

Abstract
Anti-CD20 therapy using rituximab directly targeting B cells has been approved for treatment of non-Hodgkin lymphoma, rheumatoid arthritis and anti-neutrophil cytoplasmic antibody-associated vasculitides and has led to reappreciation of B-lineage cells for anti-rheumatic treatment strategies. Moreover, blocking B-cell activating factor with belimumab, a drug that is licensed for treatment of active, seropositive systemic lupus erythematosus (SLE), represents an alternative, indirect anti-B-cell approach interfering with proper B-cell development. While these approaches apparently have no substantial impact on antibody-secreting plasma cells, challenges to improve the treatment of difficult-to-treat patients with SLE remain. In this context, anti-CD19 antibodies have the promise to directly target autoantibody-secreting plasmablasts and plasma cells as well as early B-cell differentiation stages not covered by anti-CD20 therapy. Currently known distinct expression profiles of CD19 by human plasma cell subsets, experiences with anti-CD19 therapies in malignant conditions as well as the rationale of targeting autoreactive plasma cells in patients with SLE are discussed in this review.
Introduction
B-lineage cells, in particular antibody-secreting plasma cells (PCs), are the unique source of protective and autoreactive antibodies. In systemic lupus erythematosus (SLE), B cells and autoantibodies, including immune complexes, are considered to be intimately involved in the very complex pathogenesis of SLE as well as a number of other autoimmune diseases [1-4]. The appealing concept to ameliorate disease by abrogating autoantibody production and thereby reducing autoantibody-dependent effector mechanisms has served as the main rationale for the use of B-cell-directed therapies in patients with SLE in addition to the inhibition of B-cell-mediated processes such as antigen presentation, cytokine production and activation of T cells. Ultimately, however, differentiated PCs largely resist these therapeutic approaches - and by continuing to produce autoantibodies, they appear to play an important immunopathogenic role by providing long-lived immune memory, as has been suggested by murine lupus models and data from SLE patients undergoing successful autologous stem cell transplantation (ASCT) [5]. These patients show clinical improvement together with the disappearance of autoreactive antibodies as well as normalization of T-cell and B-cell abnormalities in peripheral blood [5]. Independent of the very intense protocols of ASCT unspecifically targeting PCs, more selective therapeutic interventions are under consideration (recently reviewed in detail [6]) - such as the use of monoclonal antibodies directed against B-cell surface antigens, like CD19, that may directly target PCs beyond other anti-B-cell approaches such as anti-CD20, anti-CD22 and anti-CD52 therapies (Table 1).
Table 1.
Approaches that directly target B cells and plasma cells
| Target | Agent | Putative/intended mechanism of action | Developmental status |
|---|---|---|---|
| CD19 | MD1342 (fully human) | B-cell depletion | Phase I for RA, trial suspended |
| CD19 chimeric antigen receptors | Phase I/II for B-cell malignancies | ||
| Blinatumomab | |||
| XmAb5574/MOR208 | |||
| MEDI-551 | |||
| CD20 | Rituximab (chimeric) | Approved for use in NHL, RA and ANCA vasculitides | |
| Ocrelizumab (humanized) | Phase III for RRMS and PPMS | ||
| Veltuzumab (humanized) | Phase III for RA, phase I/II for AITP | ||
| Ofatumumab (fully human) | Phase III for RA, phase I/II for RRMS | ||
| CD22 | Epratuzumab (humanized) | Blockade of CD22 - partial depletion of B cells, inhibition of activation, proliferation, survival of B cells | Phase III for SLE |
| CD52 | Anti-CD52 or Campath-1h, Alemtuzumab | Depletion of T cells and B cells | Phase III for MS, phase II for autoimmune cytopenias, phase I for inclusion body myositis, phase I/II for RA not continueda |
AITP, Autoimmune thrombocytopenia; ANCA, anti-neutrophil cytoplasmic antibody; MS, multiple sclerosis; NHL, non-Hodgkin lymphoma; PPMS, primary progressive multiple sclerosis; RA, rheumatoid arthritis; RRMS, relapsing/remitting multiple sclerosis; SLE, systemic lupus erythematosus. Data from http://www.clinicaltrials.gov (accessed 5 January 2012). aSee [150-152].
In addition, other principles indirectly targeting these cells by depriving signals required for PC growth and differentiation are also being studied (Table 2). B-cell activating factor (BAFF), TNFα, IL-6, IL-21, granulocyte-macrophage colony-stimulating factor, IFNα as well as other cytokines and signals affect to different degrees the survival, differentiation and function of B cells [7] or PCs [8,9]. While in vivo data from such interventions are very limited, the extent of cytokine-blocking effects on PCs remains largely unknown.
Table 2.
Representative approaches that indirectly target B cells by blocking cytokine signaling
| Target | Agent | Mechanism of action (B cells) | Developmental status |
|---|---|---|---|
| BAFF/BLyS | Belimumab (Anti-BlyS) | Partial B-cell depletion | Approved for SLE |
| BAFF and APRIL | Atacicept (TACI-Ig) | Phase I/II for RA, phase II/III for SLE | |
| TNFα/lymphotoxin-β | Etanercept | Disruption of germinal centers | Approved for RA, SpA, JIA and PsA |
| IL-6 receptor | Tocilizumab | Blocks of B-cell and plasma cell differentiation | Approved for RA, JIA and Castleman's disease, phase I for SLE reported |
| Type I interferon | MEDI 545 | Blocks plasma cell production | Phase II for SLE |
| RhuMAb IFNα | Phase II for SLE |
APRIL, a proliferation-inducing ligand; BAFF, B-cell activating factor; JIA, juvenile idiopathic arthritis; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; PsA, psoriatic arthritis; SpA, spondyloarthritis; TACI, transmembrane activator and calcium modulator and cyclophilin ligand interactor.
The members of the TNF family BAFF(B-cell activating factor, or BlyS, B-lymphocyte stimulator, TNFSF13B) and APRIL (a proliferation-inducing ligand, TNFSF13A), which share receptors on B cells and PCs - that is, BAFF receptor (TNFRSF13C), BCMA (B-cell maturation antigen, TNFRSF17) and TACI (transmembrane activator and calcium modulator and cyclophilin ligand interactor, TNFRFS13B) - are also currently studied as therapeutic targets [10]. In vivo blockade of BAFF/APRIL strongly diminishes the mature PC compartment in mice [11], and inhibition of both BAFF and APRIL by TACI-Ig (Atacicept) has been studied in early trials with patients with SLE [12] and rheumatoid arthritis (RA) [13], providing evidence for reductions of autoreactive but also of total serum immunoglobulin (Ig) levels under treatment. Total Ig reductions of comparable magnitude have also been observed under anti-BAFF therapy with belimumab without affecting APRIL [14], leaving it open whether BAFF has a role in supporting survival of human PCs in vivo (via TACI signaling) or whether certain Ig produced by PCs can also be reduced by indirect targeting of their precursors (via BAFF receptor). The latter is supported by data demonstrating a moderate reduction of plasmablast numbers by ~50% in the blood of belimumabtreated SLE patients [15]. However, the impact of belimumab on systemic antibody production occurred within a limited magnitude and with variable effects on different Ig subclasses and on autoreactive versus total Ig levels [12-15]. The clinical value of belimumab in SLE [14] could also involve targeting of B-cell functions other than autoantibody production. In this regard, B-cell development at the stage of transitional B cells is affected by this treatment [16].
Based on the serum Ig data serving as a surrogate parameter for its effect on PCs, it is difficult to differentiate between the putative impact of anti-BAFF and TACI-Ig on mature PCs versus plasmablasts recently generated from activated B cells. Although no direct comparison exists, both strategies show partial effects on IgM, IgG and IgA production. These data, however, do not allow a distinction between actual reduced PC numbers or simply a reduced capacity to produce Ig.
Overall, the clinical value of direct depletion of autoantibody-producing PCs in the bone marrow (BM) has not yet been sufficiently addressed in autoimmunity. Currently available immune therapies leave these cells largely intact. Under rituximab treatment, tissue-resident and to some degree circulating ´steady-state´ PC pools [17-19] as well as specific (protective) serum Ig levels [20,21] remain largely stable. Another anti-CD20 therapy employing ocrelizumab led to some reductions in serum IgM, IgG and IgA levels, but the dynamics appeared within the normal range of Ig subclass titers [22]. Notably, the discordance between autoantibody reduction and clinical efficacy seen in SLE patients under rituximab treatment raised questions about the mechanism of action of anti-CD20 therapy and the value of more substantial PC depletion beyond anti-CD20 reach [23].
Although the presence or levels of autoantibodies represent a surrogate for successful interventions, none of the above immune interventions but ASCT [5] have been shown to completely eradicate autoantibodies along with clinical remission status. An appealing hypothesis is therefore that successful targeting of autoreactive PCs via partial or complete depletion of the PC repertoire might constitute a therapeutic option for SLE and other autoimmune diseases, and could provide ultimate proof for the concept of autoantibody-driven immunopathology.
Plasma cells and their microenvironment
PCs are destined to produce and secrete large amounts of specific antibodies, and by this means to importantly contribute to immune protection but also to autoimmunity through generation of autoreactive antibodies. In general, PCs differentiate from activated B cells - a process influenced by various factors that determine the timing and site of PC differentiation and maturation (that is, primary vs. secondary responses, splenic vs. lymph node vs. ectopic differentiation), the localization and physiological role of the PCs (that is, mucosal vs. systemic lymphoid tissues), conferring antibody availability at mucosal surfaces versus blood serum [24,25], and the quality of antibody produced by PCs (for example, T-cell-independent vs. T-cell-dependent responses, nature of antigen; recently reviewed in [7]). Mature PCs represent the terminal stage of B-cell differentiation and are molecularly characterized by radical remodeling of their transcriptional program from a paired box 5 (PAX5)/B-cell-specific activator protein (BSAP)-dictated state to a Blimp-1 regulated state [26,27].
PCs reside in the spleen, BM and the mucosal lamina propria, but are also found in lymph nodes and at sites of autoimmune inflammation and in circulation [3,7]. PCs of the BM in particular are considered to be the major source of serum Ig [28,29] and at the same time have been suggested to contain a fraction of long-lived cells [30,31], which might account for the observed stability of specific serum antibody titers over decades [7,32,33]. Such stability of Ig-producing PCs has also been demonstrated in a lupus mouse model, including autoreactive PCs producing an anti-dsDNA idiotype, D42 [34]. In many SLE patients, anti-dsDNA IgG antibodies are reduced upon B-cell-depleting therapy with rituximab. At the same time, protective IgG but also anti-Ro and anti-La antibodies remain stable and hardly change after B-cell depletion [21,35]. These observations suggest that some autoantibody specificities are produced by long-lived PCs being refractory to rituximab treatment, while other specificities partly depend on the presence of CD20+ B cells and presumably the generation of new plasmablasts or short-lived PCs [36].
As mature PCs appear as resting cells [37,38], the survival of individual PCs is critical for the maintenance of serum Ig. PCs die rapidly when isolated ex vivo due to specific pathways of apoptosis [39], unless they receive molecular signals that confer their survival. This property contributed to the concept of a molecular niche that conditionally maintains PCs in the BM and at the same time limits their numbers in healthy individuals [36,40]. Soluble and insoluble signals such IL-5, IL-6, CXCL12, TNFα, APRIL and CD44 ligation have been demonstrated to facilitate PC survival in vitro [7,8], and various cell types able to produce these factors have been identified in close proximity to PCs in the BM or spleen [9,41]. These cell types include stromal cells [42,43], eosinophils [44] or myeloid precursor cells [45], megakaryocytes [46] and dendritic cells [41,47]. Further cell types are directly (or suggested to be) implicated in PC survival; for example, basophils [48], monocytes [49], osteoclasts [15] and osteoblasts [50]. In addition, Xiang and colleagues suggested that the inhibitory Fc receptor FcγRIIb expressed by PCs may regulate their survival via apoptosis control [51]. Notably, PCs from lupus mice lacked expression of FcγRIIb and were protected from apoptosis. The implications of these findings for human SLE remain unknown. The regulation of PC survival probably employs both redundancy and synergy as regulatory principles, as suggested by the multitude of factors and cells implicated in PC survival; for example, IL-6 and CXCL12 could partly compensate for the absence of BAFF and APRIL, or vice versa. Improved understanding of the microarchitecture hosting PCs contributes to potential anti-PC approaches by depriving them of their survival factors.
