIM 612577).15 It is now well recognized that about 10% of patients with ALS also develop florid symptoms of frontotemporal lobar dementia (FTLD) and a further 30% to 50% of patients have sub threshold disturbances of frontal lobe dysfunction.6 Patients carrying TARDBP mutations can present with FTLD and subsequently develop motor neuron dysfunction later in the course of their illness (ie, FTLD–motor neuron disease [FTLD-MND]).7 However, to our knowledge, there are no reports of TARDBP mutations being associated with the related, although distinct, phenotype of ALS-FTLD (ie, a patient presenting with ALS who subsequently develops FTLD).
Figure 4.

Right lateral (A), left lateral (B), right medial (C), left medial (D), anterior (E), posterior (F), superior (G), and inferior (H) positron emission tomography/computed tomography statistical parametric maps of patient III-2 from family B. The images show the hypometabolic areas on the frontal and temporal associative cortices, the frontal mesial cortex, and the caudate nuclei (arrows). P indicates posterior; A, anterior; R, right; and L, left.
Here we describe 3 apparently unrelated families carrying p.Ala382Thr TARDBP missense mutations in whom FTLD developed after ALS in all affected members.
METHODS
SUBJECTS
A total of 36 non-SOD1and non-FUS-carrying proband cases of FALS seen at the Turin ALS center were screened for TARDBP gene mutations. A case was considered to be familial if a firstor second-degree relative had been diagnosed with ALS. Amyotrophic lateral sclerosis was diagnosed according to revised El Escorial criteria,8 and FTLD was diagnosed according to the criteria by Neary et al.9
TARDBP MUTATIONAL SCREENING
Genomic DNA was extracted from 1 mL of peripheral blood with the Biorobot MDx DSP system (Qiagen Inc, Valencia, California). All the coding exons and 100 base pairs of the flanking intron-exon boundaries of TARDBP were amplified by polymerase chain reaction using specific primers,2 sequenced using the BigDye Terminator v3.1 sequencing kit (Applied Biosystems, Inc, Carlsbad, California), and run on an ABI Prism 3100-Avant genetic analyzer (Applied Biosystems, Inc).
NEUROPSYCHOLOGICAL BATTERY
The patients underwent neuropsychological testing according to the consensus criteria for the diagnosis of frontotemporal cognitive and behavioral syndromes in ALS.10 The following tests were performed: Frontal Systems Behavior Scale (FrSBe), using the family form evaluated by a close relative (scores of ≤59 indicated normal; 60-64, borderline; and ≥65, pathological); Mini-Mental State Examination; Wisconsin Card Sorting Test; Trail Making Test A and B; Stroop Color-Word Interference Test; letter and category fluency test; Wechsler Memory Scale–Revised (form 2); Rey-Osterrieth Complex Figure Test; Token Test; Wechsler Adult Intelligence Scale–Revised; and Raven’s Coloured Progressive Matrices. Anxiety and depression were assessed with the Hospital Anxiety and Depression Scale. When applicable, test results were corrected for age and educational level.
POSITRON EMISSION TOMOGRAPHY AND COMPUTED TOMOGRAPHY
Patients fasted for at least 6 hours prior to examination. Each patient was given a dose of 185 MBq of fludeoxyglucose F 18 (FDG) (to convert to megacurie, multiply by 2.7 × 10-11) by intravenous injection 60 minutes before the scan. Between FDG injection and the brain scans, the patients remained still and relaxed in a quiet room. The positron emission tomographic (PET) and computed tomographic (CT) scans were acquired by a Discovery ST-E PET/CT system (GE Healthcare, Waukesha, Wisconsin), combining a helical multi-slice CT scanner and a designed bismuth germanate block detector PET tomograph, in 3-dimensional modality. The FDG-PET/CT images were acquired through 2 sequential scans: CT brain scan (thickness, 3.75 mm; 140 kV; 60-80 mA/s) and PET brain scan (1 field of view of 30 cm trans-axially). The PET scan was initiated immediately after the CT examination in order to use the CT data for the attenuation correction of the PET data. Data were collected in 128 × 128 matrices.
The PET/CT images have been evaluated by visual analysis and statistical mapping techniques to compare a patient’s brain PET/CT scan with those in a database of age-matched healthy subjects (Cortex ID; GE Healthcare). The resulting data with 3-dimensional stereotactic surface projection maps of the brain allow for quantitative visualization of patterns and severity of such abnormalities.
