

Abstract
The first syntheses of Jadomycin A and the carbosugar analogue of Jadomycin B have been achieved in 6 and 20 longest linear steps respectively. The key ring system of the aglycone was prepared by a 6π-electron electrocyclic ring closure and subsequent hemi-aminal ring closure. Acid sensitivity of the glycosidic bond in Jadomycin B precluded its synthesis but led to the synthesis of the carbasugar analogue.
Keywords: Jadomycin, 6π-electrocyclic ring-closure, de novo synthesis, carbasugar
Jadomycins A & B are unique members of the angucycline family of natural products with the unusual 8H-benz[b]oxazolo[3,3-f]-phenanthridine ring system. Both Jadomycins A & B are produced by the Gram-positive soil bacteria Streptomyces venezuelae ISP5230 under specific nutrient and environmental stress.[i ] They display important bioactivities, including antitumor, antimicrobial, anti-viral, as well as, aurora-B kinase inhibition and DNA-cleaving capacity.[ii,iii,iv]
There has been no total synthesis of Jadomycin A or B, although libraries of Jadomycin analogues have been produced through fermentation approaches.[v] Several approaches to the benzo[b]-phenanthridine ring system have been developed (Scheme 1).[vi,vii] From these studies one can conclude that for stereoelectronic reasons the B-ring is best constructed by a cyclization of a 2-amino-3-phenylnaphthoquinone (e.g., 6 to 5), rather than an amine cyclization onto the 2-position of an anthroquinone (e.g., 8 to 5). However, these results stand in contrast to the proposed biosynthetic route (e.g., 1/2 via 4, Figure 1).
Scheme 1.
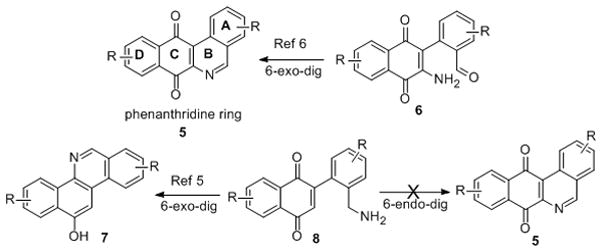
Retrosynthetic comparison for phenanthridine B-ring formation
Figure 1.
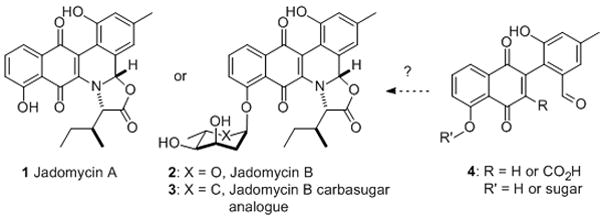
Jadomycin A, B and its analogue
Our ongoing interest in the synthesis and study of biologically active carbohydrate containing natural products,[viii] along with these biosynthetic questions, made us interested in this class of natural products. Specifically, we were interested in testing our hypothesis that by employing an 6π-electrocylic ring closure for the B-ring construction, one could skirt the stereoelectronic disadvantages of the 6-endo-dig ring closures (10 to 9).[ix] To the best of our knowledge this is the first time that switching to an electrocyclic ring closure was used to change the regioselectivity for ring closure. This may also be the biosynthetic mode of ring closure and thus implying that the B-ring cyclization step doesn’t need enzyme mediation to control the regioselectivity of cyclization.
Retrosynthetically, we envisioned 1 and 2 arising from the condensation of a protected aminoacid 11 and aldehyde 4 to form imine 10. A 6π-electron electrocyclic ring closure would form the desired benzo[b]-phenanthridine 9 after hydration and oxidation. Finally an acid catalyzed deprotection/dehydration would install the final oxazolone ring in Jadomycin (Scheme 2). Following precedent, a Stille coupling could be employed to prepare 4.[vi] In accordance with the proposed biosynthetic pathway,[v] the L-digitoxose could be incorporated earlier (e.g., 4) or on Jadomycin A, this would depend on the stability of the glycosidic bond to these acid catalyzed transformation.
