

Abstract
In bacteria, mutations affecting the major catalytic subunits of RNA polymerase (encoded by rpoB and rpoC) emerge in response to a variety of selective pressures. Here we isolated a Bacillus subtilis strain with high-level resistance to cefuroxime (CEF). Whole-genome resequencing revealed only one missense mutation affecting an invariant residue in close proximity to the C-terminal DNA-binding domain of RpoC (G1122D). Genetic reconstruction experiments demonstrate that this substitution is sufficient to confer CEF resistance. The G1122D mutation leads to elevated expression of stress-responsive regulons, including those of extracytoplasmic function (ECF) σ factors (σM, σW, and σX) and the general stress σ factor (σB). The increased CEF resistance of the rpoCG1122D strain is lost in the sigM rpoCG1122D double mutant, consistent with a major role for σM in CEF resistance. However, a sigM mutant is very sensitive to CEF, and this sensitivity is still reduced by the G1122D mutation, suggesting that other regulatory effects are also important. Indeed, the ability of the G1122D mutation to increase CEF resistance is further reduced in a triple mutant strain lacking three ECF σ factors (σM, σW, and σX), which are known from prior studies to control overlapping sets of genes. Collectively, our findings highlight the ability of mutations in RNA polymerase to confer antibiotic resistance by affecting the activity of alternative σ factors that control cell envelope stress-responsive regulons.
INTRODUCTION
Transcription in all cellular organisms is driven by a multisubunit DNA-dependent RNA polymerase (RNAP) with a conserved crab claw-like shape that embraces the template DNA (1). The bacterial RNAP holoenzyme consists minimally of the core (α2ββ′ω) enzyme and one of a family of σ factors (2–4). Gram-positive bacteria have an additional δ subunit that enhances transcriptional specificity by blocking RNAP binding at weak promoter sites (5, 6). For transcription initiation, the core enzyme must associate with one of various σ factors that recognize different promoter sequences and thus enable specific binding of RNAP to gene promoters (7). The resulting holoenzyme forms an open complex by melting the DNA near the transcription start site and then begins to synthesize RNA (8, 9). Once the initial 10 or so nucleotides of RNA have been synthesized, the σ factor is released from the core enzyme, which elongates the RNA until it encounters a termination signal (10).
Bacteria have one housekeeping σ factor, σ70, and a variable number of alternative σ factors that are activated in response to different environmental conditions (2, 7, 11). The extracytoplasmic function (ECF) σ factors, which often respond to cell envelope stress, are the largest group of alternative σ factors (11, 12). The core enzyme is highly conserved in sequence and structure from bacteria to humans, particularly among the large-subunit (the bacterial β and β′ subunits) homologs (1, 13). Four structurally conserved modules, known as the β′ jaw, β′ clamp, β lobes, and β flap, play a crucial role in proper accommodation of the σ factor and DNA within RNAP (10, 14).
RNAP is one of the most central transcriptional regulatory hubs, and mutations affecting RNAP can arise during many selections (15–19). Mutant versions of the α subunit (RpoA) are able to substantially alter the cellular phenotype (20), and mutations in β (RpoB) that lead to resistance to rifampin in Bacillus subtilis also lead to altered expression of regulons involved in growth, competence, sporulation, and germination (21). In addition, mutations of the β′ subunit (RpoC) allow regulatory adaptation for optimal growth in minimal medium (15). The substitution of one σ factor for another reprograms the transcriptional profile, and it is therefore plausible that mutations to the core enzyme that affect σ activity could effect broad and complex changes to the transcriptome.
Cephalosporins are β-lactam antibiotics that interfere with bacterial cell wall cross-linking catalyzed by transpeptidases known as penicillin-binding proteins (PBPs) (22). Three primary mechanisms lead to β-lactam resistance (23–25). The most common is the production of β-lactamases, which serve to hydrolyze the β-lactam ring. Alternatively, changes in the active site of PBPs, which reduce the affinity for β-lactams, may arise. Finally, efflux pumps may eject β-lactams from the periplasm before they reach the PBP targets. Interestingly, none of these mechanisms appears to account for the development of β-lactam resistance in B. subtilis. Previously, we have reported that the intrinsic level of resistance to cefuroxime (CEF), an expanded-spectrum β-lactam cephalosporin, is substantially reduced in cells lacking multiple ECF σ factors (26), in particular σM (27). Whereas loss of ECF σ factors leads to a significant increase in CEF sensitivity, the mechanisms that might lead to high-level CEF resistance remain unknown.
Here we employed whole-genome resequencing to identify a single point mutation (G1122D) in B. subtilis rpoC that imparts CEF resistance. Homology modeling suggests that this mutation affects an absolutely conserved G1122 residue in close proximity to the DNA-binding site of RpoC that may thereby affect processes of promoter engagement. Consistent with our prior studies, this mutation leads to elevated activity of ECF σ factors (σM, σW, and σX), which can thereby lead to CEF resistance.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All strains used in this study, unless otherwise noted, are derivatives of wild-type B. subtilis W168 (Bacillus Genetic Stock Center [BGSC] accession number 1A1), and all are listed in Table 1. Cells were routinely cultured in Luria-Bertani (LB) broth at 37°C with vigorous shaking or on LB agar plates containing 1.5% (wt/vol) Bacto agar (Difco). The following antibiotics were used for selection: spectinomycin (Spec; 100 μg/ml), tetracycline (Tet; 5 μg/ml), kanamycin (Kan; 15 μg/ml), chloramphenicol (Cat; 10 μg/ml), or macrolide-lincosamide-streptogramin B (MLS) (contains 1 μg/ml erythromycin and 25 μg/ml lincomycin). Mutations were introduced into B. subtilis by transformation as described previously (32).
Table 1.
