

Abstract
Chikungunya virus (CHIKV) is a mosquito-transmitted virus that has reemerged as a significant public health threat in the last decade. Since the 2005-2006 chikungunya fever epidemic in the Indian Ocean island of La Réunion, millions of people in more than 40 countries have been infected. Despite this, there is currently no antiviral treatment for chikungunya infection. In this study, an immunofluorescence-based screening platform was developed to identify potential inhibitors of CHIKV infection. A primary screen was performed using a highly purified natural product compound library, and 44 compounds exhibiting ≥70% inhibition of CHIKV infection were identified as positive hits. Among these, four were selected for dose-dependent inhibition assays to confirm their anti-CHIKV activity. Harringtonine, a cephalotaxine alkaloid, displayed potent inhibition of CHIKV infection (50% effective concentration [EC50] = 0.24 μM) with minimal cytotoxicity and was selected for elucidation of its antiviral mechanism. Time-of-addition studies, cotreatment assays, and direct transfection of viral genomic RNA indicated that harringtonine inhibited an early stage of the CHIKV replication cycle which occurred after viral entry into cells. In addition, quantitative reverse transcription-PCR (qRT-PCR) and Western blot analyses indicated that harringtonine affects CHIKV RNA production as well as viral protein expression. Treatment of harringtonine against Sindbis virus, a related alphavirus, suggested that harringtonine could inhibit other alphaviruses. This study suggests for the first time that harringtonine exerts its antiviral effects by inhibiting CHIKV viral protein synthesis.
INTRODUCTION
Chikungunya virus (CHIKV) is an arbovirus that causes chikungunya fever, a disease characterized by myalgia, polyarthralgia, fever, nausea, and headaches (1, 2). CHIKV belongs to the Alphavirus genus in the Togaviridae family and was first isolated in Tanzania in 1952, where it was transmitted primarily by Aedes aegypti mosquitoes (3).
In 2005, the resurgence of CHIKV in several islands in the Indian Ocean caused outbreaks of unprecedented magnitude. In the French island of La Réunion alone, one-third of its 785,000 inhabitants were infected with CHIKV, resulting in more than 250 fatalities (4, 5). Apart from the newfound pathogenicity, the La Réunion epidemic was also associated with complex clinico-pathological complications, including encephalopathy, lymphopenia, and hemorrhagic fever (6, 7). In addition, a mutation of alanine to valine (Ala226Val) was detected in the E1 envelope glycoprotein of the chikungunya viral particle, resulting in a new CHIKV strain that became more prevalent as the epidemic progressed. This mutation is believed to have resulted in a change in the primary transmission vector from Aedes aegypti to Aedes albopictus (8). The efficiency with which Aedes albopictus transmits CHIKV across varied geographical regions has led to researchers postulating the possibility of CHIKV being established almost globally (9). To date, CHIKV has infected millions of people in more than 40 countries, including India, Malaysia, Indonesia, Thailand, Singapore, the United States, and European countries (1, 10–13).
Despite the significant public health threat that CHIKV continues to pose, there is currently no antiviral treatment or vaccine against CHIKV infection. Previous studies have reported anti-CHIKV activities for some compounds in vitro. These include chloroquine (14, 15), furin inhibitors (16), arbidol (17, 18), mycophenolic acid (19), and a combination of ribavirin and alpha interferon (20). Of these, only chloroquine has been tested in vivo. Clinical trials conducted during the La Réunion outbreak found chloroquine to be ineffective as a CHIKV antiviral (21). At present, none of these compounds have been approved for antiviral treatment of CHIKV infection. For these reasons, there is an urgent need for the discovery of novel antivirals for CHIKV infection.
The CHIKV replication cycle offers a good starting point for identification of potential targets during the development of antiviral compounds. CHIKV is an enveloped virus with a single-stranded, positive-sense RNA genome. The CHIKV genome is approximately 11 kb long and consists of two open reading frames which encode the viral nonstructural proteins (nsP1, nsP2, nsP3, and nsP4) as well as the viral structural proteins, which include the capsid (C) and envelope proteins (E1, E2, E3, and 6k) (22, 23). The envelope proteins E1 and E2 form glycoprotein spikes on the viral particle surface and facilitate the initial binding of the viral particle to susceptible host cells. This is followed by entry of the CHIKV particle via clathrin-dependent endocytosis and uncoating of the viral genome (24, 25). Translation of viral RNA then produces the viral nonstructural protein complex, which replicates the CHIKV RNA genome (26). This is followed by translation of viral structural proteins and assembly of viral components within the cytoplasm (16, 27). Viral particles bud out through the plasma membrane, forming mature, infectious progeny (28, 29).
In this study, an immunofluorescence cell-based assay was developed to perform a screen for small-molecule inhibitors of CHIKV replication. The primary screen was performed on a library of 502 highly purified compounds with defined chemical structures which were derived from natural products. A number of hits from the primary screen were selected for dose-dependent inhibition studies to confirm their anti-CHIKV activity. From the results of the secondary assays, harringtonine was selected for further analysis due to its potent inhibition of CHIKV infection and novelty as an antiviral. Harringtonine is a cephalotaxine ester derived from the Japanese plum yew, Cephalotaxus harringtonia. Harringtonine is known to inhibit the first cycle of the elongation phase of eukaryotic translation (30). In this study, we took several approaches to investigate the possible mechanisms of anti-CHIKV activity of harringtonine. The results indicated that harringtonine acts on the postentry stage of the CHIKV replication cycle and strongly interferes with the process of viral protein synthesis.
MATERIALS AND METHODS
Cell lines and virus.
In this study, three cell lines were used, namely, BHK21 (baby hamster kidney) cells, C6/36 mosquito cells isolated from Aedes albopictus embryonic tissue, and human skeletal muscle myoblasts (HSMM) (Lonza, Walkersville, MD). BHK21 cells were cultured in RPMI 1640 medium containing 10% inactivated fetal calf serum (FCS) at 37°C with 5% CO2. C6/36 cells were maintained in L-15 medium containing 10% FCS at 28°C. HSMM cells were cultured in skeletal muscle cell growth medium (SkGM) (Lonza) supplied with growth factors and 10% FCS (Lonza) at 37°C with 5% CO2. This study used two strains of CHIKV, CHIKV-0708 (Singapore/07/2008, kindly provided by the National Public Health Laboratory, Ministry of Health, Singapore) and CHIKV-122508 (SGEHICHD 122508, kindly provided by the Environmental Health Institute, Singapore). CHIKV-0708 (which lacks the A226V mutation in the E1 protein) and CHIKV-122508 (which contains the A226V mutation in the E1 protein) were propagated in BHK21 cells and C6/36 cells, respectively. Sindbis virus was propagated in BHK21 cells.
Natural product library.
The primary screening of antiviral activity against CHIKV infection was performed using 502 highly purified compounds from a natural product library (BioMol, Plymouth Meeting, PA). All compounds in the library are of low molecular weight, are highly purified, and have known chemical structures. The library does not contain any extracts or mixtures. The library contains compounds from microbial, plant, and marine sources, including a range of terpenoids, peptolides, coumarins, alkaloids, flavones, and isoflavones. The complete list of compounds is available upon request at compoundlibraries@enzolifesciences.com. The library compounds were dissolved in 100% dimethyl sulfoxide (DMSO), giving a stock concentration of 10 mM.
Primary screening.
BHK21 cells were seeded at a density of 3.5 × 103 cells per well in 384-well plates (Corning, Corning, NY) and incubated overnight at 37°C with 5% CO2. The cells were infected with CHIKV-0708 at a multiplicity of infection (MOI) of 1 for 1.5 h with gentle rocking every 15 min. After the 1.5 h viral adsorption period, cells were washed twice with phosphate-buffered saline (PBS) to remove unbound viruses before addition of the library compounds. The compounds were diluted from the 10 mM stock to a final concentration of 10 μM prior to being incubated with the cells. CHIKV-infected cells were treated with 0.1% DMSO or 0.5 μg/ml (2.05 μM) of ribavirin for the negative and positive controls, respectively. CHIKV-infected cells were incubated for 24 h with the natural product compounds prior to being processed for immunofluorescence assay. Cell monolayers were fixed with 100 μl of cold absolute methanol (Sinopharm Chemical, Shanghai, China) at −20°C for 15 min before being rehydrated with 100 μl PBS. Fixed cell monolayers were incubated with 40 μl monoclonal mouse antialphavirus antibody (Santa Cruz, Santa Cruz, CA) diluted to 1:250 for 1 h at 37°C. Cells were then washed three times with PBS using the automated Embla 384 cell washer station (Molecular Devices, Sunnyvale, CA) before being incubated for another 1 h at 37°C with 40 μl of the secondary antibody, fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (Chemicon, Temecula, CA), diluted to 1:500. The cell nuclei were stained using DAPI (4′,6-diamidino-2-phenylindole) (Invitrogen, Carlsbad, CA) for 15 min at room temperature before the cells were washed in PBS again. For background controls, cells were stained with only the secondary FITC-conjugated antibody and DAPI.
