

Abstract
The Amyloid Hypothesis states that the cascade of events associated with Alzheimer's disease (AD)—formation of amyloid plaques, neurofibrillary tangles, synaptic loss, neurodegeneration, and cognitive decline—are triggered by Aβ peptide dysregulation (Kakuda et al., 2006, Sato et al., 2003, Qi-Takahara et al., 2005). Since γ-secretase is critical for Aβ production, many in the biopharmaceutical community focused on γ-secretase as a target for therapeutic approaches for Alzheimer's disease. However, pharmacological approaches to control γ-secretase activity are challenging because the enzyme has multiple, physiologically critical protein substrates. To lower amyloidogenic Aβ peptides without affecting other γ-secretase substrates, the epsilon (ε) cleavage that is essential for the activity of many substrates must be preserved. Small molecule modulators of γ-secretase activity have been discovered that spare the ε cleavage of APP and other substrates while decreasing the production of Aβ 42. Multiple chemical classes of γ-secretase modulators have been identified which differ in the pattern of Aβ peptides produced. Ideally, modulators will allow the ε cleavage of all substrates while shifting APP cleavage from Aβ 42 and other highly amyloidogenic Aβ peptides to shorter and less neurotoxic forms of the peptides without altering the total Aβ pool. Here, we compare chemically distinct modulators for effects on APP processing and in vivo activity.
1. Introduction
Gamma-secretase (γ-secretase) is required for the production of amyloid beta peptides (Aβ) and decreasing Aβ production as a disease modifying approach for the treatment of Alzheimer's disease (AD) has received intense interest. The initial focus was on the discovery of compounds that would decrease γ-secretase activity. γ-Secretase cleaves the membrane bound C-terminal domain (C99) of APP at the ε site to produce the intracellular domain, AICD. The enzyme then makes sequential cuts of the remaining intramembrane APP fragment at each turn of the alpha helix (every 3-4 amino acids) until Aβ peptides are formed and released into the extracellular space [1–3]. This protein processivity produces Aβ peptides that vary in size, from 43–34 amino acids in length [4, 5]. In Alzheimer's disease, a greater number of the longer forms of Aβ, including Aβ 42 and Aβ 43, or a high ratio of the long peptides to the shorter forms, appear to occur [6]. These longer Aβ peptides readily oligomerize, forming toxic species, as well as becoming the seeds for amyloid plaques [7, 8].
The full inhibition of γ-secretase appeared to be a sound approach. However, it was found that γ-secretase plays a broader biological role and cleaves multiple proteins to yield physiologically essential products. Thus, total inhibition results in severe adverse effects in vivo [9–11]. This played out in the clinic in the trial of the γ-secretase inhibitor, semagacestat from Eli Lilly [12–14]. Patients treated with this drug developed skin and gastrointestinal side effects that are characteristic of the inhibition of γ-secretase processing of Notch, leading to the discontinuation of the clinical trial in 2010 [13, 14].
The discovery of compounds that could decrease the production of the more amyloidogenic Aβ 42 peptide while preserving total Aβ levels and γ-secretase cleavage of other substrates led to a clinical trial of one of these newly identified, first generation gamma-secretase modulators (GSMs) [15]. The NSAID-derived, Flurizan from Myriad Genetics was tested in a Phase 3 trial in mild to moderate AD patients. However, Flurizan is a very weak modulator of γ-secretase, with an IC50 of ~250 μM [16]. In addition, this compound has very poor distribution into the central nervous system [16, 17]. Not surprisingly, the combination of these less than desirable properties resulted in no exposure in the brain, and a failed Phase III trial in June 2008 [18].
Second and now third generation GSMs have been discovered and are proceeding toward clinical trials in AD. These GSMs are significantly more potent than Flurizan and appear to have better drug-like properties. However, the majority of these compounds fall into one of two chemical classes, with little structural diversity within each of these classes and the development of some has been discontinued because of toxicities and biopharmaceutical limitations that other class members may also share [19]. A structurally unique GSM derived from a core molecule isolated from a natural product is also moving through preclinical testing [20]. While all three chemical series of GSMs share some common pharmacological properties, they differ in other fundamental ways. Here we present data contrasting the pharmacology of members of several structural series of GSMs and how modulation may avoid the pitfalls associated with γ-secretase inhibitors (GSIs).
2. Materials and Methods
2.1. Test Compounds
LY411575, GSI-953, BMS-708163, MK-GSM1, JNJ-40418677, and E-2012 were prepared according to published methods. SPI-1802 and SPI-1810 were prepared at Satori Pharmaceuticals.
2.2. Cell Culture and Compound Treatment
SUP-T1 cells (ATCC) were cultured in T75 flasks in RPMI media (Mediatech 10-041-CV) supplemented with 10% FBS and penicillin/streptomycin at 37°C in a 5% CO2 atmosphere. One hour prior to drug treatment, six well plates were seeded with 1.5 mL of media containing 2% FBS and cells at a density of 1.5 × 106 cells/mL. Test compounds in DMSO were diluted 100-fold directly into the media with the cells and incubated for 18 hours at 37°C. After treatment 100 μL aliquots of treated cells were assayed for viability with the Promega Cell Titer Glo assay system. The conditioned media and cells were further processed to measure Aβ levels and NICD levels, respectively.
CHO-2B7 cells (Mayo Clinic) are Chinese hamster ovary cells stably transfected with human βAPP 695 wt [38, 39]. The cells were cultured in Ham's F12 media (Thermo Fisher SH30026.01) supplemented with 10% FBS, 0.25 mg/mL Zeocin and penicillin/streptomycin at 37°C in a 5% CO2 atmosphere. For compound treatment, cells were plated in 96-well plates at a density of 1.0 × 105 cells/mL and allowed to grow to 100% confluence over two days. Test compounds in DMSO were diluted 100-fold directly into the media before adding to the cells. Immediately prior to adding compound-containing media to the cells, they were washed once with 1XPBS. Conditioned media from CHO-2B7 cells were collected after 5 hours of treatment and the levels of Aβ peptides were assessed as described below.
