

Abstract
Cryptococcus neoformans PKH2-01 and PKH2-02 are orthologous to mammalian PDK1 kinase genes. Although orthologs of these kinases have been extensively studied in S. cerevisiae, little is known about their function in pathogenic fungi. In this study, we show that PKH2-02 but not PKH2-01 is required for C. neoformans to tolerate cell wall, oxidative, nitrosative, and antifungal drug stress. Deletion of PKH2-02 leads to decreased basal levels of Pkc1 activity and, consequently, reduced activation of the cell wall integrity mitogen-activated protein kinase (MAPK) pathway in response to cell wall, oxidative, and nitrosative stress. PKH2-02 function also is required for tolerance of fluconazole and amphotericin B, two important drugs for the treatment of cryptococcosis. Furthermore, OSU-03012, an inhibitor of human PDK1, is synergistic and fungicidal in combination with fluconazole. Using a Galleria mellonella model of low-temperature cryptococcosis, we found that PKH2-02 is also required for virulence in a temperature-independent manner. Consistent with the hypersensitivity of the pkh2-02Δ mutant to oxidative and nitrosative stress, this mutant shows decreased survival in murine phagocytes compared to that of wild-type (WT) cells. In addition, we show that deletion of PKH2-02 affects the interaction between C. neoformans and phagocytes by decreasing its ability to suppress production of tumor necrosis factor alpha (TNF-α) and reactive oxygen species. Taken together, our studies demonstrate that Pkh2-02-mediated signaling in C. neoformans is crucial for stress tolerance, host-pathogen interactions, and both temperature-dependent and -independent virulence.
INTRODUCTION
Cryptococcus species are basidiomycetous fungi that are an important contributor to human disease worldwide (1). The vast majority of cryptococcal disease is caused by C. neoformans var. grubii, also known as serotype A, which affects patients with compromised immune function and manifests as meningoencephalitis (CEM). Recent epidemiological estimates indicate that approximately one million new cases of CEM occur each year and that 600,000 people die from the disease annually (2). HIV infection is the most common risk factor for CEM, and accordingly, CEM is one of the most common AIDS-defining opportunistic infections (3). More recently, an ongoing outbreak of C. neoformans var. gattii infections in the Pacific Northwest region of North America has emerged among apparently immunocompetent patients (4). Finally, cryptococcal disease continues to affect people who are undergoing therapy with immunomodulatory medications designed to treat other conditions (5). Thus, Cryptococcus species are an important and dynamic cause of human infectious disease.
Current antifungal therapy for cryptococcosis is based on two regimens: amphotericin preparations combined with 5-flucytosine in resource-rich regions and fluconazole in resource-limited regions (6). Both regimens have drawbacks; amphotericin B-flucytosine is expensive, toxic, and logistically impractical in regions with the highest burden of disease, while fluconazole, although safe and inexpensive, is less effective due to its fungistatic mode of action (6, 7). The most recent additions to the antifungal pharmacopeia, echinocandin 1,3-β-glucan synthase inhibitors, do not have clinically useful activity toward Cryptococcus spp., and therefore, no new classes of anticryptococcal agents have been brought to the clinic in over 20 years (8). Thus, the identification and development of new agents with activity toward Cryptococcus constitute an area of active research.
A deeper understanding of the biology and pathogenesis of Cryptococcus will be required if new approaches to treatment are to be developed. A high-yield area of research in this regard would seem to be the study of signal transduction pathways, since these networks are central to the regulation of a wide variety of biological processes in all eukaryotes. Accordingly, a number of groups have investigated the role of signal transduction pathways in the biology and pathogenesis of Cryptococcus (9–11). Among the pathways that have been studied are the cell wall integrity mitogen-activated kinase pathway (12), the calmodulin-calcineurin pathway (13), the protein kinase A pathway (14), and the hyperosmolar-glycerol (HOG) pathway (15). Many of these pathways are conserved across fungal species in general and, as such, have been extensively studied in the model yeast Saccharomyces cerevisiae. However, it is important to note that important differences between the model yeast and pathogenic yeast with respect to pathway function and/or pathway structure have been identified (16).
Recently, the Krysan laboratory reported that molecules targeting human phosphoinositide-dependent kinase 1 (hPDK1) have excellent in vitro activity toward both Candida spp. and Cryptococcus spp. and appear to target the fungal orthologs of PDK1 (17). In S. cerevisiae (18), three kinases have homology to hPDK1, and these genes have been named PKH1, -2, and -3. PKH1 and PKH2 encode a redundant pair of kinases that carry out essential functions in S. cerevisiae, as evidenced by the fact that deletion of one has no phenotype while deletion of both genes is lethal (19). PDK1 and Pkh kinases are members of the AGC superfamily of protein kinases (18). In addition to being the subject of intensive study in mammalian and human cells due to their role in human cancers (20), PDK1/Pkh kinases have been well characterized in S. cerevisiae. These studies have shown that the Pkhs regulate a wide variety of cellular functions in S. cerevisiae, including cell wall integrity (21), sphingolipid biosynthesis (22, 23), endocytosis (24), eisosome formation (25, 26), flippase activity (22), and RNA metabolism (25). In S. cerevisiae, six key Pkh substrates have been identified, i.e., the AGC kinases protein kinase A (PKA), Pkc1, Ypk1/2, and Sch9 as well as a component of the eisosome, Pil1 (26–29).
Prior to the initiation of this work, very little was known about the function of PDK1 orthologs in pathogenic fungi. PDK1/Pkh orthologs in both Candida albicans and Cryptococcus neoformans had been identified and deleted as part of large-scale genetic screening projects (30, 31). In C. neoformans, two genes with homology to hPDK1 have been identified and annotated as PKHs. As part of their large-scale gene deletion project (31), Liu et al. generated CNAG_02820 (PKH2-01) and CNAG_02915 (PKH2-02) deletion mutants. While the pkh2-01Δ mutant had no phenotypes with respect to high-temperature growth, melanin formation, or capsule generation, the pkh2-02Δ mutant was temperature sensitive and, accordingly, showed decreased virulence in a mouse model of pulmonary cryptococcosis (31).
As our work was in progress, Lee et al. reported a series of experiments investigating the role of PKHs in fluconazole susceptibility and renamed PKH2-02 as PDK1; they also suggested that because the PKH2-01 product does not have a clearly definable catalytic domain, it may not be a true PDK1/Pkh kinase (32). Deletion of C. neoformans PKH2-02 (CnPKH2-02) led to decreased fluconazole tolerance as well as hypersensitivity to high salt, rapamycin, aureobasidin A, myriocin, and phytosphingosine. In addition, Lee et al. found that activation of the cell wall integrity (CWI) mitogen-activated protein kinase (MAPK) cascade by fluconazole was dependent on PKH2-02 and that pkh2-02Δ mutants have altered sphingolipid profiles. They also provided data that suggest that PKH2-02-dependent fluconazole tolerance is mediated by phosphorylation of Ypk1 (32).
Here we report our characterization of the role of PKH2-02 in the ability of C. neoformans to tolerate a variety of stresses, including cell wall, oxidative, and nitrosative stress. Our data indicate that activation of the CWI pathway in response to cell wall, oxidative, and nitrosative stress is modulated by PKH2-02. We have also found that the role of PKH2-02 in virulence is not limited to its function in supporting high-temperature growth. Specifically, C. neoformans is known to modulate phagocyte activation and suppress tumor necrosis factor alpha (TNF-α) production in a process that we have found is dependent on PKH2-02. Furthermore, we show that C. neoformans, like other pathogenic fungi (33–35), suppresses production of reactive oxygen species (ROS) by mouse macrophage-like J774 cells and that this process is also PKH2-02 dependent. Taken together with our previous work (17), these data indicate that CnPKH2-02-mediated signaling is crucial to C. neoformans stress tolerance and host-pathogen interactions and, consequently, further support the notion that PKHs represent a highly attractive antifungal drug target.
MATERIALS AND METHODS
Strains, growth conditions, and materials.
Cryptococcus neoformans var. grubii CM018 and a derived isolate obtained from the deletion collection assembled by Liu et al. (31) were used as reference strains and are noted as wild type (WT) in this paper. The mpk1Δ, lac1Δ, and cap59Δ deletion mutants were also obtained from the deletion collection (31), and the expected phenotypes reported for those strains were confirmed prior to use in assays. Yeast strains were grown in yeast extract-peptone-dextrose (YPD) or synthetic dropout medium prepared according to standard recipes (36). All incubations were performed at 30°C unless noted otherwise. DH5α Escherichia coli cells (Invitrogen, Carlsbad, CA) were used for routine plasmid manipulations. Except for OSU-03012 (Selleck Chemicals, Houston, TX), all chemicals were obtained from Sigma (St. Louis, MO) and used as received. Oligonucleotides were synthesized by IDT (Coralville, IA).
