

Abstract
Lipoatrophy in HIV patients can result from prolonged exposure to thymidine analogues. Mitochondrial toxicity leading to dysregulated adipogenesis and increased cell death has been proposed as a leading factor in the etiology of peripheral fat loss. We hypothesized that thymidine analogues interfere with autophagy, a lysosomal degradation pathway, which is important for mitochondrial quality control, cellular survival, and adipogenesis. We assessed the effects of zidovudine (AZT), stavudine (d4T), and lamivudine (3TC) on autophagy in eukaryotic cells and adipocytes (3T3-F442A) by fluorescence microscopy and flow cytometry. The effects were compared to interventions with established genetic and pharmacological inhibitors of autophagy and correlated to assessments of cell viability, proliferation, and differentiation. AZT and d4T, but not 3TC, inhibited both constitutive and induced autophagic activity in adipocytes. This inhibition was associated with accumulation of dysfunctional mitochondria, increased reactive oxygen species (ROS) production, increased apoptosis, decreased proliferation, and impaired adipogenic conversion. Autophagy inhibition was dose and time dependent and detectable at therapeutic drug concentrations. Similar phenotypic changes were obtained when genetic or pharmacological inhibition of autophagy was employed. Our data suggest that thymidine analogues disturb adipocyte function through inhibition of autophagy. This novel mechanism potentially contributes to peripheral fat loss in HIV-infected patients.
INTRODUCTION
Highly active antiretroviral therapy (HAART) has been associated with the development of the so-called “lipodystrophy syndrome” (LD) (1–3). In cohorts with predominant use of thymidine analogues (TA), the percentage of HIV-positive patients diagnosed as lipodystrophic reached the level of almost 50% (1). LD prevalence remains a major issue in HIV medicine, given that thymidine analogues are still heavily used in resource-limited countries (3, 4) and that lipoatrophy demonstrates only little reversibility after replacement of thymidine analogues.
Peripheral fat loss as a part of the lipodystrophy syndrome was mostly related to the use of nucleoside analogues, particularly stavudine (d4T) and zidovudine (AZT) (5, 6). Subcutaneous abdominal adipose tissue from HIV-1-infected patients with peripheral lipoatrophy was characterized by an increased level of apoptosis (7, 8) and impaired expression of adipogenic markers (9). Drug-related disturbance of adipogenesis in combination with increased cell loss was hypothesized to lead to fat tissue atrophy. Using well-characterized cell lines and primary human adipocytes, we and others repeatedly confirmed AZT's and d4T's antiadipogenic properties in vitro (10–15), which could well have a clinical impact on adipogenesis in vivo (16).
Autophagy represents a cellular lysosomal degradation pathway which is crucial for cell homeostasis, differentiation, and survival (17). This process is considered an adaptive response that is invoked in order to keep cells alive under stressful conditions (17). Macroautophagy begins with the formation of a vesicular sac (isolation membrane), which elongates and encloses cytoplasmic components (e.g., mitochondria) together with parts of the cytoplasm. Ultimately, the isolation membrane closes in the form of a double-membrane vacuole autophagosome. The autophagosome fuses with a lysosome through its outer membrane, creating an autolysosome, in which the autophagosomal materials and the inner autophagosomal membrane undergo degradation. There are several established ways to experimentally modulate autophagic activity (18). Autophagy is induced by (i) a physiological stimulus such as starvation and (ii) pharmacological modulation of nutrient-sensing signaling pathways such as mTOR, mostly through the use of mTOR inhibitors, such as rapamycin and PP242. Autophagy is inhibited by pharmacological interference with (i) AP formation using PI3-kinase inhibitors such as 3-MA, wortmannin, and LY294002; (ii) autophagosome-lysosome fusion using microtubule-disrupting agents such as nocodazole and vinblastine (18); and (iii) autolysosomal degradation of autophagic substrates using ammonium chloride, chloroquine, and hydrohychloroquine.
A number of recent studies suggested a central role of adipocyte autophagy in the maintenance of adipose tissue homeostasis (19–21). Genetic and pharmacological inhibition of adipocyte autophagy has been mechanistically related to decreased adipose mass and impaired adipogenesis (19–21).
As in vivo and in vitro effects of AZT and d4T treatment of adipocyte homeostasis are reminiscent of a situation where autophagic balance is compromised, we hypothesized that some of the antiadipogenic effects of these drugs might be mediated through their impact on autophagy.
MATERIALS AND METHODS
Cell culture.
293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum (FCS) with 100 U/100 μg/ml penicillin/streptomycin. 3T3-F442A preadipocytes were kindly provided by Jacqueline Capeau (France) and cultured as previously described (14). Preadipocytes were cultured in DMEM containing 5% newborn calf serum (NCS) supplemented with 100 U/100 μg/ml penicillin/streptomycin (preadipocyte medium). Before initiation of differentiation, subconfluent preadipocytes were maintained for 2 days in preadipocyte medium supplemented with 5% FCS. Then, differentiation was performed using DMEM containing 10% FCS, 4 μg/ml pantothenic acid, 8 μg/ml biotin, and 100 U/100 μg/ml penicillin/streptomycin, supplemented with 1 μM insulin (Sigma-Aldrich, St. Louis, MO), which was added to cells 2 days after confluence (designated day 0), with subsequent incubation until day 9. AZT, d4T, and lamivudine (3TC) (Sigma-Aldrich, St. Louis, MO) were dissolved in dimethyl sulfoxide (DMSO). The drugs were used at concentrations near the therapeutic maximum concentrations of drug in serum (Cmax) of AZT (6 μM), d4T (3 μM), and 3TC (8 μM) as discussed in references 14 and 22. In some experiments targeting the investigation of dose-dependent effects, higher concentrations (5× and 30× Cmax) were used. Traditional inhibitors and activators of autophagy were used at concentrations previously reported in similar in vitro experiments (18) as follows: 3-MA, 3 to 10 mM; wortmannin (W), 30 to 100 nM; LY294002 (LY), 7 to 20 μM; nocodazole (N), 12 to 50 μM; vinblastine (V), 12 to 50 μM; rapamycin (Rapa), 5 μM; PP242, 5 μM; ammonium chloride (ACH), 10 to 20 mM; and hydrohychloroquine (HCQ) and chloroquine (CQ), 5 to 10 μM. All reagents were dissolved in DMSO except for 3-MA, ACH, CQ, and HCQ, which were dissolved in phosphate-buffered saline (PBS). The highest concentration of the solvent used in the incubation experiments (0.1% DMSO) did not affect cellular viability and preadipocyte differentiation. For experiments with starvation-induced autophagy, cells were incubated for the designated time periods in Hanks' Balanced Salt Solution (HBSS) containing 6 mM glucose (starvation medium). The extent of preadipocyte differentiation was assessed by Oil red O staining and microscopically for acquisition of a spherical shape and triacylglyceride droplet accumulation. Cell numbers and viability were determined both microscopically using trypan blue staining and by flow cytometric counting and propidium iodide staining. Relative amounts of apoptosis were determined using flow cytometry as changes of cellular forward scatter (FSC) versus side scatter (SSC) dot plots in comparison to viable control cell results as previously described (23).