H4 human neuroglioma cells (ATCC) were cultured in 10% FBS/DMEM (Media Tech) with Pen/Strep (50 units/50 μg/mL; Invitrogen). Human WT APP stably transfected CHO cells were cultured in 10% FBS/HAM'S F-12 growth media (Media Tech) supplemented with Pen/Strep and G418 (500 μg/mL; Promega). Cells were plated and grown to confluency in 96-well plates prior to dosing. Cells were washed with PBS and 100 μL of media containing DMSO alone (vehicle) or test compounds in DMSO at a final DMSO concentration of 1% (v/v). Conditioned media was collected after 18 hours of treatment and diluted 1 : 1 with MSD blocking buffer (1% BSA in MSD wash buffer).
2.3. Solid Phase Extraction
Wells of 30 mg Oasis HLB 96-well extraction plates (Waters Corporation) were activated by addition of 1 mL of methanol followed by rinsing with 1 mL of water utilizing a vacuum plate manifold. 1 mL of SUP-T1 conditioned media was added and wells were then washed sequentially with 2 mL of 10% methanol and then with 2 mL of 30% methanol. Samples were eluted into sample collection tubes by adding 250 μL of 90% methanol with 2% ammonium hydroxide to each well. Eluted samples were concentrated to dryness under vacuum without heating.
2.4. Aβ In Vitro Assay Measurement
Conditioned media was collected after 5–18 hours of treatment and diluted with 1 volume of MSD blocking buffer (1% BSA in MSD wash buffer). Alternatively, dried films of SUP-T1 conditioned media after solid phase extraction were resuspended with 1 volume of MSD blocking buffer (1% BSA in MSD wash buffer). Samples were transferred to blocked MSD Human (6E10) Aβ 3-Plex plates and incubated for 2 hours at room temperature with orbital shaking followed by washing and reading according to the manufacturer's instructions (SECTOR Imager 2400 Meso Scale Discovery, Gaithersburg MD).
2.5. NICD Assay
The remaining cells were washed twice in PBS and then lysed with Promega reporter lysis buffer containing a complete protease inhibitor cocktail (Roche) for 1 hour at 4°C. Lysates were spun at 5,000 RPM for 5 minutes and supernatants were collected. Total protein levels were measured and adjusted to 1-2 mg/mL total protein using the BCA total protein assay (Thermo Scientific). NICD levels were then measured with a cleavage specific Notch1 sandwich ELISA (Cell Signaling Technologies) according to the manufacturer's instructions.
2.6. Immunoprecipitation and Matrix-Assisted Laser Desorption/Ionization Time-of-Flight (MALDI-TOF) Mass Spectrometry
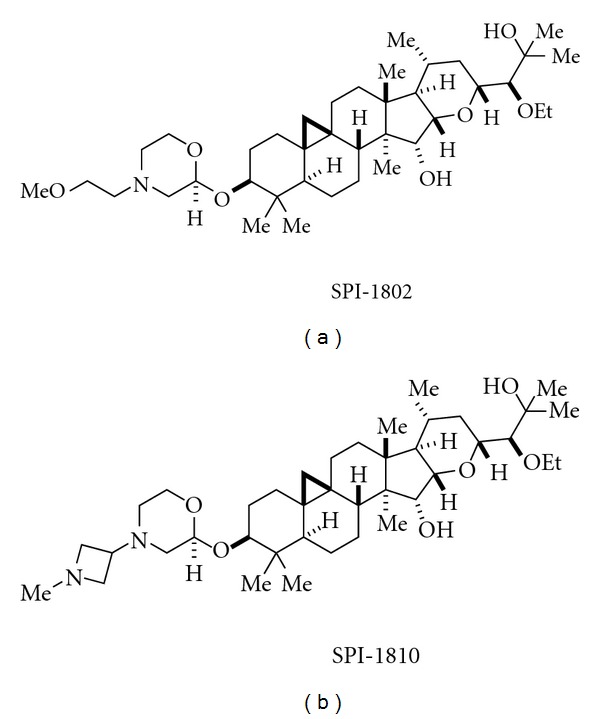
Chinese Hamster Ovary cells stably transfected with wild-type human APP were treated for 6 hrs with γ-secretase modulators at an approximate concentration of 10-fold the IC50 (Merck GSM1 at 1 μM, JNJ-40418677 at 2 μM, SPI-1802 at 3 μM, and SPI-1810 at 2 μM.) Monoclonal Aβ antibodies 6E10 (specific for amino acids 1–16 of Aβ) and 4G8 (specific for amino acids 17–24 of Aβ; Covance, Dedham, MA) were immobilized with agarose resin using the AminoLink Plus reagents (Thermo Scientific, Rockport, IL). Conditioned media from treated cells was precleared with agarose resin overnight, and the supernatant was incubated with agarose-conjugated 6E10/4G8 for 6 hrs. Immunoprecipitates were washed extensively prior to analysis.
Analyses were performed on a Shimadzu Biotech Axima TOF2 (Shimadzu Instruments) matrix-assisted-laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometer. Peptides were analyzed in positive ion linear mode. For intact peptide mass measurement the instrument was set with a mass range extending up to 6000 m/z using a pulsed extraction setting of 3500. An average mass external standard was used which consisted of angiotensin II (1047.2), P14R (1534.86) and ACTH clip 18–39 (2465.20), insulin B (3496.67), and insulin (5734.51). For sample preparation, 5 μL aliquots of Aβ containing immunoprecipitates were diluted with 10 μL of 0.1% TFA and then desalted using a C18 Zip Tip (Millipore, Corp.). Samples were directly deposited from the Zip Tip onto the MALDI sample target and then mixed with 0.5 μL of matrix solution which consisted of 5 mg/mL of alpha cyano-4-hydroxy cinnamic acid in acetonitrile: 0.1% TFA (50 : 50). Data was acquired manually using a set laser power and averaging 1500–2000 laser shots.
2.7. In Vivo Study Methods
All animal handling and procedures were conducted in full compliance to AAALAC International and NIH regulations and guidelines regarding animal care and welfare.
