

Abstract
Development of daptomycin (DAP) resistance in Enterococcus faecalis has recently been associated with mutations in genes encoding proteins with two main functions: (i) control of the cell envelope stress response to antibiotics and antimicrobial peptides (LiaFSR system) and (ii) cell membrane phospholipid metabolism (glycerophosphoryl diester phosphodiesterase and cardiolipin synthase [cls]). However, the genetic bases for DAP resistance in Enterococcus faecium are unclear. We performed whole-genome comparative analysis of a clinical strain pair, DAP-susceptible E. faecium S447 and its DAP-resistant derivative R446, which was recovered from a single patient during DAP therapy. By comparative whole-genome sequencing, DAP resistance in R446 was associated with changes in 8 genes. Two of these genes encoded proteins involved in phospholipid metabolism: (i) an R218Q substitution in Cls and (ii) an A292G reversion in a putative cyclopropane fatty acid synthase enzyme. The DAP-resistant derivative R446 also exhibited an S333L substitution in the putative histidine kinase YycG, a member of the YycFG system, which, similar to LiaFSR, has been involved in cell envelope homeostasis and DAP resistance in other Gram-positive cocci. Additional changes identified in E. faecium R446 (DAP resistant) included two putative proteins involved in transport (one for carbohydrate and one for sulfate) and three enzymes predicted to play a role in general metabolism. Exchange of the “susceptible” cls allele from S447 for the “resistant” one belonging to R446 did not affect DAP susceptibility. Our results suggest that, apart from the LiaFSR system, the essential YycFG system is likely to be an important mediator of DAP resistance in some E. faecium strains.
INTRODUCTION
The rise of Enterococcus faecium as an important nosocomial pathogen in the last few decades has led to treatment challenges in clinical settings, particularly after the emergence of multidrug-resistant isolates. The current FDA-approved therapies for vancomycin-resistant E. faecium (VRE), namely, quinupristin-dalfopristin and linezolid, have important limitations for the treatment of serious E. faecium infections due to their intrinsic bacteriostatic effects, toxicity profiles, problems with administration, and emergence of resistance (1). Daptomycin (DAP) is a lipopeptide antibiotic with potent in vitro bactericidal activity against VRE. Many clinicians use DAP as first-line off-label therapy in VRE endovascular infections despite the lack of FDA approval, and that efficacy is supported only by retrospective data (2–4). However, the main challenge when using DAP against VRE appears to be development of resistance during therapy, which has been extensively documented (5–10) (although the accepted term is “DAP nonsusceptibility,” we use the term “DAP resistance” throughout for ease of presentation).
The main mechanism of action of DAP involves interactions with the cell membrane in a calcium-dependent manner (11), and our initial genomic studies in Enterococcus faecalis (12) provided compelling evidence for the involvement of mutations in two groups of genes in the development of DAP resistance. The first group include genes encoding a three-component regulatory system (designated LiaFSR for lipid-II interacting antibiotics) that has been characterized in other Gram-positive bacteria (13–18) and is involved in the homeostasis of the cell envelope. Moreover, we recently examined the liaFSR genes in clinical E. faecium bloodstream isolates and found that a high proportion (ca. 80%) of isolates with DAP MICs of 3 to 4 μg/ml had mutations in liaFSR (19). Furthermore, 3 of 6 fully DAP-resistant clinical isolates of E. faecium exhibited point mutations in liaFSR (12), suggesting that, like in E. faecalis, the LiaFSR locus may well be important in the development of DAP resistance in E. faecium.
The second group of genes that appear to be involved in enterococcal DAP resistance encodes enzymes participating in cell membrane phospholipid metabolism, since mutations in genes causing amino acid deletions in two enzymes, a glycerophosphoryl diester phosphodiesterase (GdpD) and cardiolipin synthase (Cls), were associated with DAP resistance (12). Indeed, using an allelic replacement strategy, we were able to show that concomitant mutations in liaF and gdpD were sufficient to increase the DAP MIC above the clinical breakpoint (12). Moreover, an R218Q substitution in the putative Cls was also recently shown to influence DAP susceptibility when the mutated allele was overexpressed in a laboratory strain of E. faecalis (20).
Previous DNA sequence analyses of liaFSR in a clinical pair of DAP-susceptible and DAP-resistant E. faecium strains (S447 and R446, respectively) recovered from a single patient indicated that substitutions in LiaFSR were absent in R446 (12). In the same pair, an R218Q substitution in the putative Cls enzyme was identified in the DAP-resistant derivative R446 (12, 20). In this work, we present whole-genome comparative analysis of this strain pair and, using allelic replacements, evaluate the role of the Cls substitution in DAP resistance.
MATERIALS AND METHODS
Bacterial isolates and MIC determinations.
Bacterial strains used in this study are shown in Table 1. Vancomycin-resistant, DAP-susceptible E. faecium S447 was recovered from the urine of a patient upon admission. The DAP-resistant derivative R446 was recovered from the same patient's bloodstream after 17 days of DAP therapy (7). These isolates exhibited resistance to vancomycin mediated by the vanA gene cluster (7) and had identical pulsed-field gel electrophoresis (PFGE) patterns. Both isolates also had ampicillin MICs of >128 μg/ml. After identifying genetic changes in the putative YycG protein in the DAP-resistant derivative R446 and not in the predicted LiaFSR system (see below), we sought to determine whether mutations in YycG were also present in a clinical strain set of DAP-susceptible E. faecium isolates (TX0133a and derivatives [Table 1]) that were recovered sequentially from the bloodstream of a patient with endocarditis who failed initial DAP monotherapy and have been described before (21). TX0133a (the first isolate recovered from the patient) harbors two subpopulations of vancomycin-susceptible and vancomycin-resistant bacteria (21) (Table 1). DAP MICs were determined by Etest on cation-adjusted Mueller-Hinton agar. Results were interpreted based on breakpoints recommended by the CLSI (22).
Table 1.