In this context, PCs also reside in kidneys of SLE patients or lupus-prone NZB/W mice [52,53]. Although it is not known whether these cells have distinct requirements for their survival compared with BM PCs, their disappearance after resolution of inflammation indicates that PC survival is extrinsically regulated, including micro environmental requirements. Studies on cells and soluble factors present in ectopic sites comprising PCs may thus help to define the essential constituents of the PC niche.
Conventional therapies including glucocorticoids and cytostatic drugs such as cyclophosphamide or mycophenolate motefil have very limited impact on mature resting PCs, including autoreactive PCs [34,37,54,55]. Proteasome inhibition with bortezomib affecting B cells and PCs among various other cell types leads to improvement of murine SLE including suppression of anti-dsDNA production [56], whereas data from patients are still very limited [57]. Moreover, immunoablation by rabbit anti-thymocyte globulin (rATG) combined with ASCT [5] represents a unique strategy to abrogate autoantibody production (including protective antibodies) and shows promise for clinical efficacy in severe SLE. The combined clinical and laboratory data from these patients exemplify the critical pathogenic role of the adaptive immune system in SLE, and provide the proof of concept that immune tolerance can be re-established in these patients.
In this regard, CD19 appears to be a candidate target molecule for specific targeting of B cells and PCs, since it has promise to add to the mechanisms of rituximab (that is, through the depletion of CD20+ B cells) the depletion of CD19+CD20- cells - that is, PCs and at least some pro/pre-B cells (CD19+ CD20-CD38+CD10+CD24+IgD-), both of which are only limitedly reduced by rituximab treatment [17,18]. In contrast to CD20 with hitherto largely unknown function, CD19 is known to be contained in the B-cell receptor (BCR) complex, regulating its activation threshold [58]. In line with this, defects in CD19 expression or function elicit autoantibody production in mice and also may contribute to human autoimmunity. Dysregulated CD19 expression has been reported in patients with systemic sclerosis, SLE and anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis. To evaluate CD19 as a candidate target of therapy, we will discuss aspects of its expression on B cells and PCs with particular emphasis on SLE.
Expression and function of CD19
Expression of CD19 and its regulation
CD19 is a 95 kDa transmembrane glycoprotein of the immunoglobulin superfamily encoded by the CD19 gene that is expressed under the control of the transcription factor BSAP (encoded by PAX5) [59-61], a master regulator of B-cell identity [62,63]. No other transcriptional regulator has been identified to bind to the CD19 promoter [59], so CD19 expression probably recapitulates the activity of PAX5/BSAP. Consistently, CD19 is expressed by B lymphocytes but not by any other cell type, except for an unconfirmed report suggesting CD19 expression by some follicular dendritic cells [64].
In B cells, expression of CD19 (also referred to as B4 antigen) starts prior to the expression of CD20 (B1 antigen) at the late pro-B-cell stage together with that of CD10 [65-67]. CD19 expression is maintained throughout various B-cell differentiation and activation stages until PC differentiation, which is then associated with partial loss of CD19 expression (Figure 1).
Figure 1.
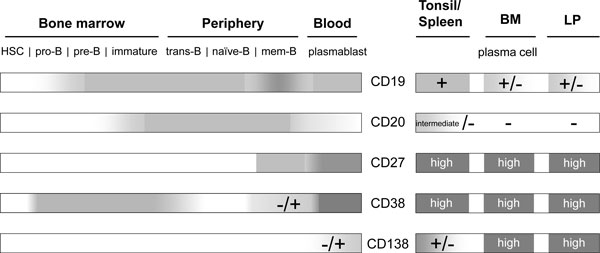
Expression of CD19 and CD20 during human B-cell development. B cells emerge from hematopoietic stem cells (HSC) and acquire maturity in the bone marrow (BM). During this process, CD19 expression starts at the stage of late pro-B cells and precedes that of CD20, starting in immature B cells. CD19 and CD20 expression is maintained during B-cell differentiation and activation in the periphery, while activation and differentiation into memory B cells may fine-tune expression levels. During activation of B cells in lymphoid organs - such as the spleen and tonsil, resulting in their differentiation into plasmablasts and finally plasma cells - CD20 expression is downregulated and subsequently lost. At the same time, CD19 expression is partially downregulated but not extinguished. Being released into circulation, only few plasmablasts express low amounts of CD20, while most have lost CD20 expression but express CD19. After successful immigration into deposits in the BM or the lamina propria (LP), all plasma cells lack CD20, while CD19 expression is maintained by one subset of tissue-resident plasma cells, while another subset acquires a CD19-/CD20- phenotype. Other surface molecules such as CD27, CD38 and CD138 do not qualify as therapeutic targets for global B-cell depletion approaches, as these are not continuously expressed throughout B-cell differentiation and their expression is shared with, for example, T lymphocytes and endothelial cells.
Regular expression of CD19 appears to require CD81, as CD81-deficient mice show a 50% decrease in CD19 expression as compared with controls [68] and a patient deficient in CD81 did not express CD19 on the B-cell surface despite the expression of intact CD19 alleles [69]. BCR deficiency and mutations in Btk are associated with higher and lower CD19 expression by circulating human B cells, respectively [70]. Mutations within one allele of CD19 itself lead to ~2.4-fold reduced surface expression of CD19 [71], while homozygous mutations in the exons encoding cytoplasmic residues of CD19 result in the generation of a truncated, dysfunctional CD19 molecule that is not recognized or is poorly recognized by standard anti-CD19 antibodies [72]. In almost all cases, dysregulated CD19 expression is associated with abnormalities of the immune system, as also suggested by the development of autoantibodies in mice carrying a human CD19 transgene leading to overexpression of CD19 [73]. Consistently, CD19 expression has also been reported to be dysregulated in peripheral B cells from autoimmune patients.
Overall, CD19 is expressed by almost all developmental and activation-dependent B-cell differentiation stages, except for certain PCs, and functions as an important regulator of specific B-cell activation by antigen (Figure 1). CD19 expression coverage exceeds that shown by CD20 expression. Whether all types of B cells will be similarly targeted by therapeutic anti-CD19 antibodies requires first experiences in patients treated with such antibodies.
Functions of CD19
On the surface of B cells, CD19 functions as a co-receptor of the BCR and contains two extracellular C2-type Ig-like domains. Through these domains, CD19 interacts with CD21 (CR2, C3d fragment receptor) and CD81, forming together with CD225 the BCR complex [74].
The intracellular domain of CD19 is involved in intracellular signaling cascades, primarily but not exclusively regulating signals downstream of the BCR and CD22 [58]. Mice either lacking endogenous CD19 expression or overexpressing a human CD19 transgene under control of the endogenous promoter revealed that CD19 is essential for the normal function of the BCR, and in particular regulates the threshold for its activation. While CD19-deficient mice show impaired B-cell responses, B cells from hCD19 transgenic mice were hyperresponsive [75,76] and produce autoantibodies. CD19 is thus a regulator of BCR signaling threshold and is implicated in the control of peripheral tolerance [77]. Human CD19 deficiency has no apparent impact on B-cell development but relates to the phenotype of CD19-/- mice, in that these patients suffer from antibody deficiency [72]. CD19 has also been suggested to have a role in regulating cell proliferation, apparently independent of B-cell identity. CD19 transfection of multiple myeloma but also other cell lines led to reduced culture growth and tumor formation when injected in mice. This effect depended on the cytoplasmic part of CD19 [78]. Overall, the currently available data suggest that CD19 plays critical roles in B-cell and PC differentiation as well as in cell turnover.
CD19 expression by B cells in autoimmunity
Evaluation of anti-CD19 therapy as a therapeutic option requires careful consideration of CD19 expression levels by B cells from patients with autoimmune diseases.
CD19 expression levels of B-cell subsets in the blood
In the blood of patients with lupus, ANCA-associated vasculitis and systemic sclerosis, but also in healthy individuals, CD27+ (memory) B cells appear to express higher levels of CD19 than CD27- (naïve) B cells [79-81], perhaps corresponding to the distinct activation threshold of memory versus naïve B cells in response to antigen. Moreover, circulating CD27high-expressing plasmablasts usually but not always show downregulated, however clearly detectable, expression of CD19 in autoimmune patients and healthy individuals [80-84]. The cell surface density of CD19 is lower than that of CD20 in normal B cells [85]. This difference probably has limited implications for the functionality and biological efficacy of anti-CD19 versus anti-CD20 treatments, because specific modifications of the antibodies (engineering) preferentially define target cell binding and clearing properties [86,87].
Disturbed expression of CD19 in SLE and other autoimmune diseases
In autoimmune patients, both reduced and enhanced CD19 expression by peripheral blood B cells has been described compared with healthy controls. Patients with systemic sclerosis displayed a ~20% increase of CD19 expression on peripheral blood B cells irrespective of whether naïve or memory B cells were analyzed [73,81]. CD19 expression levels correlated with serum IgG and IgM concentrations but not with serum anti-nuclear antibody in these patients [73].
In our analyses, average CD19 expression levels of memory and naive B cells from patients with SLE were comparable with those of healthy controls based on mean fluorescence intensities, whereas plasmablasts showed a trend towards lower CD19 levels (Figure 2). However, CD19 levels detected on patients' memory cells and also naive B cells showed higher variability than those of healthy controls. Stratification of the patient cohort according to high and low disease activity (Systemic Lupus Erythematosus Disease Activity Index (SLEDAI)) or ascending expression levels of CD19 suggested an inverse correlation of CD19 expression levels with disease activity. Moreover, CD19 levels expressed by plasmablasts, memory cells and naive B cells strongly correlated with each other (P < 0.0001). Overall, CD19 expression by plasmablasts was significantly lower as compared with that of memory cells and naive B cells in both SLE patients and controls. This suggests that CD19 expression levels in B cells are regulated via differentiation-independent mechanisms as well as differentiation-dependent mechanisms. Other studies documented about 20% reduced CD19 expression levels by naïve and memory B cells from SLE patients [73,81]. In this regard, our SLE cohort appears to comprise patients with either markedly higher or lower CD19 expression, while there were only few patients within the limited range of CD19 expression values of healthy individuals. These data reproduce both upregulation and downregulation of CD19 by blood B cells from autoimmune patients, which may correspond to the previous observations of CD19high B cells (as discussed below) or reduced CD19 expression [81].
Figure 2.

CD19 expressed by peripheral B-cell subsets from healthy donors and systemic lupus erythematosus patients. Peripheral blood mononuclear cells from 33 systemic lupus erythematosus (SLE) patients and 10 controls were stained for CD3, CD14, CD20, CD19 (clone SJ25C1) and CD27. (a) Lymphocytes including large cells were gated based on forward scatter (FSC) and side scatter (SSC) properties; cell aggregates were excluded in a dotplot depicting forward-scatter height (H) versus area (A) signals. Dead cells, T cells and monocytes were excluded from the analysis, and B cells were identified by gating on the DAPI-CD3-CD14-CD19+ population. (b) Geometric mean fluorescence intensity (MFI) values reflecting CD19 expression were compared among B-cell subsets in SLE patients vs. healthy controls (HD); that is, CD27-CD20+ naive and CD27+CD20+ memory B cells and CD27highCD20-/low plasmablasts. (c) SLE patients were subdivided into three equally sized groups according to ascending expression levels of CD19 by CD27- B cells, CD27+ B cells or plasmablasts (indicated by the triangles below the graph) and analyzed for disease activity (Systemic Lupus Erythematosus Disease Activity Index (SLEDAI)). (d) SLE patients were divided into two groups according to disease activity (SLEDAI <6 vs. SLEDAI ≥6) and analyzed for B-cell CD19 expression levels. Horizontal lines represent median values. P < 0.05 was considered to reflect a statistically significant difference according to the Wilcoxon or Mann-Whitney test used for intra-donor or inter-group comparisons, respectively. (e) CD19 expression levels by CD27- and CD27+ B cells and plasmablasts were analyzed between the individual B-cell subsets using Spearman's rank correlation, demonstrating close relations between their CD19 surface expression levels. *P < 0.05, **P < 0.01 and ***P < 0.001 classify significant values. DAPI, 4',6-diamidino-2-phenylindole.
