

Abstract
Adenylate kinase (AK) is a ubiquitous intracellular enzyme that is released into the extracellular space upon cell lysis. We have shown that AK release serves as a useful reporter of bactericidal agent activity and can be exploited for antimicrobial screening purposes. The AK assay exhibits improved sensitivity over that of growth-based assays and can detect agents that are active against bacteria in clinically relevant growth states that are difficult to screen using conventional approaches, such as small colony variants (SCV) and bacteria within established biofilms. The usefulness of the AK assay was validated by screening a library of off-patent drugs for agents that exhibit antimicrobial properties toward a variety of bacterial species, including Escherichia coli and all members of the “ESKAPE” pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species). The assay detected antibiotics within the library that were expected to be active against the organism screened. Moreover, 38 drugs with no previously reported antibacterial activity elicited AK release. Four of these were acquired, and all were verified to exhibit antimicrobial activity by standard susceptibility testing. Two of these molecules were further characterized. The antihistamine, terfenadine, was active against S. aureus planktonic, SCV population, and biofilm-associated cells. Tamoxifen, an estrogen receptor antagonist, was active toward E. faecium in vitro and also reduced E. faecium pathogenesis in a Galleria mellonella infection model. Our data demonstrate that the AK assay provides an attractive screening approach for identifying new antimicrobial agents. Further, terfenadine and tamoxifen may represent novel antimicrobial drug development scaffolds.
INTRODUCTION
Antibiotics are life-saving medications that represent one of the most important advances of modern medicine. However, the ongoing emergence of organisms that are resistant to clinically used agents indicates that, like microbial evolution, anti-infective drug development is an ongoing process. Unfortunately, regulatory and economic pressures have led to the downsizing of pharmaceutical anti-infective drug discovery efforts and consequently contributed to a void in the antibiotic development pipeline. In response to this state of affairs, the Infectious Diseases Society of America and other agencies have called for increasing efforts to identify and develop new antibiotics. A particular emphasis has been placed on developing strategies for the treatment of the so-called “ESKAPE” pathogens: Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species (1–3). This set of organisms frequently causes health care-associated bacterial infections and can escape the effects of most currently available antibiotics.
Recently, a number of authors have described the challenges inherent in modern antibiotic drug discovery that must be considered during antimicrobial development. One particularly vexing problem is that screening approaches tend to detect only the most potent antimicrobials and consequently rediscover stereotypical molecules over and over again. In response to this problem, one prominent author has proposed that “the screening paradigm should focus on seeing what had previously not been seen” (4).
The most successful and widely applied method to identify agents with antibacterial activity has been whole-cell, bacterial growth assays. In this approach, libraries of small molecules or natural products are screened for agents that limit bacterial growth. However, growth-based assays have limitations (4, 5). For example, the growth or no-growth readout has a limited dynamic range. This is likely to contribute to the aforementioned rediscovery problem because growth assays lack the sensitivity required to detect antimicrobial molecules that are present in low concentrations within complex natural product extract libraries or compounds with limited antimicrobial activity. While the latter would obviously not represent a molecule that could be directly translated to clinical use, these low-activity hits could provide structurally novel scaffolds suitable for medicinal chemistry-based optimization. In addition, traditional growth-based assays are not readily amenable to screening for agents that target bacteria within certain clinically relevant bacterial growth states, such as established biofilms and small-colony variants.
To address these limitations, we have developed a high-throughput screen (HTS)-compatible whole-cell assay to detect agents that directly kill bacteria. The assay is based on the release of intracellular adenylate kinase (AK) into culture medium as a reporter of bacterial cell death. We show that the AK assay exhibits improved sensitivity over that of conventional whole-cell growth assays and displays specificity for bactericidal agents. Further, we establish that the assay can be used to screen for agents that kill small-colony-variant bacteria and bacteria within established biofilms.
To validate the AK assay as an HTS-compatible screening platform, we screened the Prestwick library of off-patent drugs against E. coli and each of the ESKAPE pathogens. This library contains representative examples of nearly all classes of antibiotics, and we identified the bactericidal agents within the library that were expected to be active against the organism screened. Additionally, we identified 38 agents with no previously reported antibiotic activity. Traditional MIC testing confirmed the antimicrobial properties of many of these molecules, suggesting that they could be repurposed as antimicrobials or serve as lead molecules for antibiotic development. Consistent with that prediction, we showed that one of these compounds, tamoxifen, is active against E. faecium in a Galleria mellonella model of infection. Further, we showed that terfenadine is active against planktonic, small-colony variant, and biofilm-associated S. aureus. Taken together, our data demonstrate that the AK assay provides a general approach to screening for new antimicrobial agents active against a variety of pathogens during planktonic and other disease-associated growth states.
MATERIALS AND METHODS
Bacterial growth conditions.
The bacterial species and strains used in these experiments are listed in Table 1. S. aureus strain UAMS-1112 (generous gift from M. Smeltzer, University of Arkansas Medical Center) is a stable small-colony variant of the common laboratory S. aureus strain 8325-4, which harbors a hemB deletion. Unless otherwise noted, bacteria were grown for 16 h in Mueller-Hinton (MH) (Becton, Dickinson, Franklin Lakes, NJ) or brain heart infusion (BHI) (Becton, Dickinson) medium at 37°C on a rotary shaker at 225 rotations per min (rpm) and then used to inoculate (1:100) fresh medium and processed, as described below.
Table 1.
Bacterial strains used in this study
| Species | Strain | Relevant resistancea | Source or referenceb |
|---|---|---|---|
| Enterococcus faecium | 824-05 | Amp, Cf, Cp, Cl, Mp, Tmp, Sul, Erm, Kan | Clinical isolate |
| Staphylococcus aureus | USA300-0114 | Amp, Cf, Cl, Sul, Kan | 6 |
| UAMS-1 | ND | 7 | |
| UAMS-1112 | Erm | Univ. of Arkansas | |
| RN4220 | Cl | 8 | |
| Klebsiella pneumoniae | cKP1 | Amp, Cp, Sul, Erm, Van | 9 |
| Acinetobacter baumannii | 98-37-09 | Amp, Lz | 10 |
| Pseudomonas aeruginosa | PA01 | Amp, Kan, Lz, Sul, Van | 11 |
| Enterobacter cloacae | PMD1001 | Amp, Erm, Lz, Sul, Van | Clinical isolate |
| Escherichia coli | 8295 | Erm, Lz, Sul | Clinical isolate |
Abbreviations: ampicillin, Amp; colistin, Cl; ceftriaxone, Cf; ciprofloxacin, Cp; erythromycin, Erm; kanamycin, Kan; linezolid, Lz; meropenem, Mp; minocycline, Min; Sulfamethoxazole, Sul; vancomycin, Van; not determined, ND; Univ., university.
Clinical isolates were obtained from the University of Rochester School of Medicine and Dentistry.
Chemicals.
The Prestwick Chemical Library of molecules with known biological activities was acquired from Prestwick Chemical (Illkirch, France). ToxiLight BioAssay kits were obtained from Lonza (Basel, Switzerland). Terfenadine, suloctidil, clomiphene citrate, ceftriaxone, sulfamethoxazole, erythromycin, kanamycin, ciprofloxacin, rifampin, ampicillin, minocycline, tamoxifen, and trimethoprim were purchased from Sigma-Aldrich (St. Louis, MO). Meropenem, linezolid, and vancomycin were purchased from Thermo Fisher (Waltham, MA). Colistin was purchased from APP Pharmaceuticals (Schaumburg, IL).
MIC testing.
MIC testing was performed to determine the antibiotic susceptibility profile of selected bacterial strains according to Clinical and Laboratory Standards (CLSI) protocols (12). Briefly, colonies of each bacterial species were collected from MH agar plates and suspended in individual tubes of MH medium to an optical density (600 nm) of 0.8. The resulting cultures were incubated at 37°C in a rotary shaker at 225 rpm to exponential phase (∼1 × 108 CFU ml−1) and then diluted in fresh MH medium to a cell density of ∼3 × 107 CFU ml−1. Ten microliters of the diluted cultures was added to 88 μl of MH medium in individual wells of a 96-well, round-bottom plate (Corning, Inc.), and 2 μl of a stock solution of the indicated reference antibiotic or test compound (0 to 256 μg ml−1) was added to each well. The carrier solvent was either water or dimethyl sulfoxide (DMSO); final DMSO concentrations were less than or equal to 2%. Plates were incubated at 37°C for 24 h, and the MIC was defined as the lowest concentration of antibiotic in which there was no visible cell pellet in the wells.