Scheme 2.
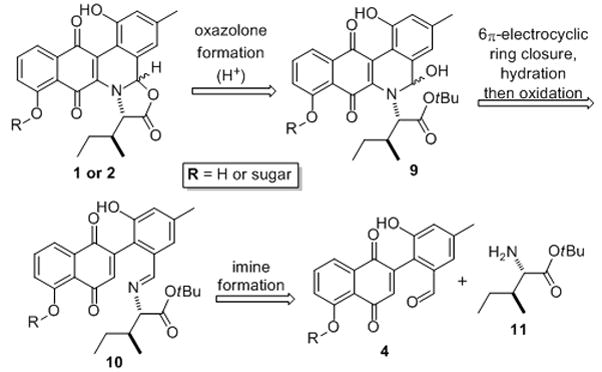
Our biomimetic approach toward the Jadomycins
Our synthesis began with commercially available phenol 12,[vi] which was then protected as MOM and BOM ethers 13a (68%) and 13b (70%). Ortho-metalation of 13a/b gave stannanes 14a (68%) and 14b (49%) in modest yields along with some recovered starting material (~30%). The other coupling partner 16 was prepared by benzylation of commercially available bromojuglone 15 (Scheme 3).[x]
Scheme 3.
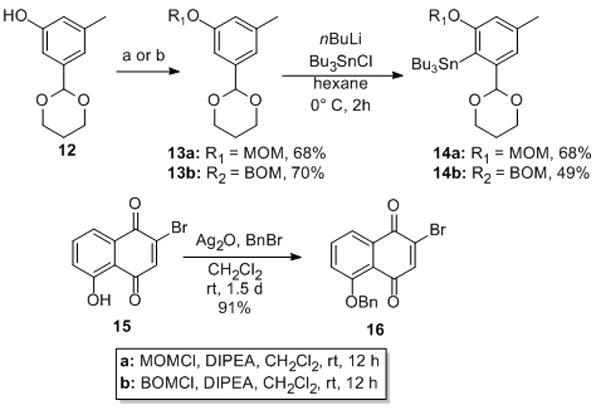
Synthesis of the desired coupling partners: MOM = methoxymethyl, BOM = benzyloxymethyl, DIPEA = N,N-diisopropylethylamine.
Coupling between stannane 14a and juglone 16 gave 17a in 73% yield (5 mol% Pd2(dba)3•CHCl3, 20 mol% PPh3 and 20 mol% CuI co-catalyst in THF, 75 °C for 12 h) (Scheme 4). Under identical conditions, unprotected juglone 15 was coupled with 14a and 14b to similar yields of 17b (76%) and 17c (54%). Selective acetal cleavage on 17a (TFA(aq)/THF, 10 min, 86%) gave the desired cyclization precursor aldehydes 19. Unfortunately, when 19 was subjected to condensation conditions with protected isoleucine 11 no products of cyclizations were detected (e.g., 20). We surmised that replacing the MOM-ether with a phenol would turn the negative ortho-ortho steric interaction into a positive hydrogen-bonding interaction.
Scheme 4.
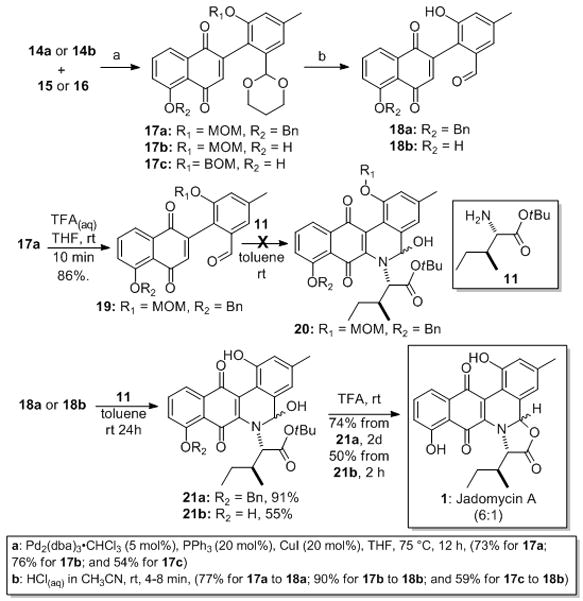
Synthesis of Jadomycin A: dba = dibenzylideneacetone, TFA = trifluoroacetic acid.