Either transgenic mice (Tg2576, 3 mos; N = 21) or wild-type Sprague Dawley rats (200–225 g; N = 8) were utilized to assess in vivo efficacy. All animals were acclimated to the test facility for a minimum of two days prior to initiation of the study. Compounds were dosed orally in 10 : 20 : 70 Ethanol/Solutol/Water via oral gavage. Samples were harvested at 6 hrs after dose for Aβ and compound exposure levels. Blood samples were collected into K2EDTA and stored on wet ice until processed to plasma by centrifugation (3500 rpm at 5°C) within 30 minutes of collection. Each brain was dissected into three parts: left and right hemispheres and cerebellum. Brain tissues were rinsed with ice cold phosphate buffered saline (without Mg2+ or Ca2+), blotted dry and weighed. Plasma and cerebella were analyzed for parent drug via LC/MS/MS. Parent drug levels were compared to a standard curve to establish the unknown levels.
2.8. Rodent Aβ Determination
This protocol is a modification of protocols described by Lanz et al. [40] and Rogers et al. [41]. Frozen hemispheres were weighed into tared homogenization tubes (MP Biomedicals#6933050 for rat; MP Biomedicals, Solon, OH) and (Simport#T501-4AT; Simport, Beloeil, Qc, Canada) containing one 5 mm stainless steel bead (Qiagen#69989) for mouse). For every gram of brain, 10 mLs of 6 M guanidine hydrochloride (wild-type rat) or 0.2% diethyl amine in 50 mM NaCl (transgenic mouse) was added to the brain-containing tubes on wet ice. Rat hemispheres were homogenized for one minute and mouse hemispheres were homogenized for 30 seconds at the 6.5 setting using the FastPrep-24 Tissue and Cell homogenizer (MP Biomedicals#116004500). Homogenates were rocked for two hours at 4°C, then precleared by ultracentrifugation at 100,000 ×g for one hour at 4°C. Precleared wild-type rat homogenates were concentrated over solid phase extraction (SPE) columns (Oasis HLB 96-well SPE plate 30 um, Waters#WAT058951; Waters Corp., Milford, MA). Briefly, SPE columns were prepared by wetting with 1 mL of 100% methanol followed by dH2O using vacuum to pull liquids through. Brain homogenates were then added to the prepared columns (1.0 mL from rat). Columns were washed twice with 10% methanol followed by two washes with 30% methanol. Labeled eluent collection tubes (Costar cluster tubes #4413; Corning Inc., Corning, NY) were placed under SPE columns and samples were eluted under very mild vacuum with 300 μL of 2% NH4OH/90% methanol. Eluents were dried to films under vacuum with no heat in a speed vacuum microcentrifuge. Films were resuspended in 150 μL of Meso Scale Discovery (MSD, Gaithersburg MD) blocking buffer (1% BSA in MSD wash buffer) for one hour at room temperature with occasional vortexing. A volume of 45 μL of precleared transgenic mouse brain homogenates were diluted into 450 μL of blocking buffer and were neutralized with 5 μL of 0.5 M Tris pH 6.8. For Aβ38, 40, and 42 measurements, MSD 96 well multispot Human/Rodent (4G8) Aβ triplex ultrasensitive ELISA plates were blocked with MSD blocking buffer for 1 hour at room temperature with orbital shaking. A volume of 25 μL of neat resuspended wild-type rat brain homogenate films or diluted transgenic mouse brain homogenates were added in duplicates to the blocked 3-plex Aβ MSD plates with SULFO-TAG 4G8 antibody (MSD). The Aβ 3-Plex plates were incubated for 2 hours at room temperature with orbital shaking followed by washing and reading according to the manufacturer's instructions (SECTOR Imager 2400, MSD). The average Aβ concentrations from duplicate measurements of each animal were converted to percent vehicle values and the treatment group averages were statistically compared by ANOVA analysis.
3. Results and Discussion
γ-Secretase is a complex enzyme with multiple substrates and multiple cleavage sites on at least some of these substrates, including APP. Complete inhibition of γ-secretase activity by targeting the ε cleavage site prevents the processing of multiple physiologically relevant proteins, leading to the severe side effects reported in AD patients [12, 13]. On the other hand, chemically modulating the enzyme with a GSM is a more precise mechanism to enhance certain cleavage events while preventing the cut that yields the amyloidogenic peptide, Aβ 42 which is linked to the pathophysiological initiation of AD (Figure 6). In preclinical toxicological testing, GSMs appear to be free of the mechanism-based toxicities attributed to the inhibition of Notch processing that have plagued the enzyme inhibitors. In vitro data demonstrate that Notch cleavage to NICD is not inhibited by any of several represented GSMs at concentrations that do not disrupt cell viability (Figure 1). Treatment with GSMs in rodents have not shown the classical Notch-related toxicities that are associated with GSIs, suggesting that the complete processing of Notch to NICD can occur in the presence of GSMs.
Figure 6.
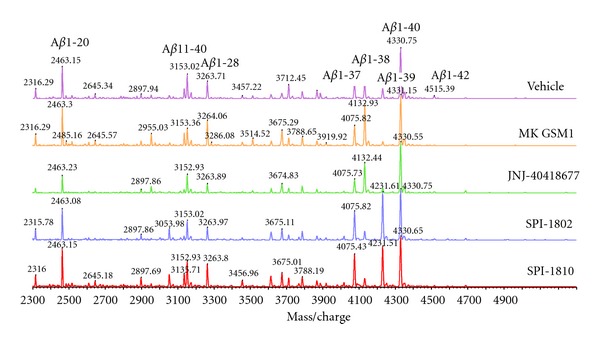
MALDI-TOF analysis of Aβ peptides from conditioned media of APP-overexpressing CHO cells treated with GSMs. The immunoprecipitated Aβ peptides were subjected to MALDI-TOF analysis to visualize individual Aβ fragments. Using a combination of two Aβ antibodies, 6E10, and 4G8 allows precipitation of full length and N- and C-terminal truncated Aβ peptides.
Figure 1.
