

Abstract
Progressively enhanced activity of a humanized tigecycline (TGC) regimen was noted over 3 days against an extended-spectrum-β-lactamase (ESBL)-producing Escherichia coli isolate and an ESBL-producing Klebsiella pneumoniae isolate. Bacterial density reduction approximated 3 log10 approaching bactericidal activity at 72 h. This level of activity has not been previously noted for compounds such as tetracyclines, normally considered bacteriostatic antimicrobials. Extended regimen studies in vivo may aid in better delineation of antimicrobial effects, producing improved correlation with clinical outcomes.
TEXT
Historically, in vitro and in vivo pharmacodynamic (PD) assessments have been conducted over 24 h. While these studies have been noted to correlate with clinical outcomes for rapidly bactericidal agents (i.e., fluoroquinolones or aminoglycosides), these PD endpoints appear more poorly correlated for agents such as the tetracyclines and related derivatives which have slower killing profiles in vitro. The tetracyclines and the class-related extended-spectrum agents, such as tigecycline (TGC), have demonstrated bacteriostatic activity in in vitro studies against a variety of bacterial strains (1). In a recent study using the endpoint of reduction in numbers of CFU after 24 h of TGC exposure against several Escherichia coli and Klebsiella pneumoniae isolates, we noted antibacterial activity in both immunocompromised and immunocompetent mice (2). The majority of doses were bacteriostatic at best. The maximum reduction in numbers of CFU was 2 log10 over this 24-h exposure period; however, these effects were noted only when exposures were well above that typically seen in humans. In addition, it was also noted that the in vivo exposures required to produce substantial reductions in numbers of CFU in the immunocompromised murine model were well in excess of that recognized to produce good clinical and microbiologic outcomes in patients (2). While the immunocompromised model appeared to have exposures discordant to that observed in humans, the immunocompetent model required exposures similar to that observed in patients. Therefore, in the current study, we sought to determine the magnitude of bacterial kill over an extended treatment period of 72 h using an exposure in immunocompetent animals that would mimic the regimen of a 100-mg loading dose with subsequent 50-mg doses every 12 h (q12h) of TGC in humans.
Two extended-spectrum-β-lactamase (ESBL)-producing clinical strains, one E. coli (E. coli strain 363; TGC MIC, 0.125 μg/ml) and one K. pneumoniae (K. pneumoniae strain 404; TGC MIC, 0.25 μg/ml), provided by Tetraphase Pharmaceuticals, Inc. (Watertown, MA), were tested in an immunocompetent thigh infection model. ICR (CD-1) mice weighing approximately 25 g were obtained from Harlan Sprague Dawley, Inc. (Indianapolis, IN). These studies were approved by and followed the guidelines of the Institutional Animal Care and Use Committee at our facility. Bacterial colonies of a fresh subculture of each isolate were suspended in sterile 0.9% sodium chloride to produce a suspension of approximately 108 CFU/ml. Final inoculum concentrations were confirmed by plating serial dilutions of inocula on Trypticase soy agar with 5% sheep blood (BD Biosciences, Sparks, MD) and incubating plates at 35°C overnight. Both hind legs of each mouse were injected with 0.1 ml of the above-described bacterial suspension (approximately 107 CFU) of one of the test isolates. The first TGC dose was administered at two hours postinoculation. Each bacterial isolate was tested once over a 72-h period.
Tigecycline powder was obtained from Pfizer, Inc. (Groton, CT), and solubilized in normal saline immediately prior to dosing. The dosing regimen that would produce a drug exposure in vivo that is similar to that in humans, quantified by the area under the concentration-time curve from 0 to 24 h (AUC0-24) in blood, was replicated (3, 4). As such, a free (unbound) TGC AUC0-24 of 1.13 (or a total—bound and unbound—TGC AUC0-24 of 5.39 with 79% protein binding), observed in humans following the regimen of a 100-mg loading dose with subsequent 50-mg doses q12h (4, 5), was simulated over each of the three 24-h intervals in the mice. The in vivo regimen utilized in this current study was 12.5 mg/kg q24h, administered via subcutaneous (s.c.) injection, which was previously determined to produce a drug exposure similar to that in humans (2, 6). This regimen was administered to mice daily for a 3-day period (72 h) or until tissue harvest. Prior to dose administration, one group of mice (n = 3) for each bacterial isolate was sacrificed and thigh tissues were harvested to assess the initial CFU level. Thighs from one group each (n = 3) of control (vehicle-dosed) and TGC-treated mice infected with E. coli 363 or K. pneumoniae 404 were harvested and processed for quantitative culture after 24, 48, and 72 h of treatment. For tissue harvest, both rear thighs were removed from each mouse and individually homogenized in 5 ml sterile normal saline. Dilutions of the thigh homogenates were plated on Trypticase soy agar with 5% sheep blood agar plates (BD Biosciences) and incubated at approximately 37°C in ambient air overnight. Efficacy was calculated as the change in bacterial density (Δlog10 numbers of CFU) obtained in the TGC-treated mice after 24, 48, and 72 h relative to that of the 0-h untreated controls for E. coli 363 and K. pneumoniae 404.