Turning to an unselective acetal cleavage conditions (2.4 M HCl in CH3CN, 4–8 min, 59–90%)[xi] acetals 17a–c were converted into aldehydes 18a/b. To our delight, when aldehyde 18a was condensed with 11 a smooth imine formation and cyclization occurred to give hemi-aminal 21a (91%), which when treated with neat TFA underwent a t-butyl ester hydrolysis, oxazolone ring formation and benzyl ether cleavage to give the natural product Jadomycin A (74%, dr = 6:1).
The discovery of the surprising acid sensitivity of the benzyl ether in 21a, led us to our optimal synthesis of Jadomycin A. Thus exposure of 18b to the previous imine formation/electrocyclic ring closure conditions cleanly gave 21b mixture of diastereomers (55%) and subsequent exposure of 21b to neat TFA gave Jadomycin A (1) in 50% yield. This optimized route allowed for the formation of Jadomycin A in 6 steps from both commercially available 12 and 15 in 17% overall yield. A plausible mechanism for this key ring closure is outlined in Scheme 5.
Scheme 5.
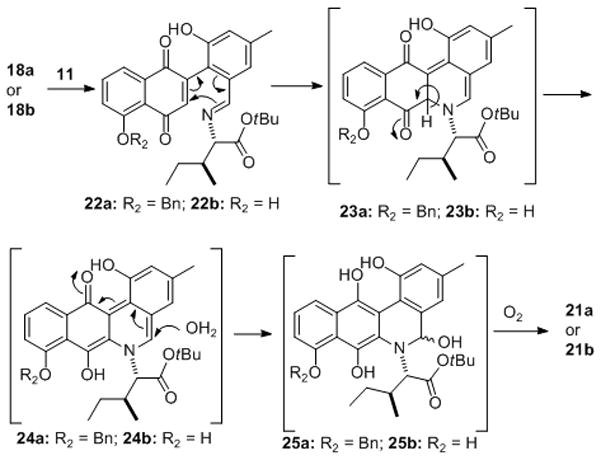
A plausible mechanism for ring closure
We next decided to undertake a chemical glycosylation using our de novo carbohydrate approach as a part of our effort to study SAR of Jadomycin B. The synthesis of glycosyl donor 27, 31, 32 and 33 were achieved by a de novo asymmetric synthesis with achiral acyl furan as the starting material.[xii] Following a procedure we previously disclosed for its enantiomer, acylfuran 26 was enantioselectively converted into t-butyl carbonate 28 in six steps via α-pyranone 27.[xiii] An NIS promoted iodo-carbonate cyclization installed the desired digitoxose stereochemistry in 29 and a radical TTMSS deiodination gave 30, which could be debenzylated to give 31.[xiv] The anomeric alcohol in 31 could be activated for a Schmidt glycosylation[xv] as an unstable trichloroacetimidate 32, which was used in situ. Simply switching the cyclic carbonate protecting group of 30 to an acetonide and debenzylation gave another potential glycosyl donor 33 (Scheme 6).[xvi]
Scheme 6.