Constructs and retroviral infection.
pBABE-puro mCherry-enhanced green fluorescent protein (EGFP)-LC3B (plasmid 22418) and pBABEpuro GFP-LC3 (plasmid 22405) generated by Jayanta Debnath (24) were purchased from Addgene. The respective sequences were inserted into retroviral constructs, which were used for cell transduction. The mCherry signal was used only for confirmatory purposes (data not shown) and was not relevant for this study. Retroviral production and cell transduction were essentially done as previously explained (25). Briefly, 10 μg retroviral vectors and calcium phosphate precipitation were used for the transfection of amphotrophic producer cells. Transfected ΦNX cells (constantly >95% GFP-positive cells) were incubated for 24 h. The medium was changed (DMEM containing 10% FCS), and retroviral supernatants were harvested 16 to 24 h later. After filtration (45-μm-pore-size filter; Schleicher & Schuell), retroviral supernatant was added to the 293T and 3T3-F442A cells preincubated for 24 h in the presence of 5 μg/ml Polybrene. After the addition of retroviral supernatant, 293T and 3T3-F442A cells were initially centrifuged at 2,300 rpm (890 × g) for 90 min at 30°C (in order to improve the efficiency of viral transduction) and then left for overnight incubation at 37°C, at the end of which the supernatant containing the viral particles was replaced by fresh medium. After 72 h of puromycin selection (Sigma, Taufkirchen, Germany) (1 μg/ml), GFP-positive cells were subjected to fluorescence-activated cell sorter (FACS) analysis (FACSAria). Cellular LC3 expression alone or in combination with GFP expression has been suggested to result in aggregate formations indistinguishable from autophagosomes (18). In order to overcome this limitation and before use of fluorescence microscopy and flow cytometry, 293T and 3T3-F442A cell lines stably expressing GFP-LC3 were selected for clones presenting GFP-LC3 expression levels giving a minimum artificial aggregation (18).
Genetic inhibition of autophagy was accomplished by short hairpin RNA (shRNA)-mediated ATG5 (shATG5) knockdown (Santa Cruz). ATG5 encodes a critical autophagic protein required for the maturation of the autophagic membrane (19). CopGFP and nonspecific shRNA were used as controls according to the instructions of the manufacturer (Santa Cruz). Transduction and knockdown were performed by using lentiviral particles with up to five distinct expression constructs. Transduction efficiency as determined by the number of GFP-positive cells after puromycin selection generally exceeded 95%.
Analysis of effects of nucleoside reverse transcriptase inhibitors (NRTIs)on cellular autophagic activity.
An essential consideration in the analysis of cellular autophagy is the fact that autophagosomes correspond to an intermediate structure of a dynamic process and that their total amount at a particular time point is a function of two variables: generation versus disappearance (18). Therefore, an increase in autophagosome presence might signify either induction or blockage of late autophagy (at a step downstream of autophagosome formation [AF]). Measurement of autophagic flux allows discrimination between these two scenarios (18).
Fluorescence microscopy analysis of NRTI effect on autophagosome formation.
GFP-LC3 was visualized using conventional fluorescence microscopy according to recently updated guidelines (18). The GFP-LC3 cytoplasmic pool was detected as a homogeneously dispersed signal, whereas GFP-LC3-II-labeled autophagosomes were visualized as formations of punctae (18). The formation of experimental artifacts as a result of potential bulky GFP-LC3 aggregates was minimized by the use of stably transduced cells in combination with appropriate clone selection (18).
For experiments with imaging of living cells, 293T and 3T3-F442A cells stably expressing GFP-LC3 were grown on single glass slides. The cells were subjected to the designated incubations at 37°C in 5% CO2. For each separate treatment condition, fluorescence images were taken from several cells belonging to a number of randomly chosen fields with a GFP filter using an Olympus IX81 instrument and analySIS (Soft Imaging System GmbH).
Flow cytometric analyses of autophagic activity.
In order to measure autophagic flux, we took advantage of a recently developed flow cytometric assay to monitor autophagy in living mammalian cells (18). This highly sensitive quantitative method is based on monitoring of the turnover of the traditional autophagosomal marker LC3 tagged with GFP using flow cytometry. Activation and inhibition of autophagic activity are correspondingly detected as a time-dependent decrease or increase in total cellular GFP signal (18). Autophagy activation intensifies GFP-LC3 delivery into autolysosomes, leading to degradation and selective disappearance of the GFP signal (18). Autophagy inhibition, on the other hand, results in a blockage of autophagic flux, leading to GFP-LC3 accumulation and therefore an increase in fluorescent signal (18). Autophagy inhibition was also studied by measuring the ability of NRTI to reverse the activator-induced disappearance of GFP-LC3 fluorescence signal (18).