E. faecium strains, MICs, and genotypes
| Strain | Source | Etest MIC (μg/ml)d |
YycG change | LiaFSR changec | Reference | |
|---|---|---|---|---|---|---|
| DAP | VAN | |||||
| S447a | Urine | 3 | 256 | No | No | 7 |
| R446a | Blood | 16 | 16 | Yes | No | 7 |
| S447ClsR218Q | Allelic replacement of cls in S447 | 3 | 256 | No | No | This work |
| TX0133ab | Blood | 3 | 256 | Yes | Yes | 21 |
| TX0133a.4 | In vitro derivative of TX0133a obtained outside the inhibition zone of a vancomycin Etest strip | 3 | 1 | Yes | Yes | 21 |
| TX0133a.1 | In vitro derivative of TX0133a obtained within the inhibition zone of a vancomycin Etest strip | 4 | 512 | No | Yes | 21 |
| TX0133bb | Blood after DAP therapy | 4 | 2 | No | Yes | 21 |
| TX0133cb | Blood after VAN therapy | 3 | 512 | No | Yes | 21 |
Clinical strain pair from a patient who developed urinary tract infection (S447) and bacteremia (R446) (7).
Clinical strain set of DAP-susceptible isolates from a different patient with E. faecium endocarditis who failed DAP monotherapy (21).
S105N substitution in LiaS.
DAP, daptomycin; VAN, vancomycin.
Genome sequencing, comparative analysis, and sequence confirmation.
Whole-genome analysis of the E. faecium strain pair (S447 and R446) (7) and TX0133a and derivatives (21) was performed by generating paired-end sequence readings on the Illumina Genome Analyser IIx at Washington University Genome Center, producing 100 base reads and assembled using Velvet (23). In order to predict coding genes, Glimmer 3 (24) and GeneMark (25) were used. Prediction of tRNA, rRNA, and noncoding RNA genes was performed by using tRNAScan (26), RNAmmer (27), and RFAM/internal (28). Gene annotation was performed by a gene annotation pipeline at Washington University Genome Center. BWA/samtools (29) was used to call single nucleotide polymorphisms (SNPs) directly from the Illumina reads, and Nucmer (an algorithm of MUMmer 3.0) was applied to identify SNPs from the contigs. The variants detected were rechecked, and the high-quality ones were included. The SNPs were annotated using ANNOVAR (30). A mutation was defined as a nucleotide change that results in a change of an amino acid in a specific position that is not present in the same position in any of the genomes publicly available (strains that are presumed to be daptomycin susceptible). PCR sequencing of both strands in each gene region was used to confirm the predicted nucleotide changes by Sanger dideoxy-terminator sequencing. Mutations were compared with enterococcal genomes available at http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi (last accessed 12 October 2012) using genome BLAST.
Allelic replacements of cls encoding cardiolipin synthase in the daptomycin-susceptible E. faecium strain S447.
In order to study the role of the cls mutation that had been previously implicated in DAP resistance in E. faecalis (20), we replaced the native gene encoding the Cls enzyme of E. faecium S447 with that of E. faecium R446, which harbors the predicted R218Q substitution. We used the p-chloro-phenylalanine (p-Chl-Phe) sensitivity counterselection system (PheS*) to deliver the mutated alleles using plasmid pHOU3, which confers resistance to chloramphenicol, as described previously (31, 32). Briefly, primers 5′-GCTCTAGAGGAGGAACATTTTTGTGGTA (forward) and 5-CGGGGATCCAGCCAGTTAAACATTTCCTTG (reverse) 500 bp upstream and downstream of the cls mutation, respectively, were used for PCR amplifications using the genomic DNA of R446 (DAP resistant) as the template. PCR products were cloned into pHOU3 using XbaI and BamHI. The plasmid constructs were electroporated into E. faecalis CK111 (31) and subsequently delivered to E. faecium S447 by conjugation (32). First-recombination integrants were selected on plates containing chloramphenicol (15 μg/ml) and vancomycin (32 μg/ml). In order to obtain the desired replacement, first-event integrants were grown on p-chloro-phenylalanine and colonies were subsequently tested by replica plating in the presence and absence of chloramphenicol (32). Chloramphenicol-susceptible mutants were characterized by PFGE and sequencing of the entire cls open reading frames (ORFs). MIC determination was performed by Etest on Mueller-Hinton agar (19).
RESULTS
Genomic analysis of DAP-susceptible (S447) and DAP-resistant (R446) E. faecium clinical strain pair.
Table 2 summarizes the changes identified in our comparative genomic analysis. Using genome analysis, we were able to confirm that, as opposed to previously described DAP-resistant E. faecalis strains, the DAP-resistant E. faecium derivative R446 had no predicted nucleotide substitutions in the genes encoding the LiaFSR system (Table 1) (12). Instead, we were able to identify an S333L substitution in the putative YycG protein (Table 2), which is predicted to be a sensor histidine kinase from the YycF/YycG two-component regulatory system (Fig. 1A). Point mutations in the latter regulatory system have been previously associated with DAP resistance in Staphylococcus aureus (33–36). The substitution was located in the predicted PER-ARNT-SIM (PAS) sensor domain (Fig. 1B). PAS domains were first found in eukaryotes and were named after homology to the Drosophila melanogaster period protein (PER), the aryl hydrocarbon receptor nuclear translocator protein (ARNT), and the Drosophila single-minded protein (SIM) (37, 38). They are widespread components of signal transduction proteins serving as universal signal sensors and interaction hubs. Our data suggest that changes in the sensing activity of the YycG histidine-kinase are likely to play a role in DAP resistance.
Table 2.