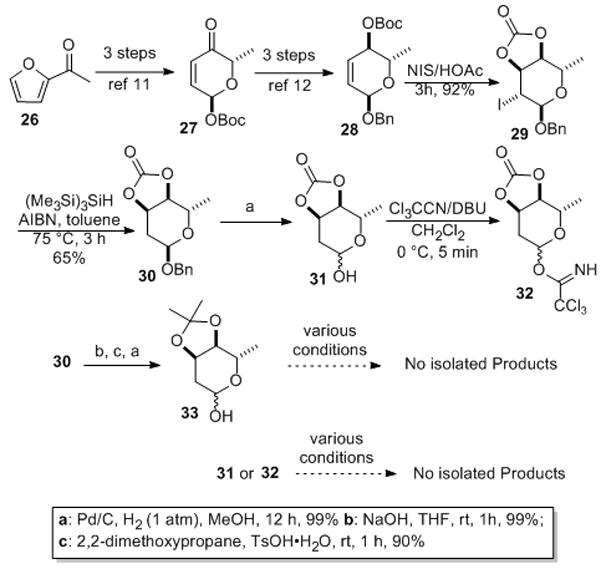
De Novo synthesis of glycosyl donors: NIS = N-iodosuccinimide, AIBN = azobisisobutyronitrile, DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene
We primarily focused on the installation of the L-digitoxose onto the aglycon employing mild Mitsunobu conditions to construct Jadomycin B (e.g., 31 or 33 and 17b/c or 1 with DIAD/PPh3, −78 °C to rt).[xvii] While these reaction conditions had TLC evidence consistent with the formation of the desired glycosidic bond (a distinctive wine-red spot),[xviii] unfortunately, none of the desired products could be isolated from these reaction mixtures. Changing of solvent and/or order of addition did not seem to have any significant affect, whereas, raising the temperature above −20 °C caused the in situ formed wine-red product to disappear. Schmidt glycosylation using 32 failed to produce the desired in situ product as determined by TLC.[xv] The acid sensitivity of the glycosylated hydroxylquinones was evident by the fact that even our non-Lewis acidic Pd-catalyzed glycosylation of 1 and 17b/c with pyranone 27 also failed. This instability of Jadomycin B precluded a practical SAR type studies and thus, we decided to investigate the synthesis of a less acid sensitive cyclitol analogue 3.
The synthesis of carbasugar glycosyl donor equivalent of L-digitoxose starts from quinic acid (Scheme 7). Following a known procedure, D-(−)-quinic acid was converted into dihydroxyketone 35 in nine steps.[xix] Boc protection followed by base mediated elimination resulted enone 36 (88%, two steps). A diastereoselective substrate controlled reduction of ketone 36 afforded 37 (94%). A Myers reductive rearrangement (NBSH/DIAD/PPh3) cleanly gave alkene 38 (87%).[ xx ] Stereoselective dihydroxylation using OsO4 and subsequent diol protection gave acetonide 39 (80%, two steps). Finally, our proposed Mitsunobu type cyclitolization precursor 40 was prepared by a low temperature LiAlH4 removal of the Boc group (97%).
Scheme 7.
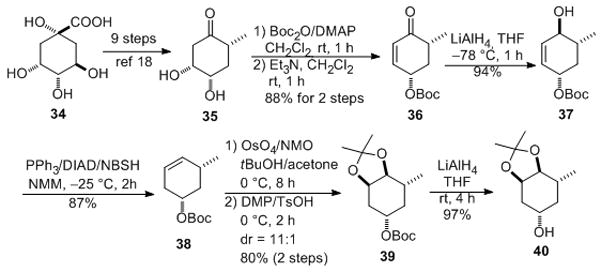
Asymmetric synthesis of a cyclitol donor: Boc = t-butyl carboxylate, DIAD = diisopropyl azodicarboxylate, NBSH = o-nitrobenzenesulfonylhydrazide, NMO = N-methylmorpholine N-oxide, DMP = 2,2-dimethoxypropane, TsOH = p-toluenesulfonic acid.
With the cyclitol donor 40 in hand, we employed the proposed Mitsunobu cyclitolization with Jadomycin A 1, unfortunately no cyclitol product 41 was observed (Scheme 8). Success occurred when we performed the Mitsunobu on oxazolone precursors (e.g., 17b and 17c). As before these reactions TLCs had the tell-tell sign of the wine-red spots, which this time did not disappear upon warming to rt. More convincingly, stable products 42a and 42b were isolated in excellent yields (84% and 91% respectively) when the reactions were run at rt. These acid stable products 42a and 42b could be globally deprotected upon exposure to aqueous acid (2.4 M, HCl/MeCN) giving moderate yield of 43 (59% from 42a, 56% from 42b).