Data acquisition.
The images for the immunofluorescence assay were obtained using an Olympus IX81 inverted fluorescence microscope (Olympus, Japan) under a magnification of ×20. Images were acquired for both the DAPI and FITC channels. The total number of cells per well was determined by counting the DAPI-stained nuclei, and the total number of CHIKV-infected cells was determined by counting the FITC-stained cytoplasm. Counting of cells was done using the CellProfiler software (www.cellprofiler.org). The average number of cells per well was calculated and compared against the mock-infected cells in 0.1% DMSO and against the CHIKV-infected cells in 0.1% DMSO. Wells that had fewer than 500 cells were excluded in the data analysis. Positive hits from the primary screen were defined as compounds displaying 70% or more inhibition of CHIKV replication relative to 0.1% DMSO-treated, CHIKV-infected controls. A number of hit compounds were then selected for secondary assays to validate their anti-CHIKV activity.
The robustness of our screening assay was determined using the Z′ factor, a statistical measurement of the distance between the standard deviation of the signal versus the noise of the assay (31). Experiments to determine the Z′ factor were conducted in 384-well plates using BHK21 cells infected with CHIKV-0708 at an MOI of 1. One hundred wells of CHIKV-0708-infected BHK21 cells were treated with 0.1% DMSO, and another 100 wells were incubated with 0.5 μg/ml (2.05 μM) ribavirin for 24 h at 37°C with 5% CO2. The experiment was carried out in triplicates. The cells were then processed for immunofluorescence assay under the same conditions as described above. The Z′ factor was computed using the equation 1 − [(3 × SDDMSO + 3 × SDribavirin)/(mean DMSO − meanribavirin)], where SD is standard deviation, DMSO represents CHIKV-infected cells treated with 0.1% DMSO, and ribavirin represents CHIKV-infected cells treated with 0.5 μg/ml (2.05 μM) ribavirin.
Validation of primary screen hits.
Hits from the primary screen were validated using dose-dependent drug-treatments on BHK21 cells. BHK21 cells were seeded into 96-well plates at a density of 1.2 × 104 cells per well and incubated overnight at 37°C with 5% CO2. Cell monolayers were then infected with CHIKV-0708 at an MOI of 1 for 1.5 h (viral adsorption). Cells were washed twice with PBS and incubated with the selected compounds over a range of concentrations (0.01 μM, 0.1 μM, 1 μM, 5 μM, and 10 μM) for 24 h at 37°C with 5% CO2. After the incubation period, cell culture supernatants were harvested for the quantification of infectious virus titer via plaque assays.
The anti-CHIKV effect of harringtonine was further validated by repeating the dose-dependent treatment on BHK21 cells infected with CHIKV-122508. Dose-dependent inhibitory studies were also carried out with homoharringtonine (Enzo Life Sciences, Farmingdale, NY), an analogue of harringtonine, on BHK21 cells infected with CHIKV-0708 or CHIKV-122508. In addition, dose-dependent treatments for harringtonine were carried out on HSMM infected with CHIKV-0708 or CHIKV-122508. All validation experiments for harringtonine and homoharringtonine were carried out in a 96-well format, and cell supernatants were harvested for plaque assays at the end of the 24-h incubation period.
For harringtonine treatment studies with Sindbis virus, BHK21 cells were seeded into 96-well plates and infected with Sindbis virus at an MOI of 1 for 1 h prior to being washed twice with PBS and incubated with various concentrations of harringtonine (0.1 μM, 1 μM, 5 μM, and 10 μM) at 37°C with 5% CO2. Cell supernatants were harvested for plaque assays at 24 h postinfection (hpi).
Plaque assay.
For quantification of virus titer, BHK21 cells were plated onto 24-well plates and incubated overnight at 37°C with 5% CO2. The supernatants from virus-infected samples were diluted in 10-fold dilutions with RPMI supplemented with 2% FCS before being used to infect the cells for 1.5 h at 37°C with gentle rocking during the adsorption period. Infected cells were washed twice with PBS and overlaid with 1% carboxymethyl cellulose (CMC) in RPMI with 2% FCS. Cell monolayers were incubated for 3 days at 37°C with 5% CO2. CMC was then removed, and cells were stained and fixed with 10% paraformaldehyde–1% crystal violet (Sigma-Aldrich Chemical, St. Louis, MO) solution for visualization and counting of plaques. Virus titers were expressed as PFU per milliliter.
Cell viability assay.
The cell viability profiles of selected drugs were assessed by alamarBlue cytotoxicity assay (Invitrogen) as recommended by the manufacturer's protocol. The cell viability assay was carried out in similarly to the assay for validation of primary screening hits, except that cells were not infected prior to drug treatment. In brief, BHK21 or HSMM cells were incubated with selected compounds at a concentration range of 0.01 μM to 10 μM in RPMI with 2% FCS for 24 h (unless otherwise stated) at 37°C with 5% CO2. Cell monolayers were then incubated with alamarBlue reagent for 2 h (BHK21) or 4 h (HSMM) prior to fluorescence detection at an excitation wavelength of 570 nm and an emission wavelength of 585 nm. Fluorescence was measured with Infinite 200 Pro multiplate reader (Tecan, Männedorf, Switzerland). All cell viability assays were conducted in triplicates. Measurements from compound-treated or 0.1% DMSO-treated cells were normalized against those from untreated cells.
Time-of-addition studies.
Time-of-addition studies were performed for harringtonine (Biomol) on CHIKV-0708-infected BHK21 cells in 96-well plates. For the pretreatment assay, cell monolayers were treated with 0.1 μM, 1 μM, or 5 μM harringtonine for 2 h at 37°C prior to being washed twice PBS and infected with CHIKV-0708 at an MOI of 1. After the 1.5-h virus adsorption period, infected cells were incubated in RPMI with 2% FCS at 37°C with 5% CO2 for 24 h before supernatants were harvested for plaque assay.
For the posttreatment assays, BHK21 cells were infected with CHIKV-0708 at an MOI of 1 for 1.5 h (viral adsorption), and harringtonine (0.1 μM, 1 μM, or 5 μM) was added at five different time points postinfection (0 h, 2 h, 6 h, 12 h, and 16 h). For time-of-addition studies, CHIKV-infected cells treated with 0.1% DMSO were used as positive controls.
For the cotreatment assay, CHIKV-122508 or CHIKV-0708 was treated with harringtonine (1 μM or 10 μM) or 0.1% DMSO for 30 min at 37°C. In order to remove excess harringtonine or DMSO solutions, viruses were subjected to centrifugal filtration in 100,000-molecular-weight centrifugal filter units (Millipore, Darmstadt, Germany) at 1,500 × g for 15 min at 4°C. Viruses were then resuspended in PBS and filtered through the same filter units a second time before being resuspended in appropriate amounts of RPMI with 2% FCS. BHK21 cells were infected with purified CHIKV at an MOI of 1 for 1.5 h prior to being incubated in maintenance medium for about 24 h at 37°C with 5% CO2. The supernatants were harvested at 24 hpi for quantification of viral titer via plaque assays.
Viral RNA transfection into BHK21 cells.
Viral RNA was extracted from CHIKV viral supernatants using the QIAamp viral RNA minikit (Qiagen GmbH, Hilden, Germany) according to the manufacturer's instructions. For transfection, BHK21 cells were seeded into 96-well plates at a density of 1.2 × 104 cells per well and incubated overnight at 37°C with 5% CO2. BHK21 cells were transfected with 100 ng of CHIKV-0708 viral RNA for 1.5 h before harringtonine treatment for 24 h. During transfection, 100 ng viral RNA was appropriately diluted in DharmaFECT cell culture reagent (Thermo Scientific, Lafayette, CO) to make up a volume of 25 μl per well and incubated at room temperature for 5 min. A 0.2-μl portion of DharmaFECT-1 transfection reagent (Thermo Scientific) was diluted in DharmaFECT cell culture reagent to make up a volume of 25 μl per well and incubated at room temperature for 5 min. The diluted viral RNA solution was then added to the diluted DharmaFECT-1 and incubated at room temperature for 30 min to allow complexing. Fifty microliters of viral RNA–DharmaFECT-1 complexes was added to each well, and cell monolayers were incubated at 37°C with 5% CO2 for 1.5 h with gentle rocking at intervals. Harringtonine diluted in RPMI with 10% FCS was added to the wells to make up final concentrations of 0.1 μM, 1 μM, and 10 μM, and cells were incubated for 24 h at 37°C with 5% CO2. After 24 h, cell supernatants were harvested for plaque assays.