Overall, the reduction of CD19 expression by SLE B cells in at least some patients appears to be a reproducible phenomenon, independent of disease activity, the presence of anti-nuclear antibodies or use of the different anti-CD19 antibodies applied to expression analyses [79,80]. Interestingly, the difference of CD19 expression by B cells between healthy controls and SLE patients has not been found on a transcriptional level and was reversible after a 2-day, nonstimulating in vitro culture [80].
These data representing an apparent bidirectional dysregulation of CD19 expression by B cells from autoimmune patients are consistent with the fine-tuning of CD19 expression in vivo, which however may not be specifically linked to autoimmunity in general.
CD19high B cells in SLE patients
Additional studies have characterized a population of CD19high-expressing B cells in patients with SLE and ANCA-associated vasculitis, representing up to 10% of the B cells in most patients and up to 30% in individual cases [79,88,89]. CD19high B cells showed markedly higher CD19 expression than B cells from healthy controls, while the remaining CD19low B cells recapitulated the reduced CD19 expression [79] as also documented by other studies [73,80,81]. Detailed flow cytometric analyses of CD19high B cells revealed increased forward-scatter signals compared with CD19low B cells. CD19high B cells mainly expressed CD27, and to a lesser degree IgD, and displayed increased expression of CD86, MHC II and CD95 [79,89] consistent with recent activation of this putative memory B-cell subset in vivo. Consistently, CD19high B cells displayed enhanced phosphorylation of signaling molecules downstream of the BCR complex, such as Syk and ERK, which distinguished them from CD19low B cells [88] and could be generated in vitro from both BCR/CpG-activated naive or memory B cells [90].
In SLE and other autoimmune and immune-deficient patients, CD19high B cells are characterized by low surface CD21 expression [70,73,79,81,89]. These cells therefore resemble the phenotype of B cells that were reported as 'exhausted tissue-like memory B cells' in patients with HIV [91] or common variable immunodeficiency [92]. In the latter, CD19high B cells were not associated with autoimmune phenomena but with splenomegaly and cytopenia [92]. The presence of CD19high/CD21low B cells is hence not unique for patients with SLE and probably corresponds to distinct functional stages of activated B cells. In support of this hypothesis, such CD19high B cells express reduced levels of CD62L (L-selectin), CXCR4 and CXCR5 [70,89,91], which may exclude these cells from regular recirculation and germinal center participation, while enhanced expression of functional CXCR3 [88] might allow for their migration to sites of inflammation in vivo.
The presence of CD19high B cells did not correlate with disease activity in SLE or ANCA-associated vasculitis [79,89], but did correlate with neurological and renal disease manifestations [79,88], shorter clinical benefit from rituximab treatment [88] as well as anti-Sm but not anti-nuclear antibody or anti-Ro/La titers [79]. Indeed, CD19high B cells from anti-Sm-positive SLE patients yielded considerable amounts of Sm-specific antibody-secreting cells 6 days after in vitro activation culture, in contrast to CD19low B cells [88], suggesting that this subset might contain a substantial part of autoreactive B cells.
In summary, abnormalities in CD19 expression appear to be related to the activation status of B cells rather than specifically linked to autoimmunity (Table 3). The combined data of CD19 expression in conjunction with changes of CD21, CD81, BCR and Btk suggest that CD19 expression is fine-tuned by BCR-mediated activation signals. Given the enhanced CD19 expression by some B cells under the condition of autoimmunity, these cells may constitute a key target. However, given that blood lymphocytes represent approximately 2% of total body lymphocytes [93,94], whether the data above are also representative for tissue-resident B cells in the spleen, lymph nodes, BM, gut-associated lymphoid tissues and inflamed sites remains open. Apparently all B-cell stages in the human blood and probably in the relevant tissues express CD19 and presumably could be targeted by an anti-CD19 antibody.
Table 3.
Human conditions associated with altered CD19 expression
| CD19 upregulation | CD19 downregulation |
|---|---|
| Systemic sclerosis (MFI) | SLE (MFI) |
| SLE (CD19high B cells) | CD19 deficiency |
| CVID, HIV (CD19high B cells) | Heterozygous mutation in CD19 |
| BCR deficiency (CD19high B cells) | CD81 deficiency |
| Mutations in Bruton's tyrosine kinase |
BCR, B-cell receptor; CVID, common variable immunodeficiency; MFI, mean fluorescence intensity; SLE, systemic lupus erythematosus.
CD19 expression by plasma cells
Importantly, a substantial fraction of antibody-secreting PCs express CD19 in contrast to CD20, which is downregulated during plasmablast differentiation in association with their relocation from secondary lymphoid tissue to PC depots in the BM or lamina propria. Splenic and tonsillar plasmablasts appear as CD19+CD20+/intermediate- expressing cells [95-97], while plasmablasts in the blood express low or no CD20 and diminished CD19 levels [80,81,83,84,96,98]. One must emphasize that PCs in the BM consist of both CD20-CD19+ and CD20-CD19- PC subsets in man [96,99] and in the mouse [100]. The bimodal distribution of CD19 expression among PCs in the BM of both healthy and autoimmune individuals (Figure 3) has important implications since CD19-directed therapies could therefore target some, but not all, PCs.
Figure 3.
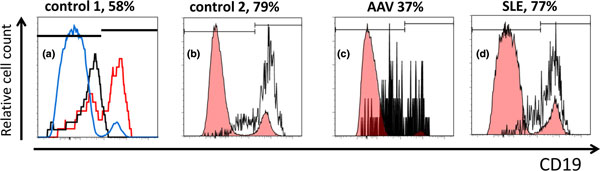
CD19+ and CD19- plasma cells detectable in bone marrow cells from autoimmune patients and controls. Bone marrow (BM) cell suspensions were obtained from iliac crest or femoral head samples from autoimmune or nonautoimmune control individuals and stained for CD38 and CD19 (clone SJ25C1), and in some cases additionally for CD138, CD3 and CD14. Dead cells were excluded by adding DAPI and gating on DAPI- cells. Plasma cells (PCs) were identified based on light scatter properties and high expression of CD38. CD38high PCs showed co-expression of CD138 and lacked that of CD3 and CD14. (a), (b) Nonautoimmune individuals. (c), (d) Patients as indicated. (a) Red histogram depicts PCs stained for CD19, black histogram shows isotype control-stained PCs, blue histogram shows lymphoid cells including CD19+ B cells for comparison. Note that control-stained and CD19-negative PCs exhibit an increased fluorescent background signal as compared with other lymphoid cells. (b) to (d) Black histograms represent PCs; red histograms depicting lymphoid cells including CD19+ B cells are shown for comparison. Markers define CD19+ and CD19- PCs, respectively. Frequencies represent proportions of CD19+ cells among PCs. AAV, ANCA-associated vasculitis; DAPI, 4',6-diamidino-2-phenylindole; SLE, systemic lupus erythematosus.
Both PC differentiation and downregulation of CD19 are tightly interrelated with the loss of PAX5/BSAP expression [101,102], and consequently BSAP binding sites in the CD19 promoter are not occupied in PCs [59]. While many PC lines and multiple myeloma cells lack CD19 expression, fractions of normal mature PCs in the human BM but also in the mucosal lamina propria express CD19 [96,99,103-105], including IgG-secreting cells in man [98] and Blimp-1+ PC in the mouse [100].
Little is known about the nature of underlying immune reactions and/or the relationship between CD19+ and CD19- PC in the BM or lamina propria, which makes it challenging to estimate the potential effects of CD19-directed therapy on humoral (auto)immunity. In the mouse, the gradual loss of CD19 on PCs seems to be associated with advanced maturity; that is, higher expression of Blimp-1, lower expression of HLA-DR and B220, and eventually a longer lifespan [100]. Neither CD19+ nor CD19- PCs from human BM expressed PAX5 mRNA, but they did express IRF4, PRDM1 and CD138 at comparable levels (n ≥4; HE Mei and T Dörner, un published data). This suggests additional factors regulating the expression of CD19 on PCs beyond transcriptional control by PAX5/BSAP.
These data are consistent with the assumption that anti-CD19 therapy would probably target significant fractions of PCs while not enhancing all PCs. More information is therefore required on the dynamics of and the relationship between CD19+ and CD19- PCs in order to estimate the impact of anti-CD19 treatment.
Plasmablasts and certain plasma cells can be targeted by anti-CD19
CD19+ cells comprise virtually all antibody-secreting cells in the peripheral blood in both healthy individuals and patients with SLE as well as other autoimmune diseases [25,83,106]. These comprise (at least) two major fractions: first, recently generated antibody-secreting cells that undergo cell division, are capable of in vitro migration towards gradients of CXCL12 [25], express high levels of HLA-DR and contain antigen-specific cells that pass the peripheral blood 1 week after a tetanus booster vaccination [83]. We termed these cells plasmablasts [7,25,36,83], whereas the remaining antibody-secreting cells were defined as PCs that were unable to proliferate and migrate in vitro [25], lack surface expression of the adhesion molecules β7-integrin and CD62L (L-selectin), express low levels of HLA-DR and do not contain antigen-specific cells after vaccination [25,83]. When analyzed 1 week after tetanus vaccination, circulating HLA-DRlow PCs phenotypically, functionally and transcriptionally showed a high degree of similarity with BM PCs [7,25], and probably reflect outcompeted mature BM PCs [83]. Both subsets can be detected by flow cytometry after gating on CD19+ cells combined with an enlarged lymphocyte gate [25,82-84].
CD19 expression by plasmablasts in autoimmune diseases has rarely been analyzed. Sato and colleagues did not detect statistically significant differences in the CD19 signals by flow cytometry when comparing plasmablasts (together with PCs using the CD27high criterion for detection) among healthy individuals and patients with SLE or systemic sclerosis [81] - while another study showed lower CD19 expression by SLE CD27high plasmablasts as compared with those from healthy controls [80], which is consistent with our data (Figure 2). As illustrated in a representative SLE patient, approximately 80% of the peripheral blood CD138+ plasmablasts/PCs express CD19 [84], indicating that a minority of circulating PCs lacks expression of this marker.
Frequencies and numbers of CD19-expressing plasmablasts/PCs are drastically increased in patients with SLE [54,82,84,107] and reflect up to 50% of all circulating CD19+ B cells [84] while containing dsDNA-specific cells [54]. The cell frequency and number correlated with disease activity and autoantibody production [82,84,106], and this correlation was even more pronounced when HLA-DRhigh plasmablasts were analyzed separately [54]. The presence of these plasmablasts therefore reflects acute, presumably pathogenic B-cell activation in patients with SLE. Elevation of peripheral blood plasmablast levels has also been recently demonstrated for Takayasu's arteriitis patients [108], but not in patients with primary Sjögren's syndrome [109], autoimmune thrombocytopenia [110], systemic sclerosis [81] and RA [19]. Further analyses focusing on CD19+ B cells in the BM demonstrated an imbalance of HLA-DRhigh plasmablasts versus HLA-DRlow PC subsets - that is, relative expansion of plasmablasts in SLE patients' BM - indicating that not only the periphery but also the major deposit sites of PC are disturbed in SLE [54].
The prominent presence of presumably pathogenic plasmablasts in SLE patients' blood and BM [3] provide a further rationale for anti-CD19 therapy since CD19+CD20-/low plasmablasts are refractory to B-cell depletion with rituximab in RA, SLE and other autoimmune syndromes [19,21,111-114]. In this context, we found that rituximab-resistant plasmablasts carry a mucosal phenotype in patients with RA and show multiple traits of their recent generation, such as cell proliferation and spontaneous in vitro migration. This finding points towards a self-sufficient, rituximab-resistant mucosal B-cell subset in man [19] that is chronically activated and gives rise to plasmablasts in circulation. In SLE patients, higher levels of plasmablasts after B-cell depletion have been reported to be associated with poorer clinical response [112]. Anti-CD19 therapy might constitute a possibility to overcome this limitation of rituximab by directly targeting these plasmablasts and certain CD19+ PCs. There is thus a possibility that especially autoantibody-positive patients could benefit from anti-CD19 therapy, but this needs to be demonstrated.