Heat-killed bacterial AK release assays.
Overnight cultures of E. coli strain 8295 or S. aureus RN4220 were used to inoculate (1:100 dilution) 25 ml of fresh MH medium and grown at 37°C on a rotary shaker at 225 rpm to exponential phase (∼1 × 108 CFU ml−1). Cells were pelleted by centrifugation (2,000 × g) and resuspended in 2.5 ml of sterile water. One milliliter of the resulting suspension was boiled for 3 min and filter sterilized (0.45-μm filter) to remove cell debris. The filtrate was serially diluted in sterile water, and 100 μl of each dilution was added to individual wells of a white-walled, 96-well plate (Corning, Inc., Corning, NY). To measure the AK activity in the supernatants at each dilution, 100 μl of ToxiLight AK reagent was added to each well, followed by incubation at room temperature for 30 min, and luminescence was measured using a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA). Cells were serially diluted and plated to enumerate CFU (CFU ml−1) before and after boiling to correlate cell lysis with viability.
AK assay 96-well format.
Overnight cultures of each bacterial species were used to inoculate (1:100 dilution) 25 ml of fresh MH medium and grown at 37°C on a rotary shaker at 225 rpm to exponential phase (∼1 × 108 CFU ml−1). Ninety-eight microliters of MH medium, 2 μl of the indicated antibiotic, and 5 × 106 bacteria were added to individual wells of a white-walled, 96-well microtiter plate. Well components were mixed by pipetting and incubated at 37°C for 3 h. The plate was equilibrated to room temperature for 30 min. Next, 100 μl of ToxiLight AK reagent was added to each well and incubated at room temperature for 30 min, and luminescence was measured using a SpectraMax M5 plate reader.
AK assay 384-well format and high-throughput screening.
Overnight cultures of each bacterial species were used to inoculate (1:100 dilution) 25 ml of fresh medium and grown at 37°C on a rotary shaker at 225 rpm to exponential phase (∼1 × 108 CFU ml−1). In a white-walled, 384-well plate, 24 μl of MH medium, 0.3 μl (50 μM) of antibiotic or compound, and 5 × 106 bacteria were added to individual wells and incubated at 37°C for 3 h. The plates were equilibrated at room temperature for 1 h. Twenty-five microliters of ToxiLight AK reagent was then added to each well, followed by incubation at room temperature for 30 min, and luminescence was measured using a SpectraMax M5 plate reader.
AK assay of established biofilms.
Biofilms were grown as previously described (13–15). Briefly, P. aeruginosa and A. baumannii were cultured overnight in Luria-Bertani medium and then used to seed 96-well, flat-bottom plates. Plates were incubated at 37°C in a humidified incubator for 48 h to allow the formation of static biofilms. Nonadherent cells were removed by aspiration and washing with sterile phosphate-buffered saline (PBS). Fresh LB medium supplemented with 0, 1×, 10×, or 100× MIC of antibiotic was added to each well and incubated overnight at 37°C. Following treatment, 100 μl of each biofilm supernatant was transferred to 96-well, white-walled plates, 100 μl of ToxiLight AK reagent was added to each well, mixtures were incubated for 30 min at room temperature, and luminescence was measured using a SpectraMax M5 plate reader. Biofilm-associated bacteria were enumerated by resuspending each biofilm in fresh PBS and plating. For S. aureus UAMS-1 biofilms, 96-well, flat-bottom plates were first coated with 100 μl of 20% human plasma in carbonate buffer overnight at 4°C. Following coating, the plasma solution was removed and cells were inoculated in each well 1:200 in 100 μl of tryptic soy broth supplemented with 3% glucose and 0.5% NaCl. Biofilms were cultured for 48 h in a humidified incubator at 37°C. Established S. aureus biofilms were washed once with PBS and then treated with a 100 μl of ToxiLight lysis buffer for 3 h, after which the amount of AK released into supernatants was measured, as described above.
Small-colony variant AK assays.
Thirty-six-hour cultures of S. aureus strain UAMS-1112 were used to inoculate (1:100 dilution) 100 ml of fresh MH medium and grown at 37°C on a rotary shaker at 225 rpm to an optical density (600 nm) of 0.1 to 0.2, corresponding to ∼1 × 106 CFU ml−1. Cells were pelleted by centrifugation and resuspended in 2 ml of fresh MH medium. Ninety-eight microliters of MH medium containing 5 × 106 bacteria and 2 μl of the indicated antibiotic were added to individual wells of a white-walled, 96-well microtiter plate. Well components were mixed by pipetting and incubated at 37°C for 3 h. The plate was equilibrated to room temperature for 30 min. Next, 100 μl of ToxiLight AK reagent was added to each well, followed by incubation at room temperature for 30 min, and luminescence was measured using a SpectraMax M5 plate reader.
Galleria mellonella model of S. aureus infection.
A Galleria mellonella model of infection was used to measure the putative antimicrobial properties of tamoxifen against E. faecium and terfenadine against S. aureus (16, 17). To do so, overnight cultures of E. faecium strain 824-05 or S. aureus strain USA300-0114 were used to inoculate (1:100 dilution) 25 ml of fresh MH medium and grown at 37°C on a rotary shaker at 225 rpm to exponential phase (∼1 × 108 CFU ml−1). Cultures were pelleted by centrifugation (2,000 × g), washed with sterile PBS, and resuspended at ∼1 × 109 CFU ml−1 in fresh PBS. Galleria mellonella larvae (Vanderhorst Wholesale, Inc.; St. Marys, OH) weighing 200 to 300 mg were inoculated with 5 μl of E. faecium or S. aureus (5 × 106 CFU) into the last left proleg using a 10-μl Hamilton syringe. Worms were then mock treated with either DMSO (negative control), vancomycin (20 mg kg−1; positive control) at 2 h and 24 h postinoculation. For E. faecium studies, groups were also treated with the test compound tamoxifen at 80, 160, or 320 mg kg−1, whereas groups were treated with the test compound terfenadine (80, 160, or 320 mg kg−1) for S. aureus studies. Treatments were administered in the same manner as infection, except that each injection was in the next left proleg moving toward the head of the worm. Larvae were housed in petri dishes in the dark at 37°C and monitored for viability at the conclusion of the study (48 h postinoculation); worms were considered dead if they did not respond to physical stimuli. In addition to mock or compound treatment of infected larvae, studies included two additional noninfected negative-control groups: one group that did not receive injections and one group that was injected with PBS to control for the impact of physical trauma. All experimental groups contained 15 worms, and each experiment was repeated three times.
RESULTS
Rationale for adenylate kinase as a reporter of bacterial cell lysis.
Adenylate kinase (AK) is a ubiquitous intracellular enzyme that catalyzes the conversion of 2 ADP ↔ ATP + AMP and is released into the extracellular space upon cell lysis. Partly because of the robust nature of the AK enzyme, its detection has been used to measure lysis of bacterial contaminants on food contact surfaces and autolysis of brewing yeast strains (18–20). Recently we showed that the AK assay is useful in the identification of antifungal agents (21, 22). The premise of the assay is that agents which disrupt cellular integrity, either directly through damage of the membrane/cell wall or indirectly following the death of the cell, will induce release of AK into the culture medium. Extracellular AK is subsequently detected by the addition of commercially available ToxiLight AK assay reporter cocktail (Lonza, Basel, Switzerland), which generates a luminescent signal by utilizing AK-generated ATP in the standard luciferase catalyzed reaction (Fig. 1). The goal of the current study was to develop the AK assay as a high-throughput screening platform for antibacterial drug discovery.
Fig 1.