Quantitative reverse transcription-PCR (qRT-PCR).
Prior to the quantification of the positive- and negative-sense chikungunya viral RNAs, a CHIKV RNA standard curve was first generated from the Colors pcDNA 6.2/C-EmGFP-GW/TOPO mammalian expression vector containing the CHIKV-122508 capsid gene cloned into the vector via TA cloning (Invitrogen). Positive- and negative-sense CHIKV RNAs were generated via a T7 promoter using the MAXIscript in vitro transcription kit (Applied Biosystems, Oslo Area, Norway) according to the manufacturer's instructions. A standard curve was then generated using serial dilutions of positive- and negative-sense viral RNAs assayed via RT-PCR. Samples were assayed in a 20-μl reaction mixture containing 10 μl of SYBR green (Fermentas, Hanover, MD), 1 μl of either the CHIKV capsid forward primer or reverse primer, 1 μl of RNA, 1 μl of reverse transcriptase, and 7 μl of nuclease-free water. Reactions were carried out in the Applied Biosystems StepOnePlus real-time PCR system (Applied Biosystems, Carlsbad, CA), beginning with a 30-min reverse transcription step at 44°C. After reverse transcription, the complementary primer was added, and this was followed by 5 min of Taq polymerase activation at 94°C and 40 amplification cycles at 94°C for 15 s each and 60°C for 30 s for fluorescence measurement during amplification. Following amplification, a melting curve analysis was performed to verify the melting temperatures of PCR products amplified by the primer pairs. The primer sequences were as follows: forward, 5′-GCGGTACCCCAACAGAAG-3′; reverse, 5′-GGTTTCTTTTTAGGTGGCTG-3′.
For sample preparation, CHIKV-infected cells were plated on 24-well plates at a density of 9 × 104 cells per well infected with CHIKV-0708 or CHIKV-122508 at an MOI of 1 for 1.5 h. After virus adsorption, cells were washed twice with PBS before treatment with harringtonine at a concentration range (0.01 μM, 0.1 μM, 1 μM, and 10 μM) and incubation at 37°C with 5% CO2 for 6 h. Mock-infected cells treated with 0.1% DMSO and CHIKV-infected cells treated with 0.1% DMSO were used as controls. Total cellular RNA extraction was carried out using the RNeasy minikit (Qiagen) according to the manufacturer's instructions. Samples were assayed via RT-PCR as described above. The copy numbers of positive-sense and negative-sense CHIKV RNAs were then derived from the cycle threshold value of the amplification plot by using the standard curve as a reference.
SDS-PAGE and Western blotting.
BHK21 cells were plated onto 6-well plates at a density of 5 × 105 cells per well before being infected with CHIKV-122508 or CHIKV-0708 at an MOI of 1 for 1.5 h. CHIKV-infected cells were treated with harringtonine at a concentration range (0.01 μM, 0.1 μM, 1 μM, and 10 μM) for 6 h or 24 h for detection of nsP3 or E2, respectively. Mock-infected cells treated with 0.1% DMSO and CHIKV-infected cells treated with 0.1% DMSO were used as controls. Cells were lysed using 500 μl CelLytic M cell lysis reagent (Sigma) containing complete protease inhibitor cocktail (1 tablet dissolved in 10 ml cell lysis reagent) (Roche, Indianapolis, IN) at 4°C for 15 min. Lysed cells were scraped and collected in microcentrifuge tubes before being subjected to three freeze-thaw cycles to ensure complete lysis of cells. Cellular debris was pelleted out by centrifuging lysed cells at 1,000 × g for 5 min. The protein-containing supernatants were separated in 10% acrylamide gels run at 100 V for 1.5 h (for detection of E2) or 2 h (for detection of nsP3). The PageRuler prestained protein ladder (Fermentas) was used as a molecular weight standard. The gels were then equilibrated in Towbin buffer (0.025 M Tris, 0.192 M glycine 20% methanol) for at least 10 min to remove the sodium dodecyl sulfate (SDS). The gels were transferred to a nitrocellulose membrane using the Bio-Rad semidry transfer system (Bio-Rad, San Francisco, CA) at 0.3 A for 1 h.
For detection of nsP3, membranes were blocked with 5% bovine serum albumin (BSA) (MP Biomedicals, Santa Ana, CA) dissolved in Tris-buffered saline–Tween 20 (TBST) overnight at 4°C on a shaker. The blots were rinsed three times with TBST before being incubated with primary anti-CHIKV nsP3 rabbit polyclonal antibody dissolved in 5% BSA at a dilution of 1:100 (the generation of the polyclonal nsP3 antibody was contracted to ProSci Inc., Poway, CA). For the loading control, separate blots containing the same samples were incubated with primary anti-β-actin mouse monoclonal antibody (Millipore, Temecula, CA) dissolved in 5% BSA at a dilution of 1:5,000. The blots were incubated with primary antibodies overnight at 4°C on a shaker. The blots were then washed six times with TBST for 10 min each time. This was followed by incubation with the secondary antibodies polyclonal goat anti-rabbit IgG(H+L)–horseradish peroxidase and polyclonal goat anti-mouse IgG(H+L)–horseradish peroxidase (Thermo Scientific, Rockford, IL) for 1 h at room temperature on an orbital shaker. Dilution of both secondary antibodies was done in 5% BSA at a ratio of 1:1,500. Membranes were then washed three times with TBST for 10 min each time. Membranes were developed by the enhanced chemiluminescence (ECL) method using SuperSignal West Pico chemiluminescent substrate (Thermo Scientific, Hudson, NH).
For detection of E2, membranes were blocked with 5% skim milk dissolved in TBST overnight at 4°C on a shaker. The blots were then rinsed three times with PBS before being incubated with primary anti-CHIKV E2 rabbit polyclonal antibody at a dilution of 1:100 (the generation of the polyclonal E2 antibody was contracted to ProSci Inc., Poway, CA). For the loading control, separate blots containing the same samples were incubated with primary anti-β-actin mouse monoclonal antibody (Millipore, Temecula, CA) at a dilution of 1:1,500. The blots were incubated with primary antibodies for 1.5 h at 37°C on an orbital shaker. Dilutions for both primary and secondary antibodies were done in PBS. The blots were then washed three times with TBST for 10 min each time and three times with PBS for 10 min each time. This was followed by incubation with the secondary antibodies alkaline phosphatase (AP)-conjugated goat anti-rabbit IgG and AP-conjugated goat anti-mouse IgG (Thermo Scientific, Rockford, IL) for 1 h at 37°C on an orbital shaker. The dilution ratio for both secondary antibodies was 1:1,500. Membranes were washed twice with TBST for 10 min each time and once with PBS for 10 min. Bands were detected using the colorimetric method by developing membranes with BCIP-NBT (5-bromo-4-chloro-3-indolylphosphate p-toluidine phosphatase and nitroblue tetrazolium chloride) substrate (Thermo Scientific, Rockford, IL).
Ultrastructural study using transmission electron microscopy (TEM).
Confluent T75 flasks of BHK21 cells were infected with CHIKV-122508 or CHIKV-0708 at an MOI of 10 for 1.5 h before incubation with 1 μM harringtonine diluted in RPMI with 2% FCS for 24 h at 37°C with 5% CO2. CHIKV-infected flasks treated with 0.1% DMSO were used as positive controls. Cell monolayers were fixed with 7.5 ml of a primary fixative consisting of 1% glutaraldehyde (Agar Scientific, Stansted, United Kingdom) at 4°C over 3 days. After primary fixation, the cell monolayers were washed and scraped off before being postfixed with 1% osmium tetroxide (Ted Pella, Redding, CA) for 2 h. A few grains of potassium ferrocyanide were added to enhance the contrast of the membranous structure within cells. After 2 h, the cell pellets were washed and dehydrated with progressively increasing concentrations of ethanol (25%, 50%, 75%, 95%, and 100%). The dehydration step was enhanced by another two rounds of absolute acetone treatment for 10 min each. Dehydrated cell pellets were then infiltrated with increasing concentrations of araldite 502 (Ted Pella) to acetone at increasing temperatures before embedding in fresh araldite for 24 h at 60°C. The embedded samples were then trimmed with an ultramicrotome (Reichert-Jung, Depew, NY) to approximately 50 to 70 nm. Cut sections were then placed on a 200-mesh copper grid before being stained with 2% uranyl acetate and postfixed with lead citrate. Stained sections were viewed using a Philips EM 208 transmission electron microscope (Philips, Eindhoven, The Netherlands) and captured digitally with a dual-view digital camera (Gatan Inc., Werrendale, CA).