Twenty-four hours after inoculation, the bacterial density of E. coli 363 increased 1.9 log10 in the untreated control animals. After the initial day of TGC exposure, a reduction of 1.4 log10 in bacterial density was quantified. Unfortunately, all of the control animals randomized to the 48- and 72-h control groups perished by 48 h. As a result, CFU data from these animals were combined into the 48-h control group as noted in Fig. 1. All TGC-treated animals survived until the designated tissue harvest interval; thus, the remaining TGC treatment groups were evaluated at 48 h and 72 h. On the 2nd (48 h) and 3rd (72 h) days of treatment, the TGC regimen produced a cumulative log10 reduction in numbers of CFU of 2.2 and 3.2, respectively (Fig. 1).
Fig 1.

Reduction in bacterial density of E. coli 363 over 72 h after administration of TGC in immunocompetent mice. Black bars, change in bacterial burden in control groups; gray bars, change in bacterial burden in TGC treatment groups relative to untreated controls 2 h postchallenge. Error bars indicate standard deviations. At 48 h, all remaining control animals perished, so the two groups of control animals (48 h and 72 h) were grouped together (n = 6).
The numbers of CFU of K. pneumoniae 404 increased by 0.9 log10 after 24 h in the untreated control animals. The growth of this strain was variable at 48 h (e.g., in the untreated control group, CFU range of +0.1 to −1.6 log10) with a net negative average number of CFU; however, sustained growth was noted at 72 h in those animals randomized to the later time point (Fig. 2). An increasing effect of the TGC treatment was observed over the treatment period, as noted by the log10 reduction in numbers of CFU of 0.9, 1.7, and 2.9 at 24 h, 48 h, and 72 h, respectively, after initiation of therapy (Fig. 2). The combined log10 reduction in numbers of CFU for both E. coli 363 and K. pneumoniae 404 averaged 1.1, 2.0, and 3.0 at 24 h, 48 h, and 72 h, respectively.
Fig 2.
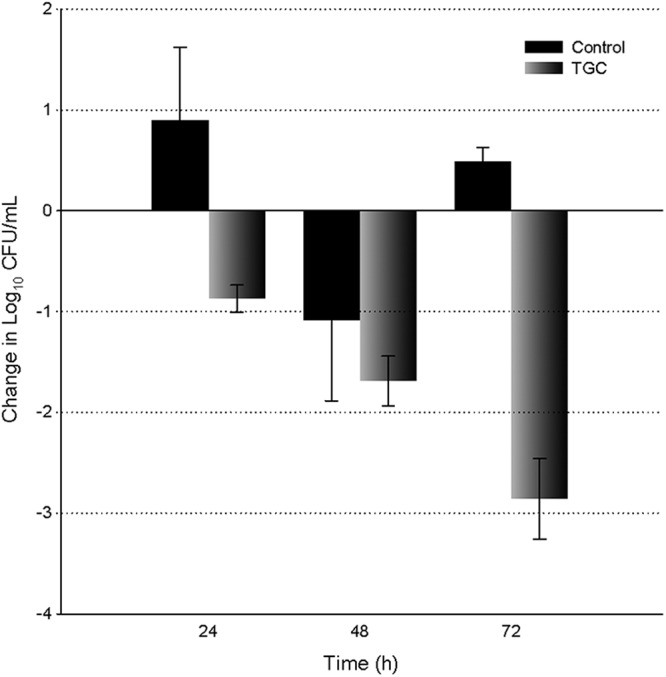
Reduction in bacterial density of K. pneumoniae 404 over 72 h after administration of TGC in immunocompetent mice. Black bars, change in bacterial burden in the control groups; gray bars, change in bacterial burden in TGC treatment groups relative to untreated controls 2 h postchallenge. Error bars indicate standard deviations.
The human simulated exposures of TGC utilized in this current investigation resulted in enhanced antibacterial activity against both isolates in vivo with each subsequent day of therapy. Exposures of TGC equivalent to a 100-mg loading dose and subsequent 50-mg doses q12h in humans exhibited antibacterial activity of about 1 log10 bacterial kill at 24 h in this infection model, as might have been anticipated (2, 6); however, this efficacy was improved on each subsequent day of dosing. After 3 days of treatment, these cumulative TGC exposures had a pronounced effect on the order of a 3-log10 reduction in numbers of CFU compared to the numbers of CFU in control animals 2 h postchallenge, displaying bactericidal activity.