Summary of amino acid changes in E. faecium R446 (DAP-resistant) isolate compared to S447 (DAP susceptible)
| Predicted gene product | Nucleotide change in R446 | Predicted amino change in R446 | Comments |
|---|---|---|---|
| Cls | C→T | Arg218→Gln | A transmembrane protein predicted to be involved in phospholipid metabolism; the mutation is within the phospholipase D domain (two conserved phospholipase D domains are usually present in these enzymes) |
| YycG | C→T | Ser333→Leu | Putative histidine kinase of an essential two-component regulatory system that is conserved in most Firmicutes and is involved in cell wall homeostasis |
| Cfa | C→G reversion | Ala292→Glya | Cyclopropane synthase, which catalyzes the conversion of a double bond in a fatty acid to a cyclopropane ring (39) |
| RrmA | A→C | Ser76→Tyr | 23S rRNA methyltransferase (70) |
| SulP | C→T | His71→Tyr | Putative sulfate transporter and anti-sigma factor antagonist (STAS) domain (71) |
| XpaC | C→G reversion | His198→Aspb | Putative protein that mediates the hydrolysis of 5-bromo-4-chloroindolyl-phosphate bonds |
| PTS-EIIA member protein | T→A reversion | Asn118→ Lysc | Member of the phosphoenolpyruvate:sugar phosphotransferase system (PTS), which plays a role in the uptake of carbohydrates (64) |
| Protein with an HD domain | A→G | Arg57→His | The HD domain defines a superfamily of enzymes that may possess phosphohydrolase activity and may be involved in nucleic acid metabolism and signal transduction (41) |
The amino acid change in R446 is an apparent reversion to the wild type, since the consensus sequence in position 292 of putative Cfa enzymes from other E. faecium strains whose genomes have been sequenced is glycine.
The amino acid change in R446 is also a reversion to the wild type, since the consensus sequence in position 198 of the predicted XpaC enzyme from other E. faecium strains whose genomes have been sequenced is aspartate.
The amino acid change in R446 is also a reversion to the wild type, since the consensus sequence in position 118 of the PTS transporters from other E. faecium strains whose genomes have been sequenced is lysine.
Fig 1.
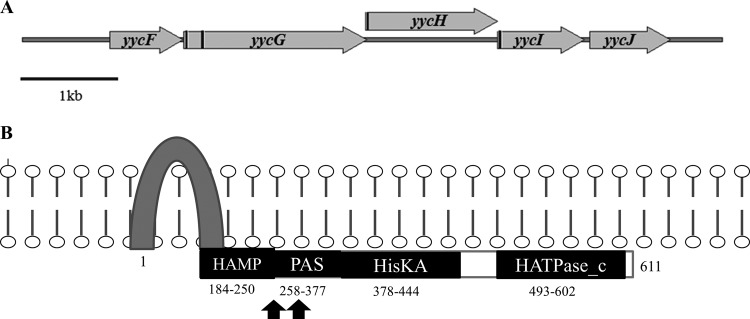
The Yyc system in E. faecium. (A) Structural organization of the orthologous yyc gene cluster in E. faecium; transmembrane coding regions are marked as black bars. Annotations are based on a published report by Türck et al. (49). (B) Predicted domain architecture of the putative YycG histidine kinase and changes found in E. faecium R446, TX0133a, and TX0133a.4, with black arrows denoting the localization of the predicted amino acid changes. Numbers indicate the corresponding amino acids spanning each domain. The HAMP domain is predicted to promote communication between the input and output domains; the PAS sensor domain was named for homology to the PER-ARNT-SIM proteins of D. melanogaster; HisKA, phosphoacceptor domain of histidine kinases; HATPase_c, histidine kinase-like ATPase. The scheme was based in part on published data from Dubrac et al. (56).
In order to evaluate if mutations in the Yyc system were also present in additional E. faecium strains, we had available a strain set of vancomycin-resistant E. faecium isolates (TX0133 series [Table 1]), three of which were sequentially recovered from the bloodstream of the same patient with endocarditis after DAP therapy (21). Of note, DAP monotherapy (6 mg/kg) against the initial isolate (TX0133a [Table 1]) failed to clear the organism from the bloodstream of the patient, although it had a DAP MIC within the susceptibility range (Table 1). Furthermore, TX0133a was previously shown to harbor two subpopulations of vancomycin-resistant and vancomycin-susceptible cells due to the loss of the vanA gene cluster (designated TX0133a.1 and TX0133a.4, respectively [Table 1]) (21). Using whole-genome analysis, we found that unlike E. faecium S447, all TX0133 isolates harbored a substitution in the predicted LiaS protein (S105N, compared to other putative LiaS proteins from E. faecium strains whose genomes are sequenced) but not in Cls. Additionally, we found that TX0133a had a prominent change in the putative YycG, which resulted in a deletion of 12 amino acids in the PAS sensor domain of the histidine kinase. Interestingly, subsequent derivative isolates of TX0133a recovered from the same patient (namely, TX0133b and TX0133c [Table 1]) had DAP MICs of 4 and 3 μg/ml, respectively, but harbored no deletions in YycG and retained the LiaS substitution.
Apart from the previously described R218Q substitution in the putative Cls (12, 20), we were also able to identify a change in a gene predicted to be involved in the synthesis of cyclic fatty acid (CFA). The predicted enzyme, designated Cfa synthase (for cyclopropane fatty acyl synthetase) belongs to the family of enzymes that catalyze the addition of a methylene group from S-adenosyl-l-methionine to a double bond of unsaturated fatty acyl chains in membrane phospholipids (39) to produce cyclic fatty acids; of note, this gene also participates in the synthesis of mycolic acids in Mycobacterium spp. Interestingly, the consensus amino acid in position 292 of predicted Cfa enzymes from those E. faecium isolates whose genomes have been sequenced is glycine. However, an alanine was identified in the parental DAP-susceptible S447 (instead of glycine). Thus, the “consensus wild-type” Gly292 was found in the DAP-resistant derivative R446, which suggests that this isolate reverted to the consensus wild-type after exposure to DAP. Interestingly, we also found a mutation in the putative Cfa (T299S) in TX0133a, which was recovered from a patient who failed DAP therapy (but not subsequent isolates [Table 1]). Taken together, our findings confirm that genes associated with cell membrane phospholipid metabolism are strongly associated with development of in vivo DAP resistance in enterococci.