Statistical analyses.
A one-way analysis of variance (ANOVA) test was conducted to evaluate the significance of the data from the drug treatment studies. For samples that showed statistical significance (P < 0.05) from ANOVA analysis, a Dunnett's posttest was carried out to compare data from treated samples against those from the 0.1% DMSO (solvent) control. Results from Dunnett's posttest were used to determine the concentrations of compounds which resulted in a statistically significant difference compared to the solvent control.
RESULTS
Development of immunofluorescence screening assay.
A primary screening assay was first developed to screen for inhibitors of CHIKV infection from a library of highly purified natural product compounds. We used immunofluorescence to detect the alphaviral envelope protein as an indication of successful CHIKV infection and replication. Ribavirin has previously been shown to inhibit CHIKV replication (20) and was selected to serve as a positive control, as well as to assess the robustness of the immunofluorescence screening assay. As shown in Fig. 1, ribavirin displayed a dose-dependent inhibition of CHIKV-0708 infection compared to the 0.2% DMSO control. A 50% effective concentration (EC50) of 0.5 μg/ml (2.05 μM) was obtained with BHK21 cells. Mock-infected cells were included as negative controls to ensure specificity of the primary antibody. Infected cells stained with only the secondary FITC antibody and DAPI were added as background controls to check for specificity of the secondary FITC antibody (Fig. 1). A Z′ factor of 0.76 was obtained for the primary screening assay, indicating that the primary screening assay was sufficiently robust. Thus, the screening platform via the immunofluorescence assay method was found to be suitable and reliable for the screening of the compound libraries and detection of potential CHIKV inhibitors.
Fig 1.

Dose-dependent inhibition of ribavirin on CHIKV infectivity shown using immunofluorescence assay. (a) Immunofluorescence detection of alphavirus envelope protein is used as an indication of CHIKV infection. CHIKV-0708 infection of BHK21 cells is compared between ribavirin-treated cells (positive control) and untreated cells. Cell nuclei are stained with DAPI (blue), and CHIKV infection is indicated by FITC (green) staining. Mock-infected cells were stained with both primary and secondary antibodies, as well as DAPI. Background control refers to infected cells that were stained with only the secondary FITC antibody and DAPI. (b) A dose-dependent reduction of viral antigen-positive cells for the different ribavirin concentrations (EC50, 0.5 μg/ml or 2.05 μM) is observed from immunofluorescence images. Error bars represent standard errors of triplicate means.
Primary screening assay.
Once the immunofluorescence-based screening platform was established, a primary screen was performed using a library of 502 highly purified natural product compounds to detect potential CHIKV inhibitors. The natural product compounds were screened at a concentration of 10 μM and analyzed to detect the percent reduction of virus antigen-positive cells compared to those with the 0.1% DMSO control. Using a criterion of ≥70% inhibition to define positive hits, a total of 44 compounds were identified as positive hits (see Table S1 in the supplemental material).
Validation of selected hits.
Of the list of hit compounds, four were selected for secondary assays to confirm their anti-CHIKV activity. The compounds chosen were harringtonine, hypocrellin A, rottlerin, and daunorubicin (see Fig. S1 in the supplemental material). These compounds were selected to include a mixture of known virus inhibitors such as hypocrellin A (32), as well as alkaloids and enzyme inhibitors with no previously reported inhibitory activities against alphaviruses, such as harringtonine and rottlerin. The anti-CHIKV activities of selected compounds were further evaluated in dose-dependent inhibition studies via plaque assays to quantify infectious viral titer.
As shown in Fig. 2, all compounds tested displayed dose-dependent inhibition of CHIKV infection. Harringtonine and hypocrellin A displayed the greatest magnitude of inhibition of CHIKV-0708 infection compared to the 0.1% DMSO control (Fig. 2a and b). The 50% effective concentration (EC50) of harringtonine (0.24 μM) was lower than that of hypocrellin A (1 μM), suggesting that harringtonine may be the most potent anti-CHIKV compound in the set. The EC50s for rottlerin and daunorubicin (Fig. 2c and d) exceeded 10 μM.
Fig 2.

Dose-dependent study of anti-CHIKV activities of selected compounds from a primary high-throughput screen as observed via plaque assay. Treatment of CHIKV-0708-infected BHK21 cells with harringtonine (a), hypocrellin A (b), rottlerin (c), and daunorubicin (d) results in a significant dose-dependent inhibition of CHIKV-0708 infection. Line graphs for cell viability experiments correspond to secondary axes. Minimal cytotoxicity is observed for all compounds tested. Error bars represent standard errors of triplicate means for all cell viability experiments as well as all dose-dependent inhibition studies.
To ensure that the inhibitory effects on CHIKV replication were not due to cell cytotoxicity induced by the compounds, cell viability assays were conducted using alamarBlue. Cell viability remained above 90% for all concentrations of the panel of compounds assayed, indicating minimal cytotoxicity (Fig. 2). Harringtonine was selected for further analysis of its anti-CHIKV activity due to its significant inhibition of CHIKV infection and minimal cytotoxicity, as well as its novel status as an antiviral.
Antiviral activities of harringtonine and its analogue homoharringtonine against CHIKV infection.
The primary screen was performed with CHIKV-0708, a strain which does not contain the alanine-to-valine mutation at position 226 (A226V) of the CHIKV E1 protein. The A226V mutation was detected during the later part of the outbreak in La Réunion and resulted in a change in mosquito species vector from Aedes aegypti to Aedes albopictus. It was thus important to confirm the dose-dependent anti-CHIKV effect of harringtonine on a strain that does contain the mutation. The dose-dependent inhibitory effect of harringtonine was observed with both CHIKV-0708 and CHIKV-122508 (Fig. 3a), indicating that harringtonine targeted a phase in the viral replication cycle that was not strain specific.
Fig 3.

Dose-dependent studies of antiviral activities of harringtonine and homoharringtonine against both CHIKV strains. Harringtonine (a) and homoharringtonine (b) exhibit a statistically significant dose-dependent inhibition of CHIKV-0708 infection at concentrations of 1 μM and above and of CHIKV-122508 infection at concentrations of 0.1 μM and above. Line graphs for cell viability experiments correspond to secondary axes. Minimal cytotoxicity is observed with treatment of BHK21 cells with harringtonine or homoharringtonine over 24 h. Statistical significance is analyzed from a one-way ANOVA test and Dunnett's posttest. ***, P < 0.001. Error bars represent standard errors of triplicate means for cell viability experiments. Error bars for dose-dependent studies represent standard errors of means from three independent experiments.
The anti-CHIKV effect of a harringtonine analogue, homoharringtonine, was also tested to compare its potency to that of harringtonine. Homoharringtonine contains an additional methyl group in its side chain, which is believed to increase stability of the compound (33) (see Fig. S2 in the supplemental material). Homoharringtonine displayed minimal cytotoxicity when used to treat BHK21 cells (Fig. 3b). Like harringtonine treatment, homoharringtonine treatment resulted in a significant dose-dependent inhibition of CHIKV infection for both strains. The results also showed evidence of a strain-specific difference in potency of the compounds. Treatment with harringtonine produced a greater magnitude of inhibition of virus titer for CHIKV-122508 (4.3 log10 inhibition) than for CHIKV-0708 (2.3 log10 inhibition) at 10 μM and 5 μM concentrations. This trend was also observed with homoharringtonine treatment for both virus strains. Significant inhibition of CHIKV-122508 infection could be observed with a 0.1 μM concentration of both compounds. In contrast, inhibition of CHIKV-0708 infection was significant at drug concentrations of 1 μM and above (Fig. 3a and b). The data suggested that CHIKV-122508 may be more susceptible to treatments with harringtonine and homoharringtonine than CHIKV-0708.
Given that harringtonine and homoharringtonine had been previously found to be inhibitors of eukaryotic protein synthesis, it was important to confirm that the concentrations of both drugs were noncytotoxic to the cells. BHK21 cells were incubated with various concentrations (0.01 μM to 100 μM) of the compounds for 3 days, with the compounds being replaced every day. Cell viability did not decrease below 90% at 1 μM, suggesting that the in vitro antiviral effects of both harringtonine and homoharringtonine against CHIKV could be obtained at concentrations that did not affect cell viability (see Fig. S3 in the supplemental material).