Flow cytometric analysis of the autophagic activity of 293T and 3T3-F442A cells was measured, as subconfluent cells stably expressing GFP-LC3 were incubated under different conditions. Cells were trypsinized, put on ice, and analyzed using either FACSCalibur or LSR II (Becton, Dickinson Biosciences) and the data of cell counts plotted as GFP fluorescence intensity.
Intracellular lipid staining.
For Oil red O (Sigma-Aldrich, Munich, Germany) staining, adherent 3T3-F442A cells were formaldehyde (10%) fixed, washed, and stained with a 0.21% (wt/vol) Oil red O solution (60% isopropanol, 40% water). Quantification of triacylglyceride content was performed after drying the cells and Oil red O extraction with 100% isopropanol followed by photometric measurement at 495 nm.
Statistics.
The statistical evaluation for comparisons of more than two groups was performed by analysis of variance (ANOVA) with Dunnett post hoc analysis. The level of significance was set at P < 0.05. All data are presented as means ± standard deviations (SD). All calculations were performed using GraphPadPrism 4.
RESULTS
AZT and d4T augment PP242-induced autophagosome accumulation.
Fluorescence microscopy analysis of cells stably expressing LC3-GFP detects autophagosomes as formation of punctae on the background of homogeneously distributed LC3-GFP patterns. Autophagy induction is detected as accumulation of autophagic punctae. Under conditions leading to autophagy activation, early inhibition of the autophagic processes can be identified by the inability of the cell to form autophagic punctae. In contrast, late autophagy inhibition is expected to result in additional autophagosome accumulation.
In order to evaluate the effects of NRTI on eukaryotic autophagic activity, we cultured 293T cells stably expressing LC3-GFP in the presence or absence of therapeutic Cmax concentrations of AZT (6 μM) and d4T (3 μM) for 24 h and 3-MA (10 mM), wortmannin (100 nM), LY294002 (20 μM), vinblastine (50 μM), or rapamycin (5 μM) for 6 h. PP242 (5 μM) was added to some cultures for the last 4 h (Fig. 1A). Fluorescence microscopy analysis of control cultures revealed homogeneously distributed LC3-GFP patterns (Fig. 1A). As expected, rapamycin and PP242 incubation induced substantial formation of autophagic punctae, indicating stimulated autophagosome formation. Coincubation with an early autophagy inhibitor such as 3-MA, wortmannin, or LY294002 abrogated PP242-mediated formation of autophagic punctae (Fig. 1A). Coincubation with AZT, d4T, or the fusion inhibitor vinblastine, on the other hand, had an additive effect on PP242-induced formation of autophagic punctae (Fig. 1A). As autophagosomes represent an intermediate structure in a dynamic process, total cellular autophagosome abundance at any point in time is a function of equilibrium between their new formation and autolysosome degradation (18). Therefore, we concluded that the increase in autophagosome presence following AZT and d4T incubation was consistent with either drug-related induction of autophagy or inhibition of autophagosome maturation (AM).
Fig 1.

AZT and d4T augment PP242-induced autophagosome accumulation. 293T cells were incubated with AZT (6 μM) and d4T (3 μM) for 24 h and 3-MA (10 mM), LY294002 (20 μM), wortmannin (100 nM), rapamycin (5 μM), or vinblastine (50 μM) for 6 h in the presence and absence of PP242 (5 μM) for the last 4 h. (A) Fluorescence microscopy analysis of formation of autophagic punctae in 293T cells stably expressing a LC3-GFP fusion protein. (B) Flow cytometry analysis of autophagic flux in 293T cells expressing LC3-GFP fusion protein incubated with vinblastine (50 μM), nocodazole (50 μM), or PP242 (5 μM) for 6 h in the presence and absence of starvation for the last 5 h (left), with a representative histogram (right). (A) Figures are representative of the results of three independent experiments. (B) Results are presented as distributions of the means and are representative of the results of three independent experiments with three replicates. **, P < 0.01.
AZT and d4T, but not 3TC, inhibit autophagic flux.
Flow cytometric monitoring of LC3 turnover is considered one of the most sensitive and reliable methods to measure autophagic flux (18). In order to validate this method for our experimental system, we incubated 293T cells stably expressing LC3-GFP with the fusion inhibitors nocodazole and vinblastine and induced autophagy either by starvation or with PP242. Flow cytometric analysis of total cellular LC3-GFP demonstrated an increase in nocodazole- and vinblastine-treated cultures corresponding to reduced autophagic flux and, consequently, reduced LC3-GFP disappearance (Fig. 1B). In contrast, PP242-treated cultures had decreased LC3-GFP signals, which would argue for increased autophagic flux (Fig. 1B). Of note, nocodazole and vinblastine treatment reversed the effect of starvation on 293T autophagic flux (Fig. 1B).
In order to extend our analysis and to better distinguish between autophagy induction and inhibition of autophagosome maturation, we cultured 293T cells stably expressing LC3-GFP in the presence or absence of increasing concentrations of AZT, d4T, and 3TC for up to 72 h with and without starvation. Incubation with starvation medium and rapamycin (5 μM) for 6 h was included as a positive control for autophagy activation and with 3-MA, wortmannin, and LY294002 for 6 h for autophagy inhibition. Our flow cytometric analysis demonstrated that AZT and d4T, but not 3TC, suppress 293T autophagic activity in a dose-dependent manner (Fig. 2A). In addition, there was an obvious dose-related reversion of starvation-mediated activation of autophagic flux in cultures coincubated with AZT and d4T, but not 3TC, similar to the effect of 3-MA, wortmannin, and LY294002 (Fig. 2B). Importantly, time-dependent reversion of starvation-induced activation of autophagic flux was readily detected at therapeutic Cmax concentrations of AZT and d4T (Fig. 2C).
Fig 2.