Construction and complementation of PKH2-02 mutants.
The pkh2-02Δ mutant from the deletion collection generated by Liu et al. (31) was utilized, and it genotype was confirmed as described below. PKH2-02 was also deleted in KN99α using standard single-gene deletion methods and biolistic transformation. A linear cassette containing flanking sequences complementary to the 5′ and 3′ untranslated regions of PKH2-02 was created by fusion PCR using the following primers (37): PKH2-02-1, GGAAGAGGAGTGAGACGTCTGAGAGGGG; PKH2-02-2, ATGGGGATATGCATGGCAGTGTGAGAGT; PKH2-02-3, ATGGCCTCCTCGCATTTCGGCcaggaaacagctatgaccatg (lowercase bases are complementary to the nourseothricin marker); PKH2-02-4, catggtcatagctgtttcctgGCCGAAATGCGAGGAGGCCAT; PKH2-02-5, cactggccgtcgttttacaacATGGCGGCAATTCACCGGGTC; PKH2-02-6, GACCCGGTGAATTGCCGCCATgttgtaaaacgacggccagtg; PKH2-02-7, TCTCTTCTACCGGCAAGGTCAAATTCGG; and PKH2-02-8, CGACTTTTACTCCAAGCTTCCCAAGGGC. The pkh2-01Δ and pkh2-02Δ mutants was obtained from the deletion collection (31). The deletion of PKH2-02 in the collection strain was confirmed using two methods. First, the absence of the PKH2-02 open reading frame (ORF) was confirmed by PCR of genomic DNA isolated from the reference strains and deletion mutants. Second, quantitative reverse transcription-PCR (RT-PCR) confirmed that there was no detectable expression of PKH2-02 in the respective deletion mutants. Primers used in this experiment were PKH2-02-qPCRF (TCAATGTCTCTTGGTGGACTGCG) and PKH2-02-qPCRR (TGTCTCTTCTTGGGGGCTGGTAAC). To ensure that phenotypes were due to PKH2-02 deletion, the mutation was complemented as follows. PKH2-02 as well as 1,000 bp upstream and downstream of the ORF was PCR amplified from genomic DNA isolated from the WT strain from the deletion collection using primers PKH2-02F (AGGAGTGAGACGTCTGAGAGG) and PKH2-02R (CTGACTGAAAAACCCACACC). The product was cloned into pJET-2 (Fermentas, Glen Burnie, MD) using the manufacturer's protocol and transformed into the pkh2-02Δ mutant using a standard biolistic protocol. Candidate transformants were selected on YPD plates containing 1.5 M NaCl, passaged 3 times on nonselective medium, and selected again on YPD with 1.5 M NaCl. Integration of the PKH2-02 gene was confirmed by PCR.
Plate-based growth assays.
Strains were grown overnight in YPD at 30°C with shaking and diluted to an optical density at 660 nm (OD660) of 1.0 in phosphate-buffered saline (PBS), and a set of 10-fold dilutions was prepared. The cell suspensions (5 μl) were spotted on YPD plates containing calcofluor white (CFW) (1.5 mg/ml), caffeine (10 mM), Congo red (5 mg/ml), and rapamycin (200 ng/ml). For oxidative and nitrosative stress phenotypes (37), strains (3 μl each dilution) were plated on yeast nitrogen base (YNB) (pH 4.0 with succinate) containing H2O2 (1 mM), diamide (1 mM), or NaNO2 (1.5 mM). All plates were incubated at 30°C for 3 to 7 days.
C. neoformans melanin production.
Strains were grown overnight in YPD and diluted to an OD660 of 1.0. Five 10-fold dilutions in PBS were spotted (5 μl) onto l-3,4-dihydroxyphenylalanine (l-DOPA) plates and incubated at 30°C for 2 to 10 days.
Antifungal susceptibility assays.
Fluconazole and amphotericin B MICs were determined using Etest strips (AB Biodisk, catalog numbers 510858 and 526348, respectively). Cells were grown overnight in YPD at 30°C with shaking and then diluted to an OD650 of 0.1 in sterile NaCl solution (0.85%). Sterile cotton swabs were used to streak cells on RPMI plates (10.4 g/liter RPMI [Sigma, catalog number R6504], 165 mM morpholinepropanesulfonic acid [MOPS], 2% dextrose, and 1.5% Bacto agar). Immediately after swabbing of cells, Etest strips containing the indicated drug were applied to plates. Plates were incubated at 35°C for 3 days.
Total cellular PKC activity.
Total cellular protein kinase C (PKC) activity was measured using the PepTag assay for nonradioactive detection of protein kinase C (Promega, Madison, WI) based on procedures previously reported for C. albicans (38). Briefly, cells were grown overnight in YPD at 30°C with shaking. Cells were then washed three times with Dulbecco's PBS (DPBS) and adjusted to 1 × 107 cells/ml. Whole-cell lysates were prepared by glass bead lysis using the extraction buffer provided in the kit. Equivalent amounts of total cellular protein were added to each reaction mixture. Reaction products were fractionated using agarose gel electrophoresis, visualized using a GEL DOC system, and quantified using the histogram function of Adobe Photoshop.
Western blot analysis.
Western blot analysis was performed as described previously (37). Briefly, cultures were grown overnight in YPD or YNB at 30°C with shaking and then diluted to an OD660 of 0.2 in 50 ml of medium. The cells were grown with shaking for 3 h and then treated with H2O2 (1 mM or 10 mM) or NaNO2 (1 mM) for 30 min or with calcofluor white (10 μg/ml) for 2 h. The cells were diluted 1:1 with ice-cold stop buffer (0.9% NaCl, 1 mM NaN3, 10 mM EDTA, and 50 mM NaF) and harvested at 652 × g at 4°C for 10 min. Cells were then washed once with 10 ml ice-cold stop buffer. The cell pellet was resuspended in 0.3 ml 1× lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 5 mM EDTA pH 8.2, 5 mM EGTA, 0.2 mM Na3VO4, 50 mM KF, 30 mM sodium pyrophosphate, 15 mM p-nitrophenylphosphate, 1× protease inhibitor cocktail [Roche, catalog number 11836170001], and 10 μl/ml each phosphatase inhibitor cocktail II and III [Sigma, catalog numbers P-5726 and P-0044]). Cells were then harvested and lysed. Total protein was determined using the Quick Start Bradford dye reagent (Bio-Rad), and 25 to 50 μg of total protein was loaded into precast 4 to 15% gradient or 10% Mini-Protean TGX (Bio-Rad) gels. Fractionated proteins were transferred to nitrocellulose membranes and blocked overnight at 4°C (5% nonfat milk in 50 mM Tris [pH 7.5], 150 mM NaCl, and 0.05% Tween 20 [TBST]). The membranes were probed using anti-active MAPK (Promega, Madison, WI) or phospho-p44/42 MAPK (thr202/Tyr204) rabbit polyclonal antibody (Cell Signaling Technology) to detect Mpk1 phosphorylation followed by anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibody (Bio-Rad). Anti-PSTAIRE or anti-Tsa1 antibody was used as a loading control. Blots were visualized with the ECL Plus Western blotting detection system (GE Healthcare).
Galleria mellonella model of cryptococcosis.
C. neoformans cells were grown overnight in YPD at 30°C. Cells were then washed three times with Dulbecco's phosphate-buffered saline (DPBS) and resuspended at a concentration of 2 × 107 cells/ml. Each G. mellonella larva was injected in the terminal pseudopod with 5 μl of the inoculum (39). Larvae were incubated at 30°C, and virulence was measured by scoring the survival of the larvae every 24 h. The survival of groups of Galleria infected with different mutants was compared by Kaplan-Meier analysis.
Survival of C. neoformans cells in J774 cells.
J774 cells were seeded at a concentration of 2 × 105 cells/ml in a 24-well plate for 18 h. Overnight yeast cultures were washed three times with DPBS, adjusted to 2 × 106 cells/ml, and opsonized with monoclonal antibody (MAb) 18B7 (kindly provided by Arturo Casadevall) for 1 h. Yeast strains were then cocultured with J774 cells for 80 min. Each well was washed with DPBS to remove unattached yeasts, and fresh medium was added. Plates were incubated for an additional 18 h. J774 cells were lysed with cold sterile water and lysates plated on YPD and incubated at 30°C for 48 h. To control for the slight growth defect of the pkh2-02Δ mutant at 37°C, the C. neoformans strains were incubated in tissue culture medium alone in parallel with the phagocyte experiments (40). The survival data were then normalized to growth in the absence of macrophages to give the survival ratio (macrophages with yeast/yeast in medium).