The antiviral effect of harringtonine on HSMM (primary human skeletal myoblasts), an in vivo target of CHIKV infection (5), was also evaluated. The dose-dependent inhibitory effect of harringtonine was recapitulated for both strains of CHIKV in HSMM (Fig. 4). As observed with CHIKV-infected BHK21 samples, treatment with harringtonine at a concentration of 0.1 μM was sufficient to result in a statistically significant inhibition of CHIKV-122508 infection but not CHIKV-0708 infection. This affirms the strain-specific difference in the potency of harringtonine in treating CHIKV infections. Minimal cytotoxicity was observed when HSMM cells were incubated with various concentrations of harringtonine for 24 h (Fig. 4), further establishing that the in vitro inhibitory effect of harringtonine on CHIKV infection could be observed at concentrations below that required to induce significant cytotoxicity.
Fig 4.

Antiviral effect of harringtonine on CHIKV infection of HSMM (primary human skeletal myoblasts), an in vivo target of CHIKV infection. Harringtonine results in a statistically significant dose-dependent inhibition of CHIKV-0708 infection at concentrations of 1 μM and above and of CHIKV-122508 infection at concentrations of 0.1 μM and above. The line graph for cell viability corresponds to the secondary axis. Minimal cytotoxicity is observed over the concentration range tested, suggesting that the antiviral activity of harringtonine is due to specific effects of harringtonine on CHIKV replication. Statistical significance is analyzed from a one-way ANOVA test and Dunnett's posttest. *, P < 0.05; ***, P < 0.001. Error bars represent standard errors of triplicate means.
Time-of-addition studies of harringtonine in CHIKV infection.
Time-of-addition studies were conducted to identify the window in the CHIKV replication cycle when harringtonine exerts its antiviral effect. Harringtonine (0.1 μM, 1 μM, and 5 μM) was added at different time points before and after infection of BHK21 cells with CHIKV (Fig. 5a). All cell culture supernatants were collected for plaque assays at 24 h postinfection (hpi).
Fig 5.

(a) BHK21 cells were treated with harringtonine at different time points before and after infection in time-of addition studies. Cell supernatants were harvested at 24 hpi for quantification of virus titer via plaque assays. (b) Time-of-addition studies indicate that harringtonine acts between 0 hpi and 6 hpi to inhibit CHIKV infection. (c) In the cotreatment assay, viruses were treated with harringtonine before being used to infect BHK21 cells. No significant inhibition of CHIKV infection is observed with harringtonine treatment with both strains of CHIKV. (d) When CHIKV-0708 genomic RNA is transfected into BHK21 cells prior to treatment with harringtonine, a significant dose-dependent inhibition of CHIKV-0708 replication is observed at harringtonine concentrations of 1 μM and above. The results suggest that harringtonine acts at a postentry step in the CHIKV replication cycle. Statistical significance is analyzed from a one-way ANOVA test and Dunnett's posttest. ***, P < 0.001. Error bars represent standard errors of triplicate means.
From the results shown in Fig. 5b, pretreatment of cells with harringtonine for 2 h prior to CHIKV infection showed a minimal inhibitory effect against viral infection except at a high concentration of 5 μM. The high harringtonine concentration of 5 μM may have allowed the compound to enter and be retained in the cells during the pretreatment period, resulting in inhibition of CHIKV infection. Figure 5b suggests that harringtonine did not inhibit the CHIKV entry process. A significant reduction of the CHIKV titer was observed when harringtonine was added at the 0- and 2-hpi time points. At 6 hpi, the antiviral activity of harringtonine was diminished at the 0.1 μM and 1 μM concentrations. Addition of harringtonine at 12 hpi onwards resulted in an almost complete loss of inhibition of CHIKV replication at the 0.1 μM and 1 μM concentrations. This suggested that harringtonine was acting at an early phase in the viral replication cycle that occurs after viral entry.
A cotreatment assay was performed to complement data from the pretreatment assay in the time-of-addition studies by determining if harringtonine affected the surface of the viral particle surface in some way to hinder viral binding and entry into cells. Cotreatment of both CHIKV strains with harringtonine (1 μM or 10 μM) failed to result in inhibition of viral infection (Fig. 5c). To further verify that harringtonine's antiviral action occurs at a postentry event, an indirect assay where 100 ng of CHIKV-0708 genomic RNA was transfected into BHK21 cells prior to harringtonine treatment was conducted. The process of direct transfection of the viral genome allowed the exclusion of CHIKV binding and entry from the process of infection. The dose-dependent inhibitory effect of harringtonine was recapitulated in this assay (Fig. 5d), confirming data from pretreatment and cotreatment assays that harringtonine acts on postentry events of CHIKV infection and is unlikely to affect the infectious entry process of CHIKV into cells.
Harringtonine affects CHIKV RNA production and protein synthesis.
Given that harringtonine was most likely not affecting CHIKV binding and entry, its effect on viral RNA production and protein synthesis was investigated. In the initial postentry stages of the CHIKV replication cycle, the positive-sense CHIKV RNA genome is used as a template for the synthesis of nonstructural proteins by the host cell translation machinery. The nonstructural proteins then complex to form the viral replicase, which synthesizes a negative-sense RNA strand from the positive-sense template. This negative-sense strand serves as a template for the synthesis of more positive-sense genome copies, as well as the subgenomic RNA. The subgenomic RNA is subsequently translated by the host cell machinery into virus structural proteins.
To investigate the effect of harringtonine on viral RNA synthesis, qRT-PCR was conducted on CHIKV-infected BHK21 cells treated with harringtonine for 6 h. Figure 6 shows that harringtonine treatment resulted in a significant dose-dependent reduction in both negative- and positive-sense RNAs for both CHIKV strains, suggesting that harringtonine inhibits a phase in the CHIKV replication cycle that occurs before RNA production. The strain-specific difference in potency of harringtonine was also detected at the RNA level.
Fig 6.

Harringtonine acts at a step in the CHIKV replication cycle that occurs before negative- and positive-sense RNA production. Harringtonine exhibits a statistically significant dose-dependent inhibition of CHIKV-0708 RNA levels at concentrations of 1 μM and above (a) and CHIKV-122508 RNA levels at concentrations of 0.1 μM and above (b). Statistical significance is analyzed from a one-way ANOVA test and Dunnett's posttest. **, P < 0.01; ***, P < 0.001. Error bars represent standard errors of triplicate means.
In order to determine the effect of harringtonine on CHIKV protein synthesis, Western blot analyses were performed. A dose-dependent reduction of CHIKV nsP3 and E2 proteins for both CHIKV strains was observed upon harringtonine treatment (Fig. 7a and b). At concentrations of 1 μM and 10 μM, minimal amounts of nsP3 and E2 proteins were detected for both CHIKV strains. The size of CHIKV E2 has been previously determined to be 50 kDa (34). The size of nsP3 is taken to be approximately 76 to 78 kDa (35). At harringtonine concentrations of 0.1 μM and 0.01 μM, nsP3 and E2 protein levels for both CHIKV strains were comparable to that for the 0.1% DMSO control. This suggested that harringtonine may inhibit CHIKV protein production, leading to a decrease in infectious virus titers as seen above. In addition to E2, the anti-CHIKV E2 antibody used also detected the precursor of E2 (pE2), a 65-kDa protein (36) containing E2 as well as E3 (Fig. 7b). The strain-specific difference in potency was less apparent in Western blot analyses. Nevertheless, there was a general trend of dose-dependent inhibition of CHIKV infection by harringtonine, supporting results obtained with the different experimental approaches reported above. β-Actin was used as a loading control in the experiment, as well as to ensure that the concentration of harringtonine used in this study did not affect the synthesis and expression of host cellular proteins (37).
Fig 7.

Western blot analyses were performed to determine the effect of harringtonine on production of CHIKV nsP3 and E2 proteins. A dose-dependent reduction of CHIKV nsP3 (a) and E2 and pE2 (b) proteins is observed for CHIKV-0708 and CHIKV-122508 samples upon harringtonine treatments of 6 h and 24 h, respectively. β-Actin is used as a loading control for each set of samples.
The host cell translation machinery is involved in translation of both nonstructural and structural viral proteins, and this indirectly affects negative- and positive-sense RNA levels during infection. Taken together, the results from Fig. 6 and 7 indicate that the antiviral effect of harringtonine may depend on its function as a protein synthesis inhibitor, directly decreasing viral protein production and indirectly decreasing viral positive- and negative-sense RNA levels. These direct and indirect effects of harringtonine may then result in a cumulative, strongly pronounced inhibitory effect on infectious virus titers.
Harringtonine inhibits Sindbis virus, a related alphavirus.