Overall, a better understanding of the homeostasis and differentiation of different mature PC subsets in healthy subjects and autoimmune patients not only related to the expression of CD19 but also to their distribution and characteristics in certain organs and organ systems is required to fully explore the potential benefits and potential side effects of, and suitable molecular targets for PC-directed therapies.
Anti-CD19 treatment approaches and effects on antibody production
Autoreactive clones of long-lived PCs have been considered an obstacle for existing therapies in autoimmunity [3], while anti-CD19 therapies have been initially developed to delete malignant B-cell clones using both native and modified anti-CD19 antibodies with or without conjugated toxins as well as bispecific antibodies targeting, for example, CD19 and CD3 or transgenic T cells carrying a T-cell receptor recognizing CD19. Although approaches in oncology differ in their rationales compared with autoimmunity, the existing data for anti-CD19 antibodies in treating malignant diseases represent a valuable mechanism of action and a safety data pool.
Native naked and conjugated anti-CD19 antibodies
Initial studies in non-Hodgkin's lymphoma patients [115,116] using CLB-CD19 with or without IL-2 demonstrated binding of anti-CD19 antibody to B cells in vivo and led to partial reduction of blood B cells, but did not consistently change total antibody levels during treatment (up to 12 weeks) and in the follow-up period.
Various anti-CD19 immunotoxins are under development for malignant diseases - for example, bound to auristatin [117], saporin [118] or ricin [119] - but have not so far entered studies in autoimmunity.
In a mouse model of anti-CD19 therapy using hCD19 transgenic mice [120] and anti-human CD19 antibodies (HB12a, HB12b and FMC63), depletion of >90% of B cells in the BM, blood, spleen and lymph nodes was observed, while peritoneal cavity B cells were reduced by only two-thirds. Notably, pro-B cells in the BM were not depleted in this system. Blood B cells remained depleted for about 12 weeks and only partially repopulated until 30 weeks after treatment. In this model, levels of total serum antibody concentrations were reduced by 95% for IgM, by 10 to 20% for IgG and by about 40% for IgA compared with baseline values. Specific IgM and IgG1 responses to T-independent antigens and primary and secondary responses to T-dependent antigens were virtually abrogated in mice treated with anti-CD19. Reduction of specific serum IgM and IgG1 concentrations also occurred in previously immunized mice treated with anti-CD19 [120], most probably reflecting that antibody-secreting cells were successfully targeted in this model.
Autoantibody production analyzed in this mouse model was critically dependent on the overexpression of CD19 [121,122], and CD19 expression was reduced over at least 10 weeks after anti-CD19 therapy. While animals treated with control antibody developed increasing levels of anti-dsDNA, anti-ssDNA and anti-histone IgM and IgG antibodies, anti-CD19-treated mice showed reductions of IgM autoantibodies within 2 weeks. IgG autoantibodies were also reduced in a delayed manner at the latest by week 10 after treatment [120]. These Ig-class-specific responses of serum antibody levels to anti-CD19 treatment indicate differential dynamics of IgM+ and IgG+ PC subsets and indicate that some but perhaps not all autoreactive PCs express CD19 in this model. However, additional studies are required in control mice and in models of SLE that are independent of genetic modification of CD19 expression.
Engineered anti-CD19 antibodies
Modifications of the amino acid sequence and the glycosylation of native anti-CD19 antibodies [86,87], as well as engineering of bispecific antibodies co-targeting other antigens (for example, CD3 [123]) to enhance target cell binding clearing properties, have generated interest in anti-CD19 immunotherapy.
In this context, the bispecific CD19×CD3 antibody (Blinatumomab, MT103, MEDI-538, bispecific T-cell engager (BiTE)) that has been designed to recruit and activate T cells in the vicinity of CD19+ tumor cells in non-Hodgkin's lymphoma patients resulted in substantial and efficacious depletion of blood B cells (residual numbers <10 cells/μl) for up to 2 months and obviously cleared malignant B cells in the BM, spleen and liver. More than 85% of the study participants showed no significant decline of serum Ig levels, reflecting that most patients retained an intact PC compartment during therapy.
The application of XmAb5574 (MOR208) in Cynomolgus monkeys did not reduce serum IgG, IgA and IgM levels over a 3-month period [124], indicating that the PC compartment remained intact either due to resistance of PCs or incomplete depletion of B cells in this model. Indeed, only 80 to 90% of CD20+ B cells in peripheral blood and 40 to 60% within the BM, spleen and lymph nodes were depleted 1 month after anti-CD19 treatment [124]. A clinical phase I trial with XmAb5574 in chronic lymphocytic leukemia patients is currently ongoing (Trial #NCT01161511). A further anti-CD19 antibody is MEDI-551, which is able to deplete B cells in suspensions of human peripheral blood mononuclear cells as well as blood and splenic B cells in hCD19/hCD20 double transgenic mice [86]. Data that might allow an evaluation of targeting PC in humans by this antibody are not yet available, but clinical trials have been initiated. The anti-CD19 antibody MDX-1342 was under investigation in RA patients in combination with methotrexate [125] but was discontinued in April 2010. Further anti-CD19 approaches such as derivates of the anti-human CD19 clone HD37 are being developed [126].
Multiple anti-CD19 antibodies are being and have been developed for therapeutic use, so far with very inconsistent experiences in preclinical models or pilot trials with regard to both the efficacy and duration of B-cell and PC depletion.
Anti-CD19 chimeric antigen receptor-expressing T cells
A different way of CD19 targeting employs transplants of autologous T lymphocytes that are isolated from the patients' blood, stimulated in vitro with, for example, anti-CD3 and IL-2, and transduced with a vector conferring expression of a chimeric antigen receptor (CAR) that is directed against CD19. Upon incubation with CD19+ cells, CAR-bearing T cells produce IFNγ and IL-2 [127,128] and lead to sustained depletion of B cells, including tumor cells in human blood (at least for 40 weeks) and in BM (at least for 36 weeks) [127]. Notably, serum Ig levels gradually dropped during this therapy with reductions of approximately 40 to 50% for IgG within 14 weeks, 85% (within 14 weeks) and 75% (until week 36) for IgA and IgM, respectively. In a subsequent study of eight non-Hodgkin's lymphoma patients, four individuals experienced profound B-cell depletion with hypogammaglobulinemia requiring Ig substitution [129] - similar to that described for treatment of chronic lymphocytic leukemia patients using CD19 CAR-expressing T cells [130]. Depletion failed in the other four patients, as has also been reported by an independent study [131], and was probably due to properties of the malignant B cells or result from variations of CD19 chimeric antigen receptor T-cell generation.
The available data on B-cell and PC targeting by anti-CD19-directed approaches in oncologic diseases do not allow firm conclusions as to what extent PC depletion occurs in autoimmunity and may differ between certain agents and techniques. Ongoing clinical studies will provide more detailed information on the efficacy and especially safety of anti-CD19 approaches, which will also permit a unique opportunity to compare those with anti-CD20 experiences and at the same time better define potential treatment options for autoimmunity.
Potential risks and benefits of anti-CD19 therapy in autoimmunity
Humoral immunity and the potential risks for infections
Apparently, targeting both B cells and PCs raises safety concerns with regard to sufficient control of environmental pathogens and infections by protective Ig generated as a first-line defense as well as during B-cell memory responses. CD19 targeting can be assumed to enlarge the window of cell depletion compared with anti-CD20 therapy, with an additional impact on pro-B cells and PCs. As the maintenance of protective and autoreactive antibody titers and the contributions of different subsets of CD19+ versus CD19- PCs and CD19+ plasmablasts are incompletely understood, one may only speculate about the impact of anti-CD19 on immune protection. Successful targeting of CD19+ PCs in patients with SLE is expected to at least substantially reduce autoantibody titers produced by plasmablasts and certain PCs. However, severe reductions of Ig levels can be managed by intravenous Ig infusions [129]. We believe one cannot exclude the possibility that patients might experience loss of or impairment of sufficient antibody production under anti-CD19 therapy, as observed in patients with common variable immunodeficiency who often show reduced numbers of PCs in the BM [132] and usually fail to mount sufficient vaccination responses [133]. The failure to sufficiently respond to vaccinations of most patients treated with anti-CD20 will probably also affect patients treated with anti-CD19 therapy. During rituximab treatment, both primary and secondary serological antibody responses are substantially impaired [134-138], while pre-existing antibody titers were largely maintained. The estimated time period of B-cell absence after anti-CD19-mediated B-cell depletion is currently unknown, while B-cell depletion with rituximab lasts usually 6 months or longer. Given the broader coverage of anti-CD19 that includes pro-B cells, it is possible that peripheral B-cell repopulation after anti-CD19 therapy differs from that after anti-CD20 therapy.
A situation of antibody deficiency has been described in SLE patients after ASCT with prior immunoablation using high-dose cyclophosphamide and rabbit antithymocyte globulin, which often leads to improvement of the disease, perhaps remission, but also caused loss of protective serum antibody titers directed to both protein and viral antigens (anti-tetanus, anti-measles, anti-diphtheria, anti-rubella) [5]. The risk of opportunistic infections therefore requires careful monitoring and potentially might represent a limitation. Given that diminishing autoimmune PCs provides substantial benefit for the patients, including long-term remission, the possibility of intravenous Ig substitution may allow balancing the risk of hypoimmunoglobulinemia and infections.
Experiences with this therapy in tumor patients [123,127,129,131] might in part be instructive for potential risks of anti-CD19 therapy in autoimmunity but can only be extrapolated with caution because of the different underlying disease and pre-existing and co-medications. Further potential side effects of anti-CD19 therapy probably overlap with various adverse events reported after B-cell depletion with rituximab, such as the occurrence of infections [139,140], suppression of specific B-cell responses [134-136] and risk to develop progressive multifocal leukoencephalopathy [141]. These aspects are of particular importance since B-cell depletion by anti-CD19 strategies appears to be broad and long-lasting [129] while B cells and antibody is required to suppress John Cunningham virus (see [141]). Overall, the safety profile of anti-CD19 therapy in autoimmunity will be of central importance but requires further data.
CD19 ligation as an additional mechanism beyond depletion
Animal model data for anti-CD19 therapy documented efficient reductions but sometimes not complete depletion of B cells [120,124]. In such cases, B cells labeled by anti-CD19 would remain in vivo, with as yet unclear consequences for the modulation of BCR signaling [75,77,121]. In vitro, anti-CD19 (HD37) lowers the threshold for anti-BCR (IgM)-induced human B-cell activation under certain conditions [142], which could confer enhanced B-cell activation and even exacerbation of autoimmunity, while other reports using different protocols reported an inhibition of BCR signaling by anti-CD19 antibody. The above notions remain a theoretical possibility since overactivation of B cells during anti-CD19 treatment has not so far been reported.
Regulatory B cells
B-cell depletion also affects B cells with immuno-suppressive potential, which might explain some rare cases of autoimmunity that were reported during rituximab therapy [143-146]. In this regard, several studies in mice indicate an important role of B cells producing IL-10 in controlling autoimmunity [147]. In vitro generated IL-10-producing B cells express CD19 [148,149] and were therefore subject to CD19-directed depletion. The phenotype and the importance of human bona fide IL-10-secreting B cells in vivo remains unknown, however, and there is no clear evidence for impaired control of immune responses after either anti-CD19 or anti-CD20 therapy in humans.
The estimated immunological side effects of envisaged B-cell depletion using CD19-directed approaches mostly resemble the experiences from rituximab treatment. While additional putative depletion of pro-B cells leading to prolonged peripheral reconstitution and reductions of major PC subsets might lead to an enhanced risk for infections, CD19 therapy has the promise to improve efficacy compared with the more restricted B-cell targeting by anti-CD20 antibodies.
MEDI 545 is in clinical development by Medimmune Inc.
Abbreviations
ANCA: anti-neutrophil cytoplasmic antibody; APRIL: a proliferation-inducing ligand; ASCT: autologous stem cell transplantation; BAFF: B-cell activating factor; BCR: B-cell receptor; Blimp-1: B-lymphocyte-induced maturation protein 1; BM: bone marrow; BSAP: B-cell-specific activator protein; Btk: Bruton's tyrosine kinase; CXCL: CXC motif ligand; CXCR: CXC motif receptor; dsDNA: double-stranded DNA; ERK: extracellular signal-related kinase; Fc: Fragment crystallizable; IFN: interferon; Ig: immunoglobulin; IL: interleukin; La: La/SSB protein; MHC: major histocompatibility complex; PAX5: paired box 5; PC: plasma cell; RA: rheumatoid arthritis; Ro: Ro/SSA protein (calreticulin); SLE: systemic lupus erythematosus; Sm: Smith protein; ssDNA: single-stranded DNA; Syk: spleen tyrosine kinase; TACI: transmembrane activator and calcium modulator and cyclophilin ligand interactor; TNF: tumor necrosis factor.