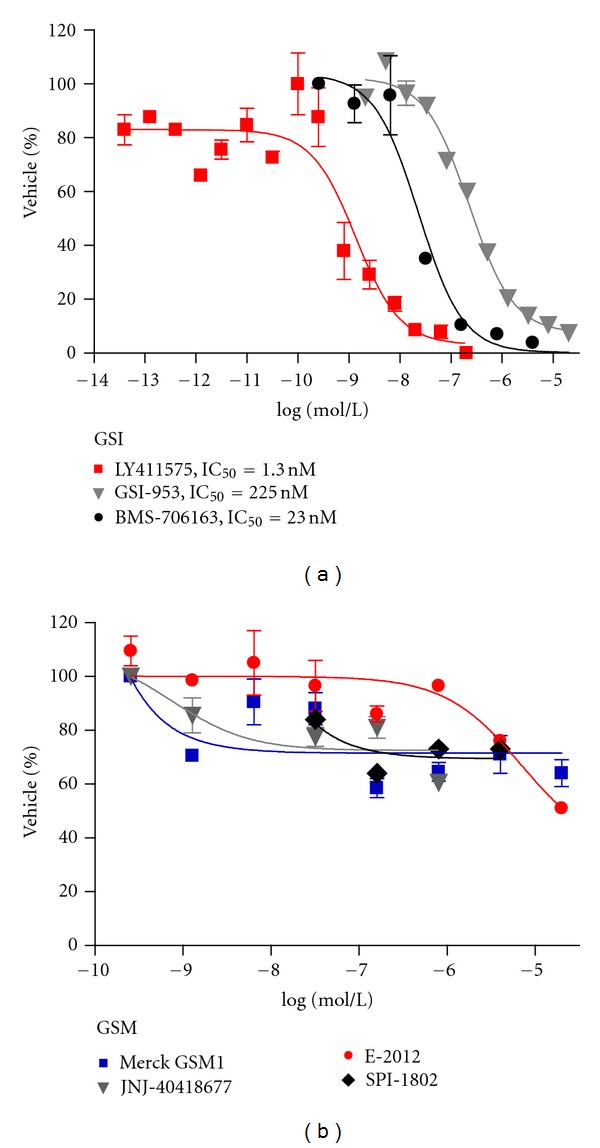
Multiple GSI and GSMs were examined in for their ability to inhibit NOTCH cleavage using the SUP-T1 cellular assay.
3.1. GSMs Do Not Show a Potency Shift with Changes in Substrate Concentration
γ-Secretase inhibitors can show a significant shift in potency in APP transfected cell-based assays depending on the level of expression of APP. Table 1 compares the IC50 for Aβ 42 lowering of different GSIs in multiple cell lines with varying levels of APP expression. In the highest expressing cell lines, the GSIs assayed here appear more potent (lower IC50s in the high expressing, transfected CHO-2B7 cell line) than when the same compounds are tested in assays using lower expressing cell lines (native H4 cell line). This potency shift seen with GSIs has been attributed to a shift in the enzyme/substrate ratio [42], where there is a higher ratio in wild-type cells versus those that overexpress APP. This higher ratio in turn requires more compounds to inhibit the enzymatic reaction. Interestingly, the potency shift due to substrate concentration that occurs with GSIs does not occur with GSMs. As seen in Table 1, the IC50 for all the GSMs shown are consistent across cell lines, regardless of the APP expression levels. While this potency shift is not well understood, these data do suggest that GSIs and GSMs affect the enzyme in fundamentally different ways.
Table 1.
GSMs do not show a potency shift with APP overexpression.
| Cell line | H4 | H4-APP | CHO-SW | CHO-7W | CHO-2B7 |
|---|---|---|---|---|---|
| Aβ 42 Levels (pg/mL) | 20 | 110 | 131 | 834 | 200 |
|
| |||||
| Inhibitors | Aβ 42 IC50 (nM) per cell line | ||||
|
| |||||
| LY411575 | 1.40 | 1.20 | 0.20 | ND | 0.05 |
| GSI-953 | 706 | 52 | 5.20 | 8.70 | 2.50 |
| BMS-708163 | 40 | 8 | 0.98 | 1.10 | 0.20 |
|
| |||||
| Modulators | Aβ 42 IC50 (nM) per cell line | ||||
|
| |||||
| GSM1 | 54 | 64 | 154 | 73 | 62 |
| JNJ-40418677 | 115 | 133 | 190 | 122 | 172 |
| E-2012 | 42 | 84 | 54 | 36 | 33 |
3.2. Structural Diversity of GSMs
Many of the second generation GSMs were inspired by the NSAIDs (Figure 2). For instance, all compounds shown in the figure were initially derived from the aryl acetic acid motif found in 1 and similar NSAIDs [21]. The similarity between these compounds makes for a crowded patent landscape, with some compounds potentially covered in multiple patent applications from different sponsors. In addition, for this structural series, the physicochemical properties indicate that all of these compounds carry an increased safety risk due to the high logP, low PSA [43–45], and high degree of aromaticity [46, 47]. Compounds 7–9 are designed to improve upon these properties, but are still considered high risk due to these same physicochemical based in silico models [27–29]. In vivo preclinical toxicity testing will ultimately be needed to assess the safety profile of these similar structures. However, the lack of progression of compounds from this class into and through clinical development suggests that this scaffold may have challenges that will continue to slow or prevent successful conduct of the studies required for registration.
Figure 2.
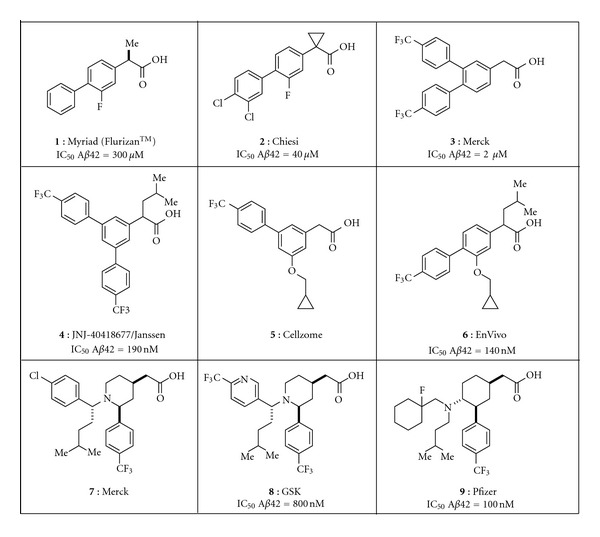
Representative NSAID-inspired GSMs. Compounds 1–6 are aryl acetic acids [21–26], compounds 7 and 8 are piperidine acetic acids [27, 28] and compound 9 is a cyclohexane acetic acid [29].