To investigate the specificity of harringtonine's antiviral action, BHK21 cells were infected with Sindbis virus (an alphavirus related to CHIKV) and treated with a range of concentrations of harringtonine. The dose-dependent antiviral effect of harringtonine was reproduced with Sindbis virus, as shown in Fig. 8. This indicates that the antiviral activity of harringtonine is not restricted to CHIKV but may extend to other alphaviruses as well.
Fig 8.
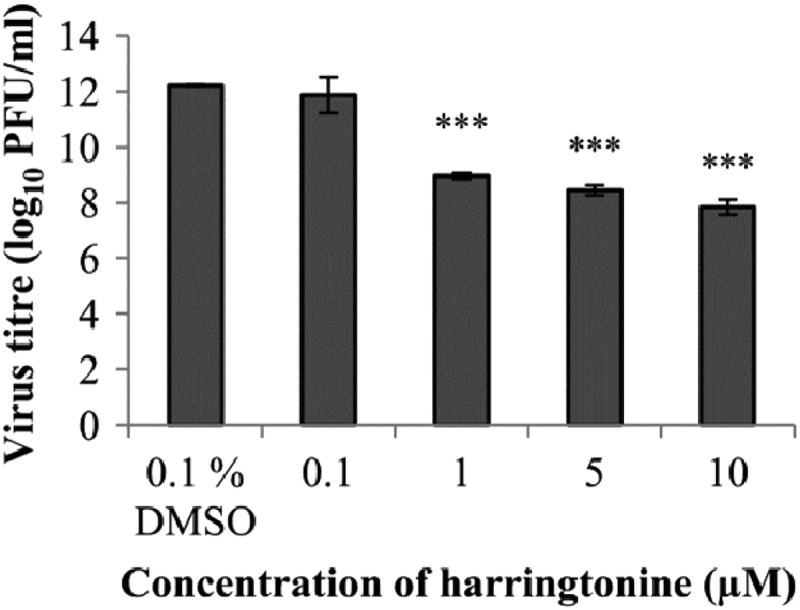
Harringtonine treatment causes significant inhibition of Sindbis virus (an alphavirus related to CHIKV) at concentrations of 1 μM and above. Error bars represent standard errors of triplicate means. Statistical significance is analyzed from a one-way ANOVA test and Dunnett's posttest. ***, P < 0.001.
Ultrastructural analyses of CHIKV-infected cells treated with harringtonine.
To visualize the effects of harringtonine treatment on CHIKV replication, transmission electron microscopy (TEM) was carried out. CHIKV-infected cells were posttreated with 1 μM harringtonine before being processed for TEM at 24 hpi. The mock-infected BHK21 cells displayed normal morphological features of healthy cells (Fig. 9a). CHIKV-infected cells that were treated with the solvent control (0.1% DMSO) displayed characteristic morphological features of CHIKV infection. CHIKV-0708-infected BHK21 cells treated with 0.1% DMSO showed the presence of cytopathic vacuoles I (CPV I), which display modified membranous structures for replication of the viral genome as well as structural proteins (Fig. 9b). Infected cells treated with the solvent control also displayed presence of cytopathic vacuoles II (CPV II) filled with large numbers of electron-dense viral particles (Fig. 9b and d). Budding of viral particles on the plasma membrane could also be observed (Fig. 9b and c).
Fig 9.

(a) Mock-infected BHK21 cells display normal morphology of healthy cells. (b) CHIKV-0708-infected BHK21 cells treated with 0.1% DMSO show characteristic features of infected cells, including CPV I and II. White arrows indicate viral nucleocapsids budding into CPV II. Black arrows indicate viral particles budding on the cell surface. (c) Higher magnification of CHIKV-0708-infected cells, showing extensive viral budding, as indicated by black arrows. (d) CHIKV-122508-infected BHK21 cells treated with 0.1% DMSO. White arrows indicate viral nucleocapsids budding into CPV II. (e and f) CHIKV-0708 (e)- and CHIKV-122508 (f)-infected cells treated with 1 μM harringtonine show restoration of normal morphology and absence of CPV I, CPV II, or viral particles. CV, cellular vesicles; CPV I, cytopathic vacuole I; CPV II, cytopathic vacuole II; ER, endoplasmic reticulum; G, Golgi apparatus; L, lysosome; M, mitochondria; N, nucleus; NM, nuclear membrane; PM, plasma membrane.
In contrast, harringtonine-treated cells displayed no late-stage virus-associated morphological changes. No CPV could be detected, and electron-dense viral particles on the surface were absent (Fig. 9e and f). This confirmed the above data showing that harringtonine inhibited CHIKV replication. Restoration of normal cellular morphology at 1 μM harringtonine was observed, affirming the possible role of harringtonine as a CHIKV antiviral. In addition, harringtonine-treated cells displayed no difference in morphology compared to the mock-infected sample (Fig. 9a). This further confirmed the cell viability data from earlier experiments, suggesting that the antiviral effects of harringtonine may occur at concentrations that are not cytotoxic to the cells.
DISCUSSION
In response to the lack of antiviral treatment for CHIKV infections, a screening platform was developed to screen for novel anti-CHIKV compounds. The use of an immunofluorescence-based screening approach developed for a 384-well format allows a large number of small-molecule compounds to be screened for the rapid identification of potential antivirals (37). The immunofluorescence-based screening platform in this study was used to screen 502 highly purified compounds from a Natural Product Library for inhibition of CHIKV infection. The screening assay was also statistically validated and met the criteria for a robust screening assay, with a Z′ factor of 0.76. The Z′ factor is a simple, dimensionless parameter that ensures that the screening platform developed has a properly implemented format with a sufficient dynamic range and an acceptable signal variability that is capable of providing useful data (31). Compounds which resulted in ≥70% inhibition of CHIKV infection were classified as hits. Our primary screen identified 44 compounds as positive hits, including mycophenolic acid, which has recently been reported to be effective in inhibiting the replication of CHIKV (19). Having mycophenolic acid independently identified as a hit in our primary screen further indicated the reliability of our primary screening assay in identifying putative anti-CHIKV inhibitors.
A number of hits from the primary screen were selected for evaluation of their anti-CHIKV activities in dose-dependent inhibition studies. These compounds were harringtonine, hypocrellin A, rottlerin, and daunorubicin. All compounds tested displayed dose-dependent inhibition of CHIKV infection, confirming that they were true positive hits. Harringtonine is a known inhibitor of the eukaryotic large ribosomal subunit that has not been previously shown to possess antiviral activities (30). Harringtonine was selected for further investigation into its antiviral mechanism due to its potent inhibition of CHIKV replication and minimal cytotoxicity, as well as its novel status as an antiviral. Dose-dependent inhibitory assays also confirmed the antiviral effects of harringtonine against a second CHIKV strain which contained the Ala226Val mutation. In addition, the anti-CHIKV effect of harringtonine was reproduced when tested in HSMM (human skeletal muscle myoblasts), a primary cell line and an in vivo target of CHIKV infection.
Homoharringtonine, an analogue of harringtonine with an additional methyl group, was also found to be inhibitory toward both strains of CHIKV with minimal cytotoxicity when tested in BHK21 cells. This confirms previous studies showing that the inhibition of eukaryotic protein synthesis is dependent on the presence of the side chain in cephalotaxine alkaloids. Cephalotaxines that do not have the side chain have been found to be only minimally active (38). Homoharringtonine, like harringtonine, possesses the ester side chain and thus was found to also be able to inhibit CHIKV infection.
Time-of-addition studies were conducted to identify the potential antiviral mechanism of harringtonine. BHK21 cells were treated with harringtonine at different time points before and after infection with CHIKV. The results suggested that harringtonine inhibited the early events of the CHIKV replication cycle after viral entry into cells. Cotreatment assays, as well as direct transfection of CHIKV genomic RNA, confirmed results from the pretreatment assay in time-of-addition studies, verifying that harringtonine did not affect CHIKV binding and entry. Harringtonine's effect on CHIKV RNA production and protein synthesis was also investigated in this study. Data from qRT-PCR and Western blot analyses indicated that harringtonine treatment resulted in a decrease in CHIKV RNA production and synthesis of nonstructural (nsP3) as well as structural (E2) proteins. Given previous literature citing that harringtonine is an inhibitor of protein synthesis (30, 39), it is most likely that harringtonine inhibits the eukaryotic large ribosomal unit, thereby suppressing the translation of nonstructural and structural proteins. The observed decrease in the levels of nsP3 proteins suggest that the antiviral function of harringtonine may begin with its inhibition of translation of nonstructural proteins, leading to a decrease in levels of replicase complexes. This is likely to result in a reduction in levels of negative-sense RNA to function as templates for synthesis of the positive-sense RNA strands. Consequently, the lowered levels of template negative-sense RNA, coupled with the decreased levels of the replicase, may result in an even more significant decrease in positive-sense RNA levels and possibly subgenomic RNA levels. If harringtonine does exert its antiviral effect by inhibiting the host translational machinery, it may be postulated that a second step of direct inhibition occurs at the stage of viral structural protein synthesis. Lowered subgenomic RNA levels, coupled with the direct inhibition of host cell translation machinery by harringtonine, may then culminate in a strong inhibition of viral structural protein production.