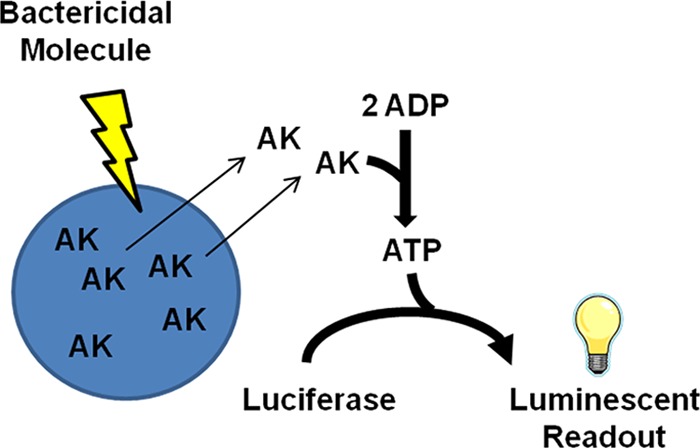
AK assay. Bactericidal agents compromise bacterial cell integrity, releasing cellular adenylate kinase. Extracellular AK is measured by the addition of a commercial ToxiLight AK cocktail containing ADP and luciferase, resulting in luminescence.
The AK assay provides a sensitive measure of bacterial lysis.
As an initial test of AK release as a reporter of bacterial cell death for Gram-positive and Gram-negative organisms, the sensitivity with which the assay measures AK in the culture supernatants of heat-killed E. coli and S. aureus was determined. Each bacterial species was grown to exponential phase, harvested, and resuspended at 1 × 109 CFU per ml in Mueller-Hinton (MH) medium. Bacterial suspensions were heat killed, and a 10-fold dilution series of supernatants was prepared; an aliquot of each heat-killed sample was plated to ensure ≥99% bacterial death. The AK activity of the dilution series was measured and compared to the AK activity of mock-treated (viable) bacteria by Student's t test. In comparison to untreated cells, a statistically significant increase in AK activity was detected at dilutions containing 1.7 × 103 or more heat-killed E. coli supernatants, compared to results for live bacteria (Fig. 2A). AK activity increased ∼433-fold and ∼1,574-fold in cultures with 1 × 107 and 1 × 108 heat-killed E. coli supernatants, respectively, compared to results for mock-treated bacteria (not shown). A comparison of AK released from heat-killed S. aureus to that of mock-treated cells revealed a statistically significant difference in AK activity was detected at dilutions containing 1.8 × 104 or more heat-killed S. aureus supernatants, with a maximum 122-fold increase in AK activity observed for 1 × 108 lysed cells (data not shown). Taken together, these results indicate that the AK assay reproducibly detects AK release following bacterial cell lysis and that the assay is extremely sensitive, allowing the identification of agents that cause lysis in 0.0001% or 0.001% of the starting inoculum of E. coli or S. aureus cells, respectively. Furthermore, the AK assay exhibited a dynamic range of nearly 3 orders of magnitude and an excellent signal-to-noise ratio.
Fig 2.
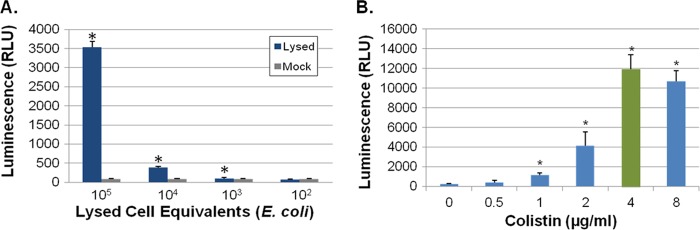
AK assay development. (A) AK assay measures of E. coli lysed cell supernatants. E. coli DH5α cells (1 × 109) were heat inactivated by boiling and diluted, and the AK assay was used to measure adenylate kinase release. Background signal is also shown (mock-treated cells). “*” indicates a significant difference between results for boiled and mock treatment (Student's t test; P ≤ 0.05). (B) AK assay results for colistin-treated A. baumannii strain 98-37-09. Cells were treated with the indicated concentration of colistin, and AK was measured; green indicates the MIC (4 μg ml−1). “*” indicates a significant increase in signal compared to that for mock treatment (0 μg ml−1; Student's t test, P ≤ 0.05).
The AK assay detects bactericidal molecules that are active against the ESKAPE pathogens.
Next, we examined the ability of the AK assay to detect the activities of bactericidal antibiotics toward E. coli and each of the ESKAPE pathogens. To do so, the MICs of six classes of bactericidal antibiotics (penicillin, cephalosporin, quinolone, glycopeptide, carbapenem, and polymyxin) were determined for each organism (Table 2). We then performed the AK assay at 0, 0.125×, 0.25×, 0.5×, 1×, and 2× MIC of each antibiotic. As discussed below, AK release was detected at antibiotic concentrations below the MIC for most antibiotics tested, suggesting that the AK assay would be more sensitive than growth-based assays for detecting bactericidal agents. As a representative example, Fig. 2B presents the AK assay measured polymyxin (colistin)-mediated killing of A. baumannii strain 98-37-09. The MIC of colistin for this strain is 4 μg ml−1, whereas AK release was robustly detected at 0.25×, 0.5×, and 1× MIC value.
Table 2.
MIC and AK measures of bacterial species and antibiotic combinationsa
| Strain and parameter | Treatment (× MIC) | Value for agent |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bactericidal |
Bacteriostatic |
|||||||||||
| Ampicillin | Ceftriaxone | Ciprofloxacin | Vancomycin | Meropenem | Colistin | Sulfamethoxazole | Minocycline | Erythromycin | Kanamycin | Linezolid | ||
| Enterococcus faecium 824-05 | ||||||||||||
| MIC | >256 | >256 | 256 | 2 | >256 | >256 | >256 | 1 | >256 | >256 | 16 | |
| Fold increase in AK signal | 0.5 | ND | ND | ND | 1 | ND | ND | ND | 3 | ND | ND | 2 |
| 1.0 | ND | ND | ND | 2 | ND | ND | ND | 4 | ND | ND | 2 | |
| Staphylococcus aureus | ||||||||||||
| USA300-0114 | ||||||||||||
| MIC | >256 | >256 | 2 | 1 | 4 | >256 | >256 | 1 | 64 | >256 | 2 | |
| Fold increase in AK signal | 0.5 | ND | ND | 3 | 11 | 13 | ND | ND | 1 | 1 | ND | 1 |
| 1.0 | ND | ND | 5 | 3 | 17 | ND | ND | 1 | 1 | ND | 1 | |
| RN4220 | ||||||||||||
| MIC | 2 | 2 | 2 | 2 | 2 | >256 | 2 | 8 | 2 | 16 | 2 | |
| Fold increase in AK signal | 0.5 | 4 | 10 | 8 | 3 | 11 | ND | 2 | 1 | 1 | 1 | 1 |
| 1.0 | 3 | 12 | 10 | 6 | 11 | ND | 3 | 1 | 1 | 1 | 1 | |
| Klebsiella pneumoniae cKP1 | ||||||||||||
| MIC | >256 | 2 | >256 | >256 | 2 | 8 | >256 | 64 | >256 | 2 | >256 | |
| Fold increase in AK signal | 0.5 | ND | 50 | ND | ND | 128 | 34 | ND | 1 | ND | 1 | ND |
| 1.0 | ND | 55 | ND | ND | 220 | 33 | ND | 1 | ND | 1 | ND | |
| Acinetobacter baumannii 98-37-09 | ||||||||||||
| MIC | 256 | 8 | 0.0625 | 64 | 2 | 4 | 8 | 1 | 16 | 2 | 256 | |
| Fold increase in AK signal | 0.5 | ND | 5 | 2 | 2 | 79 | 17 | 6 | 23 | 3 | 3 | ND |
| 1.0 | ND | 21 | 2 | 4 | 134 | 49 | 5 | 14 | 12 | 14 | ND | |
| Pseudomonas aeruginosa PA01 | ||||||||||||
| MIC | >256 | 4 | 2 | >256 | 2 | 4 | 256 | 32 | 32 | 256 | >256 | |
| Fold increase in AK signal | 0.5 | ND | 2 | 27 | ND | 45 | 5 | ND | 1 | 3 | ND | ND |
| 1.0 | ND | 14 | 182 | ND | 39 | 12 | ND | 6 | 1 | ND | ND | |
| Enterobacter cloacae PMD1001 | ||||||||||||
| MIC | >256 | 2 | <1 | >256 | 2 | 2 | >256 | 4 | 256 | 2 | >256 | |
| Fold increase in AK signal | 0.5 | ND | 20 | 6 | ND | 99 | 10 | ND | 4 | ND | 1 | ND |
| 1.0 | ND | 17 | 7 | ND | 296 | 32 | ND | 9 | ND | 1 | ND | |
| Escherichia coli 8295 | ||||||||||||
| MIC | 128 | 2 | 0.125 | 16 | 2 | 8 | >256 | 16 | 256 | 32 | >256 | |
| Fold increase in AK signal | 0.5 | 50 | 51 | 17 | 1 | 60 | 15 | ND | 4 | ND | 55 | ND |
| 1.0 | 74 | 76 | 39 | 2 | 85 | 22 | ND | 38 | ND | 70 | ND | |
| % increase expected | 0.5 | 100 | 83.3 | 83.3 | 40 | 100 | 100 | 50 | 50 | 50 | 40 | 0 |
| 1.0 | 100 | 100 | 83.3 | 60 | 100 | 100 | 100 | 62.5 | 25 | 40 | 0 | |
Shading indicates significant increase in AK signal (greater than or equal to threefold over vehicle-treated cells).