Our genomic analyses (confirmed by PCR sequencing) also revealed 5 additional mutations in R446 compared with the DAP-susceptible parental strain S447 (Table 2). Most of these genes have not been previously associated with DAP resistance, and sequence comparisons yielded predicted functions that have no obvious relationship with the DAP mechanism of action. Two of these genes encode transporters: (i) sulP, which codes for a putative sulfate transporter that has 9 predicted transmembrane helices and harbors a H71Y substitution located seven amino acids after the end of the second predicted transmembrane domain, and (ii) a gene encoding a member of the phosphoenolpyruvate-dependent sugar phosphotransferase system (PTS EIIA 2), which has an N118K “reversion to wild type” in a hydrophilic domain of the predicted protein (40) (Table 2). The remaining three genes appear to encode putative proteins involved in general metabolism (Table 2) including a putative methyltransferase (RrmA with an S77Y substitution); XpaC, which is predicted to be a 5-bromo-4-chloroindolyl phosphate hydrolysis protein that is involved in amino acid metabolism (H198D reversion); and a predicted metal-dependent phosphohydrolase that harbors an R57H substitution in the HD domain that has been implicated in nucleic acid metabolism (41) (Table 2). Interestingly, we found no mutations in the above homologous genes belonging to the TX0133 isolates except for a predicted N88D substitution in XpaC, which is present in only one other E. faecium strain whose genome has been sequenced (E. faecium E1679).
The R218Q substitution in cardiolipin synthase (Cls) does not affect DAP susceptibility.
The R218Q substitution in cardiolipin synthase was previously linked with DAP resistance in an in vitro-derived DAP-resistant E. faecalis (20) and in clinical isolates of DAP-resistant E. faecium (12). Thus, we first sought to evaluate the role of this substitution alone in the DAP-susceptible E. faecium S447. Thus, we delivered the mutated cls allele from R446 to the chromosome of S447 using the PheS* counterselection system. This strategy was chosen in order to maintain the gene in its native location in the chromosome without increasing the copy number and directly investigate the actual effect of the mutation on DAP susceptibility. As shown in Table 1, introduction of the mutated allele encoding the mutated Cls enzyme did not have any effect on DAP susceptibility in E. faecium S447, indicating that the single R218Q substitution is not sufficient to confer DAP resistance.
DISCUSSION
We previously reported that development of the DAP resistance phenotype among in vivo-selected E. faecalis was associated with two predicted amino acid substitutions (12). The first was a deletion of Ile in position 177 of the predicted transmembrane protein LiaF, a member of a three-component regulatory system involved in cell envelope responses to antibiotics and antimicrobial peptides in B. subtilis (42). The second change was a deletion of Ile in position 170 in a putative GdpD enzyme, which is likely to affect phosphatidylglycerol metabolism, a crucial component of cell membranes and involved in phospholipid metabolism (12). Additionally, a third mutation in a gene encoding a putative Cls enzyme that is also likely to participate in cell membrane phospholipid metabolism, was also identified (deletion of Lys61), although the role of this specific substitution in DAP resistance has not been studied. Palmer et al. (20) showed that a different Cls mutation (R218Q), located in the phospholipase D domain of the enzyme, affected DAP susceptibility when it was overexpressed in a laboratory strain of E. faecalis. Furthermore, the same R218Q substitution was found in other unrelated clinical isolates of DAP-resistant E. faecium (12), supporting the notion that this enzyme also contributes to DAP resistance in enterococci.
In order to determine the overall genetic basis of DAP resistance in E. faecium, we performed whole-genome comparison of a clinical strain pair of DAP-susceptible (S447) and DAP-resistant (R446) isolates of E. faecium recovered from the same patient before and after DAP therapy (7). We and others (12, 20) had shown that the DAP-resistant isolate R446 harbored the R218Q substitution in the putative Cls enzyme but lacked changes in the predicted LiaFSR system or in the GdpD enzyme; our current results confirmed those previous observations. Thus, we sought to further investigate the contributory role of the Cls R218Q substitution in DAP resistance by performing an allelic replacement of the “susceptible” cls in S447 with that belonging to the DAP-resistant isolate, R446. Interestingly, the allelic switch had no effect on DAP susceptibility in the S447 derivative, with DAP MICs being identical to that of the parental S447 (Table 1). This finding is consistent with our previous observations in E. faecalis, which indicated that a single mutation in the gene encoding another phospholipid enzyme (GdpD) did not affect DAP susceptibility (12), suggesting that amino acid changes in enzymes regulating phospholipid metabolism may not be sufficient to confer DAP resistance unless they are accompanied by additional mutations in other genes (i.e., genes controlling cell envelope homeostasis). An alternative scenario is that the mutated allele is not properly transcribed. However, since we placed the allele in its native location on the chromosome (without modifying the locus), it is unlikely that transcription was affected. Of note, our results are in conflict with those of Palmer et al. (20), who showed that the R218Q substitution in Cls was sufficient to change DAP susceptibility. The discrepancy may be due to the fact that Palmer et al. overexpressed the mutated allele in a multicopy plasmid in the background of a laboratory strain that harbors a “wild-type” cls copy in the chromosome. This differential expression may have caused important changes in cell membrane metabolism that might not be physiological, although their results suggest that increased production of the enzyme affects the susceptibility to DAP. Our mutagenesis strategy placed the mutated allele in the original chromosomal location, which represents a more accurate reflection of the change that occurred during therapy. Another important difference between our work and that of Palmer et al. is that the effect of the Cls substitution may be different in E. faecium and in E. faecalis since membrane phospholipid composition has been shown to differ in the two species (43). Indeed, we recently showed that the phospholipid content of E. faecalis membranes is more complex than that of E. faecium, with E. faecalis containing three different species of amino-containing phospholipids (only lysyl-PG was identified in E. faecium). Of note, striking reductions in PG content were associated with development of DAP resistance in both species (43).
Additionally, we found another gene difference between S447 and R446 in an enzyme that may affect phospholipid metabolism, namely, an A292G change in a cyclopropane fatty acid synthase (Cfa), an enzyme that is involved in the synthesis of cyclic fatty acids (CFA). CFA are important components of cell membrane phospholipids that may affect fluidity and stabilization of the cell membrane (44) and could well alter DAP's interaction with its main cell membrane target. The substitution found appears to be a reversion to the wild type in the DAP-resistant isolate R446, which may have led to a significant increase in cyclic fatty acid in cell membrane of R446 (43), suggesting that the change may have occurred as a compensatory response following DAP exposures (perhaps due to alterations in cardiolipin cell membrane composition or distribution). Indeed, CFA may alter the biophysical properties of the membrane and have been implicated in the response to (i) acid stress in Escherichia coli (44) and (ii) the presence of environmental toxic compounds in Pseudomonas aeruginosa (45); in addition, CFAs are essential for cell viability, drug resistance, and cell wall integrity in Mycobacterium tuberculosis (46). Interestingly, CFA deficiency has been associated with a decrease in viability in the presence of low pH and increasing concentrations of NaCl in E. faecalis (47, 48).