STANDARD PROTOCOL APPROVALS AND PATIENT CONSENTS
The study has been approved by the ethical committee of our institution. The patients provided written informed consent.
RESULTS
While performing mutational screening of TARDBP in 36 index cases of FALS, we found 3 apparently unrelated FALS patients carrying a c.1144G>A (p.Ala382Thr) missense mutation. This mutation was not found in 280 healthy subjects from our center and was not found in 956 healthy controls in 2 previous studies on TARDBP mutations in ALS.11,12 Each proband belonged to a large, apparently unrelated pedigree. Because the 3 cases showed evident clinical symptoms of FTLD, they underwent an in-depth neuropsychological battery and a brain PET/CT scan. A summary of patients’ clinical characteristics is reported in the Table.
FAMILY A
In family A (Figure 1A), the proband (II-7) is a 60-year-old woman who developed dysphagia, dysarthria, tongue hypotrophy, and emotional lability at age 58 years, followed by wasting and weakness of the upper limbs, mainly distally. She had generalized hyperreflexia, and Babinski and Hoffmann signs were present. At neurophysiological examination, she showed chronic and active denervation, confirmed by a muscle biopsy. Brain and spinal cord magnetic resonance imaging (MRI) results were negative. The diagnosis was definite ALS. One year after the onset of motor signs, she developed apathy, mental rigidity, perseverative behavior, distractibility, aspontaneity, and speech stereotypies. Her FrSBe total score was 70 (premorbid score of 49), with increased scores in all domains (apathy, disinhibition, and executive dysfunctions). Neuropsychological testing demonstrated a global frontal cognitive deterioration. The PET/CT brain study showed a reduction of glucose metabolism on the associative frontal cortex, associative parietal cortex, anterior cingulus, and left head of caudate nuclei. The reduction was more marked on the left side. The PET/CT images from a healthy member of the same family (Figure 2) compared with the PET/CT images from this patient (Figure 3) are shown.
Figure 1.
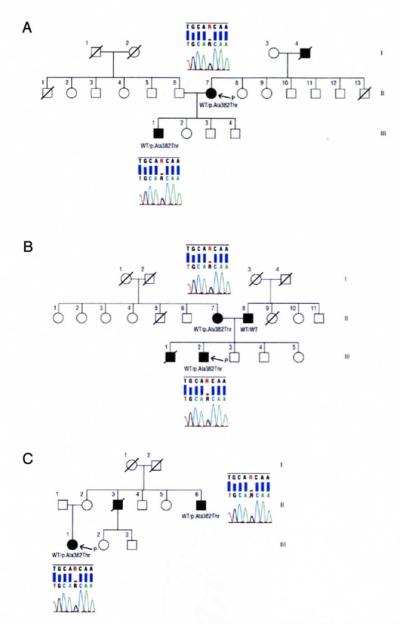
Pedigrees of family A (A), family B (B), and family C (C) with chromatograms of part of exon 6 of the TARDBP gene showing c.1144G▪A (p.Ala382Thr) mutations in all of the examined patients. Squares indicate males; circles, females; diagonal lines, deceased; open symbols, unaffected; solid symbols, affected; P, proband; and WT, wild type.
Figure 2.

Right lateral (A), left lateral (B), right medial (C), left medial (D), anterior (E), posterior (F), superior (G), and inferior (H) positron emission tomography/computed tomography statistical parametric maps of a healthy member of family A. P indicates posterior; A, anterior; R, right; and L, left.
Figure 3.

Right lateral (A), left lateral (B), right medial (C), left medial (D), anterior (E), posterior (F), superior (G), and inferior (H) positron emission tomography/computed tomography statistical parametric maps of patient II-7 from family A. The images show the reduction in fludeoxyglucose F 18 uptake on the frontal and parietal associative cortices, the anterior cingulus, and the caudate nuclei. The metabolism is reduced on the left side (white arrows). A slight reduction in fludeoxyglucose F 18 uptake on the right cerebellar cortex (crossed cerebellar diaschisis) is also evident (yellow arrow). P indicates posterior; A, anterior; R, right; and L, left.
The proband’s father (I-4) developed wasting and weakness of the distal upper limbs at age 78 years, rapidly followed by dysphagia and dysarthria. At neurological examination, he also showed diffuse hyperreflexia. At neurophysiological examination, he showed chronic and active denervation. He was diagnosed as having definite ALS. According to clinical records, he showed marked apathy and executive dysfunction, confirmed by the FrSBe total score of 75 (premorbid score of 37). He died 2 years later of respiratory failure. No DNA was available from this subject.