Competing interests
The authors declare that they have no competing interests.
Acknowledgements
This work was supported by DFG SPP ImmunoBone, by the SFB650 project TP16 from the DFG and by Project Do491/5-4, as well as by a start-up grant from the Deutsche Gesellschaft für Rheumatologie (DGRh). The authors thank Dr BF Hoyer and Dr T Alexander for the provision of samples producing the results shown in Figure 3.
Declarations
This article has been published as part of Arthritis Research & Therapy Volume 14 Supplement 5, 2012: B cells in autoimmune diseases: Part 1. The supplement was proposed by the journal and content was developed in consultation with the Editor-in-Chief. Articles have been independently prepared by the authors and have undergone the journal's standard peer review process. Publication of the supplement was supported by Medimmune.
References
- Edwards JC, Cambridge G. B-cell targeting in rheumatoid arthritis and other autoimmune diseases. Nat Rev Immunol. 2006;14:394–403. doi: 10.1038/nri1838. [DOI] [PubMed] [Google Scholar]
- Dorner T. SLE in 2011: deciphering the role of NETs and networks in SLE. Nat Rev Rheumatol. 2012;14:68–70. doi: 10.1038/nrrheum.2011.200. [DOI] [PubMed] [Google Scholar]
- Hiepe F, Dorner T, Hauser AE, Hoyer BF, Mei H, Radbruch A. Long-lived autoreactive plasma cells drive persistent autoimmune inflammation. Nat Rev Rheumatol. 2011;14:170–178. doi: 10.1038/nrrheum.2011.1. [DOI] [PubMed] [Google Scholar]
- Reininger L, Radaszkiewicz T, Kosco M, Melchers F, Rolink AG. Development of autoimmune disease in SCID mice populated with long-term 'in vitro' proliferating (NZB × NZW)F1 pre-B cells. J Exp Med. 1992;14:1343–1353. doi: 10.1084/jem.176.5.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander T, Thiel A, Rosen O, Massenkeil G, Sattler A, Kohler S, Mei H, Radtke H, Gromnica-Ihle E, Burmester GR, Arnold R, Radbruch A, Hiepe F. Depletion of autoreactive immunologic memory followed by autologous hematopoietic stem cell transplantation in patients with refractory SLE induces long-term remission through de novo generation of a juvenile and tolerant immune system. Blood. 2009;14:214–223. doi: 10.1182/blood-2008-07-168286. [DOI] [PubMed] [Google Scholar]
- Dorner T, Radbruch A, Burmester GR. B-cell-directed therapies for autoimmune disease. Nat Rev Rheumatol. 2009;14:433–441. doi: 10.1038/nrrheum.2009.141. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Mei H, Dorner T, Hiepe F, Radbruch A, Fillatreau S, Hoyer BF. Memory B and memory plasma cells. Immunol Rev. 2010;14:117–139. doi: 10.1111/j.1600-065X.2010.00938.x. [DOI] [PubMed] [Google Scholar]
- Cassese G, Arce S, Hauser AE, Lehnert K, Moewes B, Mostarac M, Muehlinghaus G, Szyska M, Radbruch A, Manz RA. Plasma cell survival is mediated by synergistic effects of cytokines and adhesion-dependent signals. J Immunol. 2003;14:1684–1690. doi: 10.4049/jimmunol.171.4.1684. [DOI] [PubMed] [Google Scholar]
- Tangye SG. Staying alive: regulation of plasma cell survival. Trends Immunol. 2011;14:595–602. doi: 10.1016/j.it.2011.09.001. [DOI] [PubMed] [Google Scholar]
- Stohl W, Scholz JL, Cancro MP. Targeting BLyS in rheumatic disease: the sometimes-bumpy road from bench to bedside. Curr Opin Rheumatol. 2011;14:305–310. doi: 10.1097/BOR.0b013e328344c15e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson MJ, Dillon SR, Castigli E, Geha RS, Xu S, Lam KP, Noelle RJ. Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFFand APRIL. J Immunol. 2008;14:3655–3659. doi: 10.4049/jimmunol.180.6.3655. [DOI] [PubMed] [Google Scholar]
- Dall'Era M, Chakravarty E, Wallace D, Genovese M, Weisman M, Kavanaugh A, Kalunian K, Dhar P, Vincent E, Pena-Rossi C, Wofsy D . Reduced B lymphocyte and immunoglobulin levels after atacicept treatment in patients with systemic lupus erythematosus: results of a multicenter, phase Ib, doubleblind, placebo-controlled, dose-escalating trial. Arthritis Rheum. 2007;14:4142–4150. doi: 10.1002/art.23047. [DOI] [PubMed] [Google Scholar]
- Genovese MC, Kinnman N, de La Bourdonnaye G, Pena Rossi C, Tak PP. Atacicept in patients with rheumatoid arthritis and an inadequate response to tumor necrosis factor antagonist therapy: results of a phase II, randomized, placebo-controlled, dose-finding trial. Arthritis Rheum. 2011;14:1793–1803. doi: 10.1002/art.30373. [DOI] [PubMed] [Google Scholar]
- Navarra SV, Guzman RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, Li EK, Thomas M, Kim HY, Leon MG, Tanasescu C, Nasonov E, Lan JL, Pineda L, Zhong ZJ, Freimuth W, Petri MA . Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;14:721–731. doi: 10.1016/S0140-6736(10)61354-2. [DOI] [PubMed] [Google Scholar]
- Geffroy-Luseau A, Jego G, Bataille R, Campion L, Pellat-Deceunynck C. Osteoclasts support the survival of human plasma cells in vitro. Int Immunol. 2008;14:775–782. doi: 10.1093/intimm/dxn035. [DOI] [PubMed] [Google Scholar]
- Jacobi AM, Huang W, Wang T, Freimuth W, Sanz I, Furie R, Mackay M, Aranow C, Diamond B, Davidson A. Effect of long-term belimumab treatment on B cells in systemic lupus erythematosus: extension of a phase II, doubleblind, placebo-controlled, dose-ranging study. Arthritis Rheum. 2010;14:201–210. doi: 10.1002/art.27189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng YK, Levarht EW, Hashemi M, Bajema IM, Toes RE, Huizinga TW, van Laar JM. Immunohistochemical analysis as a means to predict responsiveness to rituximab treatment. Arthritis Rheum. 2007;14:3909–3918. doi: 10.1002/art.22967. [DOI] [PubMed] [Google Scholar]
- Rehnberg M, Amu S, Tarkowski A, Bokarewa MI, Brisslert M. Short- and long-term effects of anti-CD20 treatment on B cell ontogeny in bone marrow of patients with rheumatoid arthritis. Arthritis Res Ther. 2009;14:R123. doi: 10.1186/ar2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei HE, Frolich D, Giesecke C, Loddenkemper C, Reiter K, Schmidt S, Feist E, Daridon C, Tony HP, Radbruch A, Dorner T. Steady state generation of mucosal IgA+ plasmablasts is not abrogated by B cell depletion therapy with rituximab. Blood. 2010;14:5181–5190. doi: 10.1182/blood-2010-01-266536. [DOI] [PubMed] [Google Scholar]
- Cambridge G, Leandro MJ, Edwards JC, Ehrenstein MR, Salden M, Bodman-Smith M, Webster AD. Serologic changes following B lymphocyte depletion therapy for rheumatoid arthritis. Arthritis Rheum. 2003;14:2146–2154. doi: 10.1002/art.11181. [DOI] [PubMed] [Google Scholar]
- Vallerskog T, Gunnarsson I, Widhe M, Risselada A, Klareskog L, van Vollenhoven R, Malmstrom V, Trollmo C. Treatment with rituximab affects both the cellular and the humoral arm of the immune system in patients with SLE. Clin Immunol. 2007;14:62–74. doi: 10.1016/j.clim.2006.08.016. [DOI] [PubMed] [Google Scholar]
- Stohl W, Gomez-Reino J, Olech E, Dudler J, Fleischmann RM, Zerbini CA, Ashrafzadeh A, Grzeschik S, Bieraugel R, Green J, Francom S, Dummer W. Safety and efficacy of ocrelizumab in combination with methotrexate in MTX-naive subjects with rheumatoid arthritis: the phase III FILM trial. Ann Rheum Dis. 2012;14:1289–1296. doi: 10.1136/annrheumdis-2011-200706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisetsky DS, Grammer AC, Ning TC, Lipsky PE. Are autoantibodies the targets of B-cell-directed therapy? Nat Rev Rheumatol. 2011;14:551–556. doi: 10.1038/nrrheum.2011.108. [DOI] [PubMed] [Google Scholar]
- Kunkel EJ, Butcher EC. Plasma-cell homing. Nat Rev Immunol. 2003;14:822–829. doi: 10.1038/nri1203. [DOI] [PubMed] [Google Scholar]
- Mei HE, Yoshida T, Sime W, Hiepe F, Thiele K, Manz RA, Radbruch A, Dorner T. Blood-borne human plasma cells in steady state are derived from mucosal immune responses. Blood. 2009;14:2461–2469. doi: 10.1182/blood-2008-04-153544. [DOI] [PubMed] [Google Scholar]
- Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat Rev Immunol. 2005;14:230–242. doi: 10.1038/nri1572. [DOI] [PubMed] [Google Scholar]
- Oracki SA, Walker JA, Hibbs ML, Corcoran LM, Tarlinton DM. Plasma cell development and survival. Immunol Rev. 2010;14:140–159. doi: 10.1111/j.1600-065X.2010.00940.x. [DOI] [PubMed] [Google Scholar]
- Benner R, Hijmans W, Haaijman JJ. The bone marrow: the major source of serum immunoglobulins, but still a neglected site of antibody formation. Clin Exp Immunol. 1981;14:1–8. [PMC free article] [PubMed] [Google Scholar]
- McMillan R, Longmire RL, Yelenosky R, Lang JE, Heath V, Craddock CG. Immunoglobulin synthesis by human lymphoid tissues: normal bone marrow as a major site of IgG production. J Immunol. 1972;14:1386–1394. [PubMed] [Google Scholar]
- Manz RA, Thiel A, Radbruch A. Lifetime of plasma cells in the bone marrow. Nature. 1997;14:133–134. doi: 10.1038/40540. [DOI] [PubMed] [Google Scholar]
- Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to longlived plasma cells. Immunity. 1998;14:363–372. doi: 10.1016/S1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
- Amanna IJ, Carlson NE, Slifka MK. Duration of humoral immunity to common viral and vaccine antigens. N Engl J Med. 2007;14:1903–1915. doi: 10.1056/NEJMoa066092. [DOI] [PubMed] [Google Scholar]
- Manz RA, Hauser AE, Hiepe F, Radbruch A. Maintenance of serum antibody levels. Annu Rev Immunol. 2005;14:367–386. doi: 10.1146/annurev.immunol.23.021704.115723. [DOI] [PubMed] [Google Scholar]
- Hoyer BF, Moser K, Hauser AE, Peddinghaus A, Voigt C, Eilat D, Radbruch A, Hiepe F, Manz RA. Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J Exp Med. 2004;14:1577–1584. doi: 10.1084/jem.20040168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambridge G, Isenberg DA, Edwards JC, Leandro MJ, Migone TS, Teodorescu M, Stohl W. B cell depletion therapy in systemic lupus erythematosus: relationships among serum B lymphocyte stimulator levels, autoantibody profile and clinical response. Ann Rheum Dis. 2008;14:1011–1016. doi: 10.1136/ard.2007.079418. [DOI] [PubMed] [Google Scholar]
- Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KG, Dorner T, Hiepe F. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat Rev Immunol. 2006;14:741–750. doi: 10.1038/nri1886. [DOI] [PubMed] [Google Scholar]
- Miller JJ, Cole LJ. Resistance of long-lived lymphocytes and plasma cells in rat lymph nodes to treatment with prednisone, cyclophosphamide, 6-mercaptopurine, and actinomycin D. J Exp Med. 1967;14:109–125. doi: 10.1084/jem.126.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roldan E, Brieva JA. Terminal differentiation of human bone marrow cells capable of spontaneous and high-rate immunoglobulin secretion: role of bone marrow stromal cells and interleukin 6. Eur J Immunol. 1991;14:2671–2677. doi: 10.1002/eji.1830211105. [DOI] [PubMed] [Google Scholar]