Bacterial strains and plasmid used in this study
| Strain or plasmid | Genotype or description | Source or referencea |
|---|---|---|
| Strains | ||
| W168 | trpC2 | BGSC accession no. 1A1 |
| HB13506 | W168 rpoCG1122D citM::Tn7(CEF-resistant isolate) | Tn7 library with W168 |
| HB13576 | W168 rpoCWT-kan | LFH PCR with W168 |
| HB13577 | W168 rpoCG1122D-kan | LFH PCR with W168 |
| HB10016 | 168 sigM::tet | 56 |
| HB13591 | W168 sigM::tet | HB10016 chr DNA with W168 |
| HB7007 | CU1065 sigX::spec | 29 |
| HB13592 | W168 sigX::spec | HB7007 chr DNA with W168 |
| HB13549 | W168 sigW::mls | 30 |
| HB13634 | W168 sigM::tet sigX::spec | HB7007 chr DNA with HB13591 |
| HB13635 | W168 sigM::tet sigW::mls sigX::spec | HB13549 chr DNA with HB13634 |
| HB553 | CU1065 sigB::cat | Laboratory stock |
| HB13551 | W168 sigB::cat | HB553 chr DNA with W168 |
| HB13552 | W168 sigB::cat sigW::mls | HB13551 chr DNA with HB13549 |
| HB13528 | W168 rocG::spec | 30 |
| HB10348 | 168 spx::mls | 27 |
| HB13599 | W168 spx::mls | HB10348 chr DNA with W168 |
| HB10381 | 168 disA::cat | 27 |
| HB13601 | W168 disA::cat | HB10381 chr DNA with W168 |
| HB13547 | W168 yuaFG::mls | 30 |
| HB13618 | W168 yuaFG::spec | ECE79 with HB13547 |
| HB13605 | W168 spx::mls disA::cat | HB10381 chr DNA with HB13599 |
| HB13619 | W168 spx::mls yuaFG::spec | HB13618 chr DNA with HB13599 |
| HB13579 | W168 sigB::cat rpoCWT-kan | HB13576 chr DNA with HB13551 |
| HB13580 | W168 sigB::cat rpoCG1122D-kan | HB13577 chr DNA with HB13551 |
| HB13581 | W168 sigW::mls rpoCWT-kan | HB13576 chr DNA with HB13549 |
| HB13582 | W168 sigW::mls rpoCG1122D-kan | HB13577 chr DNA with HB13549 |
| HB13583 | W168 sigB::cat sigW::mls rpoCWT-kan | HB13576 chr DNA with HB13552 |
| HB13584 | W168 sigB::cat sigW::mls rpoCG1122D-kan | HB13577 chr DNA with HB13552 |
| HB13593 | W168 sigM::tet rpoCWT-kan | HB13576 chr DNA with HB13591 |
| HB13594 | W168 sigM::tet rpoCG1122D-kan | HB13577 chr DNA with HB13591 |
| HB13595 | W168 sigX::spec rpoCWT-kan | HB13576 chr DNA with HB13592 |
| HB13596 | W168 sigX::spec rpoCG1122D-kan | HB13577 chr DNA with HB13592 |
| HB13636 | W168 sigM::tet sigW::mls sigX::spec rpoCWT-kan | HB13576 chr DNA with HB13635 |
| HB13637 | W168 sigM::tet sigW::mls sigX::spec rpoCG1122D-kan | HB13577 chr DNA with HB13635 |
| HB13597 | W168 rocG::spec rpoCWT-kan | HB13576 chr DNA with HB13528 |
| HB13598 | W168 rocG::spec rpoCG1122D-kan | HB13577 chr DNA with HB13528 |
| HB13602 | W168 spx::mls rpoCWT-kan | HB13576 chr DNA with HB13599 |
| HB13603 | W168 spx::mls rpoCG1122D-kan | HB13577 chr DNA with HB13599 |
| HB13614 | W168 disA::cat rpoCWT-kan | HB13576 chr DNA with HB13601 |
| HB13615 | W168 disA::cat rpoCG1122D-kan | HB13577 chr DNA with HB13601 |
| HB13621 | W168 yuaFG::spec rpoCWT-kan | HB13576 chr DNA with HB13618 |
| HB13622 | W168 yuaFG::spec rpoCG1122D-kan | HB13577 chr DNA with HB13618 |
| HB13608 | W168 spx::mls disA::cat rpoCWT-kan | HB13576 chr DNA with HB13605 |
| HB13609 | W168 spx::mls disA::cat rpoCG1122D-kan | HB13577 chr DNA with HB13605 |
| HB13623 | W168 spx::mls yuaFG::spec rpoCWT-kan | HB13576 chr DNA with HB13619 |
| HB13624 | W168 spx::mls yuaFG::spec rpoCG1122D-kan | HB13577 chr DNA with HB13619 |
| Plasmid ECE79 | pErm::Spec (cassette switching vector) | 31 |
chr, chromosomal.
Tn7 transposon mutagenesis.
The modified Tn7 transposon mutagenesis system mTn7SX was used to select mutants with increased resistance to CEF (33). The amplified Tn7 library DNA was introduced by transformation into the wild-type B. subtilis strain, and cells were spread onto LB agar plates containing 100 μg/ml Spec and 15 μg/ml CEF. To confirm that the increased CEF resistance of the Tn7 mutants is due to the transposon insertion, linkage tests were performed by transforming the chromosomal DNA of the Tn7 mutants into the wild-type strain and selecting with 100 μg/ml Spec. The 10 transformants were restreaked onto LB agar plates containing 100 μg/ml Spec and 15 μg/ml CEF in order to determine whether the CEF resistance is linked to a transposon insertion. The Tn7 insertion site was determined by PCR amplification and sequencing of the Tn7-chromosomal DNA junction (34).
Whole-genome resequencing.
Chromosomal DNA was isolated from strains W168 and HB13506 grown in LB medium to an optical density at 600 nm (OD600) of 0.4 by using the Qiagen DNeasy blood and tissue kit. The quantity and purity of DNA were determined using a NanoDrop spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE), and DNA was sequenced and analyzed by the Cornell University Life Sciences Core Laboratories Center using Illumina DNA sequencing. The sequence data were assembled with MOSAIC by using the reference sequence (35) under GenBank accession number ABQK00000000.
Genetic reconstruction.