The eldest son of the proband (III-I) developed distal wasting and weakness of the upper limbs at age 33 years, followed rapidly by wasting of the lower limbs and respiratory impairment. At neurophysiological examination, he showed chronic and active denervation of all limbs. His diagnosis was definite ALS. He was tracheostomized at age 35 years. One year after the onset of ALS, the patient showed restless pacing, distractibility, impulsivity, motor impersistence, mental rigidity, and inflexibility. His FrSBe total score was 69 (premorbid score of 32). He is still alive 48 months after the onset of ALS and carries the p.Ala382Thr missense mutation of the TARDBP gene.
FAMILY B
In family B (Figure 1B), the proband (III-2) is a 44-year-old man who developed dysarthria and dysphagia with tongue hypotrophy and emotional lability at age 43 years. He also presented with pyramidal signs of the upper and lower limbs, with Babinski and Hoffman signs. He was diagnosed as having possible ALS. Head and spinal cord MRI results were normal, with the exception of a type I Chiari malformation. One year later, he developed weakness and wasting of both hands and of the shoulder girdle. He subsequently developed behavior symptoms including apathy, irritability, distractibility, stereotyped behavior, perseveration, and avolition. The total FrSBe score of 89 was pathological, with an increase from the premorbid score of 48. Neuropsychological testing results were pathological for spatial abilities and for letter fluency, with deficits in cognitive inhibitory abilities and impulsivity. The PET/CT scan showed a reduction of metabolism in the frontal associative and frontal mesial areas, right temporal associative areas, and head of right caudatus nucleus (Figure 4).
The proband’s mother (II-7) carries the p.Ala382Thr missense mutation of the TARDBP gene; she developed hypotrophy and loss of strength in both hands at age 69 years. She had pyramidal signs in the upper and lower limbs, and the neurophysiological examination showed active and chronic denervation involving the upper and lower limbs. Head and spinal cord MRI results as well as cerebrospinal fluid examination results were normal. She was diagnosed as having probable, laboratory-supported ALS. A few months after the onset of motor impairment, she developed mental rigidity, perseverative behavior, and distractibility. Neuropsychological testing demonstrated a mild global frontal cognitive deterioration. She did not undergo PET/CT brain study. She is still alive 18 months after the onset of ALS.
The proband’s father (II-8) developed hand wasting, weakness, and generalized pyramidal signs at age 64 years, followed by spreading of muscle hypotrophy to the lower limbs. At neurophysiological examination, he showed chronic and active denervation of all limbs. Head and spinal cord MRI results and cerebrospinal fluid examination results were normal. He was diagnosed as having definite ALS. At age 67 years, he underwent noninvasive ventilation and later tracheotomy. One year after the onset of muscle deficit, he developed anxiety, depression, and agitation of psychotic character. He is still alive 55 months after the onset of ALS. He does not carry SOD1, FUS, or TARDBP mutations.
The proband’s brother (III-1) developed hypotrophy and weakness of left hand muscles at age 33 years. The disturbances progressed to involvement of proximal muscles, with the development of a clinical picture of flail arm syndrome. Pyramidal signs were evident 2 years after the onset of symptoms. The neurophysiological examination showed active and chronic denervation involving the upper and lower limbs, and cerebrospinal fluid examination results were normal. Head and spinal cord MRI results were normal, with the exception of a type I Chiari malformation. He was diagnosed as having definite ALS with a flail arm phenotype. Two years after the onset of ALS, he showed neurobehavioral disturbances and his neuropsychological testing demonstrated a global frontal cognitive deterioration. He died 58 months after the onset of ALS at age 39 years. No DNA was available from this subject.
FAMILY C
In family C (Figure 1C), the proband (III-1) is a 31-year-old woman who developed distal hypotrophy and weakness of the lower limbs at age 25 years, followed by a diffusion of the weakness of both hands 1 year later. She also manifested pyramidal signs of the upper and lower limbs, with Babinski and Hoffman signs. She was diagnosed as having definite ALS. At neurophysiological examination, she showed chronic and active denervation of all limbs.