Since our allelic swap strategy indicated that the cls mutation alone was not sufficient to affect DAP susceptibility and that the DAP-resistant isolate R446 did not exhibit mutations in liaFSR, we hypothesized that other genes involved in cell envelope homeostasis may play a role in the evolution of the DAP resistance phenotype in our E. faecium clinical strain pair. Using the whole-genome comparisons, we were able to identify and confirm an S333L substitution in the putative PAS-sensing domain of the histidine kinase YycG (Fig. 1B) of R446. yycG is a member of the yyc system, an essential cell envelope regulon that has been identified in most clinically important Gram-positive organisms with low G+C content (49). The genes encoding YycFG proteins are part of this regulon and regulate a two-component regulatory system; YycF is the response regulator and YycG is the histidine kinase sensor of the system. Apart from these two genes, the yyc gene locus in our E. faecium clinical strain pair has additional open reading frames that encode orthologs of putative proteins designated YycH, YycI, and YycJ (Fig. 1A), similar to what has been shown previously in E. faecalis (49). The E. faecium YycG homologue contains (i) a HAMP domain (for a domain present in histidine kinases, adenylyl cyclases, methyl-accepting proteins, and phosphatases), which promotes two-way conformational communication between the input and output domains of many bacterial signaling proteins (50), (ii) a PAS domain, (iii) a histidine kinase domain, and (iv) an ATPase domain (Fig. 1B). Interestingly, in nongrowing Bacillus subtilis cells, the kinase activity of YycG is inhibited by the transmembrane proteins YycH and YycJ (51–55). In actively growing B. subtilis cells, YycG is dissociated from YycH and YycI and localized in the nascent septum with the cell wall machinery (52, 53). Moreover, in S. aureus, YycG appears to coordinate cell wall homeostasis by affecting the autolysin transcription (51, 52, 56, 57) and also to respond to changes in membrane fluidity (49). Furthermore, mutations in yycG and yycH have been associated with development of both vancomycin and DAP resistance (33, 58–60) in S. aureus. Additionally, the Yyc system, unlike LiaFSR, is essential for bacterial growth. Therefore, it is tempting to speculate that the Yyc system in E. faecium is an alternative pathway to LiaFSR to orchestrate cell envelope changes triggered by the attack of antimicrobial peptides; thus, modulation of this system may be associated with development of in vivo DAP resistance in some strains of E. faecium.
In order to support the above hypothesis, we analyzed the putative Yyc system of previously characterized isolates of vancomycin-resistant E. faecium that were recovered from a single patient with endocarditis who failed DAP monotherapy (TX0133 derivatives [Table 1]) (21). Of note, the initial isolate TX0133a exhibited heterogenous resistance to vancomycin (Table 1) and initial therapy with DAP monotherapy failed to clear the patient's bloodstream, but the patient subsequently responded to the combination of DAP (8 mg/kg), ampicillin, and gentamicin (21). Interestingly, DAP MICs of all isolates were within the susceptibility range but ranged from 3 to 4 μg/ml on Mueller-Hinton (Etest). A novel mutation in LiaS (S105N substitution) was identified in all isolates belonging to the TX0133 series. Additionally, our genomic analyses of these isolates revealed that TX0133a (the first isolate recovered from the patient before exposure to DAP therapy) and TX0133a.4 (vancomycin-susceptible derivative) harbor a deletion of 12 amino acids in the PAS domain of the predicted YycG histidine kinase, whereas the remaining isolates lacked mutations in yycG (Table 1). Moreover, we were able to detect a mutation in the putative Cfa enzyme in TX0133a. Thus, our findings provide some evidence that, similar to LiaFSR, modulation of the Yyc system with changes in enzymes associated with phospholipid metabolism may be an alternative pathway for developing DAP resistance in enterococci, and this also supports our previous hypothesis that E. faecium isolates with DAP MICs between 3 and 4 μg/ml by Etest on Mueller-Hinton agar (close to the breakpoint) are likely to harbor genetic changes that may predispose to DAP failure (19). The Yyc system appears to also be essential in E. faecium since repeated attempts to produce disruptions in this system in S447 were unsuccessful (data not shown), similar to what has been previously reported in B. subtilis, S. aureus, and E. faecalis (61–63).
Five other ORFs were altered in DAP-resistant R446 compared to the DAP-susceptible S447 (Table 2). The role of these mutations is less clear, although they may reflect changes in cell metabolism and transport that optimize the global cellular response to antimicrobial lipopeptide exposures. Since the predicted functions of most of these genes are not directly associated with the main membrane target of DAP and they have not been associated with DAP resistance, they are likely to play a more minor role in development of in vivo DAP resistance in enterococci, if at all. Nonetheless, changes in the HD domain (which define proteins that may possess phosphohydrolase activity and may be involved in nucleic acid metabolism and signal transduction) (41) of a transmembrane protein were previously described after in vitro selection of DAP resistance in E. faecalis (20). Similarly, the phosphoenolpyruvate:sugar phosphotransferase system (PTS), which plays a role in the uptake of carbohydrates (64), has been associated with bacteriocin resistance in Listeria monocytogenes and E. faecalis (65–68) (Table 2).
Of note, recent whole-genome comparison of three clinically isolated strains of E. faecium obtained before and after DAP therapy also identified mutations in cls and genes encoding components of the phosphoenolpyruvate:sugar phosphotransferase system associated with DAP resistance, although no changes in yycFG and liaFSR were identified (69). However, comparison of the liaFSR and yycFG sequences of the E. faecium isolates with other E. faecium genomes publicly available was not performed, and thus it is difficult to determine if the initial isolate harbored variations from the wild-type consensus sequences of LiaFSR and/or YycFG systems. Thus, the role of genes regulating cell envelope homeostasis is less clear in this set of isolates. It is unknown whether the observed genetic changes are strain specific and/or related to conditions of DAP exposure at the site of infection (e.g., bloodstream and/or urine).