The mutated rpoC allele was introduced into the wild-type chromosome by using long-flanking homology (LFH) PCR followed by DNA transformation as described previously (36, 37). The upstream fragment, ending 34 nucleotides after the stop codon of the rpoC G1122D allele, was amplified from the HB13506 mutant using primers rpoC up-fwd (5′-TATCACACAGGGTCTTCCGC-3′) and rpoC up-rev (5′-CCTATCACCTCAAATGGTTCGCTGCTGTTTTTGCAGTCTTTCAGCA-3′). A control wild-type rpoC allele was also amplified using the same primer set. The downstream fragment starting 24 nucleotides after the stop codon of rpoC was amplified using primers rpoC do-fwd (5′-CGAGCGCCTACGAGGAATTTGTATCGTGCAAAAACAGTCTTTCAGCAG-3′) and rpoC do-rev (5′-AAGCAGTAACCTCGATTCCGT-3′). A kanamycin antibiotic resistance cassette, followed by a downstream fragment, was introduced as a selectable marker after the stop codon of the wild-type and mutant rpoC alleles by using LFH PCR. The resulting constructs were introduced by transformation into the wild-type B. subtilis strain, and the presence of the mutation was confirmed by DNA sequencing.
Antibiotic susceptibility tests.
Susceptibility testing was performed by using Etest and disk diffusion assays. Cells were grown in LB medium to an OD600 of 0.4. Aliquots (100 μl) of these cultures were mixed with 4 ml of 0.7% LB or Mueller-Hinton (MH) soft agar (kept at 50°C) and were directly poured onto LB or MH agar plates (containing 15 ml of 1.5% LB or MH agar). After 30 min at room temperature, the plates were dried for 20 min in a laminar airflow hood and were then used for the assays. The Etest assays were performed on MH agar to determine the MIC. Etest strips (bioMérieux, Durham, NC) impregnated with CEF (at concentrations ranging from 0.016 to 256 μg/ml) were applied to agar plates. The plates were incubated at 37°C and were read after 18 h. The MIC (in micrograms per milliliter) was determined directly from the scale where the ellipse edge intersects the Etest strip. Disk diffusion assays were performed as described previously (38). Whatman filter paper disks containing the antibiotics to be tested were placed on top of the LB or MH agar, and the plates were incubated at 37°C overnight. The diameters of the inhibition zones were measured after subtraction of the diameter of the filter paper disk (6.5 mm). The following chemicals and quantities were used in the disk diffusion assays: Triton X-100, 20 μl of a 25% solution; fosfomycin, 500 μg; and CEF, 30 μg or 50 μg.
RNA preparation and microarray analyses.
Total RNA was isolated from three biological replicates of HB13576 (rpoCWT-kan) and HB13577 (rpoCG1122D-kan) grown in LB medium to mid-log phase (OD600, 0.4) by using the RNeasy minikit (Qiagen), followed by DNase treatment with a Turbo DNA-free kit (Ambion). The quantity and purity of RNA were determined using a NanoDrop spectrophotometer. cDNA labeling and microarray analysis were performed as described previously (39). Two microarrays were performed in biological triplicates with a dye swap, and the GenePix Pro software package (version 6.0) was used for image processing and analysis. Each expression value is representative of four separate measurements (duplicate spots on each of two arrays). Mean values and standard deviations for the normalized microarray data sets were calculated with MS Excel. The normalized microarray data sets were filtered to remove those genes that were not expressed at levels significantly above background under either condition (sum of mean fluorescence intensities, <20). In addition, the mean and standard deviation of the fluorescence intensities were computed for each gene, and those for which the standard deviation was greater than the mean value were ignored. For the regulon-based assignment of transcriptional changes, we further analyzed genes that had fluorescence signal intensities higher than 150 for at least one RNA sample. The fold change was calculated by using the average signal intensities for HB13577 divided by those for HB13576.
RNAP homology modeling.
Sequence alignments of RNAP subunits from B. subtilis, Thermus thermophilus, and Escherichia coli were created using ClustalW (40). The homology models of the B. subtilis RNAP subunits were constructed from the crystal structure of T. thermophilus RNAP (Protein Data Bank [PDB] identification code [ID] 2O5J) and the electron microscopy (EM) structure of E. coli RNAP (PDB ID 3IYD) by using the Swiss-Model Alignment mode (41–43). The individual models were superimposed onto the E. coli RNAP initiation complex structure (PDB ID 3IYD) using the Coot program (44), and all-atom contact and geometry were checked with MolProbity (45). Structures were visualized using PyMOL (http://www.pymol.org).
Microarray data accession number.
All microarray data are available in the NCBI GEO database under accession number GSE37742.
RESULTS AND DISCUSSION
Isolation of a spontaneous mutant of B. subtilis highly resistant to CEF.
To investigate the development of cephalosporin resistance in the model bacterium B. subtilis, we initially used transposon-based genome-wide random mutagenesis (33). We introduced an amplified mTn7SX library into the wild-type B. subtilis strain by transformation and isolated a mutant (HB13506) that was able to grow on LB agar containing 15 μg/ml of CEF, an expanded-spectrum cephalosporin antibiotic. To directly compare the levels of CEF resistance of wild-type and HB13506 mutant strains, we performed disk diffusion assays. Indeed, the isolated strain exhibited remarkably reduced susceptibility to CEF (Fig. 1A). Strain HB13506 also displays a small-colony phenotype: colonies are about 4-fold smaller than wild-type colonies when grown on LB agar plates (Fig. 1B). By sequencing of chromosomal DNA flanking the transposon (33), we found that mTn7SX was inserted into citM, encoding a Mg2+ citrate transporter, but the CEF resistance and small-colony phenotypes were not linked to the transposon insertion. We therefore suspected that other spontaneous mutations were responsible for the CEF resistance of HB13506.
Fig 1.

Isolation of a slow-growing B. subtilis strain with high-level resistance to CEF. (A) Susceptibility to CEF was determined by disk diffusion assays, which were performed on LB or MH agar plates with a filter paper disk containing 30 μg CEF. Each bar represents the average zone of inhibition, expressed as the total diameter minus the diameter of the filter paper disk (6.5 mm). Three independent experiments were performed for each strain, and the standard deviation is indicated by error bars. (B) Sizes of wild-type and HB13506 mutant colonies grown on LB agar plates. Cells were streaked or spread onto plates and were incubated for 1 day at 37°C.
Identification of a point mutation in an absolutely conserved glycine residue of RpoC.