The MIC measures for each ESKAPE pathogen and each organism's corresponding fold increase in AK signal following treatment with 0.5× and 1.0× the MIC are provided in Table 2 (bactericidal agents). For four of the six bactericidal antibiotics tested, the AK assay proved to be superior to growth-based assays with respect to detecting the killing properties of bactericidal agents at sub-MIC values. More specifically, 100% of the organisms that were determined to be susceptible to the cell wall-active antibiotics ampicillin, meropenem, and colistin exhibited a significant increase in AK signal (≥3-fold over that for vehicle-treated cells) at both 0.5× and 1.0× their MICs. Eighty-three percent and 100% of ceftriaxone-susceptible species exhibited increased AK signal at 0.5× and 1× their MICs, respectively. Five of six (83%) ciprofloxacin-susceptible organisms exhibited AK signal at both 0.5× and 1.0× MIC. Interestingly, the cell wall-targeted glycopeptide, vancomycin, caused significant AK release at 0.5× and 1.0× their MICs in only 40% and 60% of susceptible species. We speculate that this may be due to the well-established slow-killing kinetics of the agent (23–25).
To evaluate the specificity of the assay for detecting bactericidal agents, we also measured AK release by each of the ESKAPE pathogens following exposure to five classes of bacteriostatic agents (sulfonamide, tetracycline, macrolide, aminoglycoside, and oxazolidine). To do this, the MIC value of each bacteriostatic antibiotic/organism pair was measured by a conventional growth-based approach. We subsequently measured AK release by each bacterial species following treatment with 0.5× or 1.0× MIC of each bacteriostatic agent. As shown in Fig. 3, S. aureus strain RN4220 was susceptible to all agents tested but bacteriostatic agents generated very low AK release, whereas bactericidal antibiotics generated robust AK detection. In most instances, a similar trend was observed for the other ESKAPE pathogens, indicating that the assay enriches for the identification of bactericidal agents, which are arguably the most valuable antibiotics because they can be used to treat patients with immunological defects or rapidly lethal infections (Table 2, bacteriostatic agents). Two exceptions were noted. Surprisingly, A. baumannii generated significant AK signal in response to all the bacteriostatic agents evaluated, suggesting that AK release may be a feature of a more general stress response in this organism. Additionally, the bacteriostatic agent minocycline generated signal by five of the eight (63%) organisms tested. In that regard, others have previously observed that tetracyclines tend to exhibit bactericidal characteristics, which is presumably the cause of minocycline-mediated AK detection (26, 27).
Fig 3.
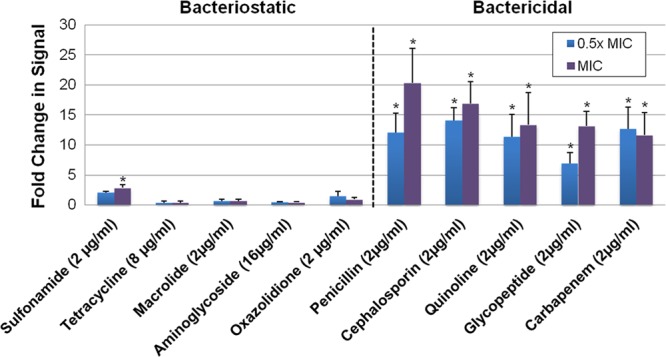
AK assay measures of S. aureus strain RN4220 treatment with bacteriostatic and bactericidal antibiotics. Standard MIC testing determined the MIC of each antibiotic class (in parentheses). Graphed are the fold changes in AK signal of cells treated with 0.5× or 1.0× the MIC value for each antibiotic, in comparison to untreated control cells; “*” indicates a significant change in signal as determined by Student's t test; P ≤ 0.05 (compared to results for untreated cells).
Taken together, these results suggest that the AK assay represents an attractive screening approach for identifying bactericidal agents at sub-MICs that may otherwise be missed by growth-based assays. Further, because the AK assay relies on bacterial killing as opposed to growth changes to generate its readout, we hypothesized that it would provide a format to develop screens that are not readily available by conventional, growth-based approaches.
Use of the AK assay to identify agents with antimicrobial activities against established biofilms and small colony variants.
Established bacterial biofilms represent a particularly problematic disease state, in part because biofilm-associated bacteria are recalcitrant to conventional antibiotic therapy. Thus, they have been a focus of antibiotic development. As a result, a number of approaches to screening for molecules with activity toward bacterial biofilms have been developed recently (28, 29). Although each has its advantages, they also have a number of limitations, including reproducibility, reliance on specialized equipment, or low throughput. We hypothesized that the AK assay would provide a solution to some of these problems because it is rapid, sensitive, and simple to perform and it detects bactericidal molecules, the type of antibiotics required to treat established biofilms.
To determine whether the AK assay could detect agents with activity against established biofilms, S. aureus strain UAMS-1, A. baumannii strain 98-37-09, and P. aeruginosa strain PAO1 static biofilms were formed in 96-well flat-bottom plates. Forty-eight hours postinoculation, one well corresponding to each organism was stained with crystal violet to verify that biofilm formation had occurred (data not shown), whereas the remaining wells were treated with 10× the MIC value with either colistin (A. baumannii biofilms) or ciprofloxacin (P. aeruginosa, and S. aureus biofilms). Following overnight antibiotic treatment, biofilm-associated bacteria were enumerated by plating, and the corresponding supernatants were analyzed by the AK assay. Plating verified that 10× MIC antibiotic treatment resulted in a significant reduction in biofilm-associated P. aeruginosa (a 3.1-log decrease), S. aureus (a 0.7-log decrease), and A. baumannii (a 1.8-log decrease) compared to findings for untreated biofilms. As shown in Fig. 4A, corresponding AK measures indicated that the assay robustly detects the mild effects of these antibiotics on each bacterial species tested, suggesting that it represents a promising approach to identify agents that exhibit bactericidal activity toward established bacterial biofilms.
Fig 4.
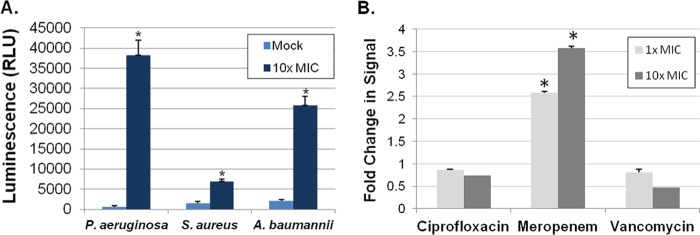
AK assay measures of antibiotic-treated biofilms and small-colony variants.(A) Graphed are AK signals generated by static biofilm-associated cells following mock or antibiotic treatment: colistin (P. aeruginosa) or ciprofloxacin (S. aureus and A. baumannii). (B) Fold change of AK measures of S. aureus SCV UAMS-1112 cells following treatment with 1× and 10× ciprofloxacin, meropenem, and vancomycin, compared to results for mock treated cells. “*” indicates a significant change in signal in comparison to results for mock-treated cells (Student's t test, P ≤ 0.05).