- Pelletier N, Casamayor-Palleja M, De Luca K, Mondiere P, Saltel F, Jurdic P, Bella C, Genestier L, Defrance T. The endoplasmic reticulum is a key component of the plasma cell death pathway. J Immunol. 2006;14:1340–1347. doi: 10.4049/jimmunol.176.3.1340. [DOI] [PubMed] [Google Scholar]
- Hofer T, Muehlinghaus G, Moser K, Yoshida T, Mei HE, Hebel K, Hauser A, Hoyer B, E OL, Dorner T, Manz RA, Hiepe F, Radbruch A. Adaptation of humoral memory. Immunol Rev. 2006;14:295–302. doi: 10.1111/j.0105-2896.2006.00380.x. [DOI] [PubMed] [Google Scholar]
- Mohr E, Serre K, Manz RA, Cunningham AF, Khan M, Hardie DL, Bird R, MacLennan IC. Dendritic cells and monocyte/macrophages that create the IL-6/APRIL-rich lymph node microenvironments where plasmablasts mature. J Immunol. 2009;14:2113–2123. doi: 10.4049/jimmunol.0802771. [DOI] [PubMed] [Google Scholar]
- Minges Wols HA, Underhill GH, Kansas GS, Witte PL. The role of bone marrow-derived stromal cells in the maintenance of plasma cell longevity. J Immunol. 2002;14:4213–4221. doi: 10.4049/jimmunol.169.8.4213. [DOI] [PubMed] [Google Scholar]
- Tokoyoda K, Egawa T, Sugiyama T, Choi BI, Nagasawa T. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity. 2004;14:707–718. doi: 10.1016/j.immuni.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Chu VT, Frohlich A, Steinhauser G, Scheel T, Roch T, Fillatreau S, Lee JJ, Lohning M, Berek C. Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat Immunol. 2011;14:151–159. doi: 10.1038/ni.1981. [DOI] [PubMed] [Google Scholar]
- Matthes T, Dunand-Sauthier I, Santiago-Raber ML, Krause KH, Donze O, Passweg J, McKee T, Huard B. Production of the plasma-cell survival factor a proliferation-inducing ligand (APRIL) peaks in myeloid precursor cells from human bone marrow. Blood. 2011;14:1838–1844. doi: 10.1182/blood-2011-01-332940. [DOI] [PubMed] [Google Scholar]
- Winter O, Moser K, Mohr E, Zotos D, Kaminski H, Szyska M, Roth K, Wong DM, Dame C, Tarlinton DM, Schulze H, MacLennan IC, Manz RA. Megakaryocytes constitute a functional component of a plasma cell niche in the bone marrow. Blood. 2010;14:1867–1875. doi: 10.1182/blood-2009-12-259457. [DOI] [PubMed] [Google Scholar]
- Rozanski CH, Arens R, Carlson LM, Nair J, Boise LH, Chanan-Khan AA, Schoenberger SP, Lee KP. Sustained antibody responses depend on CD28 function in bone marrow-resident plasma cells. J Exp Med. 2011;14:1435–1446. doi: 10.1084/jem.20110040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez Gomez M, Talke Y, Goebel N, Hermann F, Reich B, Mack M. Basophils support the survival of plasma cells in mice. J Immunol. 2010;14:7180–7185. doi: 10.4049/jimmunol.1002319. [DOI] [PubMed] [Google Scholar]
- Belnoue E, Pihlgren M, McGaha TL, Tougne C, Rochat AF, Bossen C, Schneider P, Huard B, Lambert PH, Siegrist CA. APRIL is critical for plasmablast survival in the bone marrow and poorly expressed by early-life bone marrow stromal cells. Blood. 2008;14:2755–2764. doi: 10.1182/blood-2007-09-110858. [DOI] [PubMed] [Google Scholar]
- Moser K, Tokoyoda K, Radbruch A, MacLennan I, Manz RA. Stromal niches, plasma cell differentiation and survival. Curr Opin Immunol. 2006;14:265–270. doi: 10.1016/j.coi.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Cutler AJ, Brownlie RJ, Fairfax K, Lawlor KE, Severinson E, Walker EU, Manz RA, Tarlinton DM, Smith KG. FcγRIIb controls bone marrow plasma cell persistence and apoptosis. Nat Immunol. 2007;14:419–429. doi: 10.1038/ni1440. [DOI] [PubMed] [Google Scholar]
- Hutloff A, Buchner K, Reiter K, Baelde HJ, Odendahl M, Jacobi A, Dorner T, Kroczek RA. Involvement of inducible costimulator in the exaggerated memory B cell and plasma cell generation in systemic lupus erythematosus. Arthritis Rheum. 2004;14:3211–3220. doi: 10.1002/art.20519. [DOI] [PubMed] [Google Scholar]
- Cassese G, Lindenau S, de Boer B, Arce S, Hauser A, Riemekasten G, Berek C, Hiepe F, Krenn V, Radbruch A, Manz RA. Inflamed kidneys of NZB/W mice are a major site for the homeostasis of plasma cells. Eur J Immunol. 2001;14:2726–2732. doi: 10.1002/1521-4141(200109)31:9<2726::AID-IMMU2726>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Jacobi AM, Mei H, Hoyer BF, Mumtaz IM, Thiele K, Radbruch AH, Burmester GR, Hiepe F, Dorner T. HLA-DRhigh/CD27high plasmablasts indicate active disease in patients with SLE. Ann Rheum Dis. 2010;14:305–308. doi: 10.1136/ard.2008.096495. [DOI] [PubMed] [Google Scholar]
- Bijl M, Horst G, Bootsma H, Limburg PC, Kallenberg CG. Mycophenolate mofetil prevents a clinical relapse in patients with systemic lupus erythematosus at risk. Ann Rheum Dis. 2003;14:534–539. doi: 10.1136/ard.62.6.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubert K, Meister S, Moser K, Weisel F, Maseda D, Amann K, Wiethe C, Winkler TH, Kalden JR, Manz RA, Voll RE. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat Med. 2008;14:748–755. doi: 10.1038/nm1763. [DOI] [PubMed] [Google Scholar]
- Frohlich K, Holle JU, Aries PM, Gross WL, Moosig F. Successful use of bortezomib in a patient with systemic lupus erythematosus and multiple myeloma. Ann Rheum Dis. 2011;14:1344–1345. doi: 10.1136/ard.2010.133256. [DOI] [PubMed] [Google Scholar]
- Haas KM, Tedder TF. Role of the CD19 and CD21/35 receptor complex in innate immunity, host defense and autoimmunity. Adv Exp Med Biol. 2005;14:125–139. doi: 10.1007/0-387-24180-9_16. [DOI] [PubMed] [Google Scholar]
- Kozmik Z, Wang S, Dorfler P, Adams B, Busslinger M. The promoter of the CD19 gene is a target for the B-cell-specific transcription factor BSAP. Mol Cell Biol. 1992;14:2662–2672. doi: 10.1128/mcb.12.6.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabanillas F, Burke JS, Smith TL, Moon TE, Butler JJ, Rodriguez V. Factors predicting for response and survival in adults with advanced non-Hodgkin's lymphoma. Arch Intern Med. 1978;14:413–418. doi: 10.1001/archinte.1978.03630270051019. [DOI] [PubMed] [Google Scholar]
- Nutt SL, Morrison AM, Dorfler P, Rolink A, Busslinger M. Identification of BSAP (Pax-5) target genes in early B-cell development by loss- and gain-of-function experiments. Embo J. 1998;14:2319–2333. doi: 10.1093/emboj/17.8.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt SL, Heavey B, Rolink AG, Busslinger M. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature. 1999;14:556–562. doi: 10.1038/44076. [DOI] [PubMed] [Google Scholar]
- Cobaleda C, Schebesta A, Delogu A, Busslinger M. Pax5: the guardian of B cell identity and function. Nat Immunol. 2007;14:463–470. doi: 10.1038/ni1454. [DOI] [PubMed] [Google Scholar]
- Scheuermann RH, Racila E. CD19 antigen in leukemia and lymphoma diagnosis and immunotherapy. Leuk Lymphoma. 1995;14:385–397. doi: 10.3109/10428199509059636. [DOI] [PubMed] [Google Scholar]
- Nadler LM, Anderson KC, Marti G, Bates M, Park E, Daley JF, Schlossman SF. B4, a human B lymphocyte-associated antigen expressed on normal, mitogen-activated, and malignant B lymphocytes. J Immunol. 1983;14:244–250. [PubMed] [Google Scholar]
- Anderson KC, Griffin JD, Bates MP, Slaughenhoupt BL, Schlossman SF, Nadler LM. Isolation and characterization of human B lymphocyte enriched populations. I. Purification of B cells by immune rosette depletion. J Immunol Methods. 1983;14:283–292. doi: 10.1016/0022-1759(83)90222-3. [DOI] [PubMed] [Google Scholar]
- Loken MR, Shah VO, Dattilio KL, Civin CI. Flow cytometric analysis of human bone marrow. II. Normal B lymphocyte development. Blood. 1987;14:1316–1324. [PubMed] [Google Scholar]
- Maecker HT, Levy S. Normal lymphocyte development but delayed humoral immune response in CD81-null mice. J Exp Med. 1997;14:1505–1510. doi: 10.1084/jem.185.8.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zelm MC, Smet J, Adams B, Mascart F, Schandene L, Janssen F, Ferster A, Kuo CC, Levy S, van Dongen JJ, van der Burg M. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Invest. 2010;14:1265–1274. doi: 10.1172/JCI39748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbs AK, Bosompem A, Coustan-Smith E, Tyerman G, Saulsbury FT, Conley ME. Agammaglobulinemia associated with BCR B cells and enhanced expression of CD19. Blood. 2011;14:1828–1837. doi: 10.1182/blood-2011-01-330472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artac H, Reisli I, Kara R, Pico-Knijnenburg I, Adin-Cinar S, Pekcan S, Jol-van der Zijde CM, van Tol MJ, Bakker-Jonges LE, van Dongen JJ, van der Burg M, van Zelm MC. B-cell maturation and antibody responses in individuals carrying a mutated CD19 allele. Genes Immun. 2010;14:523–530. doi: 10.1038/gene.2010.22. [DOI] [PubMed] [Google Scholar]
- van Zelm MC, Reisli I, van der Burg M, Castano D, van Noesel CJ, van Tol MJ, Woellner C, Grimbacher B, Patino PJ, van Dongen JJ, Franco JL. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med. 2006;14:1901–1912. doi: 10.1056/NEJMoa051568. [DOI] [PubMed] [Google Scholar]
- Sato S, Hasegawa M, Fujimoto M, Tedder TF, Takehara K. Quantitative genetic variation in CD19 expression correlates with autoimmunity. J Immunol. 2000;14:6635–6643. doi: 10.4049/jimmunol.165.11.6635. [DOI] [PubMed] [Google Scholar]
- Fearon DT, Carroll MC. Regulation of B lymphocyte responses to foreign and self-antigens by the CD19/CD21 complex. Annu Rev Immunol. 2000;14:393–422. doi: 10.1146/annurev.immunol.18.1.393. [DOI] [PubMed] [Google Scholar]
- Engel P, Zhou LJ, Ord DC, Sato S, Koller B, Tedder TF. Abnormal B lymphocyte development, activation, and differentiation in mice that lack or overexpress the CD19 signal transduction molecule. Immunity. 1995;14:39–50. doi: 10.1016/1074-7613(95)90157-4. [DOI] [PubMed] [Google Scholar]
- Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. 1995;14:352–355. doi: 10.1038/376352a0. [DOI] [PubMed] [Google Scholar]
- Inaoki M, Sato S, Weintraub BC, Goodnow CC, Tedder TF. CD19-regulated signaling thresholds control peripheral tolerance and autoantibody production in B lymphocytes. J Exp Med. 1997;14:1923–1931. doi: 10.1084/jem.186.11.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud MS, Fujii R, Ishikawa H, Kawano MM. Enforced CD19 expression leads to growth inhibition and reduced tumorigenicity. Blood. 1999;14:3551–3558. [PubMed] [Google Scholar]