In summary, the major pathway for development of DAP resistance in our E. faecium clinical strain pair is likely to be the result of changes in an essential two-component regulatory system (YycFG) that regulates cell envelope homeostasis and division (analogous to LiaFSR), concomitant with mutations in genes encoding enzymes involved in cell membrane phospholipid metabolism. Our findings suggest that a common strategy for development of DAP resistance in enterococci is to modulate genes that orchestrate changes in the cell envelope plus cell membrane phospholipid metabolism, although alternative genetic and biochemical pathways may exist in order to fully express the resistant phenotype.
ACKNOWLEDGMENTS
This work was supported by NIH grant R01 AI093749 (C.A.A.) from the National Institute of Allergy and Infectious Diseases (NIAID). A.S.B. is supported by NIH grant R01 AI39108, and Y.S. is supported by NIH R01 AI080714 (both from NIAID). This work was also supported in part by a John S. Dunn Foundation Collaborative Research Award. B.E.M. was supported in part by NIH grant R01 AI067861. Genome sequencing was performed under the support of grant 1U54 HG004968 from NIH to G.M.W.
We are indebted to James H. Jorgensen for providing the enterococcal isolates.
C.A.A. has received lecture fees, research support, and consulting fees from Pfizer, Inc., and Cubist Pharmaceuticals and research support from Forrest Pharmaceuticals and Theravance, Inc., and has served as speaker for Pfizer and Novartis. A.S.B. has received grant support from Astellas Pharmaceuticals, Cubist Pharmaceuticals, Lytix Biopharma, and Polymedix Pharmaceuticals. B.E.M. has grant support from Johnson & Johnson, Astellas, Palumed, Intercell, and Cubist and has served as a consultant for Astellas (Theravance), Cubist, Targanta Therapeutics Corporation, Pfizer, Rib-X, AstraZeneca, and Durata Therapeutics. None of these entities participated in any way in the design or execution of the research or in the writing of the manuscript. All other authors report no conflicts of interest.
Footnotes
Published ahead of print 31 October 2012
REFERENCES
- 1. Arias CA, Contreras GA, Murray BE. 2010. Management of multidrug-resistant enterococcal infections. Clin. Microbiol. Infect. 16:555–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cantón R, Ruiz-Garbajosa P, Chaves RL, Johnson AP. 2010. A potential role for daptomycin in enterococcal infections: what is the evidence? J. Antimicrob. Chemother. 65:1126–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Crank CW, Scheetz MH, Brielmaier B, Rose WE, Patel GP, Ritchie DJ, Segreti J. 2010. Comparison of outcomes from daptomycin or linezolid treatment for vancomycin-resistant enterococcal bloodstream infection: a retrospective, multicenter, cohort study. Clin. Ther. 32:1713–1719 [DOI] [PubMed] [Google Scholar]
- 4. Twilla JD, Finch CK, Usery JB, Gelfand MS, Hudson JQ, Broyles JE. 2012. Vancomycin-resistant Enterococcus bacteremia: an evaluation of treatment with linezolid or daptomycin. J. Hosp. Med. 7:243–248 [DOI] [PubMed] [Google Scholar]
- 5. Fraher MH, Corcoran GD, Creagh S, Feeney E. 2007. Daptomycin-resistant Enterococcus faecium in a patient with no prior exposure to daptomycin. J. Hosp. Infect. 65:376–378 [DOI] [PubMed] [Google Scholar]
- 6. Lesho EP, Wortmann GW, Craft D, Moran KA. 2006. De novo daptomycin nonsusceptibility in a clinical isolate. J. Clin. Microbiol. 44:673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lewis JS, II, Owens A, Cadena J, Sabol K, Patterson JE, Jorgensen JH. 2005. Emergence of daptomycin resistance in Enterococcus faecium during daptomycin therapy. Antimicrob. Agents Chemother. 49:1664–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Green MR, Anasetti C, Sandin RL, Rolfe NE, Greene JN. 2006. Development of daptomycin resistance in a bone marrow transplant patient with vancomycin-resistant Enterococcus durans. J. Oncol. Pharm. Pract. 12:179–181 [DOI] [PubMed] [Google Scholar]
- 9. Kanafani ZA, Federspiel JJ, Fowler VG., Jr 2007. Infective endocarditis caused by daptomycin-resistant Enterococcus faecalis: a case report. Scand. J. Infect. Dis. 39:75–77 [DOI] [PubMed] [Google Scholar]
- 10. Munoz-Price SL, Quinn LKJP. 2005. Emergence of resistance to daptomycin during treatment of vancomycin-resistant Enterococcus faecalis infection. Clin. Infect. Dis. 41:565–566 [DOI] [PubMed] [Google Scholar]
- 11. Pogliano J, Pogliano N, Silverman J. 2012. Daptomycin-mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins. J. Bacteriol. 194:4494–4504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arias CA, Panesso D, McGrath DM, Qin X, Mojica MF, Miller C, Diaz L, Tran TT, Rincon S, Barbu EM, Reyes J, Roh JH, Lobos E, Sodergren E, Pasqualini R, Arap W, Quinn JP, Shamoo Y, Murray BE, Weinstock GM. 2011. Genetic basis for in vivo daptomycin resistance in enterococci. N. Engl. J. Med. 365:892–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jordan S, Junker A, Helmann JD, Mascher T. 2006. Regulation of LiaRS-dependent gene expression in Bacillus subtilis: identification of inhibitor proteins, regulator binding sites, and target genes of a conserved cell envelope stress-sensing two-component system. J. Bacteriol. 188:5153–5166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Suntharalingam P, Senadheera MD, Mair RW, Levesque CM, Cvitkovitch DG. 2009. The LiaFSR system regulates the cell envelope stress response in Streptococcus mutans. J. Bacteriol. 191:2973–2984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wolf D, Kalamorz F, Wecke T, Juszczak A, Mader U, Homuth G, Jordan S, Kirstein J, Hoppert M, Voigt B, Hecker M, Mascher T. 2010. In-depth profiling of the LiaR response of Bacillus subtilis. J. Bacteriol. 192:4680–4693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eldholm V, Gutt B, Johnsborg O, Bruckner R, Maurer P, Hakenbeck R, Mascher T, Havarstein LS. 2010. The pneumococcal cell envelope stress-sensing system LiaFSR is activated by murein hydrolases and lipid II-interacting antibiotics. J. Bacteriol. 192:1761–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McCallum N, Meier PS, Heusser R, Berger-Bachi B. 2011. Mutational analyses of open reading frames within the vraSR operon and their roles in the cell wall stress response of Staphylococcus aureus. Antimicrob. Agents Chemother. 