A second class of GSMs that has received significant attention is summarized in Figure 3. Following initial disclosure of 10 and 11, a number of pharmaceutical companies pursued structurally related chemical series [30, 48]. Although this class offers clear distinction from the NSAID-inspired compounds above, their physicochemical properties also reside outside of the molecular space most frequently affiliated with marketed agents for oral therapy. For example, the number of aromatic rings and clogP of the representatives shown are higher than the average for oral compounds on the market [46, 47], leading to a higher probability of safety and biopharmaceutical challenges with this class. Although some groups have been successful in developing promising structural alternatives, as exemplified by 16–18, little has been reported on the development of any representatives from this general scaffold [34–36].
Figure 3.
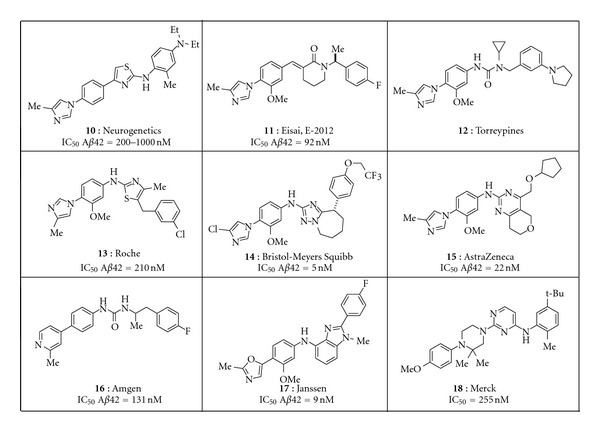
Representative aryl imidazole inspired GSMs [30–33]. Compounds 16–18 are the most unique because the aryl imidazole has been replaced by a bioisostere [34–36].
A novel and structurally distinct chemical architecture of a third class of GSMs has been reported by Satori Pharmaceuticals. This scaffold was first isolated from black cohosh, leading to the characterization of initial hit 19 (Figure 4) [20]. A combination of synthetic and medicinal chemistry optimization led to 20, which is reported to have better drug properties than 19 [20]. To date, the compounds reported by Satori also fall outside of the guidelines most typically associated with good in vivo disposition. The group notes, however, that the majority of marketed agents derived from natural products also violate these same guidelines, a trend that has led some to conclude that molecules derived via semisynthesis on natural product scaffolds define a different set of guidelines. Compounds from this class are now proceeding through preclinical development.
Figure 4.
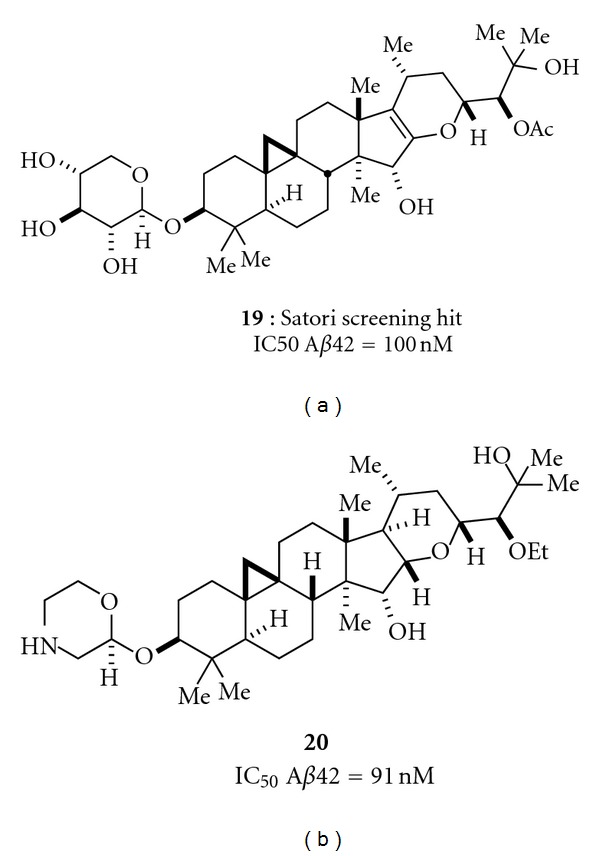
Two examples of Satori GSMs.
3.3. Aβ Peptide Profiling of Structurally Diverse GSMs
When the NSAID-type of GSMs was first described (Figure 2), the changes in Aβ peptides that were seen, a decrease in Aβ 42, an increase in Aβ 38 and little or no change on Aβ 40 or total Aβ were labeled the “modulator profile,” nomenclature that was reinforced by the pharmacology reported for the GSMs represented by the structures in Figure 3. However, since that time, additional Aβ peptide profiles have been reported for the chemically distinct scaffold disclosed by Satori Pharmaceuticals, as well as molecules more closely related to those in Figure 3. Examples of the variety of Aβ peptide profiles produced by GSMs are shown in Figure 6. Merck GSM1 and JNJ-40418677 lower Aβ 42 while increasing Aβ 38, with little or no effect on Aβ 40. Alternatively, the Satori Pharmaceutical compounds, SPI-1802 and SPI-1810 (structures shown in Figure 5) decrease both Aβ 42 and Aβ 38, but maintain total Aβ levels by increasing Aβ 37 and Aβ 39. Yet, all of these compounds can be classified as “gamma secretase modulators” based on the commonality of sparing the ε cleavage of C99 and other substrates (e.g., Notch), decreasing Aβ 42, and not affecting total Aβ levels.
Figure 5.