Another context in which the AK assay may be particularly valuable is in the identification of molecules that exhibit bactericidal activity toward bacterial small-colony variants (SCV). SCV are slow-growing populations of bacterial species that have been hypothesized to cause latent or recurrent infections and are tolerant of standard antibiotic treatment regimens. Based on the aberrant SCV growth characteristics, typical growth-based HTS assays would be difficult to employ. Accordingly, we tested the ability of the AK assay to identify agents that kill S. aureus SCV strain UAMS-1112. To do so, 1 × 106 UAMS-1112 cells were treated with 1× or 10× MIC ciprofloxacin, meropenem, or vancomycin for 3 h. Following treatment, suspensions were plated to measure the antimicrobial properties of each antibiotic, and the AK assay was performed to measure adenylate kinase release. Plating revealed that ciprofloxacin and vancomycin had no effect on SCV viability at any concentration tested (data not shown) and exhibited no change in AK release in comparison to results for mock-treated cells (Fig. 4B), supporting the observation that S. aureus small-colony variants are recalcitrant to antibiotic treatment. Meropenem treatment resulted in a 0.5-log decrease in SCV viability, which corresponded to a 2.5-fold increase in AK signal compared to that for mock-treated cells (Fig. 4B). Taken together, these results suggest that the AK assay provides the sensitivity needed to detect the slight antimicrobial effects of antibiotics, such as meropenem, toward S. aureus SCV.
Validation of AK as an HTS-compatible assay of antibacterial activity.
To test the viability of the AK assay as a general tool in HTS-based antibacterial small-molecule discovery, we optimized assay parameters, including inoculum, drug incubation time, and AK reaction time, for screening planktonic ESKAPE species as well as E. coli in a 384-well format. Based on these experiments (not shown), we developed a standardized assay as fully described in Materials and Methods. As part of this optimization process, control assays in a 384-well format were performed to measure the signal to noise and reproducibility of the assay in an HTS manner. For this, plates were seeded with E. coli or an ESKAPE pathogen. Alternating columns of the plate were then mixed with either 2% DMSO (negative control) or a bactericidal antibiotic (positive control), and AK signal was detected; a representative result for DMSO- and colistin-treated K. pneumoniae is shown in Fig. 5A. Comparisons of the variance in signal between positive- and negative-control measures indicated that the AK assay provides Z′-factor scores between 0.59 and 0.82 (depending on the specific organism), indicating that the assay is sufficiently robust for HTS (30, 31).
Fig 5.
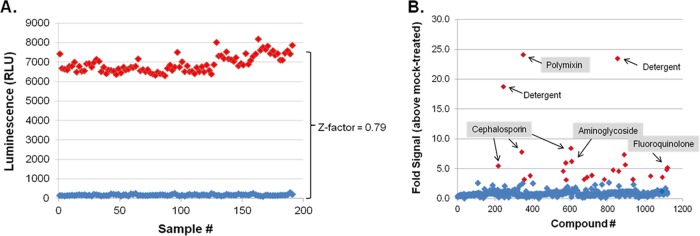
AK-based HTS development and screening. (A) Z′ factor assay results for Klebsiella pneumoniae. Three-hundred-eighty-four-well microtiter plates were seeded with K. pneumoniae, and alternating rows were mock treated (DMSO) or treated with 50 μM colistin. Following 3 h of incubation, AK release was measured and plotted. DMSO-treated well measures are shown in blue; colistin-treated wells are shown in red. (B) Prestwick library Klebsiella pneumoniae screening results. In total, 26 compounds were determined to result in a 3-fold increase in AK signal (red data points), in comparison to results for DMSO-treated cells. Included among this list were polymyxin, cephalosporins, aminoglycosides, fluoroquinolones, and detergents; the complete Prestwick screening results for K. pneumoniae and all other organisms screened are provided in Table S2 in the supplemental material.
AK-based screening of library of off-patent drugs and biologically active molecules.
To further validate the AK assay protocol, we screened E. coli and each of the ESKAPE pathogens against the Prestwick library of FDA-approved drugs and biologically active molecules (1,120 compounds, total; 50 μM final drug concentration). The Prestwick library contains representatives of nearly all classes of antibiotics currently in clinical use, making it an ideal library for testing the ability of the AK assay to detect bactericidal agents in a high-throughput screening format. Accordingly, the library was screened for antimicrobial agents that were active against planktonic E. coli and each of the ESKAPE pathogens using the AK assay; the cutoff for positive-scoring molecules was set at a 3-fold increase in extracellular AK activity, corresponding to the detection limit of statistically significant increases in AK activity for planktonic bacteria. The hit rates for the different organisms ranged from 1.4% to 4.8%; a representative example of raw screening data for Klebsiella pneumoniae is shown in Fig. 5B. Screening results for all ESKAPE pathogens are summarized in Table 3, whereas results for all compounds within the Prestwick library are provided in Table S1 in the supplemental material.
Table 3.
Prestwick Library screening results
| Compound | No. of active agents for species |
||||||
|---|---|---|---|---|---|---|---|
| E. coli | E. faecium | S. aureusa | K. pneumoniae | A. baumanniia | P. aeruginosa | E. cloacae | |
| Bactericidal | 45 | 4 | 25 | 19 | 17 | 21 | 16 |
| Bacteriostatic | 2 | 1 | 14 | 0 | 7 | 0 | 1 |
| Detergent | 2 | 2 | 2 | 2 | 2 | 1 | 0 |
| Other | 5 | 9 | 10 | 4 | 18 | 1 | 12 |
| Total | 54 | 16 | 51 | 25 | 44 | 23 | 29 |
Tetracyclines were identified.
Consistent with expectations, a detailed assessment of the hits revealed that AK screening enriches for the identification of bactericidal compounds (Table 3). More specifically, 100% of the antibiotics that were identified to be active against P. aeruginosa or K. pneumoniae represented bactericidal agents. Similarly, 96%, 94%, and 80% of the antibiotics that were active against E. coli, E. cloacae, and E. faecium, respectively, were bactericidal antibiotics. The assay identified 71% and 64% bactericidal antibiotics for A. baumannii and S. aureus.
Among the bactericidal antibiotics detected, β-lactam, cephalosporin, polymyxin, and fluoroquinolone antibiotics were active against Gram-negative pathogens (E. coli and the ESKAPE pathogens A. baumannii, P. aeruginosa, and K. pneumoniae), and penicillins, cephalosporins, quinolines, glycopeptides, and carbapenems were active toward S. aureus (see Table S1 in the supplemental material). We also determined that clofazimine, which was initially developed as an antimycobacterial agent and was recently shown to exhibit bactericidal activity toward S. aureus, was indeed active against S. aureus (32). Bacteriostatic compounds, such as clindamycin, and macrolides, such as erythromycin, were not identified as being active toward any of the species tested. Interestingly, many tetracyclines were identified as killing S. aureus strain USA300-0114 and A. baumannii strain 98-37-09. As stated above, although tetracyclines are traditionally considered to be bacteriostatic, they have been reported to display bactericidal activity (26, 27); we speculate that at high concentrations, such as those used in our proof-of-principle screen, this class of molecules exhibits bactericidal activity. In addition, the library contains membrane-active antiseptics, such as chlorhexidine, and with the exception of E. cloacae, these were also strong hits against all organisms tested. Screening results also revealed that the E. faecium strain tested proved to be resistant to most classes of antibiotics within the Prestwick library by MIC testing (Table 2), and this was also observed in the AK assay, indicating that the assay exhibits a low false-positive rate. Interestingly, the assay also detected 38 drugs with no known antimicrobial properties (denoted as “other” in Table 3) that were active against each organism; see Table S2 for a complete list of these compounds.
Antimicrobial activities of nonantibiotic drugs that induce AK release.
Recently, the exploration of the so-called “off-target” activities of previously developed drugs has emerged as an approach to identify chemical scaffolds that could be exploited for new therapeutic indications. In that regard, Prestwick library screening results revealed that 4% to 56% (depending on the organism) of the members that generated significant AK signal were compounds with no previously reported antimicrobial activity (see Table S2 in the supplemental material).