AZT and d4T inhibit basic and starvation-induced autophagic flux. (A) 293T cells stably expressing LC3-GFP were incubated in the absence or presence of AZT (6, 30, and 150 μM), d4T (3, 15, and 75 μM), or 3TC (8, 40, and 200 μM) for 24 h and with starvation medium or rapamycin (5 μM) for 6 h. (B) 293T cells stably expressing LC3-GFP were incubated with and without AZT (6, 30, and 50 μM), d4T (3, 15, and 75 μM), or 3TC (8, 40, and 200 μM) for 24 h in the absence or presence of starvation conditions. Results are presented as percent change from non-starved control results. (C) 293T cells were incubated with and without therapeutic Cmax concentrations of AZT (6 μM), d4T (3 μM), or 3TC (8 μM) for 24, 48, and 72 h in the absence or presence of starvation conditions. Results are presented as percent change from non-starved control results. (B and C) Statistical comparisons in relation to starved control results (black bars). All data are presented as means ± SD and are representative of the results of at least three independent experiments. *, P < 0.05; **, P < 0.01; ns, not statistically significant.
These results demonstrate that AZT and d4T, but not 3TC, suppress both basic and activator-induced eukaryotic cell autophagy in a dose- and time-dependent manner and that this effect is detectable at concentrations around the therapeutic concentration in HIV patients.
AZT and d4T, but not 3TC, inhibit adipocyte cell autophagy.
In order to validate our observations in adipocytes, 3T3-F442A cells stably expressing LC3-GFP were cultured in the presence or absence of therapeutic Cmax concentrations of AZT or d4T similar to the conditions mentioned above. In order to distinguish between potential drug effects on early (autophagosome formation) and late (autophagosome maturation) stages of the autophagic process, we coincubated cell cultures with 3MA, AZT, d4T, and nocodazole plus vinblastine in the presence and absence of autophagy activator PP242. Fluorescence microscopy revealed accumulation of green punctae in cultures treated with AZT, d4T, and nocodazole plus vinblastine (Fig. 3A). As expected, the early autophagy inhibitor 3MA stopped PP242-induced formation of punctae. In contrast, AZT and d4T did not reveal any decrease in the activator-induced accumulation of green punctae, which was similar to the results seen with the autophagosome-lysosome fusion inhibitors nocodazole and vinblastine (Fig. 3A).
Fig 3.

AZT and d4T inhibit adipocyte autophagy. 3T3-F442A cells were incubated with AZT (6 μM) and d4T (3 μM) for 24 h and 3-MA (10 mM) or nocodazole plus vinblastine (25 μM) for 6 h in the presence and absence of PP242 (5 μM) for the last 4 h. (A) Fluorescence microscopy analysis of formation of autophagic punctae in 3T3-F442A cells stably expressing LC3-GFP fusion protein. (B) Flow cytometry analysis of autophagic flux of 3T3-F442A cells incubated in the absence or presence of AZT (150 μM) and d4T (75 μM) for 32 h with and without PP242 for the last 6 h. (C) Flow cytometry analysis of autophagic flux of 3T3-F442A cells incubated in the absence or presence of nocodazole (50 μM), vinblastine (50 μM), or ammonium chloride (20 mM) with and without PP242 for 6 h.
For flow cytometric analysis, 3T3-F442A LC3-GFP cells were incubated in the presence or absence of increasing concentrations of AZT, d4T, and 3TC for up to 72 h. In separate experiments, incubations with nocodazole, vinblastine, and ACH were used as positive controls for late autophagy inhibition. At high concentrations, AZT and d4T had already inhibited 3T3-F442A autophagic flux after 32 h and reversed the effect of PP242-induced activation (Fig. 3B). Similar effects on the autophagic flux in adipocytes were detected in cultures treated with nocodazole, vinblastine, or ACH (Fig. 3C). After 72 h of drug incubation, AZT and d4T, but not 3TC, dose dependently inhibited autophagy in 3T3-F442A cells (Fig. 4A). This effect was paralleled by a decrease in cell proliferation (Fig. 4B) and an increase in cell death (Fig. 4C). Importantly, similar effects on 3T3-F442A cell proliferation and cell viability were observed using pharmacological (Fig. 4D) or genetic (data not shown) ATG5 knockdown inhibition of autophagy.
Fig 4.
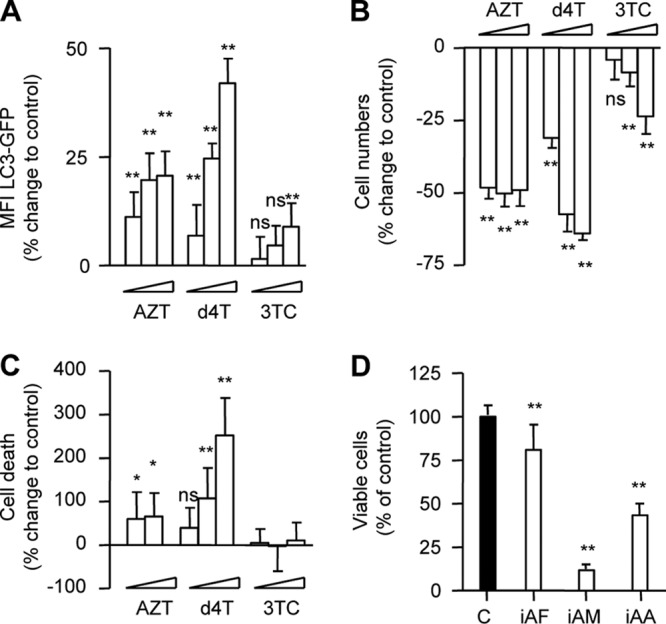
Inhibition of adipocyte autophagic flux correlates with decreased cell proliferation and viability. 3T3-F442A cells stably expressing LC3-GFP were incubated in the absence or presence of AZT (6, 30, and 150 μM), d4T (3, 15, and 75 μM), or 3TC (8, 40, and 200 μM) for 72 h. (A) Flow cytometry analysis of autophagic flux. (B) Changes in cell numbers compared to control. (C) Percentages of dead cells compared to control. (D) 3T3-F442A cells were incubated in the absence or presence of inhibitors of autophagosome formation (iAF; 3-MA [3 mM], wortmannin [30 nM], and LY294002 [7 μM]), autophagosome maturation (iAM; nocodazole [12 μM] and vinblastine [12 μM]), and autophagosome acidification (iAA; ammonium chloride [10 mM] and hydrohychloroquine and chloroquine [5 μM]) for 6 days. Data represent percentages of viable cells compared to control (= 100%). All data are presented as means ± SD and are representative of the results of at least three independent experiments with three to eight replicates. *, P < 0.05; **, P < 0.01.