The resistance phenotype of strain HB13506 was stably maintained after many transfers in the absence of CEF. We therefore performed whole-genome resequencing of the HB13506 mutant and its parental wild-type strain in order to identify the mutations conferring CEF resistance. Indeed, sequencing of whole genomes has been used successfully to directly determine novel mutations that result in increased resistance to certain antibiotics (46–49). Whole-genome sequence comparison identified a single-base substitution (G to A) at nucleotide 3365 of the rpoC gene, which encodes the RNA polymerase β′ subunit, resulting in the G1122D (codon GGT to GAT) alteration. The presence of this mutation (designated rpoCG1122D) was further confirmed by PCR and DNA sequencing (Fig. 2A).
Fig 2.

Whole-genome resequencing and single-nucleotide polymorphism identification. (A) Sanger sequencing of genomic DNA confirms a G-to-A alteration at nucleotide 3365 of rpoC in strain HB13506 (as initially identified by Illumina whole-genome resequencing). The mutated residue is indicated by an asterisk, and the altered amino acid at position 1122 is shown in red. (B) Structure-based multiple-sequence alignment of the C-terminal domain of RpoC from B. subtilis (Swiss-Prot accession no. P37871), Actinomyces odontolyticus (A7B9Q3), Clostridium botulinum (A7FZ76), Corynebacterium diphtheriae (Q6NJF6), Enterococcus faecalis (Q82Z41), Lactobacillus brevis (Q03PV0), Mycobacterium tuberculosis (A5U053), Streptococcus pneumoniae (Q97NQ8), Streptomyces coelicolor (Q8CJT1), Propionibacterium acnes (G7U6X7), Rhodococcus equi (E9T446), Acetobacter pasteurianus (C7JFQ4), Chlamydia trachomatis (O84316), E. coli (P0A8T7), Klebsiella pneumoniae (B5XYF4), Legionella pneumophila (Q5X865), Neisseria gonorrhoeae (Q5F5R6), Pseudomonas aeruginosa (Q9HWC9), Salmonella enterica serovar Typhimurium (P0A2R4), T. thermophilus (Q8RQE8), and Vibrio cholerae (Q9KV29). The absolutely conserved residues are boxed in red, and the highly conserved residues are represented by red letters. The secondary structure was adopted from the crystal structure of T. thermophilus RNAP (PDB ID 2O5J) (43). The conserved motifs were identified by using the NCBI Conserved Domain database (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) and by structural analysis of T. thermophilus RNAP (13).
To determine whether the mutated residue is related to any known structural or functional domains, we performed a multiple-sequence alignment of RpoC proteins from 11 Gram-positive (including B. subtilis) and 10 Gram-negative bacterial genera by using ClustalW and ESPript (40, 50). The results show that the G1122D mutation is within a highly conserved C-terminal region of RpoC (Fig. 2B). Analysis of conserved motifs in the C terminus of B. subtilis RpoC, using the NCBI Conserved Domain database and structural analysis of T. thermophilus RNAP (13, 28), demonstrates that G1122 lies adjacent to the DNA-binding sites and β′ clamp. The RNAP clamp, with mobile domains on the β and β′ subunits, is part of the crab claw structure that opens and closes the active-site channel (51). Strikingly, we found that the G1122 residue of B. subtilis RpoC is absolutely conserved in all 21 RpoC proteins. We note that a recent study demonstrated that a high proportion (∼30%) of the absolutely conserved residues in both the β and the β′ subunits are Gly. Some of these Gly residues are known to have distinct functional roles (13), but to our knowledge, no mutations in residue G1122 (or its equivalent) have been reported previously for any bacterial RNAP.
The rpoCG1122D allele is sufficient to confer high-level CEF resistance.
To determine whether the G1122D substitution in rpoC is sufficient for CEF resistance, we moved the mutant allele into the parental wild-type strain as outlined in Fig. 3. A kanamycin antibiotic resistance cassette was introduced as a selectable marker after the stop codon of the control wild-type and mutated rpoC alleles, respectively. The correct allelic replacements were confirmed by DNA sequencing. Determination of the MICs of the reconstructed HB13576 (rpoCWT-kan) and HB13577 (rpoCG1122D-kan) strains, by use of the Etest assay, revealed an increase from 5 to 20 μg/ml on MH agar by the introduction of the rpoC G1122D point mutation (Fig. 4A). The reconstructed mutant also exhibited a lower growth rate than the wild type in the absence of CEF and a remarkable reduction in susceptibility to CEF, as measured using a Bioscreen C growth analyzer (Fig. 4B). These results indicate that both the CEF resistance and slow-growth phenotypes of HB13506 are replicated in the reconstructed mutant strain HB13577 and are therefore due to the G1122D substitution in RpoC.
Fig 3.
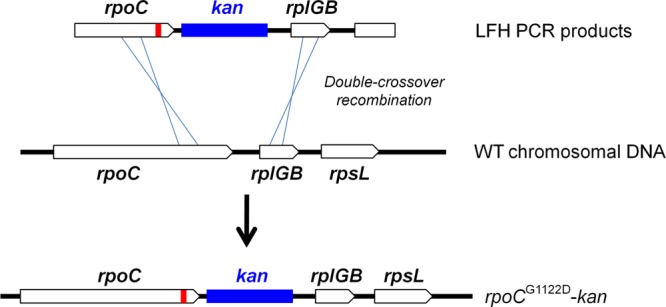
Genetic reconstruction of the mutant rpoC allele in the wild-type background. The schematic diagram shows the strategy for genetic reconstruction. The G1122D mutation is indicated by a thick red vertical line. A mutant allele of rpoC was amplified from the HB13506 mutant, and then a kanamycin antibiotic resistance cassette was introduced after the stop codon, followed by a 752-bp downstream fragment, using LFH PCR. The mutant allele was moved into the parental wild-type strain by transformation and was confirmed by DNA sequencing.
Fig 4.

The G1122D substitution alone is responsible for both the CEF resistance and slow-growth phenotypes. (A) Determination of MICs of CEF by use of an Etest assay. Etest strips (bioMérieux) with CEF concentrations of 0.016 to 256 μg/ml were applied to MH agar plates, and then the plates were incubated at 37°C for 18 h. The MIC (in micrograms per milliliter) was determined by identifying where bacterial growth intersects the Etest strip. (B) Growth assays, in the absence or presence of CEF, were performed in MH medium by using a Bioscreen C growth analyzer. The data are representative of at least three independent experiments.