ROS production by primed J774 cells.
Overnight yeast cultures were washed three times with Hanks balanced salt solution (HBSS) and adjusted to a cell density of 1× 108 cells/ml. Yeast were then opsonized utilizing MAb 18B7 for 1 h and added to J774 cells (multiplicity of infection [MOI], 5:1) in white, opaque-bottom 96-well microtiter plates (Corning Incorporated, Corning, NY). Following a previously published protocol (34), ROS production was measured by luminal-enhanced chemiluminescence using the Superluminol kit (World Precision Instruments, Inc., Sarasota, FL). Briefly, a mixture containing luminal, signal enhancer, and either HBSS or phorbol myristate acetate (PMA) was added to the wells. Luminescence was measured using a 1-s integration time at intervals of 45 s over a 3-h incubation time at 37°C using a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA).
TNF-α secretion by J774 murine phagocytes.
Supernatants were collected from the macrophage survival assay plates (described above) and assayed for TNF-α production with the Ready-Set-Go mouse tumor necrosis factor alpha kit (eBioscience, San Diego, CA) according to the manufacturer's protocol.
Time-kill assays.
C. neoformans cells from overnight cultures were washed twice with DPBS, adjusted to a density of 2 × 107 CFU/ml in YPD (50 ml), and treated with either fluconazole (2 μg/ml), OSU-03012 (2 μg/ml), or a combination of fluconazole and OSU-0312 at the same concentrations. Cultures were incubated at 37°C and 10-fold dilutions plated on YPD at time points 0 h, 3 h, 6 h, 24 h, and 30 h. Plates were incubated at 30°C for 48 h and scored for CFU/ml.
Nitrite production by J774 cells.
J774 cells were seeded at a density of 2 × 105 cells/ml and stimulated with lipopolysaccharide (LPS) and IFN-γ in a 24-well plate for 22 h. Yeast cells were then added to the wells at 2 × 106 cells/ml. After 2 h, supernatants were collected and processed. Nitrite accumulation, an indicator of NO production, was measured using the Griess reagent (Promega catalog number G2930). Briefly, 50-μl aliquots of 24-h culture supernatants were mixed with an equal amount of Griess reagent and incubated at room temperature for 15 min. The absorbance at 540 nm was measured using the SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA).
RESULTS
C. neoformans PDK1 ortholog Pkh2-02 is required for cell wall integrity.
Recently, we reported that chemical inhibitors of human PDK1 have potent activity toward C. neoformans and appear to target the PDK1 ortholog Pkh2-02 (17). The function of PDK1 orthologs (S. cerevisiae PKH1/2 [ScPKH1/2] products) in the model yeast S. cerevisiae has been extensively investigated, but no focused studies of PDK1 orthologs in human fungal pathogens had been reported prior to the initiation of our studies. In addition, the Lodge laboratory identified PKH2-02 in a screen of the C. neoformans deletion collection (31) for mutants that are hypersensitive to oxidative and nitrosative stress (P. S. Michener, K. J. Gerik, and J. K. Lodge, unpublished results). Therefore, we disrupted the C. neoformans var. grubii PDK1 ortholog PKH2-02 in the KN99α background and obtained similar deletion mutants from the collection generated by Liu et al. (31). PCR of genomic DNA confirmed the absence of PKH2-02 from both mutants. In addition, Quantitative RT-PCR confirmed that the PKH2-02 transcript was present in the parental strain but absent from the deletion mutant (Δ/ΔCT for WT versus pkh2-02Δ mutant, 29.7). In the context of their large-sale screen of deletion mutants, Liu et al. (31) found that the pkh2-02Δ mutant showed a growth defect at 37°C, and we confirmed this finding (Fig. 1A), while the pkh2-01Δ mutant showed no growth defect at elevated temperature (data not shown). Lee et al. also observed identical phenotypes for the CM108-derived pkh2-01Δ and pkh2-02Δ mutants (32). In the KN99α background, the pkh2-02Δ mutant was also sensitive to high-temperature growth, but the phenotype did not manifest until 39°C (data not shown). For consistency, we focused the remainder of our studies on strains derived from CM018. Reintegration of PKH2-02 restored the high-temperature tolerance as well as all phenotypes discussed below (Fig. 1A).
Fig 1.

PKH2-02 affects high-temperature growth and is required for cellular integrity. (A) A 10-fold dilution series of the indicated strains was spotted on YPD plates or YPD containing SDS (0.6%) and incubated at 30°C or 37°C for 3 days. (B) Cells from an overnight culture were incubated with trypan blue (0.4%) at a 1:1 dilution for 15 min. One hundred cells were counted per field and scored. There was statistical significance between the WT and the pkh2-02Δ mutant (P = 0.0005) but not between the WT and the pkh2-02::PKH2 strain (chi-square test, P > 0.05). (D) The indicated strains were streaked on plates containing YPD or YPD supplemented with 1 M sorbitol and incubated at 37°C for 3 days.
Temperature-sensitive growth is a nonspecific phenotype that can result from a variety of cellular defects. In S. cerevisiae, Pkh kinases are involved in both plasma membrane- and cell wall-related processes, both of which affect cellular integrity (21–23). Supporting a role for PKH2-02 in cellular integrity is the fact that the deletion mutant is exquisitely sensitive to the detergent SDS (Fig. 1A) and shows increased trypan blue staining (12) in stationary phase (Fig. 1B). Mutations that affect cellular integrity by disrupting cell wall stress responses or cell wall biosynthesis are generally suppressed by the addition of an osmotic support such as sorbitol to the growth medium (41). As shown in Fig. 1C, the temperature-sensitive growth of the pkh2-02Δ mutant was suppressed by the addition of sorbitol to the medium, suggesting that PKH2-02 may be involved in cell wall integrity.
In S. cerevisiae, the Pkhs are known to regulate cell wall integrity by phosphorylation of Pkc1 (21, 28). We therefore examined the effect of cell wall stressors on the growth of C. neoformans pkh2-01Δ and pkh2-02Δ mutants. As shown in Fig. 2A, the pkh2-02Δ mutant is hypersensitive to the chitin/glucan interactor Congo red. In contrast, the pkh2-02Δ mutant showed only a slight growth defect in the presence of the chitin-binding dye calcofluor white. Additionally, many yeast cell wall mutants are hypersensitive to caffeine (41), and consistent with that correlation, the pkh2-02Δ mutant is dramatically more susceptible to caffeine than the wild type (Fig. 2A). Caffeine has recently been proposed to act as an inhibitor of target of rapamycin (TOR) in yeast (42), and we therefore tested the tolerance of the pkh2-02Δ mutant to the specific TOR inhibitor rapamycin. Consistent with our expectations, the pkh2-02Δ mutant is hypersensitive to rapamycin (Fig. 2A) compared to congenic wild-type strains. This is also consistent with the observations of Lee et al. (32) and with the fact that the PDK1/Pkh kinases have been linked to the TOR pathway in both mammalian and S. cerevisiae systems.
Fig 2.

PKH2-02 is involved in cell wall integrity and activation of the cell wall integrity MAPK pathway. (A) A 10-fold dilution series of the indicated strains was spotted on YPD plates containing Congo red (5 mg/ml), calcofluor white (1.5 mg/ml), caffeine (10 mM), and rapamycin (200 ng/ml). (B) The indicated strains were processed for total cellular Pkc1 activity using the Promega PepTag kit as described in Materials and Methods. (C) Cell lysates from log-phase cells treated with either solvent (water) or CFW (10 μg/ml) were analyzed by Western blotting with phospho-specific antibodies for Mpk1 phosphorylation; anti-PSTAIRE is a loading control.
One of the well-characterized ScPkh substrates is ScPkc1 (21, 28), a key kinase in the cell wall integrity (CWI) signaling cascade and other cellular processes. The CWI pathway is conserved across many fungi (41) and is also an important regulator of the cell wall stress response in C. neoformans (12, 37, 44). We therefore hypothesized that Pkc1 activity may be reduced in pkh2-02Δ mutants compared to in the WT. To test this hypothesis, we compared the ability of total cellular lysates from WT, pkh2-02Δ, and pkh2-02Δ::PKH2-02 strains to phosphorylate the commercially available, conserved PKC substrate Pkc-tide in vitro. As shown in Fig. 2B, total cellular Pkc1 activity is reduced in the pkh2-02Δ mutant and is restored by reintroduction of the PKH2-02 allele. To test further the role of the Pkhs in CWI signaling, we also examined the effect of the deletion mutations on phosphorylation of Mpk1, the terminal MAPK of the CWI pathway (12, 37, 41, 44). In the absence of cell wall stress, Mpk1 phosphorylation is markedly reduced in the pkh2-02Δ mutant compared to the WT and the revertant (Fig. 2C), a finding consistent with the data reported by Lee et al. (32). The addition of CFW to the cultures increased Mpk1 phosphorylation in both the WT and the pkh2-02Δ::PKH2-02 revertant mutant (37, 44), but only a barely detectable amount of phosphorylated Mpk1 was observed in the pkh2-02Δ mutant (Fig. 2C). Thus, it appears that PKH2-02 is required for full activation of Mpk1 and, in turn, cell wall integrity.