Studies with both CHIKV strains revealed the possibility of a strain-specific difference in potency for both harringtonine and homoharringtonine, with CHIKV-122508 possibly being more susceptible to treatment with the two compounds than CHIKV-0708. The strain-specific difference in the potency of harringtonine could be detected early in the CHIKV replication cycle, at the level of negative-sense RNA production. Apart from the A226V mutation in E1, other sequence changes are present in CHIKV-122508 (data not shown). Hence, the strain-specific difference in potency, if present, may be due to one or more of these mutations. It is possible that even with harringtonine treatment, the levels of replicase complex in CHIKV-0708-infected cells remain high enough to allow sufficient synthesis of the negative-sense RNA strands, thereby reducing the cumulative downstream effect of harringtonine treatment. CHIKV-0708 may also exhibit strain-specific properties that affect its interaction with the eukaryotic ribosome, indirectly reducing the inhibitory effect of harringtonine against the ribosome.
The use of harringtonine against Sindbis virus, an alphavirus related to CHIKV, also resulted in a significant dose-dependent inhibition. This suggested that harringtonine's mode of antiviral action is not specific to CHIKV. However, given previous literature suggesting that harringtonine is ineffective in inhibiting growth of encephalomyocarditis virus (40), it is still plausible that the antiviral action of harringtonine may be restricted to a number of classes of viruses.
Even though harringtonine targets a host component in the CHIKV replication cycle, this may not necessarily limit its therapeutic potential. There is a scarcity of antiviral therapeutics for RNA viruses other than HIV (41). Among the drugs that have been licensed by the U.S. FDA, ribavirin is an example of a drug which is known to act by targeting a host component. Ribavirin is used for the treatment of hepatitis C virus and pediatric respiratory syncytial virus infections (19). Ribavirin is known to deplete intracellular GTP pools by inhibiting a host enzyme, IMP dehydrogenase. This has been postulated to cause an accumulation of errors during viral genome replication and failure to sustain the production of infectious virions during subsequent replication cycles (42, 43). Furthermore, recently discovered compounds such as arbidol and mycophenolic acid which have shown anti-CHIKV activity in vitro also target host components in the CHIKV replication cycle. This suggests that harringtonine may be considered a potential anti-CHIKV therapeutic despite targeting a host component. A suitable murine model of CHIKV infection is currently being developed to test the in vivo efficacy of harringtonine against CHIKV infection.
While this study postulates that harringtonine acts as a CHIKV antiviral by inhibiting the host protein translation machinery, it is possible that harringtonine may inhibit CHIKV infection by additional, complementary mechanisms. Recent studies have suggested that focusing on one aspect of a drug's action may be insufficient to explain its therapeutic effects (44). While the lethal mutagenesis theory has been a convincing explanation for inhibition of polioviruses (45), additional mechanisms have been proposed for the antiviral action of ribavirin against other viruses. These include inhibition of viral RNA capping (46), inhibition of viral RNA polymerase (47), inhibition of viral helicase activity (48), and direct incorporation of ribavirin into viral RNA in the place of guanine and adenine (42), as well as direct inhibition of viral RNA synthesis (49). Additional studies are required to allow an in-depth exploration of the molecular basis of harringtonine's anti-CHIKV action. The inhibition of CHIKV replication observed when 5 μM harringtonine was added at a late time point, such as 16 hpi, in time-of-addition studies may suggest an additional mechanism of inhibition at high concentrations of harringtonine, affecting later phases of the CHIKV replication cycle. Despite the fact that homoharringtonine is a more potent inhibitor of protein synthesis (44), there was a slightly greater magnitude of inhibition of CHIKV replication upon harringtonine treatment than upon homoharringtonine treatment at 10 μM, suggesting that additional inhibitory mechanisms may indeed be present to effect harringtonine's antiviral activity.
Supplementary Material
ACKNOWLEDGMENTS
This study was funded by a BMRC A*STAR grant (R182-000-158-305) and an NUS start-up grant (R182-000-165-133).
Footnotes
Published ahead of print 22 October 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01467-12.
REFERENCES
- 1. Hapuarachchi HC, Bandara KB, Sumanadasa SD, Hapugoda MD, Lai YL, Lee KS, Tan LK, Lin RT, Ng LF, Bucht G, Abeyewickreme W, Ng LC. 2010. Re-emergence of chikungunya virus in South-east Asia: virological evidence from Sri Lanka and Singapore. J. Gen. Virol. 91:1067–1076 [DOI] [PubMed] [Google Scholar]
- 2. Sharma S, Dash PK, Santhosh SR, Shukla J, Parida M, Rao PV. 2010. Development of a quantitative competitive reverse transcription polymerase chain reaction (QC-RT-PCR) for detection and quantitation of chikungunya virus. Mol. Biotechnol. 45:49–55 [DOI] [PubMed] [Google Scholar]
- 3. Sudeep AB, Parashar D. 2008. Chikungunya: an overview. J. Biosci. 33:443–449 [DOI] [PubMed] [Google Scholar]
- 4. Schuffenecker I, Iteman I, Michault A, Murri S, Frangeul L, Vaney MC, Lavenir R, Pardigon N, Reynes JM, Pettinelli F, Biscornet L, Diancourt L, Michel S, Duquerroy S, Guigon G, Frenkiel MP, Bréhin AC, Cubito N, Despres P, Kunst F, Rey FA, Zeller H, Brisse S. 2006. Genome microevolution of chikungunya viruses causing the Indian Ocean outbreak. PLoS Med. 3:e263 doi:10.1371/journal.pmed.0030263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sourisseau M, Schilte C, Casartelli N, Trouillet C, Guivel-Benhassine F, Rudnicka D, Sol-Foulon N, Le Roux K, Prevost MC, Fsihi H, Frenkiel MP, Blanchet F, Afonso PV, Ceccaldi PE, Ozden S, Gessain A, Schuffenecker I, Verhasselt B, Zamborlini A, Saib A, Rey FA, Arenzana-Seisdedos F, Despres P, Michault A, Albert ML, Schwartz O. 2007. Characterization of reemerging chikungunya virus. PLoS Pathog. 3:e89 doi:10.1371/journal.ppat.0030089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mavalankar D, Shastri P, Bandyopadhyay T, Parmar J, Ramani KV. 2008. Increased mortality rate associated with chikungunya epidemic, Ahmedabad, India. Emerg. Infect. Dis. 14:412–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Robin S, Ramful D, Le Seach F, Jaffar-Bandjee MC, Rigou G, Alessandri JL. 2008. Neurologic manifestations of pediatric chikungunya infection. J. Child. Neurol. 23:1028–1035 [DOI] [PubMed] [Google Scholar]
- 8. Tsetsarkin KA, Vanlandingham DL, McGee CE, Higgs S. 2007. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 3:e201 doi:10.1371/journal.ppat.0030201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Charrel RN, de Lamballerie X, Raoult D. 2007. Chikungunya outbreaks—the globalization of vector-borne diseases. N. Engl. J. Med. 356:769–771 [DOI] [PubMed] [Google Scholar]
- 10. Hochedez P, Hausfater P, Jaureguiberry S, Gay F, Datry A, Danis M, Bricaire F, Bossi P. 2007. Cases of chikungunya fever imported from the islands of the South West Indian Ocean to Paris, France. Euro Surveill. 12:679 [Google Scholar]
- 11. Lim PL, Oh HM, Ooi EE. 2009. Chikungunya in Singapore: imported cases among travelers visiting friends and relatives. J. Travel Med. 16:289–291 [DOI] [PubMed] [Google Scholar]
- 12. Parola P, de Lamballerie X, Jourdan J, Rovery C, Vaillant V, Minodier P, Brouqui P, Flahault A, Raoult D, Charrel RN. 2006. Novel chikungunya virus variant in travelers returning from Indian Ocean islands. Emerg. Infect. Dis. 12:1493–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pfeffer M, Loscher T. 2006. Cases of chikungunya imported into Europe. Euro Surveill. 11:2922. [PubMed] [Google Scholar]