To determine whether the 38 nonantibiotics identified in the screen have potential for repurposing as anti-infectives, we further evaluated 4 nonantibiotic drugs that were commercially available by two secondary assays. First, dose-response assays were performed to validate that they induced AK activity, and all were reconfirmed (data not shown). Second, the in vitro antimicrobial activity for each drug was measured by standard MIC testing. With the exception of one drug/organism pair, all drugs exhibited in vitro activity toward each organism. More specifically, tamoxifen, suloctidil, and clomiphene exhibited MICs of 8 μg ml−1 against E. faecium. S. aureus and A. baumannii were susceptible to terfenadine (16-μg ml−1 MIC and 64-μg ml−1 MIC, respectively). Suloctidil was also detected to be active against P. aeruginosa by both primary and confirmatory AK screens, but the drug did not elicit an antimicrobial response by MIC measures. Some strains of P. aeruginosa secrete AK at high cell density, and we speculate that suloctidil may trigger a similar response (33).
Terfenadine and tamoxifen, which exhibited antimicrobial properties toward planktonic S. aureus and E. faecium cells, respectively, were characterized further. Terfenadine was evaluated for activity against S. aureus biofilms and small-colony variants using the AK assays described above. Treatment of 48-h S. aureus strain UAMS-1 biofilms with 10×-MIC terfenadine elicited a modest 2.7-fold increase in AK release (Fig. 6A); plating-based viability assays determined that this correlated with a 1.1-log reduction in biofilm cell viability (data not shown), which was comparable to the activity of ciprofloxacin under the same assay conditions (Fig. 4A). Similarly, treatment of S. aureus small-colony-variant UAMS-1112 cells with 10× MIC terfenadine elicited a 3.3-fold increase in AK signal in comparison to that for mock-treated cells (Fig. 6A). Conventional MIC testing subsequently verified that terfenadine is active against UAMS-1112 at 2 μg ml−1 (data not shown). Because we have not yet established an E. faecium biofilm AK assay and stable SCV cells were not available, tamoxifen was not evaluated for activity against enterococci.
Fig 6.
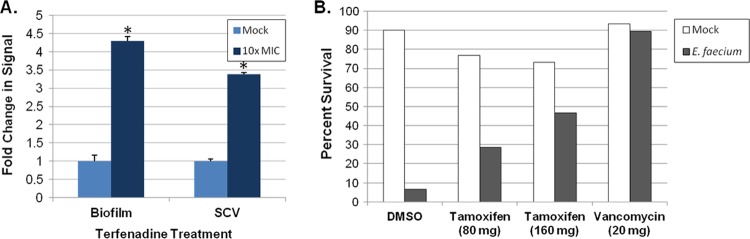
Antimicrobial properties of terfenadine and tamoxifen. (A) Fold changes in AK signal of terfenadine-treated (10× MIC) S. aureus strain UAMS-1 static biofilms and the SCV strain UAMS-1112, compared to those for mock (DMSO)-treated populations, are plotted. “*” indicates a significant increase in signal over that with mock-treated cells (Student's t test, P ≤ 0.05). (B) Plotted are the percent survival of G. mellonella larvae at 48 h post-E. faecium inoculation. Groups of larvae (n = 45) were treated at 2 h and 24 h with either PBS (mock), DMSO, 80 mg kg−1 tamoxifen, 160 mg kg−1 tamoxifen, or 20 mg kg−1 vancomycin.
Next, the in vivo antimicrobial properties of terfenadine and tamoxifen were evaluated using a Galleria mellonella model of S. aureus and E. faecium infection, respectively. For terfenadine, groups of larvae (n = 45) were infected with 1.0 × 106 S. aureus USA300-1114 cells. Worms were then treated at 2 h and 24 h with a range of terfenadine concentrations (20 to 160 mg kg−1), vehicle (DMSO; negative control), or 20 mg kg−1 vancomycin (positive control), and larval survival was assessed 48 h postinoculation. For tamoxifen studies, experiments were performed exactly as described above except that larvae were inoculated with 1.4 × 106 E. faecium strain 824-05 cells and larvae were treated with either 80 or 160 mg kg−1 tamoxifen. Terfenadine-treated larvae did not reproducibly exhibit increased survival relative to vehicle-treated larvae (data not shown). However, tamoxifen treatment of E. faecium-infected larvae resulted in a dose-dependent increase in survival. As shown in Fig. 6B, treatment with 80 or 160 mg kg−1 resulted in a 4.2-fold- or 7-fold-higher survival, respectively, than was found with vehicle-treated controls.
Taken together, these results indicate that terfenadine exhibits modest in vitro activity toward S. aureus planktonic, SCV, and biofilm populations and that tamoxifen is active toward planktonic E. faecium and against the organism in a simple animal model of infection. As discussed below, these data suggest that tamoxifen and terfenadine may represent attractive new chemical scaffolds for antibacterial optimization.
DISCUSSION
As the search for novel antimicrobials continues, so must the development of methods used for identifying compounds with therapeutic potential. In that regard, we developed a simple set of AK reporter-based assays for the identification of antibacterial agents. The AK assay robustly detects the antimicrobial properties of molecules below their measured MIC values and with a dynamic range of nearly 3 orders of magnitude. Moreover, it provides antimicrobial property measures in a simple add-and-read format, enriches for bactericidal compounds, and is ostensibly applicable to any bacterial species or clinical isolate of interest. While others have developed highly sensitive enzyme-based reporter systems for antibacterial screening purposes, these assays typically rely on β-galactosidase, luciferase, or green fluorescent protein expression to measure bacterial survival and thus require genetically engineered screening strains for screening (34–36). However, these systems do allow the identification of bacteriostatic agents, which by comparison would not be identified using the AK assay. Nonetheless, based on its ease of use in an add-and-read format, as well as adaptability to virtually any species/clinical isolate of interest, we feel the AK assay developed here provides an attractive new approach for antimicrobial screening.
As a means of validating the AK assay as an antimicrobial HTS tool, each of the ESKAPE bacterial pathogens was screened with the Prestwick library of drugs with known biological activities for antimicrobial agents. As expected, screening revealed that the assay identified known antibiotics within the library that corresponded with the susceptibility of the organism used and enriched for the detection of bactericidal antibiotics. Similarly, agents known to have no activity against a particular organism were negative in the screen. For example, we screened with a clinical isolate of E. faecium that is resistant to ampicillin, gentamicin, and vancomycin, and none of those agents was active in the AK assay at supra-MIC levels. We did not expect the AK assay to show signal with bacteriostatic agents, and in general this was the case. The exception to this trend was the fact that S. aureus and A. baumannii screens also detected bacteriostatic antibiotics, most of which belonged to the tetracycline class. As discussed above, others have shown that high levels of tetracyclines can induce cell death, and we suspect this is the reason for our observations.
Recently the concept of repurposing previously developed drugs for new therapeutic indications has emerged as an approach to expediting the development of new therapies (37). The advantages of such a strategy are twofold. In the ideal scenario, a new activity for a drug is identified and is manifest at doses currently in use, allowing the direct testing of the drug in clinical trials for its new indication. A more likely scenario is that a new activity is found for a scaffold with good pharmacokinetic and toxicology properties which can serve as a lead compound for optimization. In that regard, Prestwick library screening revealed that each bacterial species was susceptible to members of the library with no previously reported antimicrobial activity.
We further studied the antimicrobial properties of two of these drugs in more detail: terfenadine and tamoxifen. Terfenadine (Fig. 7A) is a nonsedating antihistamine based on a 4-substituted piperidine scaffold that is also known to inhibit the hERG, leading to prolonged QT syndrome (38). The latter property was the reason that terfenadine was removed from the market. Recently, molecules (Fig. 7B) based on this scaffold have emerged as nonfluoroquinolone inhibitors of type II bacterial topoisomerases (NBTIs) and have been the subject of increasing study, with particular emphasis being placed on separating the antibacterial activity from hERG inhibition (39). Based on the structural similarity between terfenadine and these molecules, we suspect that terfenadine is acting as a topoisomerase inhibitor. The majority of the NBTIs reported as part of medicinal chemistry projects are 4-amino-substituted piperidines (39); our results suggest that 4-alkyl-substituted piperidines are also active. Furthermore, we found that terfenadine has activity against S. aureus small-colony variants and biofilms, properties not previously reported for this scaffold.