AZT and d4T, but not 3TC, induce dysfunctional mitochondrion accumulation and ROS production.
Genetic inhibition of eukaryotic cell autophagy has been associated with impaired clearance and accumulation of dysfunctional mitochondria in combination with increased reactive oxygen species (ROS) production (26–29). To assess dysfunctional mitochondrion accumulation and ROS production, 3T3-F442A cells were incubated in the presence or absence of increasing concentrations of AZT, d4T, and 3TC for up to 6 days. We opted for this incubation period as approximately equivalent to the minimum time reported to be necessary for mitochondrial turnover under physiological conditions (30). Consistent with inhibition of autophagy, AZT and d4T incubation was associated with a substantial mitochondrion accumulation (Fig. 5A). This effect was again dose dependent and was readily observable at therapeutically relevant drug concentrations (Fig. 5A). In contrast, 3TC induced a minor increase in cellular mitochondrion abundance only at very high concentrations (Fig. 5A). Importantly, we were able to recapitulate the increase in mitochondrial mass using genetic ATG5 knockdown inhibition of 3T3-F422A autophagy (Fig. 5B).
Fig 5.

AZT and d4T induce accumulation of dysfunctional mitochondria and increased ROS production. 3T3-F442A cells were incubated in the absence or presence of AZT (6, 30, and 150 μM), d4T (3, 15, and 75 μM), or 3TC (8, 40, and 200 μM) for up to 6 days. (A) Flow cytometry analysis of total mitochondrial mass using MitoTracker Green (MTR Green). (B) 3T3-F442A cells transduced with lentiviral particles expressing shATG5 or a nonspecific shRNA control (shC). Flow cytometry analysis of total mitochondrial mass using MTR green (left) with representative histogram (right) immediately after puromycin selection. (C) Flow cytometry analysis of ROS production of 3T3-F442A cells incubated in the absence or presence of AZT (6 and 150 μM), d4T (3 and 75 μM), or 3TC (8 and 200 μM) for 72 h performed using chloromethyl-2′,7′-dichlorofluorescein diacetate (CM-H2DCFDA). (D) 3T3-F442A cells transduced with lentiviral particles expressing shATG5 or a nonspecific shRNA control. Flow cytometry analysis of ROS production using CM-H2DCFDA immediately after puromycin selection (left), with representative histogram (right). Different scaling between panels A and B as well as between panels C and D resulted from different settings in flow cytometry experiments. (E) 3T3-F442A cells were incubated with drugs as described for panel A or inhibitors of autophagosome formation as indicated in Fig. 4D, and Oil red O staining at the end of the adipogenic conversion was quantified in comparison to control results. (F) 3T3-F442A cells transduced with lentiviral particles expressing shATG5 or a nonspecific shRNA control were induced to differentiate, and Oil red O staining was quantified at the end of the adipogenic conversion. All data are presented as means ± SD and are representative of the results of at least three independent experiments with three to eight replicates. *, P < 0.05; **, P < 0.01.
AZT- and d4T-treated cells had evidence of an increased production of ROS (Fig. 5C) in line with the concept that a certain level of basic cellular autophagy is necessary for the clearance of dysfunctional mitochondria. Again, the effects were recapitulated using genetic ATG5 knockdown inhibition of 3T3-F422A autophagy (Fig. 5D).
Pharmacological and genetic inhibition of autophagy recapitulates the effects of AZT and d4T on adipogenesis.
We finally analyzed the impact of autophagy inhibition on phenotypic maturation and intracellular lipid accumulation in 3T3-F442A cells. We used the above-mentioned NRTI, ATG5-specific shRNA, or pharmacological inhibitors of early autophagosome formation (iAF; 3-MA LY294002 and wortmannin), inhibitors of autophagosome maturation (iAM; nocodazole and vinblastine), or inhibitors of autophagosome acidification (iAA; ammonium chloride, chloroquine, and hydroxychloroquine). AZT and d4T, but not 3TC, incubation delayed acquisition of a mature phenotype and the rate of intracellular lipid accumulation (Fig. 5E) in a manner similar to that of genetic ATG5 knockdown (Fig. 5F) and pharmacological inhibition (Fig. 5E). Taken together, these observations are consistent with the idea that autophagy is critical for adipocyte function (19–21) and that autophagy inhibition by TA contributes to compromised adipogenesis (9–15).
DISCUSSION
HAART has been associated with the development of LD characterized by severe adverse events such as fat redistribution, dyslipidemia, insulin resistance, and diabetes mellitus (1–3). Peripheral fat wasting as a part of this syndrome has been mostly related to the use of d4T and AZT (5, 6, 31).
Subcutaneous abdominal adipose tissue from HIV-1-infected patients with peripheral lipoatrophy on NRTI (predominantly d4T) treatment has been characterized with impaired expression of adipogenic markers (9) and higher levels of apoptosis (8, 32). As a result, compromised adipogenesis with enhanced rates of cell death was proposed as one of the pathogenic mechanisms of NRTI-mediated lipoatrophy (9). Furthermore, impaired differentiation with increased levels of apoptosis has been repeatedly confirmed in vitro as a result of AZT and d4T incubation (10–13, 15).