- 14. Inglot AD. 1969. Comparison of the antiviral activity in vitro of some non-steroidal anti-inflammatory drugs. J. Gen. Virol. 4:203–214 [DOI] [PubMed] [Google Scholar]
- 15. Shimizu Y, Yamamoto S, Homma M, Ishida N. 1972. Effect of chloroquine on the growth of animal viruses. Arch. Gesamte Virusforsch. 36:93–104 [DOI] [PubMed] [Google Scholar]
- 16. Ozden S, Lucas-Hourani M, Ceccaldi PE, Basak A, Valentine M, Benjannet S, Hamelin J, Jacob Y, Mamchaoui K, Mouly V, Desprès P, Gessain A, Butler-Browne G, Chrétien M, Tangy F, Vidalain PO, Seidah NG. 2008. Inhibition of chikungunya virus infection in cultured human muscle cells by furin inhibitors: impairment of the maturation of the E2 surface glycoprotein. J. Biol. Chem. 283:21899–21908 [DOI] [PubMed] [Google Scholar]
- 17. Boriskin YS, Leneva IA, Pecheur EI, Polyak SJ. 2008. Arbidol: a broad-spectrum antiviral compound that blocks viral fusion. Curr. Med. Chem. 15:997–1005 [DOI] [PubMed] [Google Scholar]
- 18. Delogu I, Pastorino B, Baronti C, Nougairède A, Bonnet E, de Lamballerie X. 2011. In vitro antiviral activity of arbidol against chikungunya virus and characteristics of a selected resistant mutant. Antiviral Res. 90:99–107 [DOI] [PubMed] [Google Scholar]
- 19. Khan M, Dhanwani R, Patro IK, Rao PV, Parida MM. 2011. Cellular IMPDH enzyme activity is a potential target for the inhibition of chikungunya virus replication and virus induced apoptosis in cultured mammalian cells. Antiviral Res. 89:1–8 [DOI] [PubMed] [Google Scholar]
- 20. Briolant S, Garin D, Scaramozzino N, Jouan A, Crance JM. 2004. In vitro inhibition of chikungunya and Semliki Forest viruses replication by antiviral compounds: synergistic effect of interferon-alpha and ribavirin combination. Antiviral Res. 61:111–117 [DOI] [PubMed] [Google Scholar]
- 21. de Lamballerie X, Ninove L, Charrel RN. 2009. Antiviral treatment of chikungunya virus infection. Infect. Disord. Drug Targets. 9:101–104 [DOI] [PubMed] [Google Scholar]
- 22. Bréhin AC, Casademont I, Frenkiel MP, Julier C, Sakuntabhai A, Despres P. 2009. The large form of human 29,59-Oligoadenylate Synthetase (OAS3) exerts antiviral effect against chikungunya virus. Virology 384:216–222 [DOI] [PubMed] [Google Scholar]
- 23. Sreekumar E, Issac A, Nair S, Hariharan R, Janki MB, Arathy DS, Regu R, Mathew T, Anoop M, Niyas KP, Pillai MR. 2010. Genetic characterization of 2006-2008 isolates of chikungunya virus from Kerala, South India, by whole genome sequence analysis. Virus Genes 40:14–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Helenius A, Kartenbeck J, Simons K, Fries E. 1980. On the entry of Semliki Forest virus into BHK-21 cells. J. Cell Biol. 84:404–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marsh M, Bolzau E, Helenius A. 1983. Penetration of Semliki Forest virus from acidic prelysosomal vacuoles. Cell 32:931–940 [DOI] [PubMed] [Google Scholar]
- 26. Barton DJ, Sawicki SG, Sawicki DL. 1991. Solubilization and immunoprecipitation of alphavirus replication complexes. J. Virol. 65:1496–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Strauss JH, Strauss EG. 1990. Alphavirus proteinases. Semin. Virol. 1:347–356 [Google Scholar]
- 28. Ekstrom M, Liljestrom P, Garoff H. 1994. Membrane protein lateral interactions control Semliki Forest virus budding. EMBO J. 13:1058–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Perera R, Owen KE, Tellinghuisen TL, Gorbalenya AE, Kuhn RJ. 2001. Alphavirus nucleocapsid protein contains a putative coiled coil alpha-helix important for core assembly. J. Virol. 75:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fresno M, Jimenez A, Vazquez D. 1977. Inhibition of translation in eukaryotic systems by harringtonine. Eur. J. Biochem. 72:323–330 [DOI] [PubMed] [Google Scholar]
- 31. Zhang JH, Chung TD, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4:67–73 [DOI] [PubMed] [Google Scholar]
- 32. Hudson JB, Zhou J, Chen J, Harris L, Yip L, Towers GH. 1994. Hypocrellin, from Hypocrella bambuase, is phototoxic to human immunodeficiency virus. Photochem. Photobiol. 60:253–255 [DOI] [PubMed] [Google Scholar]
- 33. Jiang TL, Liu RH, Salmon SE. 1983. Comparative in vitro antitumor activity of homoharringtonine and harringtonine against clonogenic human tumor cells. Invest. New Drugs 1:21–25 [DOI] [PubMed] [Google Scholar]
- 34. Cavrini F, Gaibani P, Pierro AM, Rossini G, Landini MP, Sambri V. 2009. Chikungunya: an emerging and spreading arthropod-borne viral disease. J. Infect. Dev. Ctries. 3:744–752 [DOI] [PubMed] [Google Scholar]
- 35. Li GP, La Starza MW, Hardy WR, Strauss JH, Rice CM. 1990. Phosphorylation of Sindbis virus nsP3 in vivo and in vitro. Virology 179:416–427 [DOI] [PubMed] [Google Scholar]
- 36. Simizu B, Yamamoto K, Hashimoto K, Ogata T. 1984. Structural proteins of chikungunya virus. J. Virol. 51:254–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Low JS, Wu KX, Chen KC, Ng ML, Chu JJH. 2011. Narasin, a novel antiviral compound that blocks dengue virus protein expression. Antivir. Ther. 16:1203–1218 [DOI] [PubMed] [Google Scholar]
- 38. Dyke SF, Quessy SN. 1981. Erythrina and related alkaloids, p 85 In Manske RHF, Rodrigo RGA. (ed), The alkaloids, vol 18 Academic Press, Inc., New York, NY [Google Scholar]
- 39. Gurel G, Blaha G, Moore PB, Steitz TA. 2009. U2504 determines the species specificity of the A-site cleft antibiotics: the structures of tiamulin, homoharringtonine, and bruceantin bound to the ribosome. J. Mol. Biol. 389:146–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ramabhadran TV, Thach RE. 1980. Specificity of protein synthesis inhibitors in the inhibition of encephalomyocarditis virus replication. J. Virol. 34:293–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Leyssen P, De Clercq E, Neyts J. 2008. Molecular strategies to inhibit the replication of RNA viruses. Antiviral Res. 78:9–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Crotty S, Cameron CE, Andino R. 2001. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc. Natl. Acad. Sci. U. S. A. 98:6895–6900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ruiz-Jarabo CM, Arias A, Baranowski E, Escarmís C, Domingo E. 2000. Memory in viral quasispecies. J. Virol. 74:3543–3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Efferth T, Sauerbrey A, Halatsch ME, Ross DD, Gebhart E. 2003. Molecular modes of action of cephalotaxine and homoharringtonine from the coniferous tree Cephalotaxus hainanensis in human tumor cell lines. Naunyn Schmiedebergs Arch. Pharmacol. 367:56–67 [DOI] [PubMed] [Google Scholar]
- 45. Vignuzzi M, Stone JK, Andino R. 2005. Ribavirin and lethal mutagenesis of poliovirus: molecular mechanisms, resistance and biological implications. Virus Res. 107:173–181 [DOI] [PubMed] [Google Scholar]
- 46. Benarroch D, Egloff MP, Mulard L, Guerreiro C, Romette JL, Canard B. 2004. A structural basis for the inhibition of the ns5 dengue virus mRNA 2′-O-methyltransferase domain by ribavirin 5′-triphosphate. J. Biol. Chem. 279:35638–35643 [DOI] [PubMed] [Google Scholar]
- 47. Bougie I, Bisaillon M. 2003. Initial binding of the broad spectrum antiviral nucleoside ribavirin to the hepatitis C virus RNA polymerase. J. Biol. Chem. 278:52471–52478 [DOI] [PubMed] [Google Scholar]
- 48. Rankin JT, Jr, Eppes SB, Antczak JB, Joklik WK. 1989. Studies on the mechanism of the antiviral activity of ribavirin against reovirus. Virology 168:147–158 [DOI] [PubMed] [Google Scholar]
- 49. Ruiz-Jarabo CM, Ly C, Domingo E, de la Torre JC. 2003. Lethal mutagenesis of the prototypic arenavirus lymphocytic choriomeningitis virus (LCMV). Virology 308:37–47 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.