RpoCG1122D facilitates transcriptional reprogramming by σ factors.
To better understand the molecular basis of the CEF resistance conferred by the G1122D substitution, the transcriptome of the rpoCG1122D mutant was assessed using DNA microarrays. There were genome-wide differences in expression patterns between the mutant (HB13577) and wild-type (HB13576) strains. Of the 175 genes upregulated >2.5-fold in the rpoCG1122D mutant, we found that 68 (approximately 40%) belong to ECF σ factor (σM, σW, and σX) and σB regulons (Fig. 5A). These results are in good agreement with the recently observed role of ECF σ factors in CEF resistance (26, 27, 30). The remaining 60% of genes with altered expression are not associated with known cell envelope stress regulons, although this group includes both RocR-regulated (rocA, rocB, rocE, rocF, and rocG) and Fur-regulated (yfkM and feuABC-ybbA operon) genes.
Fig 5.

Influence of the G1122D substitution on the transcription of the σ factor regulons. (A) Microarray transcriptional analysis of strains HB13576 (rpoCWT-kan) and HB13577 (rpoCG1122D-kan). RNA was extracted from cells grown in LB medium to an OD600 of 0.4. Analysis was focused on genes whose expression changed at least 2.5-fold. Data shown are mean fluorescence intensities from two independent experiments. Each comparison was performed with dye-swaps on each of three biological replicates. (B) Disk diffusion assays were performed on LB agar plates with fosfomycin and Triton X-100. Three independent experiments were performed for each strain. Error bars indicate standard deviations.
Previously, σW was shown to be a major determinant for resistance to antibiotics that impair cell wall synthesis, such as fosfomycin, and detergents that disrupt membrane function, such as Triton X-100 (38, 52). Since the microarray data suggest a general upregulation of the σW regulon, we performed disk diffusion assays under the same conditions as those used for the microarray analysis. As predicted, an increase in resistance to fosfomycin and Triton X-100 was noted for the rpoCG1122D mutant compared to controls (Fig. 5B). These results support the hypothesis that the RpoCG1122D substitution affects the global transcription profile, and thereby antibiotic resistance, by facilitating the transcriptional reprogramming of RNAP.
RpoCG1122D-induced CEF resistance is due primarily to increased σM activity.
We used disk diffusion assays to examine the individual contributions of alternative σ factors to the enhanced CEF resistance. Consistent with the known roles of σM, σW, and σX in CEF resistance (26, 27, 30), the disruption of sigM led to the greatest increase in CEF sensitivity, with more-modest effects attributed to disruption of sigW and sigX (Fig. 6A). Significantly, inactivation of σM in the rpoCG1122D strain decreased resistance to a level lower than that normally seen in the wild-type parental strain. Although this sigM rpoCG1122D mutant strain is somewhat more sensitive than the wild type to CEF (Fig. 6A), it retains the slow-growth phenotype. Thus, growth rate reduction is not, by itself, sufficient to confer CEF resistance and is likely only one result of the broad changes in the transcriptome profile of the rpoCG1122D mutant.
Fig 6.

The ECF σ factors (σM, σW, and σX) are involved in CEF resistance conferred by the rpoC point mutation. (A) Determination of the CEF susceptibilities of several B. subtilis strains. The dotted horizontal line indicates the level of CEF susceptibility of strain HB13576 (rpoCWT-kan). (B) Effect of the rpoCG1122D allele on the CEF susceptibility of the ΔMWX triple mutant strain. Disk diffusion assays were performed on MH agar plates with filter paper disks containing three different amounts of CEF (2, 10, and 50 μg). Three independent experiments were performed for each strain. Error bars indicate standard deviations. (C) Determination of MICs for CEF using an Etest assay. Etest strips (bioMérieux) with CEF concentrations of 0.016 to 256 μg/ml were applied to MH agar plates, and the plates were then incubated at 37°C for 18 h. The MICs (in micrograms per milliliter), shown to the right of each Etest strip, were determined by identifying where bacterial growth intersects the Etest strip.
In contrast to the results obtained with the ECF σ factors, the CEF resistance of strains lacking σB or RocG was relatively unaffected in either the wild-type or the rpoCG1122D mutant background (Fig. 6A). The RocG glutamate dehydrogenase has recently been linked to intrinsic CEF resistance in B. subtilis (30) but does not appear to be responsible for the increased resistance of the rpoCG1122D strain (Fig. 6A). These results suggest that the G1122D substitution directs the cell toward CEF resistance by altering the transcription of the σM, σW, and σX (ECF σ factor) regulons.
It has been reported previously that these ECF σ factors (σM, σW, and σX) have overlapping promoter selectivities (53–57). As a result, increased sensitivity to some antibiotics is revealed only when two or more ECF σ factors are inactivated (26, 38). Indeed, deletion of all three ECF σ factor genes (sigM, sigW, and sigX) (ΔMWX) renders B. subtilis more sensitive to CEF than the single deletions (26). We therefore hypothesized that much (or even all) of the CEF resistance noted in the rpoCG1122D mutant was due to increased activity of σM, σW, and σX. In support of this hypothesis, there was comparatively little change in CEF susceptibility when the rpoC point mutation was introduced into the ΔMWX triple mutant strain, as measured either by the zone of inhibition (Fig. 6B) or by Etest assays (Fig. 6C). In the Etest assays, the rpoCG1122D mutation led to a 4-fold increase in the MIC for the wild type and increased the MICs for the sigW and sigX strains between 3- and 5-fold. In contrast, a <3-fold effect was noted for the sigM single mutant and a <2-fold effect for the ΔMWX triple mutant strain (Fig. 6C). We conclude that the increased level of CEF resistance in the rpoCG1122D mutant is mediated mainly by σM, with additional roles played by both σW and σX.
Inactivation of the spx, disA, and yuaFG genes reduces CEF resistance.