55:1391–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mehta S, Cuirolo AX, Plata KB, Riosa S, Silverman JA, Rubio A, Rosato RR, Rosato AE. 2012. VraSR two-component regulatory system contributes to mprF-mediated decreased susceptibility to daptomycin in in vivo-selected clinical strains of methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 56:92–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Munita JM, Panesso D, Diaz L, Truc TT, Reyes J, Wanger A, Murray BE, Arias CA. 2012. Correlation between mutations in liaFSR of Enterococcus faecium and minimal inhibitory concentration of daptomycin: revisiting daptomycin breakpoints. Antimicrob. Agents Chemother. 56:4354–4359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Palmer KL, Daniel A, Hardy C, Silverman J, Gilmore MS. 2011. Genetic basis for daptomycin resistance in enterococci. Antimicrob. Agents Chemother. 55:3345–3356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arias CA, Torres HA, Singh KV, Panesso D, Moore J, Wanger A, Murray BE. 2007. Failure of daptomycin monotherapy for endocarditis caused by an Enterococcus faecium strain with vancomycin-resistant and vancomycin-susceptible subpopulations and evidence of in vivo loss of the vanA gene cluster. Clin. Infect. Dis. 45:1343–1346 [DOI] [PubMed] [Google Scholar]
- 22. CLSI 2011. Performance standards for antimicrobial susceptibility testing: twenty-first informational supplement. CLSI document MS100-S21. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 23. Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Delcher AL, Bratke KA, Powers EC, Salzberg SL. 2007. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23:673–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Besemer J, Borodovsky M. 1999. Heuristic approach to deriving models for gene finding. Nucleic Acids Res. 27:3911–3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. 2007. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35:3100–3108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A. 2005. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33:D121–D124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang K, Li M, Hakonarson H. 2010. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38:e164 doi:10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kristich CJ, Chandler JR, Dunny GM. 2007. Development of a host-genotype-independent counterselectable marker and a high-frequency conjugative delivery system and their use in genetic analysis of Enterococcus faecalis. Plasmid 57:131–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Panesso D, Montealegre MC, Rincon S, Mojica MF, Rice LB, Singh KV, Murray BE, Arias CA. 2011. The hylEfm gene in pHylEfm of Enterococcus faecium is not required in pathogenesis of murine peritonitis. BMC Microbiol. 11:20 doi:10.1186/1471-2180-11-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mishra NN, McKinnell J, Yeaman MR, Rubio A, Nast CC, Chen L, Kreiswirth BN, Bayer AS. 2011. In vitro cross-resistance to daptomycin and host defense cationic antimicrobial peptides in clinical methicillin-resistant Staphylococcus aureus isolates. Antimicrob. Agents Chemother. 55:4012–4018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Peleg AY, Miyakis S, Ward DV, Earl AM, Rubio A, Cameron DR, Pillai S, Moellering RC, Eliopoulos GM. 2012. Whole genome characterization of the mechanisms of daptomycin resistance in clinical and laboratory derived isolates of Staphylococcus aureus. PLoS One 7:e28316 doi:10.1371/journal.pone.0028316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Friedman L, Alder JD, Silverman JA. 2006. Genetic changes that correlate with reduced susceptibility to daptomycin in Staphylococcus aureus. Antimicrob. Agents Chemother. 50:2137–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Howell A, Dubrac S, Andersen KK, Noone D, Fert J, Msadek T, Devine K. 2003. Genes controlled by the essential YycG/YycF two-component system of Bacillus subtilis revealed through a novel hybrid regulator approach. Mol. Microbiol. 49:1639–1655 [DOI] [PubMed] [Google Scholar]
- 37. Hefti MH, Francoijs KJ, de Vries SC, Dixon R, Vervoort J. 2004. The PAS fold. A redefinition of the PAS domain based upon structural prediction. Eur. J. Biochem. 271:1198–1208 [DOI] [PubMed] [Google Scholar]
- 38. Moglich A, Ayers RA, Moffat K. 2009. Structure and signaling mechanism of Per-ARNT-Sim domains. Structure 17:1282–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Grogan DW, Cronan JE., Jr 1997. Cyclopropane ring formation in membrane lipids of bacteria. Microbiol. Mol. Biol. Rev. 61:429–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Deutscher J, Francke C, Postma PW. 2006. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol. Mol. Biol. Rev. 70:939–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aravind L, Koonin EV. 1998. The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem. Sci. 23:469–472 [DOI] [PubMed] [Google Scholar]
- 42. Mascher T, Zimmer SL, Smith TA, Helmann JD. 2004. Antibiotic-inducible promoter regulated by the cell envelope stress-sensing two-component system LiaRS of Bacillus subtilis. Antimicrob. Agents Chemother. 48:2888–2896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mishra NN, Bayer AS, Tran TT, Shamoo Y, Mileykovskaya E, Dowhan W, Guan Z, Arias CA. 2012. Daptomycin resistance in enterococci is associated with distinct alterations of cell membrane phospholipid content. PLoS One 7:e43958 doi:10.1371/journal.pone.0043958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chang YY, Cronan JE., Jr 1999. Membrane cyclopropane fatty acid content is a major factor in acid resistance of Escherichia coli. Mol. Microbiol. 33:249–259 [DOI] [PubMed] [Google Scholar]