Pkh2-02 is required for tolerance to membrane-targeted antifungal drugs.
Recent work in S. cerevisiae has highlighted the role of the ScPkh kinases in a wide variety of plasma membrane-related biological processes (22–24, 26, 27, 45). Consistent with these findings, Lee et al. demonstrated that pkh2-02Δ mutants have an altered sphingolipid content compared to the WT and show decreased Mpk1 phosphorylation in response to fluconazole, a membrane-targeted antifungal drug (32). Since the plasma membrane and the cell wall are the two most important targets for antifungal drugs currently in clinical use, we hypothesized that PKH2-02 may affect the ability of C. neoformans to tolerate antifungal drug exposure. The two most important drugs in the treatment of cryptococcosis are amphotericin B and fluconazole (1, 6). We therefore determined the susceptibility of WT and pkh2-02Δ cells to these two drugs using the Etest method. As shown in Fig. 3A, the pkh2-02Δ mutant is 2- to 3-fold more sensitive (MIC = 0.9) to fluconazole than the WT (MIC = 16). This is consistent with the spot dilution assay-based analysis of pkh2-02Δ fluconazole sensitivity recently reported by Lee et al. (32). We also found that pkh2-02Δ cells are more susceptible to amphotericin B (Fig. 3B), the gold standard therapy for CEM. In contrast, we found no difference in the susceptibilities of WT and pkh2-02Δ cells to caspofungin (data not shown), an echinocandin class antifungal to which C. neoformans is quite resistant (8).
Fig 3.
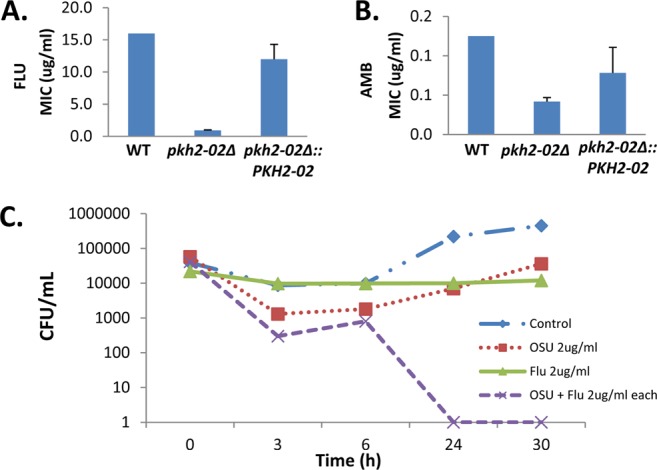
Pkh2-02 is required for tolerance of membrane-targeted antifungal drugs. (A and B) Etest strips containing fluconazole or amphotericin B were applied to YPD plates swabbed with WT, pkh2-02Δ, and pkh2Δ::PKH2-02 strains. Plates were incubated at 35°C for 3 days. Bars represent the mean MIC from at least two independent experiments performed in triplicate. Error bars indicate standard deviations. (C) Cultures containing 2 × 107 CFU/ml of WT cells in YPD were treated with fluconazole (Flu) (2 μg/ml), OSU-03012 (OSU) (2 μg/ml), or a combination of both drugs at the same concentration. Cultures were incubated at 37°C and plated, and the number of colony-forming units (CFU)/ml was determined at the indicated time points.
As mentioned above, one of our laboratories has recently demonstrated that molecules which inhibit hPDK1 also have antifungal activity and appear to target fungal Pkh kinases as part of their mechanism of action (17). Based on the results from Fig. 3A, we hypothesized that hPDK1 inhibitors could be synergistic with fluconazole. To test this hypothesis, we performed time-kill assays with fluconazole and the hPDK1 inhibitor OSU-03012 (Fig. 3C). The combination of fungistatic concentrations of OSU-03012 and fluconazole resulted in a >3-log10 decrease in the starting inoculum of C. neoformans serotype A, indicating that the addition of OSU-03012 to fluconazole results in a fungicidal combination. These data strongly suggest that Pkh activity is required for tolerance of plasma membrane-targeted antifungal drugs.
Pkh2-02 is required for nitrosative and oxidative stress tolerance.
A key characteristic of C. neoformans is its ability to survive and replicate within the phagolysosomes of macrophages (46). This ability is, in turn, dependent on the organism's tolerance of oxidative and nitrosative stresses (47). Since Pkc1 activity is required for oxidative and nitrosative stress tolerance in C. neoformans (37) and is reduced in pkh2-02Δ mutants (Fig. 2B), we hypothesized that Pkh2-02 may be required for growth in the presence of oxidative and/or nitrosative stress. As shown in Fig. 4A, the pkh2-02Δ mutant is exquisitely sensitive to H2O2, NaNO2, and diamide compared to the WT. To further explore the role of Pkh2-02 in oxidative and nitrosative stress tolerance, we examined the effects of NaNO2 and H2O2 treatment on the phosphorylation of Mpk1. As shown in Fig. 4B and C, Mpk1 phosphorylation is decreased in the pkh2-02Δ mutant relative to both the WT and the pkh2-02Δ::PKH2-02 complement. These observations strongly suggest that Pkh2-02 is required for the activation of the CWI pathway in response to nitrosative and oxidative stress.
Fig 4.

PKH2-02 is required for oxidative and nitrosative stress tolerance. (A) A 10-fold dilution series of the indicated strains was spotted on YNB (pH 4.0) supplemented with H2O2 (1.5 mM), NaNO2 (1.5 mM), or diamide (1 mM). (B and C) Cell lysates from log-phase cells exposed to either NaNO2 (1 mM) or H2O2 (1 mM or 10 mM) were analyzed for Mpk1 phosphorylation utilizing phospho-p44/42 MAPK. Anti-PSTAIRE and anti-Tsa1 are loading controls.
Pkh2-02 is required for temperature-independent virulence in a Galleria model of cryptococcosis.
As reported by Liu et al. (31), the pkh2-02Δ mutant showed infectivity defects in a large-scale competitive fitness assay using a murine model of pulmonary cryptococcosis. Since this strain also showed a growth defect at 37°C, the infectivity defect of the pkh2-02Δ mutant was reasonably attributed to this well-known Cryptococcus virulence trait. Similar findings were also reported by Lee et al. using a tail vein inoculation model of disseminated cryptococcosis (32). Because our studies as well as those reported by Lee et al. (32) and Liu et al. (31) indicated that loss of PKH2-02 affects a number of important cellular processes that could also influence pathogenesis, we hypothesized that pkh2-02Δ mutants might display virulence defects independent of high-temperature growth. To test this hypothesis, we took advantage of the ability of C. neoformans to cause disease in Galleria mellonella at temperatures below mammalian body temperature (37°C) as a means to further dissect the role of PKH2-02 in C. neoformans pathogenesis (48). As shown in Fig. 5, infection of G. mellonella caterpillars with 1 × 105 CFU/larva leads more than ∼40% mortality by 11 days postinfection, whereas the pkh2-02Δ mutant is avirulent (P = 0.04 by the Kaplan-Meier test). The complemented mutant (Fig. 5) was not statistically different from the WT (P > 0.05).
Fig 5.