Based on these data, modulation of γ-secretase is more accurately defined as a shift of the Aβ pool to shorter, but variable length, Aβ peptides. A physiological role for Aβ peptides has not been discovered, but in vitro studies have demonstrated that shorter peptides are incapable of aggregation and oligomerization [19] and may even prevent the oligomerization of Aβ 42 by binding to it, suggesting that preserving the total pool of Aβ may be beneficial.
The molecular mechanism by which GSMs modulate γ-secretase activity is not completely understood. The variety of Aβ peptide profiles that result from treatment of cells with different γ-secretase modulators suggests that the enzyme has numerous allosteric modulatory sites. These are not necessarily completely unique sites and published data suggests the sites may be overlapping [49]. The modulator binding sites thus far identified all seem to reside on the presenilin component of γ-secretase [50].
3.4. In Vivo Activity of GSMs
In vivo, GSMs exhibit the same Aβ profile as observed in vitro with reductions in Aβ 42 and increases or decreases of other Aβ peptides and no effect on total Aβ levels. However, the amount of compound required to generate these effects has been surprisingly high when compared to the in vitro potency of the molecules. For example, the Eisai GSM E-2012 has an in vitro IC50 of 33 nM in a cell-based assay. In vivo, 4.9 μM plasma concentrations of the compound were required to reduce brain Aβ by 25%. Similarly, Merck GSM1, JNJ-40418677, and SPI-1810 also require high plasma levels in order to achieve 25% lowering (Table 2), a factor that does not appear to be readily attributable to plasma protein binding. These high compound levels are not required for in vivo efficacy with GSIs which further suggests that the interaction with γ-secretase by a GSM is different from that of a GSI. Why high concentrations of some GSMs are required to modulate γ-secretase activity in vivo is not completely understood. Less than ideal pharmacokinetic properties, such as low free fraction or poor blood brain barrier permeability may contribute to the need for high plasma concentrations of GSMs to see efficacy in the brain in vivo. In addition, there are data indicating that GSMs can bind to both active and inactive forms of presenilin since the binding site of the modulators is available even prior to endoproteolysis that creates the active form of γ-secretase [51, 52]. Conversely, data indicate that GSIs require complex formation prior to binding [53], which may mean more binding sites are available for GSMs than for GSIs.
Table 2.
GSM potency versus plasma exposure in mice.
| E-2012 | JNJ-40418677 | GSM1 | SPI-1810 | |
|---|---|---|---|---|
| In vitro Aβ 42 IC50 (nM) CHO-2B7 cells | 33 | 172 | 62 | 114 |
| Plasma exposure (μM) | 4923 | 7764 | 2744 | 14638 |
| Plasma fold over Aβ 42 IC50 for 25% reduction | 149 | 45 | 44 | 128 |
| Brain exposure (μM) | 2749 | 7497 | 8681 | 20368 |
| Brain fold over Aβ 42 IC50 for 25% reduction | 83 | 44 | 140 | 179 |
4. Conclusions
Small molecule modulators of γ-secretase are now in the early stages of clinical testing. In preclinical toxicology studies, these modulators are free of the mechanism-based toxicities that have been seen with GSIs, most of which appear to be due to inhibition of Notch processing by directly targeting the ε cleavage site. Both in vitro and in vivo data support the conclusion that GSMs do not interfere with Notch processing, but instead, via slight shifts in the cleavage site on APP, lower Aβ 42 production to produce the other normal Aβ peptide products (Figures 6 and 7). Thus, modulation of γ-secretase may represent the most selective approach to treating Alzheimer's disease via a decrease in production of Aβ 42, more selective that an inhibitor of either beta or γ-secretase. Modulators do differ both in chemical structure and in their effects on APP processing, producing the desired decrease in Aβ 42 and a variety of changes in other Aβ peptides. Understanding the molecular basis of these varying profiles may shed further light on the biology of γ-secretase.
Figure 7.
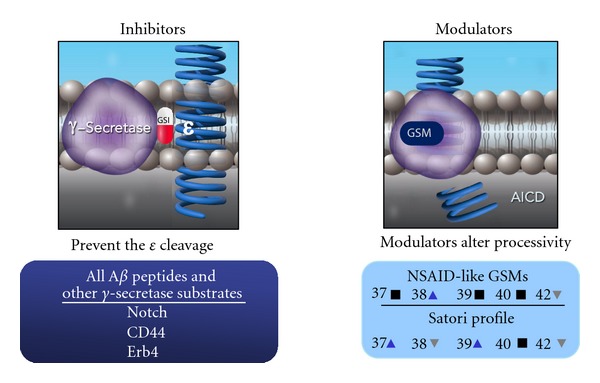
Diagram highlighting the differences between inhibitors and modulators.
Disclosures
All authors are or were employees of Satori Pharmaceuticals, Inc.
Acknowledgments
A portion of this work was presented at the AAIC Annual meetings in 2011 and 2012. The authors would like to thank Dr. John Leszyk from the University of Massachusetts for MALDI-TOF Aβ profile analysis and Kelly Ames, Business Manager at Satori Pharmaceuticals, Inc.