Brain and spinal cord MRI results were normal. She had no bulbar signs or symptoms. Three years after the onset of motor symptoms, she developed disinhibition, emotional lability, distractibility, aspontaneity, and speech stereotypy. Her total FrSBe score of 62 was borderline pathological, with an increase from the premorbid score of 34. Neuropsychological testing showed deficits in visuospatial abilities and impulsivity. She is alive 72 months after the onset of symptoms. Her PET/CT scan is shown in Figure 5.
Figure 5.

Right lateral (A), left lateral (B), right medial (C), left medial (D), anterior (E), posterior (F), superior (G), and inferior (H) positron emission tomography/computed tomography statistical parametric maps of patient III-1 from family C. The images show the hypometabolic areas on the left frontal and temporal associative cortices (arrows). The lesions are less prominent than those of the patients shown in Figure 3 and Figure 4. P indicates posterior; A, anterior; R, right; and L, left.
The proband’s mother (II-2) is aged 65 years and is neurologically normal. She carries the p.Ala382Thr missense mutation of the TARDBP gene. She refused to undergo neuropsychological assessment and PET/CT scan.
One of the proband’s maternal uncles (II-3) developed wasting and weakness of the hands, rapidly spreading to involve his lower limbs, with pyramidal signs at age 43 years. The diagnosis of definite ALS was confirmed by neurophysiological examination and by head and cervical MRI scan. Bulbar signs developed 1 year later, followed by an overt FTLD. The patient died 30 months after the onset of ALS. No DNA from this subject was available.
Another maternal uncle (II-6) developed weakness of the lower limbs with slight distal atrophy and hyperreflexia and spastic tetraparesis at age 64 years. Neurophysiological examination showed diffuse chronic and active denervation. He was diagnosed as having probable ALS. One year later, he presented with dysphagia and dysarthria. At that time, symptoms of FTLD were present with apathy and executive dysfunction. He is still alive 62 months after the onset of ALS. He carries the p.Ala382Thr missense mutation of the TARDBP gene.
COMMENT
We have described 3 families with ALS carrying the p.Ala382Thr missense mutation of the TARDBP gene in whom symptoms of ALS were followed by the onset of overt FTLD symptoms meeting the criteria for FTLD by Neary et al.9 Detailed questioning of the informants within each family failed to reveal a genealogical link between the 3 families. However, it is still possible that the families share a common haplotype and that the p.Ala382Thr mutation arose from a single mutational event many generations ago. The 3 index cases showed both behavioral and cognitive changes of FTLD as demonstrated by the neuropsychological examination and had PET/CT abnormalities, mainly in the frontal associative areas. This report expands the clinical phenotype associated with mutations in the TARDBP gene to include ALS-FTLD, which is considered to be a separate clinical entity from FTLD with MND.
It has been shown that patients with FTLD with and without ALS show neuronal ubiquitin-positive inclusions containing a hyperphosphorylated, ubiquitinated, and cleaved form of TAR DNA-binding protein 43.13 More recently, mutations in the TARDBP gene coding for TAR DNA-binding protein 43 have been found in individuals with ALS.14,15 However, no FALS cases with TARDBP mutations described in these and other articles had personal or familial histories of FTLD.11,12,14,21 A patient with FALS and the p.Gly294Val missense mutation of the TARDBP gene was affected by Alzheimer-type dementia presenting with anxiety, depression, and progressive memory loss 3 years before the onset of bulbar ALS.12 Both his siblings, affected by ALS, had no signs of cognitive impairment.
Interestingly, TARDBP mutations have not been found in a large series of Italian patients with familial and sporadic FTLD, including 4 with FTLD and MND.22 This finding could support the concept that ALS-FTLD and FTLD are different disorders, but it could simply be related to the fact that TARDBP mutations are rare in this subset of patients. In fact, recently, 2 unrelated cases carrying the c.Gly295Ser missense mutation of the TARDBP gene with behavioral FTLD and semantic dementia later developing MND have been described.7 However, in the case with a positive family history for MND (family F389), FTLD did not segregate with MND because both the affected parent and 1 sibling had MND without clinical symptoms of dementia. The other patient (family F242) was an apparently sporadic case.
The p.Ala382Thr missense mutation has been previously described in ALS, in 4 familial and 4 sporadic cases of French and Italian origin.11,12 In Italy, the p.Ala382Thr mutation seems to be the most frequent mutation of the TARDBP gene.12 However, none of the 8 p.Ala382Thr mutation carriers described in previous articles had cognitive or behavioral impairment.11,12 Therefore, apparently this specific mutation of the TARDBP gene is not always associated with cognitive impairment. The underlying reasons for the variable phenotypic presentations of the p.Ala382Thr mutation are unclear.