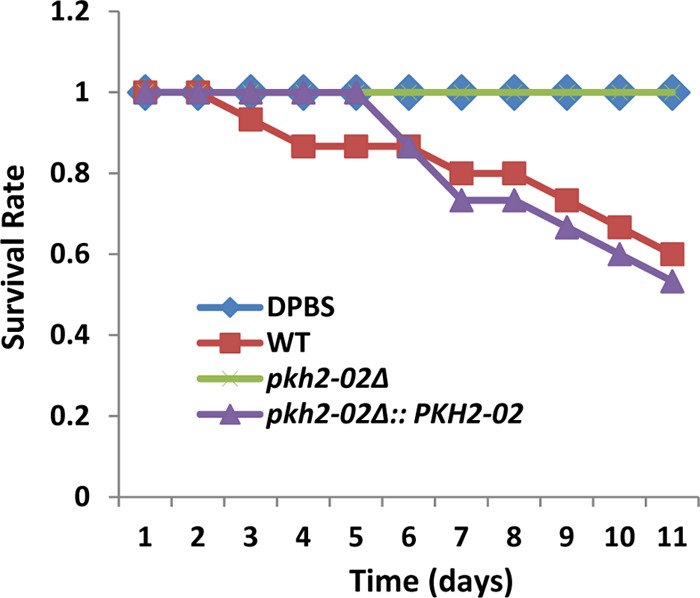
PKH2-02 is required for virulence in the G. mellonella model. G. mellonella larvae were injected in the terminal pseudopod with WT, pkh2-02Δ, and pkh2-02Δ::PKH2-02 strains at a concentration of 2 × 105 CFU/larva and incubated at 30°C. Survival was scored every 24 h. The survival difference between the WT and pkh2-02Δ strains was statistically significant (P < 0.05 by the Kaplan-Meier test).
To explore the mechanistic basis for the high-temperature growth-independent role of PKH2-02 in pathogenesis, we examined the ability of the pkh2-02Δ mutant to form capsule and melanin, two other important C. neoformans virulence factors (49). Consistent with the findings of Liu et al. (31), we found no difference in capsule formation between the pkh2-02Δ mutant and the WT using a variety of assays, including India ink staining, anticapsular antibody binding, and plate-based assays (data not shown). In the CM018 background, the pkh2-02Δ mutant showed a very mild defect in melanin formation on medium containing l-DOPA (Fig. 6A). However, this defect is not present in the KN99α background. As is evident from the plates shown in Fig. 6A, the CM018 reference strain does not melanize as strongly as KN99α under our assay conditions. We have tested the phenotype multiple times, and it is complemented by reintroduction of PKH2-02 (Fig. 6A). Neither Liu et al. (31) nor Lee et al. (32) reported a defect in melanization for the pkh2-02Δ mutant. However, the defect is quite minor and may not have been evident under the conditions used in the other laboratories. We interpret these results to indicate that Pkh2-02 has, at most, a minor role in melanin formation because its effect is apparent only when the strain background itself is relatively inefficient at melanization. As such, it seems likely that there are uncharacterized mutations in the CM018 background that are required to reveal the minor role of Pkh2-02 in melanization.
Fig 6.

Deletion of PKH2-02 affects melanization and survival in phagocytes. (A) Ten-fold dilutions of the indicated strains were spotted onto l-DOPA plates, incubated at 30°C for 5 days, and photographed. (B) Macrophage survival studies were performed utilizing J774 cells as detailed in Materials and Methods. Data were normalized to growth in the absence of macrophages to obtain the survival ratio. The bars indicate means from two independent experiments performed in triplicate, and error bars indicated standard deviations. Differences between strains were analyzed by Student's t test, and the P value is indicated over the bracket.
The pathogenesis of C. neoformans is intimately related to the ability of the organism to survive and replicate within the phagolysosome (46, 50). Thus, the hypersensitivity of the pkh2-02Δ mutant to oxidative and nitrosative stress suggested that its temperature-independent pathogenesis defect may be related to decreased replication or survival in the phagolysosome (47). To test this possibility, we compared the ability of the pkh2-02Δ mutant to survive in the murine macrophage-like cell line J774, a well-studied model for the interaction of C. neoformans with macrophages. C. neoformans cells were opsonized with anticapsular antibody prior to addition to otherwise unactivated J774 cells (51). To control for the decreased growth rate of the pkh2-02Δ mutant at 37°C, identical wells lacking J774 cells were processed in parallel, and growth of the C. neoformans cells in J774 cells was normalized to growth in medium alone (47). Under these conditions, WT cells replicate within J774 cells more efficiently than in medium alone while pkh2-02Δ cells show a statistically significant decrease (2-fold decrease; P < 0.05 by Student's t test) in growth in the presence of J774 cells (Fig. 6B). It is important to note that we could detect no difference in the ability of J774 cells to phagocytose WT and pkh2-02Δ cells (data not shown). These data strongly suggest that PKH2-02 is involved in regulating processes required for C. neoformans to survive within phagocytes and that the inability to replicate within macrophages contributes to the temperature-independent virulence defects displayed by the pkh2-02Δ mutant. The in vitro hypersensitivity to oxidative and nitrosative stress displayed by the pkh2-02Δ mutant coupled with the decreased activation of the CWI pathway in response to these stresses likely contributes to the observed defects in macrophage survival.
Phagocytes exposed to the pkh2-02Δ mutant produce increased levels of TNF-α and reactive oxygen species compared to those exposed to the wild type.
Our results indicate that Pkh2-02 regulates processes that are important for both cell wall and plasma membrane homeostasis. Since cell surface structures such as these represent the point of contact between the host and pathogen, we hypothesized that interactions between Cryptococcus and the phagocyte may also be altered in pkh2-02Δ mutants. In addition to tolerance of the oxidative and nitrosative stresses present in the phagolysosome, Cryptococcus cells are able to modulate the activation of phagocytes as part of an immune evasion strategy (46). For example, primed phagocytes challenged with Cryptococcus secrete lower levels of TNF-α than unchallenged primed phagocytes (52). To test the effect of deletion of PKH2-02 on this immune evasion property of Cryptococcus, we compared the amounts of TNF-α secreted by J774 cells that had been cocultured with either the WT or the pkh2-02Δ mutant. As previously reported (52), addition of WT cells to primed J774 phagocytes significantly reduced the amount of TNF-α secreted into the medium (Fig. 7A). This suppression was abolished in the pkh2-02Δ mutant, and the amount of TNF-α secreted into the medium of a coculture of the pkh2-02Δ mutant was indistinguishable from that for unchallenged J774 cells (Fig. 7A). Reintroduction of the PKH2-02 allele restored the ability of the mutant to suppress TNF-α production by J774 cells. Thus, PKH2-02 appears to impact processes that are required for C. neoformans to modulate the innate immune response.
Fig 7.

Primed phagocytes challenged with the pkh2-02Δ mutant produce increased levels of TNF-α and ROS relative to those challenged with the wild type. (A) The indicated strains were opsonized with MAb 18B7 for 1 h and cocultured with J774 cells that had been prestimulated with LPS and IFN-γ for 24 h. Supernatants were collected and analyzed for the presence of TNF-α as described in Materials and Methods. (B) Opsonized C. neoformans cells were cocultured with LPS- and IFN-γ-prestimulated J774 cells for 2 h. The nitrite concentration in the supernatants was determined using the Griess reagent kit. (C and D) The indicated strains were opsonized and cocultured with PMA-stimulated J774 cells. ROS were measured by luminal-enhanced chemiluminescence as described in Materials and Methods. For all panels, the solid bars indicate mean values and error bars indicate the standard deviation for at least two independent experiments performed in triplicate. Differences between strains were analyzed by Student's t test with statistical significance set at P < 0.05. NS, not significant.
It has also been shown previously that primed murine phagocytes do not induce nitric oxide synthase in response to C. neoformans (53). We therefore asked if deletion of PKH2-02 affected the amount of nitrite produced by murine phagocytes. As shown in Fig. 7B, nitrite concentrations in the coculture supernatants of J774 cells with WT and pkh2-02Δ mutant cells were essentially identical. A second important component of the phagocyte response to microbial pathogens is the generation of reactive oxygen species (ROS). Interestingly, the production of ROS by phagocytes challenged with Cryptococcus has not previously been reported to our knowledge. We therefore compared the amount of ROS generated by primed J774 cells in the presence and absence of WT and pkh2-02Δ mutant cells. Opsonized C. neoformans cells were added to J774 cells primed with PMA to activate ROS production using a modification of a procedure previously developed to assay the interaction of Candida albicans with J774 cells (33–35). The addition of WT C. neoformans significantly decreased the amount of ROS production induced by PMA relative to that for activated J774 cells that were not exposed to C. neoformans (Fig. 7C). In contrast, the pkh2-02Δ mutant induced significantly more ROS production by J774 cells than the WT (P < 0.01), while cells of the complemented mutant were not significantly different from the WT. These data indicate that C. neoformans also modulates the ability of phagocytes to generate ROS and that Pkh2-02-regulated processes are required for this modulation to occur normally.
Since the modulation of ROS production in primed J774 cells by C. neoformans has not been studied previously, we examined its dependence on the production of capsule and melanin using the acapsular cap59Δ mutant and the melanin-deficient lac1Δ mutant. As shown in Fig. 7D, neither mutant was significantly different from the WT with respect to ROS production. Similarly, a lack of CWI pathway signaling in the pkh2-02Δ strains does not explain its increased ROS production, because the mpk1Δ mutant is indistinguishable from the WT. Further studies will be required to identify the mechanistic basis of the ability of C. neoformans to suppress ROS production in phagocytes. Taken together, our results suggest that a combination of decreased tolerance to the oxidative/nitrosative environment of the phagolysosome and increased production of ROS may contribute to the temperature-independent virulence defects displayed by the pkh2-02Δ mutant.