The lysosomal degradation pathway of autophagy has been implicated in the regulation of cellular survival, development, and differentiation (17). During the process, double-membrane vesicles called autophagosomes are formed in which organelles and cytoplasm are sequestrated for later fusion with lysosomes and subsequent content degradation (17). Recently, adipocyte autophagy was implicated in the maintenance of adipose tissue homeostasis (19–21). Genetic and pharmacological inhibition of adipocyte autophagy was associated with decreased lipid accumulation, impaired production of adipogenic markers, and compromised adipogenic conversion in vitro that translated to decreased white adipose tissue mass in several in vivo adipocyte-specific knockout mouse models (19–21). In some of those studies, the autophagy-dependent impairment of adipogenic conversion was accompanied by excessive cellular apoptosis (19). Nonadipocyte studies, on the other hand, established a mechanistic link between defective autophagy, dysfunctional mitochondrion accumulation with increased ROS formation, and initiation of the intrinsic pathway of apoptosis (26–29).
Here we report that AZT and d4T, but not 3TC, have a direct suppressive effect on both basic and pharmacologically and physiologically activated autophagy. These effects were dose and time dependent and were observed at therapeutic Cmaxs. The fact that AZT and d4T did not interfere with early autophagy stages of autophagosome formation and the strong inhibitory effect on autophagic flux in combination with substantial autophagosome accumulation suggest a mechanism of autophagy inhibition downstream of autophagosome formation.
There are certain limitations concerning in vitro simulation in clinical situations. HAART-related adverse effects generally require prolonged (several-month) treatment (1, 3, 33), and specific pharmacokinetic profiles could influence clinical toxicity (34). However, similar experimental conditions have been repeatedly used in previous in vitro analyses of NRTI effects on adipogenesis and provided results in agreement with clinical data (10–13, 15). Consistent with this, our results revealed that only d4T and AZT (5, 6) suppressed adipocyte autophagy, proliferation, differentiation, and viability, whereas 3TC had no such effects. Further studies are required to assess the impact of other antiretroviral drugs on autophagy. Finally, the pharmacological inhibition of autophagy by using compounds such as vinblastine, chloroquine, and others may lead to off-target effects. Thus, these data have to be interpreted with caution and should be seen in conjunction with our results following the more specific shRNA-mediated suppression of autophagy.
d4T- and AZT-mediated inhibition of adipocyte autophagic activity correlated with their previously well-established potency to compromise the acquisition of mature adipocyte phenotype, reduce intracellular lipid accumulation, and impair the expression of several adipogenic markers such as CCAAT/enhancer-binding protein (C/EBP) alpha, peroxisome proliferator-activated receptor (PPAR) gamma, lipid-binding protein 2 (aP2), fatty acid synthase (FAS), and acetyl-coenzyme A carboxylase (ACC) (10, 12–15, 35, 36). A more direct mechanistic link might be demonstrated through the ability of autophagy activation to counteract AZT and d4T antiadipogenic effects. However, certain technical limitations did not permit the performance of such experiments. One particular limitation was the lack of a specific autophagy activator which could be used throughout the long-term (8-day) period of in vitro preadipocyte differentiation. The currently available activators of autophagy such as PP242, rapamycin, and starvation exert several off-target effects, act on a broad range of cellular processes, such as protein synthesis and cellular metabolism, and, in the long term, appear toxic and suppress differentiation (18). However, antiadipogenic effects as a result of genetic or pharmacological suppression of adipocyte autophagy have been documented in vitro and in vivo (19–21). We demonstrated that specific genetic ATG5 knockdown inhibition of 3T3-F422A autophagy completely reiterated effects of thymidine analogues on mitochondrial homeostasis and adipogenesis. Furthermore, in vivo adipocyte-specific knockout mouse models demonstrated decreased white adipose tissue mass (19–21). We speculate that impaired adipogenesis induced by thymidine analogues represents a combined effect of direct mitochondrial toxicity (3, 31, 37) and compromised cellular autophagy. In that regard, in addition to compromised expression of adipogenic markers (9), long-term NRTI treatment has been associated with severe mitochondrial toxicity (3, 31, 37) and adipose tissue mitochondrial dysfunction was already detected after 2 weeks of AZT and d4T treatment of healthy volunteers (38).
Autophagy is the only cellular pathway responsible for the clearance of dysfunctional mitochondria (29). It has been demonstrated that genetic inhibition of yeast autophagy results in impaired clearance and accumulation of dysfunctional mitochondria (29). Growth arrest, reduced oxidative phosphorylation, and oxygen consumption in combination with increased ROS have been documented in these autophagy-deficient cells (29). Interestingly, analogous alterations have been reported in murine and human cells after exposure to AZT and d4T (10–13, 15). Our in vitro observations regarding the effects on adipocyte autophagy of the two (deoxy)thymidine analogues stavudine and zidovudine contrasted with the deoxycytidine analogue lamivudine are in line with reports demonstrating increased mitochondrial mass in fat tissue of HAART-treated HIV-positive patients (6, 32, 39) and provide a potential mechanism that contributed to the generation and accumulation of dysfunctional mitochondria. The presence of dysfunctional mitochondria relates to the release of cell death-promoting factors and initiation of apoptosis (40), which explains the observed decrease in cellular viability in vitro and might be one potential explanation for the high levels of apoptosis observed in subcutaneous abdominal adipose tissue from lipoatrophic HIV patients (7, 8).
Given the strong association between autophagy and aging (17), our results may be relevant for the recently discovered association between NRTI treatment and accelerated mitochondrial aging with accumulation of mitochondrial DNA (mtDNA) mutations in HIV patients (41), as defective autophagy per se has been implicated in generation of dysfunctional mitochondria, increased ROS production, and accumulation of mtDNA mutations (29). Finally, d4T and AZT, but not 3TC, have been shown to trigger premature senescence program in human skin fibroblast and 3T3-F442A preadipocytes (11) and high expression of senescence makers has been documented in subcutaneous adipose tissue samples from lipodystrophic HIV-positive patients on NRTI-containing regiments (11).