- Culton DA, Nicholas MW, Bunch DO, Zhen QL, Kepler TB, Dooley MA, Mohan C, Nachman PH, Clarke SH. Similar CD19 dysregulation in two autoantibody-associated autoimmune diseases suggests a shared mechanism of B-cell tolerance loss. J Clin Immunol. 2007;14:53–68. doi: 10.1007/s10875-006-9051-1. [DOI] [PubMed] [Google Scholar]
- Korganow AS, Knapp AM, Nehme-Schuster H, Soulas-Sprauel P, Poindron V, Pasquali JL, Martin T. Peripheral B cell abnormalities in patients with systemic lupus erythematosus in quiescent phase: decreased memory B cells and membrane CD19 expression. J Autoimmun. 2010;14:426–434. doi: 10.1016/j.jaut.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Sato S, Fujimoto M, Hasegawa M, Takehara K. Altered blood B lymphocyte homeostasis in systemic sclerosis: expanded naive B cells and diminished but activated memory B cells. Arthritis Rheum. 2004;14:1918–1927. doi: 10.1002/art.20274. [DOI] [PubMed] [Google Scholar]
- Odendahl M, Keitzer R, Wahn U, Hiepe F, Radbruch A, Dorner T, Bunikowski R. Perturbations of peripheral B lymphocyte homoeostasis in children with systemic lupus erythematosus. Ann Rheum Dis. 2003;14:851–858. doi: 10.1136/ard.62.9.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odendahl M, Mei H, Hoyer BF, Jacobi AM, Hansen A, Muehlinghaus G, Berek C, Hiepe F, Manz R, Radbruch A, Dorner T. Generation of migratory antigen-specific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood. 2005;14:1614–1621. doi: 10.1182/blood-2004-07-2507. [DOI] [PubMed] [Google Scholar]
- Odendahl M, Jacobi A, Hansen A, Feist E, Hiepe F, Burmester GR, Lip sky PE, Radbruch A, Dorner T. Disturbed peripheral B lymphocyte homeostasis in systemic lupus erythematosus. J Immunol. 2000;14:5970–5979. doi: 10.4049/jimmunol.165.10.5970. [DOI] [PubMed] [Google Scholar]
- Ginaldi L, De Martinis M, Matutes E, Farahat N, Morilla R, Catovsky D. Levels of expression of CD19 and CD20 in chronic B cell leukaemias. J Clin Pathol. 1998;14:364–369. doi: 10.1136/jcp.51.5.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst R, Wang Y, Gallagher S, Mittereder N, Kuta E, Damschroder M, Woods R, Rowe DC, Cheng L, Cook K, Evans K, Sims GP, Pfarr DS, Bowen MA, Dall'Acqua W, Shlomchik M, Tedder TF, Kiener P, Jallal B, Wu H, Coyle AJ. B-cell depletion in vitro and in vivo with an afucosylated anti-CD19 antibody. J Pharmacol Exp Ther. 2010;14:213–222. doi: 10.1124/jpet.110.168062. [DOI] [PubMed] [Google Scholar]
- Horton HM, Bernett MJ, Pong E, Peipp M, Karki S, Chu SY, Richards J O, Vostiar I, Joyce PF, Repp R, Desjarlais JR, Zhukovsky EA. Potent in vitro and in vivo activity of an Fc-engineered anti-CD19 monoclonal antibody against lymphoma and leukemia. Cancer Res. 2008;14:8049–8057. doi: 10.1158/0008-5472.CAN-08-2268. [DOI] [PubMed] [Google Scholar]
- Nicholas MW, Dooley MA, Hogan SL, Anolik J, Looney J, Sanz I, Clarke SH. A novel subset of memory B cells is enriched in autoreactivity and correlates with adverse outcomes in SLE. Clin Immunol. 2008;14:189–201. doi: 10.1016/j.clim.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehr C, Eibel H, Masilamani M, Illges H, Schlesier M, Peter HH, Warnatz K. A new CD21low B cell population in the peripheral blood of patients with SLE. Clin Immunol. 2004;14:161–171. doi: 10.1016/j.clim.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Huggins J, Pellegrin T, Felgar RE, Wei C, Brown M, Zheng B, Milner EC, Bernstein SH, Sanz I, Zand MS. CpG DNA activation and plasma-cell differentiation of CD27-naive human B cells. Blood. 2007;14:1611–1619. doi: 10.1182/blood-2006-03-008441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir S, Fauci AS. B cells in HIV infection and disease. Nat Rev Immunol. 2009;14:235–245. doi: 10.1038/nri2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnatz K, Wehr C, Drager R, Schmidt S, Eibel H, Schlesier M, Peter HH. Expansion of CD19(hi)CD21(lo/neg) B cells in common variable immunodeficiency (CVID) patients with autoimmune cytopenia. Immunobiology. 2002;14:502–513. doi: 10.1078/0171-2985-00198. [DOI] [PubMed] [Google Scholar]
- Trepel F. Number and distribution of lymphocytes in man. A critical analysis. Klin Wochenschr. 1974;14:511–515. doi: 10.1007/BF01468720. [DOI] [PubMed] [Google Scholar]
- Westermann J, Pabst R. Lymphocyte subsets in the blood: a diagnostic window on the lymphoid system? Immunol Today. 1990;14:406–410. doi: 10.1016/0167-5699(90)90160-b. [DOI] [PubMed] [Google Scholar]
- Ellyard JI, Avery DT, Phan TG, Hare NJ, Hodgkin PD, Tangye SG. Antigen-selected, immunoglobulin-secreting cells persist in human spleen and bone marrow. Blood. 2004;14:3805–3812. doi: 10.1182/blood-2003-09-3109. [DOI] [PubMed] [Google Scholar]
- Medina F, Segundo C, Campos-Caro A, Gonzalez-Garcia I, Brieva JA. The heterogeneity shown by human plasma cells from tonsil, blood, and bone marrow reveals graded stages of increasing maturity, but local profiles of adhesion molecule expression. Blood. 2002;14:2154–2161. doi: 10.1182/blood.V99.6.2154. [DOI] [PubMed] [Google Scholar]
- Withers DR, Fiorini C, Fischer RT, Ettinger R, Lipsky PE, Grammer A C. T cell-dependent survival of CD20+ and CD20- plasma cells in human secondary lymphoid tissue. Blood. 2007;14:4856–4864. doi: 10.1182/blood-2006-08-043414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arce S, Luger E, Muehlinghaus G, Cassese G, Hauser A, Horst A, Lehnert K, Odendahl M, Honemann D, Heller KD, Kleinschmidt H, Berek C, Dorner T, Krenn V, Hiepe F, Bargou R, Radbruch A, Manz RA. CD38low IgG-secreting cells are precursors of various CD38high-expressing plasma cell populations. J Leukoc Biol. 2004;14:1022–1028. doi: 10.1189/jlb.0603279. [DOI] [PubMed] [Google Scholar]
- Harada H, Kawano MM, Huang N, Harada Y, Iwato K, Tanabe O, Tanaka H, Sakai A, Asaoku H, Kuramoto A. Phenotypic difference of normal plasma cells from mature myeloma cells. Blood. 1993;14:2658–2663. [PubMed] [Google Scholar]
- Kallies A, Hasbold J, Tarlinton DM, Dietrich W, Corcoran LM, Hodgkin PD, Nutt SL. Plasma cell ontogeny defined by quantitative changes in blimp-1 expression. J Exp Med. 2004;14:967–977. doi: 10.1084/jem.20040973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delogu A, Schebesta A, Sun Q, Aschenbrenner K, Perlot T, Busslinger M. Gene repression by Pax5 in B cells is essential for blood cell homeostasis and is reversed in plasma cells. Immunity. 2006;14:269–281. doi: 10.1016/j.immuni.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Nera KP, Kohonen P, Narvi E, Peippo A, Mustonen L, Terho P, Koskela K, Buerstedde JM, Lassila O. Loss of Pax5 promotes plasma cell differentiation. Immunity. 2006;14:283–293. doi: 10.1016/j.immuni.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Medina F, Segundo C, Campos-Caro A, Salcedo I, Garcia-Poley A, Brieva JA. Isolation, maturational level, and functional capacity of human colon lamina propria plasma cells. Gut. 2003;14:383–389. doi: 10.1136/gut.52.3.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Niro R, Mesin L, Raki M, Zheng NY, Lund-Johansen F, Lundin KE, Charpilienne A, Poncet D, Wilson PC, Sollid LM. Rapid generation of rotavirus-specific human monoclonal antibodies from small-intestinal mucosa. J Immunol. 2010;14:5377–5383. doi: 10.4049/jimmunol.1001587. [DOI] [PubMed] [Google Scholar]
- Terstappen LW, Johnsen S, Segers-Nolten IM, Loken MR. Identification and characterization of plasma cells in normal human bone marrow by high-resolution flow cytometry. Blood. 1990;14:1739–1747. [PubMed] [Google Scholar]
- Jacobi AM, Odendahl M, Reiter K, Bruns A, Burmester GR, Radbruch A , Valet G, Lipsky PE, Dorner T. Correlation between circulating CD27high plasma cells and disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2003;14:1332–1342. doi: 10.1002/art.10949. [DOI] [PubMed] [Google Scholar]
- Arce E, Jackson DG, Gill MA, Bennett LB, Banchereau J, Pascual V. Increased frequency of pre-germinal center B cells and plasma cell precursors in the blood of children with systemic lupus erythematosus. J Immunol. 2001;14:2361–2369. doi: 10.4049/jimmunol.167.4.2361. [DOI] [PubMed] [Google Scholar]
- Hoyer BF, Mumtaz IM, Loddenkemper K, Bruns A, Sengler C, Hermann K G, Maza S, Keitzer R, Burmester GR, Buttgereit F, Radbruch A, Hiepe F. Takayasu-arteritis is characterised by disturbances of B cell homeostasis and responds to B cell depletion therapy with rituximab. Ann Rheum Dis. 2012;14:75–79. doi: 10.1136/ard.2011.153007. [DOI] [PubMed] [Google Scholar]
- Hansen A, Odendahl M, Reiter K, Jacobi AM, Feist E, Scholze J, Burmester GR, Lipsky PE, Dorner T. Diminished peripheral blood memory B cells and accumulation of memory B cells in the salivary glands of patients with Sjogren's syndrome. Arthritis Rheum. 2002;14:2160–2171. doi: 10.1002/art.10445. [DOI] [PubMed] [Google Scholar]
- Martinez-Gamboa L, Mei H, Loddenkemper C, Ballmer B, Hansen A, Lipsky PE, Emmerich F, Radbruch A, Salama A, Dorner T. Role of the spleen in peripheral memory B-cell homeostasis in patients with autoimmune thrombocytopenia purpura. Clin Immunol. 2009;14:199–212. doi: 10.1016/j.clim.2008.09.009. [DOI] [PubMed] [Google Scholar]
- Dass S, Rawstron AC, Vital EM, Henshaw K, McGonagle D, Emery P. Highly sensitive B cell analysis predicts response to rituximab therapy in rheumatoid arthritis. Arthritis Rheum. 2008;14:2993–2999. doi: 10.1002/art.23902. [DOI] [PubMed] [Google Scholar]
- Vital EM, Dass S, Buch MH, Henshaw K, Pease CT, Martin MF, Ponchel F, Rawstron AC, Emery P. B cell biomarkers of rituximab responses in systemic lupus erythematosus. Arthritis Rheum. 2011;14:3038–3047. doi: 10.1002/art.30466. [DOI] [PubMed] [Google Scholar]
- Kneitz C, Wilhelm M, Tony HP. Effective B cell depletion with rituximab in the treatment of autoimmune diseases. Immunobiology. 2002;14:519–527. doi: 10.1078/0171-2985-00200. [DOI] [PubMed] [Google Scholar]
- Pers JO, Devauchelle V, Daridon C, Bendaoud B, Le Berre R, Bordron A, Hutin P, Renaudineau Y, Dueymes M, Loisel S, Berthou C, Saraux A, Youinou P. BAFF-modulated repopulation of B lymphocytes in the blood and salivary glands of rituximab-treated patients with Sjogren's syndrome. Arthritis Rheum. 2007;14:1464–1477. doi: 10.1002/art.22603. [DOI] [PubMed] [Google Scholar]