DISCUSSION
Protein kinase-based signaling pathways are crucial mediators of processes through which fungi adapt to environmental stresses, survive within the host, cause disease, and tolerate antifungal drugs (9–11). Recently, we identified fungal orthologs of human PDK1 kinases as an attractive target for antifungal drug development (17). In order to better understand the function of these kinases in pathogenic fungi, we have carried out a genetics-based study to characterize C. neoformans mutants lacking the hPDK1 ortholog Pkh2-02. As discussed above, while our work was in its final stages, Lee et al. reported that PKH2-02 plays a role in fluconazole tolerance and affects sphingolipid metabolism (32). Between our work and that of Lee et al. and Liu et al. (31), PKH2-02 has been deleted in three different genetic backgrounds of serotype A C. neoformans var. grubii. Overall, the phenotypes that have been examined in all three backgrounds have been highly concordant. However, there appears to be some variation in the penetrance of high-temperature tolerance and melanization depending on the specific genetic background. There do not appear to be overlapping ORFs in the chromosomal region of PKH2-02 that could have been differentially disrupted by the deletion strategies used to generate the mutants. Another explanation for the background variations observed for the PKH2-02 mutants is that PKH2-01 may be differentially regulated in the backgrounds and, thus, more or less able to compensate for loss of PKH2-02. However, as Lee et al. reported (32), PKH2-01 lacks the canonical sequences of a PDK1 and thus may not be biochemically related. Furthermore, we measured the expression of PKH2-01 in wild-type and pkh2-02Δ mutant strains from the CM108 and KN99α backgrounds and could find no differences (data not shown). Because the Pkh kinases appear to function in a wide range of cellular processes, we suspect that the slight variation in phenotypes may be related to uncharacterized background mutations.
ScPkhs are known to be involved in cell wall integrity signaling (7, 21), and therefore, we were interested in determining whether Pkhs also play a role in cell wall integrity. The pkh2-02Δ mutant displays two phenotypes that suggest a role for the Pkhs in cell wall integrity. First, the growth defect displayed by the pkh2-02Δ mutant at elevated temperature is suppressed by the addition of sorbitol to the growth medium (Fig. 1D). Osmoremedial temperature sensitivity is a classic phenotype for mutants with cell wall defects in all fungal species. Second, pkh2-02Δ cells are hypersensitive to the effects of Congo red, a dye that appears to interfere with cell wall chitin and glucan architecture (Fig. 2A). Interestingly, the pkh2-02Δ mutant shows only modest growth defects in the presence of calcofluor white (Fig. 2A), a dye that interferes with chitin and is typically more toxic to cell wall mutants (41). However, previous work in the Lodge laboratory has shown that deletion of components of the CWI MAPK signaling pathway in C. neoformans leads to hypersensitivity to Congo red but not to calcofluor white (12). Similarly, CWI pathway mutants also display osmoremedial temperature sensitivity. Thus, with respect to cell wall defects, pkh2-02Δ mutants share similar phenotypes with CWI pathway mutants, consistent with previous results in S. cerevisiae indicating that fungal PDK1 orthologs function in the CWI pathway (19, 21, 28).
The CWI MAPK signaling pathway is highly conserved in fungi (45). At the top of this cascade is Pkc1, an ACG kinase that is a substrate of ScPkhs; the terminal MAPK is presumed to be Mpk1 (21, 41, 44). As shown in Fig. 2B and C, cellular Pkc1 activity and Mpk1 phosphorylation are decreased in pkh2-02Δ mutants, further supporting a role for PKH2-02 in the regulation of the CWI pathway and the notion that, as in S. cerevisiae, Pkc1 is a substrate of Pkh2-02. Indeed, Pkc1 contains a sequence that matches the consensus PDK1/Pkh phosphorylation motif (TFCGTPEF) identified in human and S. cerevisiae substrates (18). Lee et al. found that Pkh2-02 is involved in fluconazole-triggered phosphorylation of Mpk1 as well (32). However, it is important to note that both our results and those of Lee et al. indicate that low levels of Mpk1 phosphorylation occur in the absence of PKH2-02 (Fig. 2C). Thus, PKH2-02 is required for full activation of the CWI pathway but is not absolutely required for its activity. This is also consistent with the fact that cells lacking Pkh2-02 are viable. Deletion of PKC1 in C. neoformans (37) results in more severe phenotypes than those observed in cells lacking Pkh2-02. Therefore, if Pkh2-02 phosphorylation were absolutely required for Pkc1 activity, then deletion of PKH2-02 should yield a more severe phenotype than that observed.
The TOR pathway is also an important signaling network with which ScPkh1/2 kinases interact. Both we and Lee et al. (32) have found that the pkh2-02Δ mutant is hypersensitive to rapamycin, indicating that this interaction is operative in C. neoformans as well. We also found that caffeine, which appears to inhibit TOR as part of its mechanism of action, is more toxic to pkh2-02Δ cells than to WT cells. In S. cerevisiae, mutants with mutations of the ScPKH1/2 substrate ScYPK1 are hypersensitive to rapamycin (53), and similarly, Lee et al. also showed that deletion of YPK1, a likely substrate of PKH2-02, increases sensitivity to rapamycin (32). Furthermore, this group also showed that a threonine-to-alanine mutation in the putative Pkh2-02 phosphorylation site of YPK1 also caused rapamycin sensitivity (32). Taken together, these observations are consistent with the idea that the hypersensitivity of the pkh2-02Δ mutant to rapamycin is due to decreased activation of Ypk1.
In addition to regulating cell wall integrity, Pkc1 plays an important role the ability of C. neoformans to withstand oxidative and nitrosative stress (37). Since C. neoformans replicates within the phagolysosomes of macrophages, oxidative and nitrosative stress tolerance is an important feature of its physiology and pathogenesis (47). Deletion of PKC1 results in oxidative and nitrosative stress sensitivity, and the CWI pathway is activated by H2O2 and NO as evidenced by phosphorylation of Mpk1. However, the downstream components of the CWI pathway are dispensable for resistance to these stresses (37). Similar to the pkc1Δ mutant, the pkh2-02Δ mutant is sensitive to both H2O2 and NO and fails to activate the CWI pathway in response to oxidative and nitrosative stress. These data are paralleled by results that show that Pkh2-02 is necessary for the suppression of ROS production in the macrophage-like cell line in an Mpk1-independent manner. Taken together, these data suggest that responses to oxidative stress, including resistance to and suppression of the oxidative burst, are mediated by Pkc1 but not the remainder of the CWI pathway. Although a pkc1Δ mutant is available (37), it requires an osmotic support for viability and therefore could not be directly tested in parallel experiments. Taken together, these results suggest that PKH2-02 may play an important role in macrophage survival and, hence, pathogenesis.
Previous studies by both Liu et al. (31) and Lee et al. (32) indicated that pkh2-02Δ mutants have decreased virulence in mouse models of cryptococcosis. However, as both groups noted, this effect is most likely due to the high-temperature growth defect displayed by pkh2-02Δ cells. Using the Galleria mellonella model at low temperature (48), we found that the mutant is also avirulent at reduced temperature, indicating that Pkh2-02 regulates processes important for virulence independent of high-temperature growth. Furthermore, our data indicate that the pkh2-02Δ mutant is unable to replicate within phagocytes after controlling for slower growth of the mutant at 37°C.
A reasonable explanation for these results is that the pkh2-02Δ mutant is unable to tolerate the oxidative environment of the phagocyte. It is likely that multiple defect in pkh2-02Δ cells contribute to this inability to survive in the phagolysosome. First, Lee et al. have shown that lipid metabolism (32) and, consequently, the plasma membrane are altered. Defects in membrane structure and function are likely to decrease cellular integrity in the face of oxidative stress. Second, we have shown that activation of CWI pathway-mediated stress responses to nitrosative and oxidative stress is blunted in the pkh2-02Δ mutant, indicating that compensatory adaptations to oxidative stress are defective in the mutants. In principle, two other characteristics of C. neoformans that contribute to its virulence and survival in macrophages are capsule and melanin production. We were unable to detect any alterations in capsule formation between the pkh2-02Δ mutant and WT strains, which is similar to the findings of Lee et al. (32) and Liu et al. (31).