In conclusion, our experiments demonstrate a dose- and time-dependent inhibitory effect of AZT and d4T on adipocyte autophagy at therapeutic Cmax drug concentrations. These effects correlated with increased cell death, reduced proliferation, and impaired differentiation. Given that autophagy is involved in the regulation of adipose mass and differentiation (19–21) as well as in aging (17) and considering the peripheral fat wasting (5, 6, 31) and premature aging phenotype of some HIV patients (42), these findings could be relevant to better understanding and the avoidance of the long-term toxicity of antiretroviral drugs.
ACKNOWLEDGMENTS
This work was supported in part by the German Research Foundation (KFO 250, TP1) and an unrestricted grant from Abbott. M.L. was supported by the German National Academic Foundation (LE-953/5-1 and LE-953/6-1). G.M.N.B. was supported by the Excellence Cluster EXC 62/1.
Footnotes
Published ahead of print 12 November 2012
REFERENCES
- 1. Carr A, Cooper DA. 2000. Adverse effects of antiretroviral therapy. Lancet 356:1423–1430 [DOI] [PubMed] [Google Scholar]
- 2. Carr A, Samaras K, Burton S, Law M, Freund J, Chisholm DJ, Cooper DA. 1998. A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin resistance in patients receiving HIV protease inhibitors. AIDS 12:F51–F58 [DOI] [PubMed] [Google Scholar]
- 3. Stankov MV, Behrens GM. 2007. HIV-therapy associated lipodystrophy: experimental and clinical evidence for the pathogenesis and treatment. Endocr. Metab. Immune Disord. Drug Targets 7:237–249 [DOI] [PubMed] [Google Scholar]
- 4. Nguyen A, Calmy A, Schiffer V, Bernasconi E, Battegay M, Opravil M, Evison JM, Tarr PE, Schmid P, Perneger T, Hirschel B. 2008. Lipodystrophy and weight changes: data from the Swiss HIV Cohort Study, 2000–2006. HIV Med. 9:142–150 [DOI] [PubMed] [Google Scholar]
- 5. Nolan D, Hammond E, James I, McKinnon E, Mallal S. 2003. Contribution of nucleoside-analogue reverse transcriptase inhibitor therapy to lipoatrophy from the population to the cellular level. Antivir. Ther. 8:617–626 [PubMed] [Google Scholar]
- 6. Nolan D, Hammond E, Martin A, Taylor L, Herrmann S, McKinnon E, Metcalf C, Latham B, Mallal S. 2003. Mitochondrial DNA depletion and morphologic changes in adipocytes associated with nucleoside reverse transcriptase inhibitor therapy. AIDS 17:1329–1338 [DOI] [PubMed] [Google Scholar]
- 7. Domingo P, Matias-Guiu X, Pujol RM, Domingo JC, Arroyo JA, Sambeat MA, Vazquez G. 2001. Switching to nevirapine decreases insulin levels but does not improve subcutaneous adipocyte apoptosis in patients with highly active antiretroviral therapy-associated lipodystrophy. J. Infect. Dis. 184:1197–1201 [DOI] [PubMed] [Google Scholar]
- 8. Domingo P, Matias-Guiu X, Pujol RM, Francia E, Lagarda E, Sambeat MA, Vázquez G. 1999. Subcutaneous adipocyte apoptosis in HIV-1 protease inhibitor-associated lipodystrophy. AIDS 13:2261–2267 [DOI] [PubMed] [Google Scholar]
- 9. Bastard JP, Caron M, Vidal H, Jan V, Auclair M, Vigouroux C, Luboinski J, Laville M, Maachi M, Girard PM, Rozenbaum W, Levan P, Capeau J. 2002. Association between altered expression of adipogenic factor SREBP1 in lipoatrophic adipose tissue from HIV-1-infected patients and abnormal adipocyte differentiation and insulin resistance. Lancet 359:1026–1031 [DOI] [PubMed] [Google Scholar]
- 10. Caron M, Auclair M, Lagathu C, Lombes A, Walker UA, Kornprobst M, Capeau J. 2004. The HIV-1 nucleoside reverse transcriptase inhibitors stavudine and zidovudine alter adipocyte functions in vitro. AIDS 18:2127–2136 [DOI] [PubMed] [Google Scholar]
- 11. Caron M, Auclairt M, Vissian A, Vigouroux C, Capeau J. 2008. Contribution of mitochondrial dysfunction and oxidative stress to cellular premature senescence induced by antiretroviral thymidine analogues. Antivir. Ther. 13:27–38 [PubMed] [Google Scholar]
- 12. Stankov MV, Lucke T, Das AM, Schmidt RE, Behrens GM. 2007. Relationship of mitochondrial DNA depletion and respiratory chain activity in preadipocytes treated with nucleoside reverse transcriptase inhibitors. Antivir. Ther. 12:205–216 [PubMed] [Google Scholar]
- 13. Stankov MV, Lucke T, Das AM, Schmidt RE, Behrens GM. 2010. Mitochondrial DNA depletion and respiratory chain activity in primary human subcutaneous adipocytes treated with nucleoside analogue reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 54:280–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stankov MV, Schmidt RE, Behrens GM. 2008. Zidovudine impairs adipogenic differentiation through inhibition of clonal expansion. Antimicrob. Agents Chemother. 52:2882–2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stankov MV, Schmidt RE, Behrens GM. 2009. Combined effect of C-reactive protein and stavudine on adipogenesis. Antivir. Ther. 14:819–829 [DOI] [PubMed] [Google Scholar]
- 16. Spalding KL, Arner E, Westermark PO, Bernard S, Buchholz BA, Bergmann O, Blomqvist L, Hoffstedt J, Naslund E, Britton T, Concha H, Hassan M, Ryden M, Frisen J, Arner P. 2008. Dynamics of fat cell turnover in humans. Nature 453:783–787 [DOI] [PubMed] [Google Scholar]