In conclusion, although rare, an association between ALS and FTLD in patients carrying a mutation of the TARDBP gene can be found. In our families, cognitive and behavioral symptoms co-segregated with ALS.
Funding/Support
This work was supported by grants from the Fondazione Vialli e Mauro for ALS Research Onlus, Federazione Italiana Giuoco Calcio, and Ministero della Salute (Ricerca Sanitaria Finalizzata 2007) (Dr Chiò); by the Ministero della Salute (Ricerca Sanitaria Finalizzata 2007) (Dr Restagno); and by grant Z01 AG000949-02 from the Intramural Research Program and National Institute on Aging, National Institutes of Health.
Footnotes
Financial Disclosure: None reported.
REFERENCES
- 1.Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):5962. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 2.Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):16681672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):12051208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 4.Vance C, Rogelj B, Hortobágyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):12081211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chow CY, Landers JE, Bergren SK, et al. Deleterious variants of FIG4, a phosphoinositide phosphataseg, in patients with ALS. Am J Hum Genet. 2009;84(1):8588. doi: 10.1016/j.ajhg.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lomen-Hoerth C, Murphy J, Langmore S, Kramer JH, Olney RK, Miller B. Are amyotrophic lateral sclerosis patients cognitively normal? Neurology. 2003;60(7):10941097. doi: 10.1212/01.wnl.0000055861.95202.8d. [DOI] [PubMed] [Google Scholar]
- 7.Benajiba L, Le Ber I, Camuzat A, et al. French Clinical and Genetic Research Network on Frontotemporal Lobar Degeneration/Frontotemporal Lobar Degeneration with Motoneuron Disease, TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol. 2009;65(4):470473. doi: 10.1002/ana.21612. [DOI] [PubMed] [Google Scholar]
- 8.Brooks BR, Miller RG, Swash M, Munsat TL. World Federation of Neurology Research Group on Motor Neuron Diseases, El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 9.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):15461554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 10.Strong MJ, Grace GM, Freedman M, et al. Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10(3):131146. doi: 10.1080/17482960802654364. [DOI] [PubMed] [Google Scholar]
- 11.Kabashi E, Valdmanis PN, Dion P, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40(5):572574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 12.Corrado L, Ratti A, Gellera C, et al. High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Hum Mutat. 2009;30(4):688694. doi: 10.1002/humu.20950. [DOI] [PubMed] [Google Scholar]
- 13.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 14.Yokoseki A, Shiga A, Tan CF, et al. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol. 2008;63(4):538542. doi: 10.1002/ana.21392. [DOI] [PubMed] [Google Scholar]
- 15.Gitcho MA, Baloh RH, Chakraverty S, et al. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63(4):535538. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rutherford NJ, Zhang YJ, Baker M, et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4(9):e1000193. doi: 10.1371/journal.pgen.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Deerlin VM, Leverenz JB, Bekris LM, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7(5):409416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Del Bo R, Ghezzi S, Corti S, et al. TARDBP (TDP-43) sequence analysis in pa-tients with familial and sporadic ALS: identification of two novel mutations. Eur J Neurol. 2009;16(6):727732. doi: 10.1111/j.1468-1331.2009.02574.x. [DOI] [PubMed] [Google Scholar]
- 19.Daoud H, Valdmanis PN, Kabashi E, et al. Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. J Med Genet. 2009;46(2):112114. doi: 10.1136/jmg.2008.062463. [DOI] [PubMed] [Google Scholar]
- 20.Kühnlein P, Sperfeld AD, Vanmassenhove B, et al. Two German kindreds with familial amyotrophic lateral sclerosis due to TARDBP mutations. Arch Neurol. 2008;65(9):11851189. doi: 10.1001/archneur.65.9.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamada M, Maruyama H, Tanaka E, et al. Screening for TARDBP mutations in Japanese familial amyotrophic lateral sclerosis. J Neurol Sci. 2009;284(1-2):6971. doi: 10.1016/j.jns.2009.04.017. [DOI] [PubMed] [Google Scholar]
- 22.Gallone S, Giordana MT, Scarpini E, et al. Absence of TARDBP gene mutations in an Italian series of patients with frontotemporal lobar degeneration. Dement Geriatr Cogn Disord. 2009;28(3):239243. doi: 10.1159/000241876. [DOI] [PubMed] [Google Scholar]