On l-DOPA-containing medium, the pkh2-02Δ mutant in the CM018 background used in the deletion collection shows a very minor defect in melanization, whereas the KN99α-derived pkh2-02Δ mutant showed melanization similar to that of the WT. We carried out our virulence and phagocyte interaction studies in the CM018 background, and it is possible that reduced melanization contributes to the decreased survival of the pkh2-02Δ mutant in phagocytes and decreased virulence. Since the melanization defect is quite minor, however, it is unlikely to be the sole explanation for the sensitivity to oxidative stress and phagocytes displayed by the pkh2-02Δ mutant. Taking these findings together, it appears that Pkh2-02 regulates a variety of cellular processes that both directly and indirectly affect the ability of C. neoformans to adapt to the harsh environment of the phagolysosome.
C. neoformans also employs a number of immune evasion strategies to survive and replicate within phagocytes (46, 49, 52, 54). One of these mechanisms is based the ability to suppress TNF-α secretion by phagocytes (52). Similarly, C. neoformans suppresses the ability of phagocytes to produce antimicrobial nitric oxide species (54) and, as we have shown here, ROS. We have found that Pkh2-02 is required for the ability of C. neoformans to suppress TNF-α secretion and ROS production but not nitrite formation (52, 54). Previous studies have shown that C. neoformans capsular polysaccharide glucuronoxylomannan (GXM) contributes significantly to the ability of C. neoformans to suppress TNF-α and nitrite production. For example, primed phagocytes challenged with acapsular mutants of C. neoformans produce increased levels of both TNF-α and nitrite compared to WT cells. However, it is important to note that acapsular mutants still induce less TNF-α and nitrite production than the WT (52, 54), suggesting that capsule-independent mechanisms may play a role in this immune evasion strategy. Since neither we nor others have found that pkh2-02Δ mutants display readily detectable alterations in the amount of capsule produced, it is possible that Pkh2-02 function is required for capsule-independent mechanisms by which C. neoformans modulates TNF-α production. It is also possible that loss of Pkh2-02 function leads to more subtle changes in capsule structure that are required for GXM to interact with the phagocyte and carry out its immunomodulatory functions.
One of the hallmark functions of phagocytes is the generation of ROS in response to microbes. To our knowledge, the effect of C. neoformans on this important function of phagocytes had not been previously studied. Similar to its effect on TNF-α and nitrite production (52, 54), here we have shown that C. neoformans suppresses the production of ROS by stimulated murine phagocytes in a capsule- and melanin-independent fashion. Additional work will be required to fully define the mechanisms of this immunomodulatory strategy of C. neoformans. The ability of other pathogenic fungi such as C. albicans and Histoplasma capsulatum to detoxify ROS through cell surface-localized superoxide dismutases has been reported recently (33–35). Because Pkh2-02 affects cell surface-associated processes, it is possible that loss of Pkh2-02 activity disrupts the localization, secretion, or activity of Sod1, the cell surface-localized superoxide dismutase in C. neoformans (43, 55). Experiments directed toward testing these hypotheses are in progress and will be reported in due course.
In summary, our results as well as those recently reported by others indicate that the C. neoformans PDK1 ortholog Pkh2-02 is a key signaling kinase that affects a wide range of physiologic and pathogenic processes in this important human pathogen. Certainly, more work will be required to dissect the cellular and molecular mechanisms by which this kinase carries out its functions. Accordingly, it seems likely that such experiments will provide important insights into the biology and pathogenesis of C. neoformans.
ACKNOWLEDGMENTS
We thank Arturo Casavedall (Albert Einstein College of Medicine) for anticapsular antibody, Rajendra Uphadya for generating the Tsa1 antibody utilized in this studies, Melanie Wellington (University of Rochester) for helpful advice regarding reactive oxygen assays, and John Panepinto (SUNY—Buffalo) for assistance with cryptococcal transformations.
This work was supported in part by National Institute of Allergy and Infectious Disease grants R01AI097142-01A1 (D.J.K.), R01AI091422 (D.J.K.), 2T32AI007464-16 (Y.C.-R.), R01AI050184 (J.K.L.), and R01HL088905 (J.K.L.).
Footnotes
Published ahead of print 19 October 2012
REFERENCES
- 1. Chayakulkeeree M, Perfect JR. 2006. Cryptococcosis. Infect. Dis. Clin. N. Amer. 20:507–544 [DOI] [PubMed] [Google Scholar]
- 2. Park BJ, Wannemuehler KA, Marston BJ, Govender N, Pappas PG, Chiller TM. 2009. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 23:525–530 [DOI] [PubMed] [Google Scholar]
- 3. Bicanic T, Wood R, Bekker L-G, Darder M, Meintjes G, Harrison TS. 2005. Antiretroviral roll-out, antifungal roll-back: access to treatment for cryptococcal meningitis. Infect. Lancet 5:530–531 [DOI] [PubMed] [Google Scholar]
- 4. Kronstad JW, Attarian R, Cadieux B, Choi J, D'Souza CA, Griffiths EJ, Geddes JM, Hu G, Jung WH, Kreschmer JW, Saikia S, Wang J. 2011. Expanding pathogenesis: Cryptococcus breaks out of the opportunistic box. Nat. Rev. Microbiol. 9:193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith JA, Kauffman CA. 2012. Pulmonary fungal infections. Respirology 17:913–926 [DOI] [PubMed] [Google Scholar]
- 6. Sloan DJ, Dedicoat MJ, Lalloo DG. 2009. Treatment of cryptococcal meningitis in resource limited settings. Curr. Opin. Infect. Dis. 22:455–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bicanic T, Muzoora C, Brouwer AE, Meintjes G, Longley N, Taseera K, Rebe K, Loyse A, Jarvis J, Bekker LG, Wood R, Limmathurotsakul D, Chierakul W, Stepniewska K, White NJ, Jaffar S, Harrison TS. 2009. Independent association between rate of clearance of infection and clinical outcome of HIV-associated cryptococcal meningitis: analysis of a combined cohort of 262 patients. Clin. Infect. Dis. 49:702–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spanakis EK, Aperis G, Mylonakis E. 2006. New agents for the treatment of fungal infections: clinical efficacy and gaps in coverage. Clin. Infect. Dis. 43:1060–1068 [DOI] [PubMed] [Google Scholar]
- 9. Brown SM, Campbell LT, Lodge JK. 2007. Cryptococcus neoformans: a fungus under stress. Curr. Opin. Microbiol. 10:320–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cowen LE, Steinbach WJ. 2008. Stress, drugs, and evolution: the role of cellular signaling in the fungal drug resistance. Eukaryot. Cell 7:747–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kozubowski L, Lee SC, Heitman J. 2009. Signaling pathway in the pathogenesis of Cryptococcus. Cell. Microbiol. 11:370–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gerik KJ, Donlin MJ, Soto CE, Banks AM, Maligie MA, Selitrennikoff CP, Lodge JK. 2005. Cell wall integrity is dependent on the Pck1 signal transduction pathway in Cryptococcus neoformans. Mol. Microbiol. 58:393–408 [DOI] [PubMed] [Google Scholar]
- 13. Kraus PR, Heitman J. 2003. Coping with stress: calmodulin and calcineurin in model and pathogenic fungi. Biochem. Biophys. Res. Commun. 311:1151–1157 [DOI] [PubMed] [Google Scholar]
- 14. Kronstad JW, Hu G, Choi J. 2011. The cAMP/protein kinase A pathway and virulence in Cryptococcus neoformans. Microbiology 39:143–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bahn YS, Kojima K, Cox GM, Heitman J. 2005. Specialization of the HOG pathway and its impact on differentiation and virulence of Cryptococcus neoformans. Mol. Biol. Cell 16:2285–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Idnurm A, Bahn YS, Nielsen K, Lin X, Fraser JA, Heitman J. 2005. Deciphering the model pathogenic fungus Cryptococcus neoformans. Nat. Rev. Microbiol. 3:753–764 [DOI] [PubMed] [Google Scholar]
- 17. Baxter BK, DiDone L, Oga D, Schor S, Krysan DJ. 2011. Identification, in vitro activity, and mode of action of phosphoinositide-dependent-1 kinase inhibitors as antifungal molecules. ACS Chem. Biol. 6:502–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Casamayor A, Torrance PD, Kobayashi T, Thorner J, Alessi DR. 1999. Functional counterparts of mammalian protein kinase PDK1 and SGK in budding yeast. Curr. Biol. 9:186–197 [DOI] [PubMed] [Google Scholar]