- Hekman A, Honselaar A, Vuist WM, Sein JJ, Rodenhuis S, ten Bokkel Huinink WW, Somers R, Rumke P, Melief CJ. Initial experience with treatment of human B cell lymphoma with anti-CD19 monoclonal antibody. Cancer Immunol Immunother CII. 1991;14:364–372. doi: 10.1007/BF01741331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlasveld LT, Hekman A, Vyth-Dreese FA, Melief CJ, Sein JJ, Voordouw AC, Dellemijn TA, Rankin EM. Treatment of low-grade non-Hodgkin's lymphoma with continuous infusion of low-dose recombinant interleukin-2 in combination with the B-cell-specific monoclonal antibody CLB-CD19. Cancer Immunol Immunother CII. 1995;14:37–47. doi: 10.1007/BF01517234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber HP, Kung-Sutherland M, Stone I, Morris-Tilden C, Miyamoto J , McCormick R, Alley SC, Okeley N, Hayes B, Hernandez-Ilizaliturri FJ, McDonagh CF, Carter PJ, Benjamin D, Grewal IS. Potent antitumor activity of the anti-CD19 auristatin antibody drug conjugate hBU12-vcMMAE against rituximab-sensitive and -resistant lymphomas. Blood. 2009;14:4352–4361. doi: 10.1182/blood-2008-09-179143. [DOI] [PubMed] [Google Scholar]
- Flavell DJ, Flavell SU, Boehm DA, Emery L, Noss A, Ling NR, Richardson PR, Hardie D, Wright DH. Preclinical studies with the anti-CD19-saporin immunotoxin BU12-SAPORIN for the treatment of human-B-cell tumours. Br J Cancer. 1995;14:1373–1379. doi: 10.1038/bjc.1995.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossbard ML, Multani PS, Freedman AS, O'Day S, Gribben JG, Rhuda C, Neuberg D, Nadler LM. A phase II study of adjuvant therapy with anti-B4-blocked ricin after autologous bone marrow transplantation for patients with relapsed B-cell non-Hodgkin's lymphoma. Clin Cancer Res. 1999;14:2392–2398. [PubMed] [Google Scholar]
- Yazawa N, Hamaguchi Y, Poe JC, Tedder TF. Immunotherapy using unconjugated CD19 monoclonal antibodies in animal models for B lymphocyte malignancies and autoimmune disease. Proc Natl Acad Sci USA. 2005;14:15178–15183. doi: 10.1073/pnas.0505539102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Ono N, Steeber DA, Pisetsky DS, Tedder TF. CD19 regulates B lymphocyte signaling thresholds critical for the development of B-1 lineage cells and autoimmunity. J Immunol. 1996;14:4371–4378. [PubMed] [Google Scholar]
- Tedder TF, Inaoki M, Sato S. The CD19-CD21 complex regulates signal transduction thresholds governing humoral immunity and autoimmunity. Immunity. 1997;14:107–118. doi: 10.1016/S1074-7613(00)80418-5. [DOI] [PubMed] [Google Scholar]
- Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, Noppeney R, Viardot A, Hess G, Schuler M, Einsele H, Brandl C, Wolf A, Kirchinger P, Klappers P, Schmidt M, Riethmuller G, Reinhardt C, Baeuerle PA, Kufer P. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;14:974–977. doi: 10.1126/science.1158545. [DOI] [PubMed] [Google Scholar]
- Zalevsky J, Leung IW, Karki S, Chu SY, Zhukovsky EA, Desjarlais JR , Carmichael DF, Lawrence CE. The impact of Fc engineering on an anti-CD19 antibody: increased Fcγ receptor affinity enhances B-cell clearing in nonhuman primates. Blood. 2009;14:3735–3743. doi: 10.1182/blood-2008-10-182048. [DOI] [PubMed] [Google Scholar]
- Cardarelli PM, Rao-Naik C, Chen S, Huang H, Pham A, Moldovan-Loomis MC, Pan C, Preston B, Passmore D, Liu J, Kuhne MR, Witte A, Blanset D, King DJ. A nonfucosylated human antibody to CD19 with potent B-cell depletive activity for therapy of B-cell malignancies. Cancer Immunol Immunother CII. 2010;14:257–265. doi: 10.1007/s00262-009-0746-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XY, Pop LM, Tsai L, Pop IV, Vitetta ES. Chimeric, divalent and tetravalent anti-CD19 monoclonal antibodies with potent in vitro and in vivo antitumor activity against human B-cell lymphoma and pre-B acute lymphoblastic leukemia cell lines. Int J Cancer. 2011;14:497–506. doi: 10.1002/ijc.25695. [DOI] [PubMed] [Google Scholar]
- Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M , Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, Morgan RA, Rosenberg SA. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;14:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, Feldman SA, Zhao Y, Xu H, Black MA, Morgan RA, Wilson WH, Rosenberg SA. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J Immunother. 2009;14:689–702. doi: 10.1097/CJI.0b013e3181ac6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, Yang JC, Kammula US, Devillier L, Carpenter R, Nathan DA, Morgan RA, Laurencot C, Rosenberg SA. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;14:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;14:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, Kamble RT , Bollard CM, Gee AP, Mei Z, Liu H, Grilley B, Rooney CM, Heslop HE, Brenner MK, Dotti G. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;14:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochtrop ML, Goldacker S, May AM, Rizzi M, Draeger R, Hauschke D, Stehfest C, Warnatz K, Goebel H, Technau-Ihling K, Werner M, Salzer U, Eibel H, Schlesier M, Peter HH. T and B lymphocyte abnormalities in bone marrow biopsies of common variable immunodeficiency. Blood. 2011;14:309–318. doi: 10.1182/blood-2010-11-321695. [DOI] [PubMed] [Google Scholar]
- Goldacker S, Draeger R, Warnatz K, Huzly D, Salzer U, Thiel J, Eibel H, Schlesier M, Peter HH. Active vaccination in patients with common variable immunodeficiency (CVID) Clin Immunol. 2007;14:294–303. doi: 10.1016/j.clim.2007.04.011. [DOI] [PubMed] [Google Scholar]
- van der Kolk LE, Baars JW, Prins MH, van Oers MH. Rituximab treatment results in impaired secondary humoral immune responsiveness. Blood. 2002;14:2257–2259. [PubMed] [Google Scholar]
- van Assen S, Holvast A, Benne CA, Posthumus MD, van Leeuwen MA, Voskuyl AE, Blom M, Risselada AP, de Haan A, Westra J, Kallenberg CG, Bijl M. Humoral responses after influenza vaccination are severely reduced in patients with rheumatoid arthritis treated with rituximab. Arthritis Rheum. 2010;14:75–81. doi: 10.1002/art.25033. [DOI] [PubMed] [Google Scholar]
- Gelinck LB, Teng YK, Rimmelzwaan GF, van den Bemt BJ, Kroon FP, van Laar JM. Poor serological responses upon influenza vaccination in patients with rheumatoid arthritis treated with rituximab. Ann Rheum Dis. 2007;14:1402–1403. doi: 10.1136/ard.2007.071878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingham CO, Looney RJ, Deodhar A, Halsey N, Greenwald M, Codding C, Trzaskoma B, Martin F, Agarwal S, Kelman A. Immunization responses in rheumatoid arthritis patients treated with rituximab: results from a controlled clinical trial. Arthritis Rheum. 2010;14:64–74. doi: 10.1002/art.25034. [DOI] [PubMed] [Google Scholar]
- Rehnberg M, Brisslert M, Amu S, Zendjanchi K, Hawi G, Bokarewa MI. Vaccination response to protein and carbohydrate antigens in patients with rheumatoid arthritis after rituximab treatment. Arthritis Res Ther. 2010;14:R111. doi: 10.1186/ar3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;14:2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, Keystone EC, Loveless JE, Burmester GR, Cravets MW, Hessey EW, Shaw T, Totoritis MC. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: results of a multicenter, randomized, doubleblind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;14:2793–2806. doi: 10.1002/art.22025. [DOI] [PubMed] [Google Scholar]
- Carson KR, Evens AM, Richey EA, Habermann TM, Focosi D, Seymour JF, Laubach J, Bawn SD, Gordon LI, Winter JN, Furman RR, Vose JM, Zelenetz AD, Mamtani R, Raisch DW, Dorshimer GW, Rosen ST, Muro K, Gottardi-Littell NR, Talley RL, Sartor O, Green D, Major EO, Bennett CL. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood. 2009;14:4834–4840. doi: 10.1182/blood-2008-10-186999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RH, Fearon DT. CD19: lowering the threshold for antigen receptor stimulation of B lymphocytes. Science. 1992;14:105–107. doi: 10.1126/science.1373518. [DOI] [PubMed] [Google Scholar]
- Dass S, Vital EM, Emery P. Development of psoriasis after B cell depletion with rituximab. Arthritis Rheum. 2007;14:2715–2718. doi: 10.1002/art.22811. [DOI] [PubMed] [Google Scholar]
- Mielke F, Schneider-Obermeyer J, Dorner T. Onset of psoriasis with psoriatic arthropathy during rituximab treatment of non-Hodgkin lymphoma. Ann Rheum Dis. 2008;14:1056–1057. doi: 10.1136/ard.2007.080929. [DOI] [PubMed] [Google Scholar]
- Goetz M, Atreya R, Ghalibafian M, Galle PR, Neurath MF. Exacerbation of ulcerative colitis after rituximab salvage therapy. Inflamm Bowel Dis. 2007;14:1365–1368. doi: 10.1002/ibd.20215. [DOI] [PubMed] [Google Scholar]
- Benedetti L, Franciotta D, Vigo T, Grandis M, Fiorina E, Ghiglione E , Roccatagliata L, Mancardi GL, Uccelli A, Schenone A. Relapses after treatment with rituximab in a patient with multiple sclerosis and anti myelin-associated glycoprotein polyneuropathy. Arch Neurol. 2007;14:1531–1533. doi: 10.1001/archneur.64.10.1531. [DOI] [PubMed] [Google Scholar]
- Fillatreau S, Gray D, Anderton SM. Not always the bad guys: B cells as regulators of autoimmune pathology. Nat Rev Immunol. 2008;14:391–397. doi: 10.1038/nri2315. [DOI] [PubMed] [Google Scholar]
- Blair PA, Norena LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, Mauri C. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity. 2010;14:129–140. doi: 10.1016/j.immuni.2009.11.009. [DOI] [PubMed] [Google Scholar]
- Iwata Y, Matsushita T, Horikawa M, Dilillo DJ, Yanaba K, Venturi GM, Szabolcs PM, Bernstein SH, Magro CM, Williams AD, Hall RP, St Clair EW, Tedder TF. Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood. 2011;14:530–541. doi: 10.1182/blood-2010-07-294249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzi AR, Clarke AM, Wooldridge T, Waldmann H, Hale G, Symmons D, Hazleman BL, Isaacs JD. Morbidity and mortality in rheumatoid arthritis patients with prolonged therapy-induced lymphopenia: twelve-year outcomes. Arthritis Rheum. 2008;14:370–375. doi: 10.1002/art.23122. [DOI] [PubMed] [Google Scholar]
- Isaacs JD, Manna VK, Rapson N, Bulpitt KJ, Hazleman BL, Matteson EL, St Clair EW, Schnitzer TJ, Johnston JM. CAMPATH-1H in rheumatoid arthritis--an intravenous dose-ranging study. Br J Rheumatol. 1996;14:231–240. doi: 10.1093/rheumatology/35.3.231. [DOI] [PubMed] [Google Scholar]
- Isaacs JD, Greer S, Sharma S, Symmons D, Smith M, Johnston J, Waldmann H, Hale G, Hazleman BL. Morbidity and mortality in rheumatoid arthritis patients with prolonged and profound therapy-induced lymphopenia. Arthritis Rheum. 2001;14:1998–2008. doi: 10.1002/1529-0131(200109)44:9<1998::AID-ART348>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]