Fig 7.
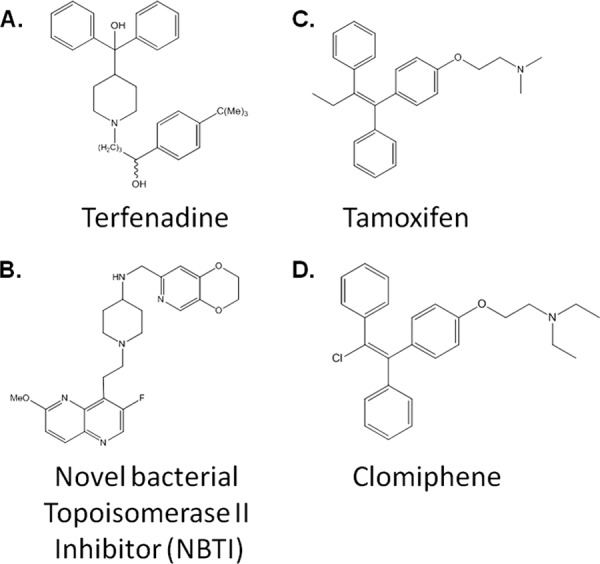
Structural features of terfenadine, tamoxifen, NS novel bacterial topoisomerase II inhibitor, and clomiphene. The chemical structure of each molecule is shown.
Our screen of E. faecium identified two structurally related nonsteroid estrogen receptor antagonists, tamoxifen and clomiphene (Fig. 7C and D). Tamoxifen is used to treat some forms of estrogen-receptor-positive breast cancer, while clomiphene is used in fertility treatment regimens (40). These two compounds are members of the triarylethylene class of estrogen receptors. This class of compounds has been previously shown to have antifungal activity by a number of groups (41, 42). Tamoxifen has been the most widely studied of these and has been found to have a number of additional targets in mammalian cells, including calmodulin and protein kinase C (43). In addition, tamoxifen is thought to directly interact with lipids in membranes (44). Although neither tamoxifen nor any of its analogs has been reported to have activity against traditionally pathogenic bacteria, its interaction with the membranes of Bacillus stearothermophilus has been studied as model for the effects of tamoxifen on mammalian membranes (44). Interestingly, tamoxifen at concentrations similar to its MIC toward E. faecium (5 μg ml−1) induces a number of ultrastructural changes in the membrane of B. stearothermophilus, including the appearance of a symmetric membrane and evidence of membrane fractures leading to the leakage of cytoplasmic contents into the extracellular space. Although more work will be required to further characterize the mode of action of tamoxifen's activity against E. faecium, these findings are consistent with our observation that it causes bacterial cell lysis.
Since the number of agents with activity toward enterococcus is quite limited, we investigated tamoxifen's in vivo activity using a Galleria model of enterococcus infection. Although it was not as active as vancomycin, tamoxifen did impart a survival benefit, indicating that it has in vivo antimicrobial activity. High-dose tamoxifen therapy has been used in experimental treatment of refractory human cancers, and dosing results in micromolar serum concentrations of tamoxifen corresponding to the levels of drug associated with antienterococcal activity observed in our studies (45). Similarly, tamoxifen has also been shown to have activity against Candida albicans in a mouse model of candidiasis that generates serum levels that approximate those levels required for efficacy in our assays (41). The MIC of tamoxifen toward C. albicans is actually 4-fold higher than its MIC toward E. faecium, suggesting that further investigation of this class of molecules as antienterococcal agents may be fruitful. Additionally, many analogs of tamoxifen have been synthesized as part of structure-activity studies designed to optimize its estrogen receptor activity; our data suggest that exploration of these molecules for optimal antienterococcal activity is warranted.
In addition to providing a powerful new HTS approach to identify antimicrobial agents active against planktonic bacteria, we show that the AK assay is easily amenable to screening bacteria in disease states that cannot be readily screened via conventional approaches. In that regard, the AK assay is capable of measuring the killing properties of bactericidal agents administered to biofilms formed by both Gram-negative and Gram-positive representatives of the ESKAPE pathogens. Similarly, the AK assay can detect the bactericidal properties of antibiotics toward a phenotypically stable S. aureus small-colony variant. We predict that these features can be exploited to develop corresponding high-throughput screening assays for the identification of agents that kill established biofilms and small-colony variants or provide powerful secondary assays aimed at characterizing the potential antimicrobial properties of molecules of interest.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by a grant from the National Institute of Allergy and Infectious Diseases, 1R01AI075033 (to D.K.).
Footnotes
Published ahead of print 1 October 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01640-12.
REFERENCES
- 1. Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48:1–12 [DOI] [PubMed] [Google Scholar]
- 2. Rice LB. 2008. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J. Infect. Dis. 197:1079–1081 [DOI] [PubMed] [Google Scholar]
- 3. Talbot GH, Bradley J, Edwards JE, Jr, Gilbert D, Scheld M, Bartlett JG. 2006. Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin. Infect. Dis. 42:657–668 [DOI] [PubMed] [Google Scholar]
- 4. Silver LL. 2011. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 24:71–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. 2007. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 6:29–40 [DOI] [PubMed] [Google Scholar]
- 6. Kazakova SV, Hageman JC, Matava M, Srinivasan A, Phelan L, Garfinkel B, Boo T, McAllister S, Anderson J, Jensen B, Dodson D, Lonsway D, McDougal LK, Arduino M, Fraser VJ, Killgore G, Tenover FC, Cody S, Jernigan DB. 2005. A clone of methicillin-resistant Staphylococcus aureus among professional football players. N. Engl. J. Med. 352:468–475 [DOI] [PubMed] [Google Scholar]
- 7. Gillaspy AF, Hickmon SG, Skinner RA, Thomas JR, Nelson CL, Smeltzer MS. 1995. Role of the accessory gene regulator (agr) in pathogenesis of staphylococcal osteomyelitis. Infect. Immun. 63:3373–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Peng HL, Novick RP, Kreiswirth B, Kornblum J, Schlievert P. 1988. Cloning, characterization, and sequencing of an accessory gene regulator (agr) in Staphylococcus aureus. J. Bacteriol. 170:4365–4372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pomakova DK, Hsiao CB, Beanan JM, Olson R, MacDonald U, Keynan Y, Russo TA. 2012. Clinical and phenotypic differences between classic and hypervirulent Klebsiella pneumonia: an emerging and under-recognized pathogenic variant. Eur. J. Clin. Microbiol. Infect. Dis. 31:981–989 [DOI] [PubMed] [Google Scholar]
- 10. Jacobs AC, Hood I, Boyd KL, Olson PD, Morrison JM, Carson S, Sayood K, Iwen PC, Skaar EP, Dunman PM. 2010. Inactivation of phospholipase D diminishes Acinetobacter baumannii pathogenesis. Infect. Immun. 78:1952–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Holloway BW. 1955. Genetic recombination in Pseudomonas aeruginosa. J. Gen. Microbiol. 13:572–581 [DOI] [PubMed] [Google Scholar]
- 12. Hindler JF. 2010. Antimicrobial susceptibility testing, section 5. Clinical microbiology procedures handbook, vol 1 American Society for Microbiology, Washington, DC [Google Scholar]
- 13. Beenken KE, Blevins JS, Smeltzer MS. 2003. Mutation of sarA in Staphylococcus aureus limits biofilm formation. Infect. Immun. 71:4206–4211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Musken M, Di Fiore S, Romling U, Haussler S. 2010. A 96-well-plate-based optical method for the quantitative and qualitative evaluation of Pseudomonas aeruginosa biofilm formation and its application to susceptibility testing. Nat. Protoc. 5:1460–1469 [DOI] [PubMed] [Google Scholar]