Scheme 8.
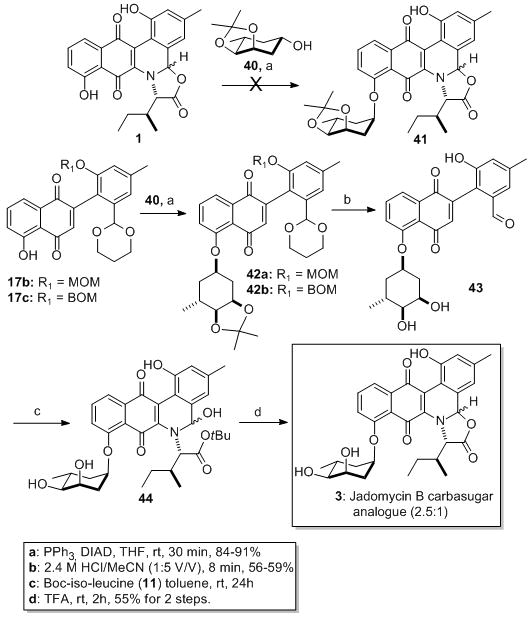
Synthesis of Jadomycin B carbasugar analogue
As before with the aglycone, the cyclitol analogues 43 could be annulated with Boc-isoleucine 11. Thus, condensation of aldehyde 43 with amino-ester 11 followed by a 6π-electrocyclic ring closure resulted in the formation of the desired ring system 44. Finally acid catalyzed trans-esterification leads to the formation of carbasugar analogue of Jadomycin B as an inseparable mixture of 2.5:1 diastereomers (Scheme 8).
In conclusion, the first total synthesis of Jadomycin A was accomplished in 6 longest linear steps in 17% overall yield. A cyclitol analogue of Jadomycin B has been successfully achieved in 20 longest linear steps in a 5% overall yield.[xxi] A 6π-electrocyclic ring closure was used to obtain the Jadomycin ring system, which should be compatible with the incorporation of various other amino acids for rapid synthesis of analogues. This synthesis supports the conclusions by Rix et al. that the isoleucine incorporation and consequent formation of the oxazolophenanthridine system occur non-enzymatically.[vb] The application of this methodology for further analogue synthesis and biological testing is ongoing.
Footnotes
We are grateful to the NIH (GM088839) and NSF (CHE-0749451) for their generous support of our research program. A dissertation fellowship to M. S. from WVU’s Office of Graduate Study and Life is greatly appreciated.
Supporting information for this article is available on the WWW under http://www.angewandte.org
References
- i.Doull JL, Ayer SW, Singh AK, Thibault P. J Antibiot. 1993;46:869. doi: 10.7164/antibiotics.46.869. [DOI] [PubMed] [Google Scholar]
- ii.Doull JL, Singh AK, Hoare M, Ayer SW. J Ind Microbiol. 1994;13:120. doi: 10.1007/BF01584109. [DOI] [PubMed] [Google Scholar]
- iii.Cottreau KM, Spencer C, Wentzell JR, Graham CL, Borissow CN, Jakeman DL. Org Lett. 2010;12:1172. doi: 10.1021/ol902907r. [DOI] [PubMed] [Google Scholar]
- iv.Krohn K, Rohr J. J Top Curr Chem. 1997;188:127. [Google Scholar]
- v.It has been suggested that the L-digitoxose sugar is attached to the aglycon by the enzyme JadS both before and after the oxazolone ring formation, see: Jakeman DL, Borissow CN, Graham CL, Timmons SC, Reid TR, Syvitski RT. Chem Commun. 2006;35:3738. doi: 10.1039/b608847c.Rix U, Wang C, Chen Y, Lipata FM, Rix LLR, Greenwell LM, Vining LC, Yang K, Rohr J. Chem Bio Chem. 2005;6:838. doi: 10.1002/cbic.200400395.