- 19. Roelants FM, Torrance PD, Bezman N, Thorner J. 2002. Pkh1 and Pkh2 differentially phosphorylate and activate Ypk1 and Ykr2 and define proteinase modules required for cell wall integrity. Mol. Biol. Cell 13:3005–3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peifer C, Alessi DR. 2008. Small molecule inhibitors of PDK1. ChemMedChem 3:1810–1838 [DOI] [PubMed] [Google Scholar]
- 21. Inagaki M, Schmelzle T, Yamaguchi K, Irie K, Hall MN, Matsumoto K. 1999. PDK1 homologs activate Pkc1-mitogen-activated protein kinase pathway in yeast. Mol. Cell. Biol. 19:8344–8352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Roelants FM, Breslow DK, Muir A, Weissman J, Thorner J. 2011. Protein kinase Ypk1 phosphorylates regulatory proteins Orm1 and Orm2 to control sphingolipid homeostasis in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 108:19222–19227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun Y, Miao Y, Yamane Y, Zhang C, Shokat KM, Takematsu H, Kozutsumi Y, Drubin DG. 2012. Orm protein phosphoregulation mediates transient sphigolipid biosynthesis response to heat stress via the Pkh-Ypk and Cdc55-PP2A pathways. Mol. Biol. Cell 23:2388–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Friant S, Lombardi R, Schmelzle T, Hall MN, Riezman H. 2001. Sphingoid base signaling via Pkh kinases is required for endocytosis in yeast. EMBO J. 20:6783–6792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lou G, Costanzo M, Boone C, Dickson RC. 2011. Nutrients and the Pkh1/2 and Pkc1 protein kinases control mRNA decay and P-body assembly in yeast. J. Biol. Chem. 18:8759–8770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Walther TC, Aguilar PS, Frolich F, Chu F, Moreira K, Burlingame AL, Walter P. 2007. Pkh-kinases control eisosome assembly and organization. EMBO J. 26:4946–4955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luo G, Gruhler A, Liu Y, Jensen ON, Dickson RC. 2008. The sphingolipid long chain base-Pkh1/2-Ypk1/2 signaling pathway regulates eisosome assembly and turnover. J. Biol. Chem. 283:10433–10444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Roelants FM, Torrance PD, Thorner J. 2004. Differential roles of PDK1- and PDK2-phosphorylation sites in the yeast AGC kinases Ypk1, Pck1, and Sch9. Microbiology 286:22017–22027 [DOI] [PubMed] [Google Scholar]
- 29. Voordeckers K, Kimpe M, Haesendonckx S, Louwet W, Versele M, Thevelein JM. 2011. Yeast phosphoinositide-dependent protein kinase-1 (PDK1) orthologs Pkh1-1 differentially regulate phosphorylation of protein kinase A (PKA) and the protein kinase B(PKB)/S6K ortholog Sch9. J. Biol. Chem. 286:22017–22207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blankenship JR, Fanning S, Hamaker JJ, Mitchell AP. 2010. An extensive circuitry for cell wall regulation in Candida albicans. PLoS Pathog. 5:e1000752 doi:10.1371/journal.ppat.1000752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu OW, Chun CD, Chow ED, Chen C, Madhani H, Nobel SM. 2008. Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell 135:174–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee H, Khanal Lamichhane A, Garraffo HM, Kwon-Chung KJ, Chang YC. 2012. Involvement of PDK1, Pkc, and Tor signaling pathways in basal fluconazole tolerance in Cryptococcus neoformans. Mol. Microbiol. 84:130–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Frohner IE, Bourgeois L, Yatsyk K, Majer O, Kuchler K. 2009. Candida albicans cell surface superoxide dismutases degrade host-derived reactive oxygen species to escape innate immunity. Mol. Microbiol. 71:240–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wellington M, Dolan K, Krysan DJ. 2009. Live Candida albicans suppresses production of reactive oxygen species in phagocytes. Infect. Immun. 77:405–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Youseff BH, Holbrook ED, Smolnycki KA, Rappleye CA. 2012. Extracellular superoxide dismutase protects Histoplasma yeast cells from host-derived oxidative stress. PLoS Pathog. 8:e1002713 doi:10.1371/journal.ppat.1002713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Burke DJ, Dawson D, Stearns T. 2000. Methods in yeast genetics. Cold Spring Harbor Laboratory Press, Woodbury, NY [Google Scholar]
- 37. Gerik KJ, Bhimireddy SR, Ryerse JS, Specht CA, Lodge JK. 2008. Pkc1 is essential for protection against both oxidative and nitrosative stresses, cell integrity, and normal manifestation of virulence factors in the pathogenic fungus Cryptococcus neoformans. Eukaryot. Cell 7:1685–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bharucha N, Chabrier-Roselló Y, Xu T, Johnson C, Sobczynski S, Song QCJ, Dobry Eckwahl MJ, Anderson CP, Benjamin AJ, Kumar A, Krysan DJ. 2011. A large-scale complex haploinsufficiency-based genetic interaction screen in Candida albicans: analysis of the RAM Network during morphogenesis. PLoS Genet. 7:e1002058 doi:10.1371/journal.pgen.1002058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mylonakis E, Moreno R, El Khoury JB, Idnurm A, Heitman J, Calderwood SB, Ausubel FM, Diener A. 2005. Galleria mellonella as a model system to study Cryptococcus neoformans pathogenesis. Infect. Immun. 73:3842–3850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Geunes-Boyer S, Oliver TN, Janbon G, Lodge JK, Heitman J, Perfect JR, Wright JR. 2009. Surfactant protein D increases phagocytosis of hypocapsular Cryptococcus neoformans by murine macrophages and enhances fungal survival. Infect. Immun. 77:2783–2794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Levin D. 2005. Cell wall integrity signaling in S. cerevisiae. Microbiol. Mol. Biol. Rev. 69:262–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reinke A, Chen JC, Aronova S, Powers T. 2006. Caffeine targets TOR complex 1 and provides evidence for a regulatory link FRB and kinase domains of Tor1p. J. Biol. Chem. 281:31616–31626 [DOI] [PubMed] [Google Scholar]
- 43.Reference deleted. [Google Scholar]
- 44. Kraus PR, Fox DS, Cox GM, Heitman J. 2003. The Cryptococcus neoformans MAP kinase Mpk1 regulates cell integrity response to antifungal drugs and loss calcineurin function. Mol. Microbiol. 48:1377–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Roelants FM, Baltz AG, Trott AE, Thorner J. 2010. A protein kinase network regulates the function of aminophospholipid flippases. Proc. Natl. Acad. Sci. U. S. A. 107:34–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Voelz K, May RC. 2010. Cryptococcal interactions with the host immune system. Eukaryot. Cell 9:835–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Missall TA, Pusateri ME, Donlin MJ, Chambers KT, Corbett JA, Lodge JK. 2006. Posttranslational, translational, and transcriptional responses to nitric oxide stress in Cryptococcus neoformans: implications for virulence. Eukaryot. Cell 5:518–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kozubowski L, Heitman J. 2010. Septins enforce morphogenetic events during sexual reproduction and contribute to virulence of Cryptococcus neoformans. Mol. Microbiol. 75:358–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Casadevall A, Steenbergen JN, Nosanchuk JD. 2003. “Ready-made” virulence and “dual use” virulence factors in pathogenic environmental fungi-the paradigm of Cryptococcus neoformans. Curr. Opin. Microbiol. 6:332–337 [DOI] [PubMed] [Google Scholar]
- 50. Casadevall A. 2010. Cryptococci at the brain gate: break and enter or use a Trojan horse? J. Clin. Invest. 120:1389–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nicola AM, Casadevall A. 2012. In vitro measurement of phagocytosis and killing of Cryptococcus neoformans by macrophages. Methods Mol. Biol. 844:189–197 [DOI] [PubMed] [Google Scholar]
- 52. Vecchiarelli A, Retini C, Pietrella D, Monari C, Tascini C, Beccari T, Kozel TR. 1995. Downregulation by Cryptococcal polysaccharide of tumor necrosis factor alpha and interleukin-1β secretion from human monocytes. Infect. Immun. 63:2919–2923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gelperin D, Horton L, DeChant A, Hensold J, Lemmon SK. 2002. Loss of ypk1 function causes rapamycin sensitivity inhibition of translation, and synthetic lethality in 14-3-3-deficient yeast. Genetics 161:1453–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Naslund PK, Miller WC, Granger DL. 1995. Cryptococcus neoformans fails to induce nitric oxide synthase in primed murine macrophage-like cells. Infect. Immun. 63:1298–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Siafakas AR, Wright LC, Sorrell TC, Djordjevic JT. 2006. Lipid rafts in Cryptococcus neoformans concentrate the virulence determinants phospholipase B and Cu/Zn superoxide dismutase. Eukaryot. Cell 5:488–498 [DOI] [PMC free article] [PubMed] [Google Scholar]