- 17. Levine B, Kroemer G. 2008. Autophagy in the pathogenesis of disease. Cell 132:27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mizushima N, Yoshimori T, Levine B. 2010. Methods in mammalian autophagy research. Cell 140:313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baerga R, Zhang Y, Chen PH, Goldman S, Jin S. 2009. Targeted deletion of autophagy-related 5 (atg5) impairs adipogenesis in a cellular model and in mice. Autophagy 5:1118–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ. 2009. Autophagy regulates adipose mass and differentiation in mice. J. Clin. Invest. 119:3329–3339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S. 2009. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc. Natl. Acad. Sci. U. S. A. 106:19860–19865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Walker UA, Setzer B, Venhoff N. 2002. Increased long-term mitochondrial toxicity in combinations of nucleoside analogue reverse-transcriptase inhibitors. AIDS 16:2165–2173 [DOI] [PubMed] [Google Scholar]
- 23. Williams O. 2004. Flow cytometry-based methods for apoptosis detection in lymphoid cells. Methods Mol. Biol. 282:31–42 [DOI] [PubMed] [Google Scholar]
- 24. Fung C, Lock R, Gao S, Salas E, Debnath J. 2008. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 19:797–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Diessenbacher P, Hupe M, Sprick MR, Kerstan A, Geserick P, Haas TL, Wachter T, Neumann M, Walczak H, Silke J, Leverkus M. 2008. NF-kappaB inhibition reveals differential mechanisms of TNF versus TRAIL-induced apoptosis upstream or at the level of caspase-8 activation independent of cIAP2. J. Invest. Dermatol. 128:1134–1147 [DOI] [PubMed] [Google Scholar]
- 26. Kanki T, Klionsky DJ. 2010. The molecular mechanism of mitochondria autophagy in yeast. Mol. Microbiol. 75:795–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. 2011. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12:222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wallace DC. 2005. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 39:359–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang Y, Qi H, Taylor R, Xu W, Liu LF, Jin S. 2007. The role of autophagy in mitochondria maintenance: characterization of mitochondrial functions in autophagy-deficient S. cerevisiae strains. Autophagy 3:337–346 [DOI] [PubMed] [Google Scholar]
- 30. Gottlieb RA, Carreira RS. 2010. Autophagy in health and disease. 5. Mitophagy as a way of life. Am. J. Physiol. Cell Physiol. 299:C203–C210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gougeon ML, Penicaud L, Fromenty B, Leclercq P, Viard JP, Capeau J. 2004. Adipocytes targets and actors in the pathogenesis of HIV-associated lipodystrophy and metabolic alterations. Antivir. Ther. 9:161–177 [PubMed] [Google Scholar]
- 32. Lloreta J, Domingo P, Pujol RM, Arroyo JA, Baixeras N, Matias-Guiu X, Gilaberte M, Sambeat MA, Serrano S. 2002. Ultrastructural features of highly active antiretroviral therapy-associated partial lipodystrophy. Virchows Arch. 441:599–604 [DOI] [PubMed] [Google Scholar]
- 33. Mallal SA, John M, Moore CB, James IR, McKinnon EJ. 2000. Contribution of nucleoside analogue reverse transcriptase inhibitors to subcutaneous fat wasting in patients with HIV infection. AIDS 14:1309–1316 [DOI] [PubMed] [Google Scholar]
- 34. Kakuda TN. 2000. Pharmacology of nucleoside and nucleotide reverse transcriptase inhibitor-induced mitochondrial toxicity. Clin. Ther. 22:685–708 [DOI] [PubMed] [Google Scholar]
- 35. Lagathu C, Bastard JP, Auclair M, Maachi M, Kornprobst M, Capeau J, Caron M. 2004. Antiretroviral drugs with adverse effects on adipocyte lipid metabolism and survival alter the expression and secretion of proinflammatory cytokines and adiponectin in vitro. Antivir. Ther. 9:911–920 [PubMed] [Google Scholar]
- 36. Walker UA, Auclair M, Lebrecht D, Kornprobst M, Capeau J, Caron M. 2006. Uridine abrogates the adverse effects of antiretroviral pyrimidine analogues on adipose cell functions. Antivir. Ther. 11:25–34 [PubMed] [Google Scholar]
- 37. Côté HC. 2005. Possible ways nucleoside analogues can affect mitochondrial DNA content and gene expression during HIV therapy. Antivir. Ther. 10(Suppl 2):M3–M11 [PubMed] [Google Scholar]
- 38. Mallon PW, Unemori P, Sedwell R, Morey A, Rafferty M, Williams K, Chisholm D, Samaras K, Emery S, Kelleher A, Cooper DA, Carr A. 2005. In vivo, nucleoside reverse-transcriptase inhibitors alter expression of both mitochondrial and lipid metabolism genes in the absence of depletion of mitochondrial DNA. J. Infect. Dis. 191:1686–1696 [DOI] [PubMed] [Google Scholar]
- 39. Pace CS, Martin AM, Hammond EL, Mamotte CD, Nolan DA, Mallal SA. 2003. Mitochondrial proliferation, DNA depletion and adipocyte differentiation in subcutaneous adipose tissue of HIV-positive HAART recipients. Antivir. Ther. 8:323–331 [PubMed] [Google Scholar]
- 40. Wallace DC. 1999. Mitochondrial diseases in man and mouse. Science 283:1482–1488 [DOI] [PubMed] [Google Scholar]
- 41. Payne BA, Wilson IJ, Hateley CA, Horvath R, Santibanez-Koref M, Samuels DC, Price DA, Chinnery PF. 2011. Mitochondrial aging is accelerated by anti-retroviral therapy through the clonal expansion of mtDNA mutations. Nat. Genet. 43:806–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Caron-Debarle M, Lagathu C, Boccara F, Vigouroux C, Capeau J. 2010. HIV-associated lipodystrophy: from fat injury to premature aging. Trends Mol. Med. 16:218–229 [DOI] [PubMed] [Google Scholar]