References
- 1.Kakuda N, Funamoto S, Yagishita S, et al. Equimolar production of amyloid β-protein and amyloid precursor protein intracellular domain from β-carboxyl-terminal fragment by γ-secretase. Journal of Biological Chemistry. 2006;281(21):14776–14786. doi: 10.1074/jbc.M513453200. [DOI] [PubMed] [Google Scholar]
- 2.Sato T, Dohmae N, Qi Y, et al. Potential link between amyloid β-protein 42 and C-terminal fragment γ 49-99 of β-amyloid precursor protein. Journal of Biological Chemistry. 2003;278(27):24294–20301. doi: 10.1074/jbc.M211161200. [DOI] [PubMed] [Google Scholar]
- 3.Qi-Takahara Y, Morishima-Kawashima M, Tanimura Y, et al. Longer forms of amyloid β protein: implications for the mechanism of intramembrane cleavage by γ-secretase. Journal of Neuroscience. 2005;25(2):436–445. doi: 10.1523/JNEUROSCI.1575-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takami M, Nagashima Y, Sano Y, et al. γ-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of β-carboxyl terminal fragment. Journal of Neuroscience. 2009;29(41):13042–13052. doi: 10.1523/JNEUROSCI.2362-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Golde TE, Ran Y, Felsenstein KM. Shifting a complex debate on γ-secretase cleavage and Alzheimer's disease. The EMBO Journal. 2012;31(10):2237–2239. doi: 10.1038/emboj.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suzuki N, Cheung TT, Cai XD, et al. An increased percentage of long amyloid β protein secreted by familial amyloid β protein precursor (βAPP717) mutants. Science. 1994;264(5163):1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 7.Saito T, Suemoto T, Brouwers N, et al. Potent amyloidogenicity and pathogenicity of Aβ 243. Nature Neuroscience. 2011;14(8):1023–1032. doi: 10.1038/nn.2858. [DOI] [PubMed] [Google Scholar]
- 8.Jarrett JT, Berger EP, Lansbury PT., Jr. The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry. 1993;32(18):4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 9.Milano J, McKay J, Dagenais C, et al. Modulation of Notch processing by γ-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicological Sciences. 2004;82(1):341–358. doi: 10.1093/toxsci/kfh254. [DOI] [PubMed] [Google Scholar]
- 10.Searfoss GH, Jordan WH, Calligaro DO, et al. Adipsin, a biomarker of gastrointestinal toxicity mediated by a functional γ-secretase inhibitor. Journal of Biological Chemistry. 2003;278(46):46107–46116. doi: 10.1074/jbc.M307757200. [DOI] [PubMed] [Google Scholar]
- 11.Wong GT, Manfra D, Poulet FM, et al. Chronic treatment with the γ-secretase inhibitor LY-411,575 inhibits γ-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. Journal of Biological Chemistry. 2004;279(13):12876–12882. doi: 10.1074/jbc.M311652200. [DOI] [PubMed] [Google Scholar]
- 12.Lilly E. Lilly Halts Development of Semagacestat for Alzheimer's Disease Based on Preliminary Results of Phase III Clinical Trials. 2010, http://newsroom.lilly.com/releasedetail.cfm?releaseid=499794.
- 13.Selkoe DJ. Resolving controversies on the path to Alzheimer's therapeutics. Nature Medicine. 2011;17(9):1060–1065. doi: 10.1038/nm.2460. [DOI] [PubMed] [Google Scholar]
- 14.Imbimbo BP, Panza F, Frisardi V, et al. Therapeutic intervention for Alzheimer’s disease with γ-secretase inhibitors: still a viable option? Expert Opinion on Investigational Drugs. 2011;20(3):325–341. doi: 10.1517/13543784.2011.550572. [DOI] [PubMed] [Google Scholar]
- 15.Weggen S, Eriksen JL, Das P, et al. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature. 2001;414(6860):212–216. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- 16.Golde TE, Schneider LS, Koo EH. Anti-Aβ therapeutics in alzheimer’s disease: the need for a paradigm shift. Neuron. 2011;69(2):203–213. doi: 10.1016/j.neuron.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Imbimbo BP. Why did tarenflurbil fail in alzheimer’s disease? Journal of Alzheimer’s Disease. 2009;17(4):757–760. doi: 10.3233/JAD-2009-1092. [DOI] [PubMed] [Google Scholar]
- 18.Green RC, Schneider LS, Amato DA, et al. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. Journal of the American Medical Association. 2009;302(23):2557–2564. doi: 10.1001/jama.2009.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bulic B, Ness J, Hahn S, Rennhack A, Jumpertz T, Weggen S. Chemical biology, molecular mechanism and clinical perspective of γ-secretase modulators in Alzheimer's disease. Current Neuropharmacology. 2011;9(4):598–622. doi: 10.2174/157015911798376352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hubbs JL, Fuller NO, Austin WF, et al. Optimization of a natural product-based class of gamma-secretase modulators. Journal of Medicinal Chemistry. 2012;55(21):9270–9282. doi: 10.1021/jm300976b. [DOI] [PubMed] [Google Scholar]
- 21.Koo EHM, Golde TE, Galasko DR. Nonsteroidal antiinflammatory drug (NSAID) and NSAID derivative amyloid Aβ42 polypeptide-lowering agents for the treatment of Alzheimer's disease, and screening methods. pp. 73, Mayo Foundation for Medical Education and Research, USA, 2001.
- 22.Raveglia L, Peretto I, Radaelli S, Imbimbo BP, Rizzi A, Villetti G. Preparation of 1-phenylalkanecarboxylic acid derivatives for the treatment of neurodegenerative diseases. pp. 91, Chiesi Farmaceutici S.p.A., Italy, 2004.
- 23.Blurton P, et al. Arylacetic acids and related compounds and their preparation, pharmaceutical compositions and their use for treatment of diseases associated with the deposition of β-amyloid peptides in the brain such as Alzheimer’s disease. pp. 58, Merck Sharp & Dohme Limited, UK, 2006.
- 24.Ho CY. Preparation of Biphenyl Derivatives as γ-secretAse Modulators. Belg, Germany: Janssen Pharmaceutica; 2009. [Google Scholar]
- 25.Wilson F, Reid A, Reader V, et al. Preparation of biphenylacetic acids as γ-secretase modulators for the treatment of Alzheimer's disease. pp. 57, Cellzome, Germany, 2006.
- 26.Shapiro G, Chesworth R. 1,3,4-Trisubstituted benzenes as γ-secretase inhibitors and their preparation and use in the treatment of neurodegenerative diseases. pp. 165, Envivo Pharmaceuticals, USA, 2009.
- 27.Kulagowski JJ, Madin A, Ridgill PM, Seward ME. Preparation of piperidines and related compounds for treatment of Alzheimer's disease. pp. 178, Merck Sharp & Dohme Limited, UK, 2006.
- 28.Hall A, Elliott RL, Giblin GMP, et al. Piperidine-derived γ-secretase modulators. Bioorganic and Medicinal Chemistry Letters. 2010;20(3):1306–1311. doi: 10.1016/j.bmcl.2009.08.072. [DOI] [PubMed] [Google Scholar]
- 29.Am ECW, et al. Aminocyclohexanes and aminotetrahydropyrans as γ-secretase modulators and their preparation and use for thetreatment of neurological and psychiatric diseases. pp. 91, Pfizer, USA, 2011.
