

Abstract
The actinomycete Amycolatopsis sp. strain ATCC 39116 is capable of synthesizing large amounts of vanillin from ferulic acid, which is a natural cell wall component of higher plants. The desired intermediate vanillin is subject to undesired catabolism caused by the metabolic activity of a hitherto unknown vanillin dehydrogenase (VDHATCC 39116). In order to prevent the oxidation of vanillin to vanillic acid and thereby to obtain higher yields and concentrations of vanillin, the responsible vanillin dehydrogenase in Amycolatopsis sp. ATCC 39116 was investigated for the first time by using data from our genome sequence analysis and further bioinformatic approaches. The vdh gene was heterologously expressed in Escherichia coli, and the encoded vanillin dehydrogenase was characterized in detail. VDHATCC 39116 was purified to apparent electrophoretic homogeneity and exhibited NAD+-dependent activity toward vanillin, coniferylaldehyde, cinnamaldehyde, and benzaldehyde. The enzyme showed its highest level of activity toward vanillin at pH 8.0 and at a temperature of 44°C. In a next step, a precise vdh deletion mutant of Amycolatopsis sp. ATCC 39116 was generated. The mutant lost its ability to grow on vanillin and did not show vanillin dehydrogenase activity. A 2.3-times-higher vanillin concentration and a substantially reduced amount of vanillic acid occurred with the Amycolatopsis sp. ATCC 39116 Δvdh::Kmr mutant when ferulic acid was provided for biotransformation in a cultivation experiment on a 2-liter-bioreactor scale. Based on these results and taking further metabolic engineering into account, the Amycolatopsis sp. ATCC 39116 Δvdh::Kmr mutant represents an optimized and industrially applicable platform for the biotechnological production of natural vanillin.
INTRODUCTION
Vanillin is one of the most important and widely used flavor compounds in the food and fragrance industry. The extraction of natural vanillin from cured seed pods of the orchid Vanilla planifolia is highly cost- and labor-intensive and does not satisfy the global demand of the continuously increasing market. Therefore, new sources for the synthesis of vanillin are of great interest for the industry. More than 99% of the industrially produced vanillin is chemically synthesized from petrochemicals or lignin derivatives (1). Due to the increasing interest in “natural” and “healthy” ingredients, especially in the food industry, alternative sources for natural vanillin originating from biotechnical processes were intensively investigated (2). Vanillin can be labeled natural if it is extracted from natural sources or “obtained by appropriate physical, enzymatic or microbiological processes from material of vegetable, animal or microbiological origin” (3).
The most attractive approaches for microbial vanillin synthesis are the biotransformation of cheap natural aromatic compounds, such as ferulic acid [3-(4-hydroxy-3-methoxy-phenyl)prop-2-enoic acid], or de novo biosynthesis from primary metabolites like glucose (4). The bioconversion of ferulic acid into vanillin was demonstrated previously for various microorganisms, including bacteria like Pseudomonas and Rhodococcus (5–8) and fungi such as the basidiomycete Pycnoporus cinnabarinus (9). Additionally, several genes encoding the responsible enzymes in some of these organisms were heterologously expressed in Escherichia coli and enabled the corresponding strains to convert ferulic acid into vanillin (10, 11). However, high concentrations of vanillin are attended by serious problems for the cells due to the toxicity of this highly reactive aromatic aldehyde, which is detoxified in the course of further catabolism or leads to cell death. Therefore, most processes did not achieve industrial applicability because of insufficient vanillin yields.
In order to overcome this drawback, screenings for vanillin-tolerant bacterial strains, which are able to convert ferulic acid and thereby accumulate large amounts of the biotransformation product vanillin, were performed (12, 13). Rabenhorst et al. (13) reached a vanillin concentration of 11.5 g/liter after the biotransformation of 19.9 g/liter ferulic acid using Amycolatopsis sp. strain HR167, corresponding to a molar yield of 77.8%. With Streptomyces setonii ATCC 39116, which meanwhile has been reclassified Amycolatopsis sp. strain ATCC 39116, Muheim et al. (12) achieved a final vanillin concentration of 13.9 g/liter with a molar yield of 75%. In contrast to most other strains used, both approaches are applicable to industrial processes, although they also face the problem of further vanillin degradation to vanillic acid and other unwanted degradation products (14, 15).
For an improved vanillin production process using the actinomycete Amycolatopsis sp. ATCC 39116, whose 16S rRNA gene sequence is identical to that of Amycolatopsis sp. HR167, the whole genome was sequenced to identify enzymes involved in vanillin catabolism, as previously done for other vanillin-producing strains (6, 8, 16–18). In order to promote a more efficient accumulation of vanillin by preventing its oxidation to vanillic acid, enzymes with homology to characterized vanillin dehydrogenases (VDHs) of other bacteria were screened. After the identification of a set of enzymes with homologies to vanillin and other aldehyde dehydrogenases, the related genes were heterologously expressed in E. coli, and the encoded enzymes were tested for their activity on vanillin. The enzyme exhibiting VDH activity showed the highest level of homology to an aldehyde dehydrogenase of the Nocardioidaceae bacterium Broad-1 (GenBank accession no. ZP_08197264) and was therefore designated VDHATCC 39116. In a next step, a precise deletion mutant was generated, and the impact of VDHATCC 39116 on the catabolism of vanillin was examined. First attempts were made to employ the deletion mutant in fermentation processes as a novel production platform for natural vanillin.