The ECF σ factors σM and σX contribute to CEF resistance by three distinct pathways involving the B. subtilis genes spx, disA, and abh (27). It has also been reported that overexpression of yuaFG rescues the CEF-sensitive phenotype of the sigW mutant (30). We used disk diffusion assays to determine if these genes are involved in CEF resistance in the G1122D mutant strain. As predicted, inactivation of either spx, disA, or yuaFG reduced CEF resistance levels, although the effects of disA and yuaFG were only slight compared to that of spx. In contrast, the abh null mutation had no effect in this genetic background (data not shown). The spx disA and spx yuaFG multiple mutant strains displayed further increases in CEF sensitivity, consistent with the hypothesis that these genes act in parallel pathways (Fig. 7). These results suggest that spx, disA, and yuaFG are among the genes controlled by ECF σ factors that contribute to CEF resistance in the rpoCG1122D mutant, but these are unlikely to be the only relevant resistance determinants.
Fig 7.
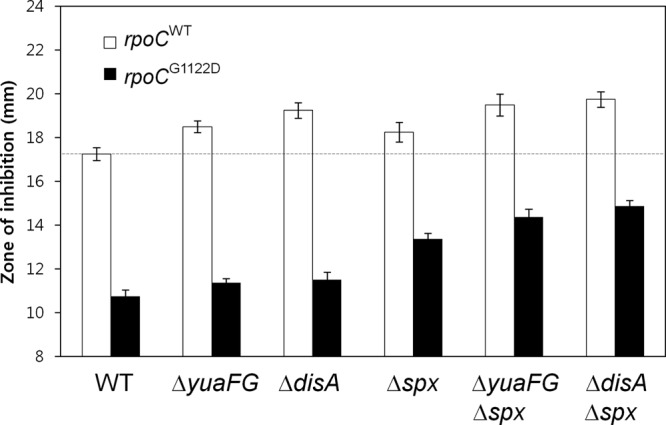
Involvement of spx, disA, and yuaFG genes in CEF resistance conferred by the rpoC point mutation. CEF susceptibility was determined by disk diffusion assays, which were performed on MH agar plates with a filter paper disk containing 50 μg CEF. The dotted horizontal line indicates the level of CEF susceptibility in the HB13576 (rpoCWT-kan) strain. Three independent experiments were performed for each strain. Error bars indicate standard deviations.
Structural homology models of B. subtilis RNAP.
To understand the possible structural effects of the G1122D substitution in the β′ subunit, and to explore whether this change might directly affect σ factor-RNAP complex formation, we performed homology modeling of the B. subtilis RNAP subunits (41, 58). The models generated were superimposed onto the EM structure of the E. coli RNAP transcription initiation complex (PDB ID 3IYD) (42), because no high-resolution crystal structure of a bacterial RNAP-promoter complex is available. The absolutely conserved β′-G1122 residue (Fig. 2B) is positioned at the DNA entrance channel in the α17-helix (numbered as for the β′ subunit of T. thermophilus RNAP) containing the DNA-binding K1125 residue (Fig. 8A).
Fig 8.

Modeling of B. subtilis RNAP. (A) Model structure of the B. subtilis RNAP transcription initiation complex. RNAP subunits are colored as follows: αI, slate; αII, pink; β, yellow; β′, cyan; ω, green; σ70, gray. The β clamp and β′ clamp are shown in magenta and blue, respectively. (B) Wild-type RNAP. The β′-G1122 residue is located at the DNA entrance channel and in close proximity to the DNA-binding residue β′-K1125, which interacts with the phosphate backbone of downstream DNA within 3.5 Å. (C) Mutant RNAP (β′-G1122D). The side chain of the substituted D1122 interacts with β′-K1125 through a salt bridge within 3.5 Å. Modeling suggests that the G1122D substitution could affect DNA binding by K1125.
Since σ factors are known to interact with many regions of the core enzyme (59), we asked whether the β′-G1122 residue has any interaction with the σ factor. However, G1122 is located ∼50 Å from the σ factor. Moreover, the distance of closest approach between G1122 and the β′ clamp is ∼10 Å; thus, they also are not likely to interact directly. We therefore undertook a closer inspection of the surroundings of the β′-G1122 residue. In the modeling of wild-type RNAP, β′-G1122 does not directly interact with the DNA recognition helix or neighbor molecules, whereas the ζN atom of β′-K1125 interacts with the phosphate backbone of promoter DNA within 3.5 Å (Fig. 8B). In the modeling of mutant RNAP (β′-G1122D), however, the δO atom of D1122 makes a salt bridge with the ζN atom in the DNA-binding residue β′-K1125 within 3.5 Å (Fig. 8C). Thus, the G1122D substitution may affect interactions between K1125 and the DNA backbone, and these changes may differentially affect the activities of the various holoenzyme forms.
Conclusions.
Our data demonstrate that a single point mutation in the absolutely conserved G1122 residue of the RNAP β′ subunit is sufficient to impart CEF resistance. The slow-growth phenotype is also directly linked to the G1122D substitution but is by itself insufficient to confer CEF resistance. A recent study reported that mutations in the β subunit (rpoB) of RNAP enhance cephalosporin resistance in enterococci (60). However, so far no mutations affecting cephalosporin resistance have been identified in the bacterial rpoC gene. Moreover, we are unaware of any mutations reported at the residue equivalent to G1122 of B. subtilis RpoC in any bacteria.
The present study indicates that the rpoCG1122D substitution increases the activity of ECF σ factors (σM, σW, and σX) and thereby contributes directly to CEF resistance in B. subtilis. As reported elsewhere (26, 27, 30) and confirmed here, these three ECF σ factors are known to control genes that contribute to CEF resistance, including spx (σMWX regulon), disA (σM regulon), and yuaFG (σW regulon). In summary, we have identified novel phenotypes associated with the alteration of an invariant β′ Gly residue (G1122) and provide a mechanistic explanation of the contribution of RNAP mutations to global transcriptional changes and thereby to the emergence of antibiotic resistance.
ACKNOWLEDGMENTS
We thank Pete Chandrangsu for helpful discussions and suggestions regarding homology modeling.
This work was supported by a grant from the National Institutes of Health (GM047446).