- 15. Tomaras AP, Dorsey CW, Edelmann RE, Actis LA. 2003. Attachment to and biofilm formation on abiotic surfaces by Acinetobacter baumannii: involvement of a novel chaperone-usher pili assembly system. Microbiology 149:3473–3484 [DOI] [PubMed] [Google Scholar]
- 16. Lebreton F, Le Bras F, Reffuveille F, Ladjouzi R, Giard JC, Leclercq R, Cattoir V. 2011. Galleria mellonella as a model for studying Enterococcus faecium host persistence. J. Mol. Microbiol. Biotechnol. 21:191–196 [DOI] [PubMed] [Google Scholar]
- 17. Peleg AY, Monga D, Pillai S, Mylonakis E, Moellering RC, Jr, Eliopoulos GM. 2009. Reduced susceptibility to vancomycin influences pathogenicity in Staphylococcus aureus infection. J. Infect. Dis. 199:532–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cameron-Clarke A, Hulse GA, Clifton L, Cantrell IC. 2003. The use of adenylate kinase measurement to determine the causes of lysis in lager yeast. J. Am. Soc. Brew. Chem. 61:152–156 [Google Scholar]
- 19. Corbitt AJ, Bennion N, Forsythe SJ. 2000. Adenylate kinase amplification of ATP bioluminescence for hygiene monitoring in the food and beverage industry. Lett. Appl. Microbiol. 30:443–447 [DOI] [PubMed] [Google Scholar]
- 20. Squirrell D, Murphy M. 1997. Rapid detection of very low numbers of micro-organism using adenylate kinase as a cell marker, p 107–113 In Stanley P, Smither R, Simpson W. (ed), A practical guide to industrial uses of ATP-luminescence in rapid microbiology. Cara Technology Limited, Leatherhead, United Kingdom [Google Scholar]
- 21. DiDone L, Scrimale T, Baxter BK, Krysan DJ. 2010. A high-throughput assay of yeast cell lysis for drug discovery and genetic analysis. Nat. Protoc. 5:1107–1114 [DOI] [PubMed] [Google Scholar]
- 22. Krysan DJ, Didone L. 2008. A high-throughput screening assay for small molecules that disrupt yeast cell integrity. J. Biomol. Screen. 13:657–664 [DOI] [PubMed] [Google Scholar]
- 23. Arhin FF, McKay GA, Beaulieu S, Sarmiento I, Parr TR, Jr, Moeck G. 2009. Time-kill kinetics of oritavancin and comparator agents against Streptococcus pyogenes. Int. J. Antimicrob. Agents 34:550–554 [DOI] [PubMed] [Google Scholar]
- 24. McKay GA, Beaulieu S, Arhin FF, Belley A, Sarmiento I, Parr T, Jr, Moeck G. 2009. Time-kill kinetics of oritavancin and comparator agents against Staphylococcus aureus, Enterococcus faecalis and Enterococcus faecium. J. Antimicrob. Chemother. 63:1191–1199 [DOI] [PubMed] [Google Scholar]
- 25. Nielsen EI, Viberg A, Lowdin E, Cars O, Karlsson MO, Sandstrom M. 2007. Semimechanistic pharmacokinetic/pharmacodynamic model for assessment of activity of antibacterial agents from time-kill curve experiments. Antimicrob. Agents Chemother. 51:128–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carleton J, Phair JP. 1972. The slow bactericidal effect of tetracycline and minocycline on wall-defective staphylococcus. J. Infect. Dis. 126:457–459 [DOI] [PubMed] [Google Scholar]
- 27. Li RC, Lee SW, Kong CH. 1997. Correlation between bactericidal activity and postantibiotic effect for five antibiotics with different mechanisms of action. J. Antimicrob. Chemother. 40:39–45 [DOI] [PubMed] [Google Scholar]
- 28. Benoit MR, Conant CG, Ionescu-Zanetti C, Schwartz M, Matin A. 2010. New device for high-throughput viability screening of flow biofilms. Appl. Environ. Microbiol. 76:4136–4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Perez LM, Alvarez BL, Codony F, Fittipaldi M, Adrados B, Penuela G, Morato J. 2010. A new microtitre plate screening method for evaluating the viability of aerobic respiring bacteria in high surface biofilms. Lett. Appl. Microbiol. 51:331–337 [DOI] [PubMed] [Google Scholar]
- 30. An WF, Tolliday N. 2010. Cell-based assays for high-throughput screening. Mol. Biotechnol. 45:180–186 [DOI] [PubMed] [Google Scholar]
- 31. Zhang JH, Chung TD, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4:67–73 [DOI] [PubMed] [Google Scholar]
- 32. Oliva B, O'Neill AJ, Miller K, Stubbings W, Chopra I. 2004. Anti-staphylococcal activity and mode of action of clofazimine. J. Antimicrob. Chemother. 53:435–440 [DOI] [PubMed] [Google Scholar]
- 33. Markaryan A, Zaborina O, Punj V, Chakrabarty AM. 2001. Adenylate kinase as a virulence factor of Pseudomonas aeruginosa. J. Bacteriol. 183:3345–3352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cooksey RC, Crawford JT, Jacobs WR, Jr, Shinnick TM. 1993. A rapid method for screening antimicrobial agents for activities against a strain of Mycobacterium tuberculosis expressing firefly luciferase. Antimicrob. Agents Chemother. 37:1348–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kirsch DR, Lai MH, McCullough J, Gillum AM. 1991. The use of beta-galactosidase gene fusions to screen for antibacterial antibiotics. J. Antibiot. (Tokyo) 44:210–217 [DOI] [PubMed] [Google Scholar]
- 36. Srivastava R, Deb DK, Srivastava KK, Locht C, Srivastava BS. 1998. Green fluorescent protein as a reporter in rapid screening of antituberculosis compounds in vitro and in macrophages. Biochem. Biophys. Res. Commun. 253:431–436 [DOI] [PubMed] [Google Scholar]
- 37. Wermuth CG. 2006. Selective optimization of side activities: the SOSA approach. Drug Discov. Today 11:160–164 [DOI] [PubMed] [Google Scholar]
- 38. Woosley RL, Chen Y, Freiman JP, Gillis RA. 1993. Mechanism of the cardiotoxic actions of terfenadine. JAMA 269:1532–1536 [PubMed] [Google Scholar]
- 39. Reck F, Alm R, Brassil P, Newman J, Dejonge B, Eyermann CJ, Breault G, Breen J, Comita-Prevoir J, Cronin M, Davis H, Ehmann D, Galullo V, Geng B, Grebe T, Morningstar M, Walker P, Hayter B, Fisher S. 2011. Novel N-linked aminopiperidine inhibitors of bacterial topoisomerase type II: broad-spectrum antibacterial agents with reduced hERG activity. J. Med. Chem. 54:7834–7847 [DOI] [PubMed] [Google Scholar]
- 40. Morello KC, Wurz GT, DeGregorio MW. 2003. Pharmacokinetics of selective estrogen receptor modulators. Clin. Pharmacokinet. 42:361–372 [DOI] [PubMed] [Google Scholar]
- 41. Dolan K, Montgomery S., Buchheit B, Didone L, Wellington M, Krysan DJ. 2009. Antifungal activity of tamoxifen: in vitro and in vivo activities and mechanistic characterization. Antimicrob. Agents Chemother. 53:3337–3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wiseman H, Cannon M, Arnstein HR. 1989. Observation and significance of growth inhibition of Saccharomyces cerevisiae (A224A) by the anti-oestrogen drug tamoxifen. Biochem. Soc. Trans. 17:1038–1039 [DOI] [PubMed] [Google Scholar]
- 43. Rowlands MG, Budworth J, Jarman M, Hardcastle IR, McCague R, Gescher A. 1995. Comparison between inhibition of protein kinase C and antagonism of calmodulin by tamoxifen analogues. Biochem. Pharmacol. 50:723–726 [DOI] [PubMed] [Google Scholar]
- 44. Luxo C, Jurado AS, Madeira VM, Silva MT. 2003. Tamoxifen induces ultrastructural alterations in membranes of Bacillus stearothermophilus. Toxicol. In Vitro 17:623–628 [DOI] [PubMed] [Google Scholar]
- 45. Robins HI, Won M, Seiferheld WF, Schultz CJ, Choucair AK, Brachman DG, Demas WF, Mehta MP. 2006. Phase 2 trial of radiation plus high-dose tamoxifen for glioblastoma multiforme: RTOG protocol BR-0021. Neuro Oncol. 8:47–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.