- vi.de Frutos O, Atienza C, Echavarren AM. Eur J Org Chem. 2001:163. [Google Scholar]
- vii.Akagi Y, Yamada S, Etomi N, Kumamoto T, Nakanishi W, Ishikawa T. Tetrahedron Lett. 2010;51:1338. [Google Scholar]
- viii.(a) Guo H, O’Doherty GA. Angew Chem, Int Ed Engl. 2007;46:5206. doi: 10.1002/anie.200701354. [DOI] [PubMed] [Google Scholar]; (b) Zhou M, O’Doherty GA. J Org Chem. 2007;72:2485. doi: 10.1021/jo062534+. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhou M, O’Doherty GA. Org Lett. 2006;8:4339. doi: 10.1021/ol061683b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Balachari D, O’Doherty GA. Org Lett. 2000;2:4033. doi: 10.1021/ol006662a. [DOI] [PubMed] [Google Scholar]; (e) Guo H, O’Doherty GA. Org Lett. 2005;7:3921. doi: 10.1021/ol051383e. [DOI] [PubMed] [Google Scholar]; (f) Zhou M, O’Doherty GA. Org Lett. 2008;10:2283. doi: 10.1021/ol800697k. [DOI] [PubMed] [Google Scholar]
- ix.For examples of diastereoselective aza-triene electrocyclic ring closures in synthesis, see: Li G, Hsung RP, Slafer BW, Sagamanova IK. Org Lett. 2008;10:4991. doi: 10.1021/ol802068q.
- x.Jung ME, Hagenah JA. J Org Chem. 1987;52:1889. [Google Scholar]
- xi.We found that most Bronsted/Lewis acid conditions for MOM/BOM-groups removal lead to decomposition of starting material. We experienced similar problem with the hydrogenolysis of the BOM-group. After some careful study we found a small window of opportunity with the short-term exposure (4–8 min) of both the MOM- and BOM-ether to conc. HCl/acetonitrile (v/v 1:5).
- xii.Babu RS, O’Doherty GA. J Am Chem Soc. 2003;125:12406. doi: 10.1021/ja037097k. [DOI] [PubMed] [Google Scholar]
- xiii.Guo H, O’Doherty GA. J Org Chem. 2008;73:5211. doi: 10.1021/jo800691v. [DOI] [PubMed] [Google Scholar]
- xiv.Shan M, Xing Y, O’Doherty GA. J Org Chem. 2009;74:5961. doi: 10.1021/jo9009722. [DOI] [PubMed] [Google Scholar]
- xv.Mueller T, Schneider R, Schmidt RR. Tetrahedron Lett. 1994;35:4763. [Google Scholar]
- xvi.Babu RS, Zhou M, O’Doherty GA. J Am Chem Soc. 2004;126:3428. doi: 10.1021/ja039400n. [DOI] [PubMed] [Google Scholar]
- xvii.Mitsunobu O, Yamada Y. Bull Chem Soc Japan. 1967;40:2380. [Google Scholar]; b) Mitsunobu O. Synthesis. 1981:1. [Google Scholar]
- xviii.Jakeman reported similar color changes, see: Jakeman DL, Dupuis SN, Graham CL. Pure Appl Chem. 2009;81:1041.
- xix.Shan M, O’Doherty GA. Org Lett. 2008;10:3381. doi: 10.1021/ol801106r. [DOI] [PubMed] [Google Scholar]
- xx.Myers AG, Zheng B. J Am Chem Soc. 1996;118:4492. [Google Scholar]; b) Myers AG, Zheng B. Tetrahedron Lett. 1996;37:4841. [Google Scholar]; c) Myers AG, Zheng B, Movassaghi M. J Org Chem. 1997;62:7507. doi: 10.1021/jo9710137. [DOI] [PubMed] [Google Scholar]; d) Haukaas MH, O’Doherty GA. Org Lett. 2002;4:1771. doi: 10.1021/ol025844x. [DOI] [PubMed] [Google Scholar]; e) Zhou M, O’Doherty GA. Org Lett. 2006;8:4339. doi: 10.1021/ol061683b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxi.The route is 20 longest linear steps from quinic acid but only 9 steps from known starting materials, 36.