Footnotes
Published ahead of print 15 October 2012
REFERENCES
- 1. Cramer P. 2002. Multisubunit RNA polymerases. Curr. Opin. Struct. Biol. 12:89–97 [DOI] [PubMed] [Google Scholar]
- 2. Gruber TM, Gross CA. 2003. Multiple sigma subunits and the partitioning of bacterial transcription space. Annu. Rev. Microbiol. 57:441–466 [DOI] [PubMed] [Google Scholar]
- 3. Murakami KS, Masuda S, Darst SA. 2002. Structural basis of transcription initiation: RNA polymerase holoenzyme at 4 Å resolution. Science 296:1280–1284 [DOI] [PubMed] [Google Scholar]
- 4. Vassylyev DG, Sekine S, Laptenko O, Lee J, Vassylyeva MN, Borukhov S, Yokoyama S. 2002. Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 Å resolution. Nature 417:712–719 [DOI] [PubMed] [Google Scholar]
- 5. Juang YL, Helmann JD. 1994. The delta subunit of Bacillus subtilis RNA polymerase. An allosteric effector of the initiation and core-recycling phases of transcription. J. Mol. Biol. 239:1–14 [DOI] [PubMed] [Google Scholar]
- 6. Lopez de Saro FJ, Woody AY, Helmann JD. 1995. Structural analysis of the Bacillus subtilis delta factor: a protein polyanion which displaces RNA from RNA polymerase. J. Mol. Biol. 252:189–202 [DOI] [PubMed] [Google Scholar]
- 7. Paget MS, Helmann JD. 2003. The σ70 family of σ factors. Genome Biol. 4:203 doi:10.1186/gb-2003-4-1-203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Borukhov S, Severinov K. 2002. Role of the RNA polymerase sigma subunit in transcription initiation. Res. Microbiol. 153:557–562 [DOI] [PubMed] [Google Scholar]
- 9. deHaseth PL, Helmann JD. 1995. Open complex formation by Escherichia coli RNA polymerase: the mechanism of polymerase-induced strand separation of double helical DNA. Mol. Microbiol. 16:817–824 [DOI] [PubMed] [Google Scholar]
- 10. Saecker RM, Record MT, Jr, Dehaseth PL. 2011. Mechanism of bacterial transcription initiation: RNA polymerase-promoter binding, isomerization to initiation-competent open complexes, and initiation of RNA synthesis. J. Mol. Biol. 412:754–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Helmann JD. 2002. The extracytoplasmic function (ECF) sigma factors. Adv. Microb. Physiol. 46:47–110 [DOI] [PubMed] [Google Scholar]
- 12. Staron A, Sofia HJ, Dietrich S, Ulrich LE, Liesegang H, Mascher T. 2009. The third pillar of bacterial signal transduction: classification of the extracytoplasmic function (ECF) σ factor protein family. Mol. Microbiol. 74:557–581 [DOI] [PubMed] [Google Scholar]
- 13. Lane WJ, Darst SA. 2010. Molecular evolution of multisubunit RNA polymerases: structural analysis. J. Mol. Biol. 395:686–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chakraborty A, Wang D, Ebright YW, Korlann Y, Kortkhonjia E, Kim T, Chowdhury S, Wigneshweraraj S, Irschik H, Jansen R, Nixon BT, Knight J, Weiss S, Ebright RH. 2012. Opening and closing of the bacterial RNA polymerase clamp. Science 337:591–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Conrad TM, Frazier M, Joyce AR, Cho BK, Knight EM, Lewis NE, Landick R, Palsson BO. 2010. RNA polymerase mutants found through adaptive evolution reprogram Escherichia coli for optimal growth in minimal media. Proc. Natl. Acad. Sci. U. S. A. 107:20500–20505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cui L, Isii T, Fukuda M, Ochiai T, Neoh HM, Camargo IL, Watanabe Y, Shoji M, Hishinuma T, Hiramatsu K. 2010. An RpoB mutation confers dual heteroresistance to daptomycin and vancomycin in Staphylococcus aureus. Antimicrob. Agents Chemother. 54:5222–5233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Friedman L, Alder JD, Silverman JA. 2006. Genetic changes that correlate with reduced susceptibility to daptomycin in Staphylococcus aureus. Antimicrob. Agents Chemother. 50:2137–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Murphy H, Cashel M. 2003. Isolation of RNA polymerase suppressors of a (p)ppGpp deficiency. Methods Enzymol. 371:596–601 [DOI] [PubMed] [Google Scholar]
- 19. Watanabe Y, Cui L, Katayama Y, Kozue K, Hiramatsu K. 2011. Impact of rpoB mutations on reduced vancomycin susceptibility in Staphylococcus aureus. J. Clin. Microbiol. 49:2680–2684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klein-Marcuschamer D, Santos CN, Yu H, Stephanopoulos G. 2009. Mutagenesis of the bacterial RNA polymerase alpha subunit for improvement of complex phenotypes. Appl. Environ. Microbiol. 75:2705–2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maughan H, Galeano B, Nicholson WL. 2004. Novel rpoB mutations conferring rifampin resistance on Bacillus subtilis: global effects on growth, competence, sporulation, and germination. J. Bacteriol. 186:2481–2486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. 2008. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32:234–258 [DOI] [PubMed] [Google Scholar]
- 23. Poole K. 2004. Resistance to beta-lactam antibiotics. Cell. Mol. Life Sci. 61:2200–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Spratt BG. 1994. Resistance to antibiotics mediated by target alterations. Science 264:388–393 [DOI] [PubMed] [Google Scholar]
- 25. Wilke MS, Lovering AL, Strynadka NC. 2005. Beta-lactam antibiotic resistance: a current structural perspective. Curr. Opin. Microbiol. 8:525–533 [DOI] [PubMed] [Google Scholar]
- 26. Luo Y, Asai K, Sadaie Y, Helmann JD. 2010. Transcriptomic and phenotypic characterization of a Bacillus subtilis strain without extracytoplasmic function sigma factors. J. Bacteriol. 192:5736–5745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luo Y, Helmann JD. 2012. Analysis of the role of Bacillus subtilis σM in beta-lactam resistance reveals an essential role for c-di-AMP in peptidoglycan homeostasis. Mol. Microbiol. 83:623–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH. 2011. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39:D225–D229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang X, Decatur A, Sorokin A, Helmann JD. 1997. The Bacillus subtilis σX protein is an extracytoplasmic function σ factor contributing to survival at high temperature. J. Bacteriol. 179:2915–2921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee YH, Kingston AW, Helmann JD. 2012. Glutamate dehydrogenase affects resistance to cell wall antibiotics in Bacillus subtilis. J. Bacteriol. 194:993–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Steinmetz M, Richter R. 1994. Plasmids designed to alter the antibiotic resistance expressed by insertion mutations in Bacillus subtilis, through in vivo recombination. Gene 142:79–83 [DOI] [PubMed] [Google Scholar]