- 45. Pini CV, Bernal P, Godoy P, Ramos JL, Segura A. 2009. Cyclopropane fatty acids are involved in organic solvent tolerance but not in acid stress resistance in Pseudomonas putida DOT-T1E. Microb. Biotechnol. 2:253–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Barkan D, Liu Z, Sacchettini JC, Glickman MS. 2009. Mycolic acid cyclopropanation is essential for viability, drug resistance, and cell wall integrity of Mycobacterium tuberculosis. Chem. Biol. 16:499–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jungkind DL, Wood RC. 1974. Physiological differences between cyclopropane fatty acid-deficient mutants and the parent strain of Streptococcus faecalis. Biochim. Biophys. Acta 337:298–310 [DOI] [PubMed] [Google Scholar]
- 48. Jungkind DL, Wood RC. 1974. Factors involved in the synthesis of cyclopropane fatty acids by Streptococcus faecalis. Biochim. Biophys. Acta 337:286–297 [DOI] [PubMed] [Google Scholar]
- 49. Türck M, Bierbaum G. 2012. Purification and activity testing of the full-length YycFGHI proteins of Staphylococcus aureus. PLoS One 7:e30403 doi:10.1371/journal.pone.0030403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou Q, Ames P, Parkinson JS. 2011. Biphasic control logic of HAMP domain signalling in the Escherichia coli serine chemoreceptor. Mol. Microbiol. 80:596–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fukushima T, Furihata I, Emmins R, Daniel RA, Hoch JA, Szurmant H. 2011. A role for the essential YycG sensor histidine kinase in sensing cell division. Mol. Microbiol. 79:503–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fukushima T, Szurmant H, Kim EJ, Perego M, Hoch JA. 2008. A sensor histidine kinase co-ordinates cell wall architecture with cell division in Bacillus subtilis. Mol. Microbiol. 69:621–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Szurmant H, Bu L, Brooks CL, III, Hoch JA. 2008. An essential sensor histidine kinase controlled by transmembrane helix interactions with its auxiliary proteins. Proc. Natl. Acad. Sci. U. S. A. 105:5891–5896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Szurmant H, Mohan MA, Imus PM, Hoch JA. 2007. YycH and YycI interact to regulate the essential YycFG two-component system in Bacillus subtilis. J. Bacteriol. 189:3280–3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Szurmant H, White RA, Hoch JA. 2007. Sensor complexes regulating two-component signal transduction. Curr. Opin. Struct. Biol. 17:706–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dubrac S, Bisicchia P, Devine KM, Msadek T. 2008. A matter of life and death: cell wall homeostasis and the WalKR (YycGF) essential signal transduction pathway. Mol. Microbiol. 70:1307–1322 [DOI] [PubMed] [Google Scholar]
- 57. Winkler ME, Hoch JA. 2008. Essentiality, bypass, and targeting of the YycFG (VicRK) two-component regulatory system in gram-positive bacteria. J. Bacteriol. 190:2645–2648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gardete S, Kim C, Hartmann BM, Mwangi M, Roux CM, Dunman PM, Chambers HF, Tomasz A. 2012. Genetic pathway in acquisition and loss of vancomycin resistance in a methicillin resistant Staphylococcus aureus (MRSA) strain of clonal type USA300. PLoS Pathog. 8:e1002505 doi:10.1371/journal.ppat.1002505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mwangi MM, Wu SW, Zhou Y, Sieradzki K, de Lencastre H, Richardson P, Bruce D, Rubin E, Myers E, Siggia ED, Tomasz A. 2007. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc. Natl. Acad. Sci. U. S. A. 104:9451–9456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jansen A, Turck M, Szekat C, Nagel M, Clever I, Bierbaum G. 2007. Role of insertion elements and yycFG in the development of decreased susceptibility to vancomycin in Staphylococcus aureus. Int. J. Med. Microbiol. 297:205–215 [DOI] [PubMed] [Google Scholar]
- 61. Fabret C, Hoch JA. 1998. A two-component signal transduction system essential for growth of Bacillus subtilis: implications for anti-infective therapy. J. Bacteriol. 180:6375–6383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Martin PK, Li T, Sun D, Biek DP, Schmid MB. 1999. Role in cell permeability of an essential two-component system in Staphylococcus aureus. J. Bacteriol. 181:3666–3673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hancock LE, Perego M. 2004. Systematic inactivation and phenotypic characterization of two-component signal transduction systems of Enterococcus faecalis V583. J. Bacteriol. 186:7951–7958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Romano AH, Saier MH, Jr, Harriott OT, Reizer J. 1990. Physiological studies on regulation of glycerol utilization by the phosphoenolpyruvate:sugar phosphotransferase system in Enterococcus faecalis. J. Bacteriol. 172:6741–6748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gravesen A, Warthoe P, Knochel S, Thirstrup K. 2000. Restriction fragment differential display of pediocin-resistant Listeria monocytogenes 412 mutants shows consistent overexpression of a putative beta-glucoside-specific PTS system. Microbiology 146:1381–1389 [DOI] [PubMed] [Google Scholar]
- 66. Vadyvaloo V, Snoep JL, Hastings JW, Rautenbach M. 2004. Physiological implications of class IIa bacteriocin resistance in Listeria monocytogenes strains. Microbiology 150:335–340 [DOI] [PubMed] [Google Scholar]
- 67. Dalet K, Cenatiempo Y, Cossart P, Hechard Y. 2001. A sigma(54)-dependent PTS permease of the mannose family is responsible for sensitivity of Listeria monocytogenes to mesentericin Y105. Microbiology 147:3263–3269 [DOI] [PubMed] [Google Scholar]
- 68. Hechard Y, Pelletier C, Cenatiempo Y, Frere J. 2001. Analysis of sigma(54)-dependent genes in Enterococcus faecalis: a mannose PTS permease (EII(Man)) is involved in sensitivity to a bacteriocin, mesentericin Y105. Microbiology 147:1575–1580 [DOI] [PubMed] [Google Scholar]
- 69. Humphries RM, Kelesidis T, Tewhey R, Rose WE, Schork N, Nizet V, Sakoulas G. 2012. Genotypic and phenotypic evaluation of the evolution of high-level daptomycin nonsusceptibility in vancomycin-resistant Enterococcus faecium. Antimicrob. Agents Chemother. 56:6051–6053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gustafsson C, Persson BC. 1998. Identification of the rrmA gene encoding the 23S rRNA m1G745 methyltransferase in Escherichia coli and characterization of an m1G745-deficient mutant. J. Bacteriol. 180:359–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sharma AK, Rigby AC, Alper SL. 2011. STAS domain structure and function. Cell Physiol. Biochem. 28:407–422 [DOI] [PMC free article] [PubMed] [Google Scholar]