- 30.Cheng S, Comer DD, Mao L, Balow GP, Pleynet D. Aryl compounds and uses in modulating amyloid β . pp. 178, Neurogenetics, USA 2004.
- 31.Baumann K, et al. Preparation of thiazolyl-substituted imidazolylphenylamine derivatives and related compounds as modulators of amyloid beta. pp. 32, Hoffmann-La Roche, USA, 2008.
- 32.Marcin LR, et al. Preparation of bicyclic compounds, especially bicyclic triazoles, for the reduction of beta-amyloid protein production. pp. 242, Bristol-Myers Squibb Company, USA, 2010.
- 33.Forsblom R, Paulsen K, Waldman M, Rotticci D, Santangelo E. Preparation of 2-aminopyrimidines as amyloid beta modulators. pp. 227, AstraZeneca AB, Sweden, 2010.
- 34.Biswas K, Chen JJ, Falsey JR, et al. Preparation of urea compounds as gamma secretase modulators. pp. 116, Amgen, USA, 2009.
- 35.Gijsen HJM, Velter AI, MacDonald GJ, et al. Novel substituted bicyclic heterocyclic compounds as gamma secretase modulators and their preparation and use in the treatment of diseases. pp. 164, Ortho-Mcneil-Janssen Pharmaceuticals, USA, 2010.
- 36.Blurton P, et al. Preparation of heteroarylpiperazine derivatives for use in treatment of Alzheimer's disease. pp.68, Merck & Co., Merck Sharp & Dohme Limited, USA, 2008.
- 37.Findeis MA, Schroeder F, McKee TD, et al. Discovery of a novel pharmacological and structural class of gamma secretase modulators derived from the extract of actaea racemosa. ACS Chemical Neuroscience. 2012;3(11):941–951. doi: 10.1021/cn3000857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haugabook SJ, Yager DM, Eckman EA, Golde TE, Younkin SG, Eckman CB. High throughput screens for the identification of compounds that alter the accumulation of the Alzheimer’s amyloid β peptide (Aβ) Journal of Neuroscience Methods. 2001;108(2):171–179. doi: 10.1016/s0165-0270(01)00388-0. [DOI] [PubMed] [Google Scholar]
- 39.Murphy MP, Uljon SN, Fraser PE, et al. Presenilin 1 regulates pharmacologically distinct γ-secretase activities: implications for the role of presenilin in γ-secretase cleavage. Journal of Biological Chemistry. 2000;275(34):26277–26284. doi: 10.1074/jbc.M002812200. [DOI] [PubMed] [Google Scholar]
- 40.Lanz TA, Karmilowicz MJ, Wood KM, et al. Concentration-dependent modulation of amyloid-β in vivo and in vitro using the γ-secretase inhibitor, LY-450139. Journal of Pharmacology and Experimental Therapeutics. 2006;319(2):924–933. doi: 10.1124/jpet.106.110700. [DOI] [PubMed] [Google Scholar]
- 41.Rogers K, et al. Optimization of a natural product-based class of gamma secretase modulators. International Conference on Alzheimer's Disease; 2009; Vienna, Austria. [Google Scholar]
- 42.Burton CR, Meredith JE, Barten DM, et al. The amyloid-β rise and γ-secretase inhibitor potency depend on the level of substrate expression. Journal of Biological Chemistry. 2008;283(34):22992–23003. doi: 10.1074/jbc.M804175200. [DOI] [PubMed] [Google Scholar]
- 43.Hughes JD, Blagg J, Price DA, et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorganic and Medicinal Chemistry Letters. 2008;18(17):4872–4875. doi: 10.1016/j.bmcl.2008.07.071. [DOI] [PubMed] [Google Scholar]
- 44.Wager TT, Hou X, Verhoest PR, Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chemical Neuroscience. 2010;1(6):435–449. doi: 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wager TT, Chandrasekaran RY, Hou X, et al. Defining desirable central nervous system drug space through the alignment of molecular properties, in vitro ADME, and safety attributes. ACS Chemical Neuroscience. 2010;1(6):420–434. doi: 10.1021/cn100007x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lovering F, Bikker J, Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. Journal of Medicinal Chemistry. 2009;52(21):6752–6756. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- 47.Ritchie TJ, Macdonald SJF. The impact of aromatic ring count on compound developability—are too many aromatic rings a liability in drug design? Drug Discovery Today. 2009;14(21-22):1011–1020. doi: 10.1016/j.drudis.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 48.Kimura T, Kawano K, Doi E, et al. Preparation of cinnamide, 3-benzylidenepiperidin-2-one, phenylpropynamide compounds as amyloid β production inhibitors. pp. 679, Eisai, Japan, 2005.
- 49.Ebke A, Luebbers T, Fukumori A, et al. Novel γ-secretase enzyme modulators directly target presenilin protein. Journal of Biological Chemistry. 2011;286:37181–37186. doi: 10.1074/jbc.C111.276972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolfe MS. gamma-Secretase inhibitors and modulators for Alzheimer's disease. Journal of Neurochemistry. 2012;120(supplement 1):89–98. doi: 10.1111/j.1471-4159.2011.07501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ohki Y, Higo T, Uemura K, et al. Phenylpiperidine-type γ-secretase modulators target the transmembrane domain 1 of presenilin 1. The EMBO Journal. 2011;30(23):4815–4824. doi: 10.1038/emboj.2011.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weggen S, Beher D. Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal-dominant Alzheimer's disease. Alzheimer's Research and Therapy. 2012;4(2) doi: 10.1186/alzrt107.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takeo K, Watanabe N, Tomita T, Iwatsubo T. Contribution of the γ-secretase subunits to the formation of catalytic pore of presenilin 1 protein. Journal of Biological Chemistry. 2012;287(31):25834–25843. doi: 10.1074/jbc.M111.336347. [DOI] [PMC free article] [PubMed] [Google Scholar]