- 32. Harwood CR, Cutting SM. 1990. Molecular biological methods for Bacillus. Wiley, Chichester, United Kingdom [Google Scholar]
- 33. Bordi C, Butcher BG, Shi Q, Hachmann AB, Peters JE, Helmann JD. 2008. In vitro mutagenesis of Bacillus subtilis by using a modified Tn7 transposon with an outward-facing inducible promoter. Appl. Environ. Microbiol. 74:3419–3425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Peters JE, Craig NL. 2000. Tn7 transposes proximal to DNA double-strand breaks and into regions where chromosomal DNA replication terminates. Mol. Cell 6:573–582 [DOI] [PubMed] [Google Scholar]
- 35. Srivatsan A, Han Y, Peng J, Tehranchi AK, Gibbs R, Wang JD, Chen R. 2008. High-precision, whole-genome sequencing of laboratory strains facilitates genetic studies. PLoS Genet. 4:e1000139 doi:10.1371/journal.pgen.1000139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mascher T, Margulis NG, Wang T, Ye RW, Helmann JD. 2003. Cell wall stress responses in Bacillus subtilis: the regulatory network of the bacitracin stimulon. Mol. Microbiol. 50:1591–1604 [DOI] [PubMed] [Google Scholar]
- 37. Wach A. 1996. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast 12:259–265 [DOI] [PubMed] [Google Scholar]
- 38. Mascher T, Hachmann AB, Helmann JD. 2007. Regulatory overlap and functional redundancy among Bacillus subtilis extracytoplasmic function σ factors. J. Bacteriol. 189:6919–6927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hachmann AB, Angert ER, Helmann JD. 2009. Genetic analysis of factors affecting susceptibility of Bacillus subtilis to daptomycin. Antimicrob. Agents Chemother. 53:1598–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- 41. Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. 2009. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 4:1–13 [DOI] [PubMed] [Google Scholar]
- 42. Hudson BP, Quispe J, Lara-Gonzalez S, Kim Y, Berman HM, Arnold E, Ebright RH, Lawson CL. 2009. Three-dimensional EM structure of an intact activator-dependent transcription initiation complex. Proc. Natl. Acad. Sci. U. S. A. 106:19830–19835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vassylyev DG, Vassylyeva MN, Zhang J, Palangat M, Artsimovitch I, Landick R. 2007. Structural basis for substrate loading in bacterial RNA polymerase. Nature 448:163–168 [DOI] [PubMed] [Google Scholar]
- 44. Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 45. Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 66:12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hachmann AB, Sevim E, Gaballa A, Popham DL, Antelmann H, Helmann JD. 2011. Reduction in membrane phosphatidylglycerol content leads to daptomycin resistance in Bacillus subtilis. Antimicrob. Agents Chemother. 55:4326–4337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nakata K, Koh MM, Tsuchido T, Matsumura Y. 2010. All genomic mutations in the antimicrobial surfactant-resistant mutant, Escherichia coli OW66, are involved in cell resistance to surfactant. Appl. Microbiol. Biotechnol. 87:1895–1905 [DOI] [PubMed] [Google Scholar]
- 48. Palmer KL, Daniel A, Hardy C, Silverman J, Gilmore MS. 2011. Genetic basis for daptomycin resistance in enterococci. Antimicrob. Agents Chemother. 55:3345–3356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Toprak E, Veres A, Michel JB, Chait R, Hartl DL, Kishony R. 2012. Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat. Genet. 44:101–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gouet P, Courcelle E, Stuart DI, Metoz F. 1999. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15:305–308 [DOI] [PubMed] [Google Scholar]
- 51. Darst SA. 2001. Bacterial RNA polymerase. Curr. Opin. Struct. Biol. 11:155–162 [DOI] [PubMed] [Google Scholar]
- 52. Cao M, Bernat BA, Wang Z, Armstrong RN, Helmann JD. 2001. FosB, a cysteine-dependent fosfomycin resistance protein under the control of σW, an extracytoplasmic-function σ factor in Bacillus subtilis. J. Bacteriol. 183:2380–2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cao M, Helmann JD. 2002. Regulation of the Bacillus subtilis bcrC bacitracin resistance gene by two extracytoplasmic function σ factors. J. Bacteriol. 184:6123–6129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Eiamphungporn W, Helmann JD. 2008. The Bacillus subtilis σM regulon and its contribution to cell envelope stress responses. Mol. Microbiol. 67:830–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Eiamphungporn W, Helmann JD. 2009. Extracytoplasmic function σ factors regulate expression of the Bacillus subtilis yabE gene via a cis-acting antisense RNA. J. Bacteriol. 191:1101–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Luo Y, Helmann JD. 2009. Extracytoplasmic function σ factors with overlapping promoter specificity regulate sublancin production in Bacillus subtilis. J. Bacteriol. 191:4951–4958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Qiu J, Helmann JD. 2001. The −10 region is a key promoter specificity determinant for the Bacillus subtilis extracytoplasmic-function σ factors σX and σW. J. Bacteriol. 183:1921–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Blundell TL, Sibanda BL, Sternberg MJ, Thornton JM. 1987. Knowledge-based prediction of protein structures and the design of novel molecules. Nature 326:347–352 [DOI] [PubMed] [Google Scholar]
- 59. Gruber TM, Markov D, Sharp MM, Young BA, Lu CZ, Zhong HJ, Artsimovitch I, Geszvain KM, Arthur TM, Burgess RR, Landick R, Severinov K, Gross CA. 2001. Binding of the initiation factor σ70 to core RNA polymerase is a multistep process. Mol. Cell 8:21–31 [DOI] [PubMed] [Google Scholar]
- 60. Kristich CJ, Little JL. 2012. Mutations in the beta subunit of RNA polymerase alter intrinsic cephalosporin resistance in enterococci. Antimicrob. Agents Chemother. 56:2022–2027 [DOI] [PMC free article] [PubMed] [Google Scholar]