Based on in vitro time-kill studies as well as in vivo assessments, the ribosomally mediated antimicrobials, such as macrolides, tetracyclines, and oxazolidinones, have been considered bacteriostatic. To this point, we also noted in vivo bacteriostatic activity after humanized doses of clarithromycin versus Streptococcus pneumoniae after 24 h (7). While the 24-h data derived in the current study point to a similar bacteriostatic endpoint, continued TGC exposures produced bactericidal activity after 72 h. Furthermore, in a recent study, we noted that 24 h of humanized dose regimens of tedizolid and linezolid produced approximately 1- to 2-log10 reductions in numbers of CFU in an in vivo immunocompetent lung infection model against methicillin-resistant Staphylococcus aureus (8). When we administered these humanized regimens of tedizolid and linezolid over a 72-h period in vivo, both compounds produced bactericidal effects against methicillin-susceptible and methicillin-resistant Staphylococcus aureus in an immunocompetent thigh infection model (7).
These studies provide insight into the in vivo potency and the magnitude of antimicrobial effects of tigecycline over an extended treatment period of 72 h in an immunocompetent host against two ESBL-producing Enterobacteriaceae. Notably, we observed an enhanced cumulative reduction in bacterial density over 72 h. While this compound is not typically regarded as bactericidal in vitro (i.e., producing a 3-log10 reduction in numbers of CFU), these in vivo results suggest that a bactericidal endpoint can be achieved over the 3-day treatment regimen with humanized doses used in this current study. The in vivo killing profile of tigecycline observed here indicates that assessments of the PD profile beyond 24 h may provide a more predictive value when considering the potential clinical efficacy of this compound or other structurally related investigational compounds in the management of infections in humans.
ACKNOWLEDGMENTS
This work was funded by a grant from Tetraphase Pharmaceuticals, Inc.
David P. Nicolau has acted as a consultant for and received research grants from Tetraphase Pharmaceuticals and Pfizer. Pamela R. Tessier has no conflict of interest to disclose.
Footnotes
Published ahead of print 31 October 2012
REFERENCES
- 1. Agwuh KN, MacGowan A. 2006. Pharmacokinetics and pharmacodynamics of the tetracyclines including glycylcyclines. J. Antimicrob. Chemother. 58:256–265 [DOI] [PubMed] [Google Scholar]
- 2. Nicasio AM, Crandon JL, Nicolau DP. 2009. In vivo pharmacodynamic profile of tigecycline against phenotypically diverse Escherichia coli and Klebsiella pneumoniae isolates. Antimicrob. Agents Chemother. 53:2756–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ambrose PG, Meagher AK, Passarell JA, Van Wart SA, Cirincione BB, Bhavnani SM, Ellis-Grosse E. 2009. Application of patient population-derived pharmacokinetic-pharmacodynamic relationships to tigecycline breakpoint determination for staphylococci and streptococci. Diagn. Microbiol. Infect. Dis. 63:155–159 [DOI] [PubMed] [Google Scholar]
- 4. Muralidharan G, Micalizzi M, Speth J, Raible D, Troy S. 2005. Pharmacokinetics of tigecycline after single and multiple doses in healthy subjects. Antimicrob. Agents Chemother. 49:220–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meagher AK, Passarell JA, Cirincione BB, Van Wart SA, Liolios K, Babinchak T, Ellis-Grosse EJ, Ambrose PG. 2007. Exposure-response analyses of tigecycline efficacy in patients with complicated skin and skin-structure infections. Antimicrob. Agents Chemother. 51:1939–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Crandon JL, Banevicius MA, Nicolau DP. 2009. Pharmacodynamics of tigecycline against phenotypically diverse Staphylococcus aureus isolates in a murine thigh model. Antimicrob. Agents Chemother. 53:1165–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maglio DM, Captiano B, Banevicius MA, Tessier PA, Nightingale CH, Nicolau DP. 2004. Efficacy of clarithromycin against Streptococcus pneumoniae expressing mef(A)-mediated resistance. Int. J. Antimicrob. Agents 23:498–501 [DOI] [PubMed] [Google Scholar]
- 8. Tessier PR, Keel RA, Hagihara M, Crandon JL, Nicolau DP. 2012. Comparative in vivo efficacy of epithelial lining fluid exposures of tedizolid, linezolid, and vancomycin for methicillin-resistant Staphylococcus aureus in a mouse pneumonia model. Antimicrob. Agents Chemother. 56:2342–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Keel RA, Tessier PR, Crandon JL, Nicolau DP. 2012. Comparative efficacies of human simulated exposures of tedizolid and linezolid against Staphylococcus aureus in the murine thigh infection model. Antimicrob. Agents Chemother. 56:4403–4407 [DOI] [PMC free article] [PubMed] [Google Scholar]