MATERIALS AND METHODS
Bacterial strains, plasmids, and cultivation conditions.
All bacterial strains and plasmids used in this study are listed in Table 1. Cells of Escherichia coli were grown in lysogeny broth (LB) medium at 30°C (19). Cells of Amycolatopsis sp. ATCC 39116 were grown at 42°C in Caso medium (Merck, Darmstadt, Germany) or in a modified mineral salts medium (20). Carbon sources were provided from stock solutions, which were sterilized by filtration before use, as indicated. For the selection of plasmid-harboring strains, the following antibiotics were added to the medium: kanamycin at 50 μg/ml for E. coli and 100 μg/ml for Amycolatopsis sp. ATCC 39116 and ampicillin at 100 μg/ml for E. coli. Cell growth was measured photometrically by using a Klett-Summerson photometer equipped with filter no. 54 (520 to 580 nm) or by measuring the optical density at 400 nm (Amycolatopsis sp. ATCC 39116) or 600 nm (E. coli).
Table 1.
Bacterial strains and plasmids used in the present study
| Bacterial strain or plasmid | Description | Source or reference |
|---|---|---|
| Bacterial strains | ||
| Amycolatopsis sp. ATCC 39116 | Wild type, kanamycin sensitive | American Type Culture Collection |
| Amycolatopsis sp. ATCC 39116 Δvdh::Kmr | vdh deletion mutant, kanamycin resistant | This study |
| E. coli Mach-1 T1 | F− ϕ80(lacZ)ΔM15 ΔlacX74 hsdR(rK− mK+) ΔrecA1398 endA1 tonA | Invitrogen GmbH, Karlsruhe, Germany |
| E. coli BL21(DE3) | F− ompT hsdSB(rB− mB−) gal dcm (DE3) | Novagen Inc., Madison, WI |
| Plasmids | ||
| pET23a | T7 promoter, His6 tag, T7 terminator, ColE1 pBR322 ori, Ampr, f1 ori | Novagen Inc., Madison, WI |
| pET23a::vdh | pET23a harboring vdh (NdeI/XhoI) coding for VDHATCC 39116 colinear to the T7 promoter | This study |
| pET23a::vdhHis | pET23a harboring vdh (NdeI/XhoI) without the native stop codon coding for VDHATCC 39116 with a C-terminal His6 tag, colinear to the T7 promoter | This study |
Biotransformation experiments.
For biotransformation experiments with E. coli or Amycolatopsis sp. ATCC 39116, cells were harvested during the stationary growth phase, washed twice with phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2HPO4 [pH 7.5]), and resuspended in an appropriate volume of the same buffer or mineral salts medium (20). The experiments were carried out with 50 ml mineral salts medium in 250-ml Erlenmeyer flasks (E. coli) or in a 2-liter bioreactor with a total volume of 1,000 ml PBS (Amycolatopsis sp. ATCC 39116). Ferulic acid or vanillin was added from sterile stock solutions, as indicated.
Determination of metabolic intermediates.
Excreted intermediates of ferulic acid metabolism were analyzed by high-performance liquid chromatography (HPLC), using an UltiMate 3000 HPLC system (Dionex, Idstein, Germany) without prior extraction. Culture supernatants were obtained after centrifugation (5 min at 16,000 × g), and catalytic reactions were stopped by the acidification of the withdrawn samples to pH 2 by the addition of trichloroacetic acid to a concentration of 6.1 mM. Intermediates were separated by reverse-phase chromatography using an Acclaim 120 C8 column (particle size of 5 μm and column size of 250 by 2.1 mm) with a gradient of 0.1% (vol/vol) formic acid (eluant A) and acetonitrile (eluant B) in a range from 26 to 100% (vol/vol) eluant B. The run started with a flow rate of 0.1 ml/min, which was raised to 0.3 ml/min after 16 min when eluant B reached 100%. For quantification, all intermediates were calibrated with external standards. The compounds were identified by their retention times, and their absorptions at a specific wavelength were determined by using an MWD-3000 multiple-wavelength detector (259 nm, 280 nm, 285 nm, and 340 nm; Dionex, Idstein, Germany). Data analyses and quantifications were performed with the associated Chromeleon 6.8 Chromatography Data Systems software (Dionex, Idstein, Germany).
DNA isolation and manipulation.
Genomic DNA of Amycolatopsis sp. ATCC 39116 was isolated with the DNeasy blood and tissue kit (Qiagen GmbH, Hilden, Germany). Plasmid DNA was isolated from E. coli by using peqGOLD Plasmid MiniPrep kit I (PeqLab GmbH, Erlangen, Germany). Plasmid isolation from Amycolatopsis sp. ATCC 39116 was performed by using the Qiagen plasmid purification minikit (Qiagen GmbH, Hilden, Germany). DNA was digested with restriction endonucleases (Fermentas GmbH, St. Leon-Rot, Germany) under the conditions described by the manufacturer. Phusion high-fidelity DNA polymerase and other DNA-manipulating enzymes (Fermentas GmbH, St. Leon-Rot, Germany) were used according to the instructions provided by the manufacturer. Oligonucleotides were purchased from Eurofins MWG Synthesis GmbH (Ebersberg, Germany). DNA fragments were isolated from agarose gels or reaction mixtures by using the peqGOLD gel extraction kit (PeqLab GmbH, Erlangen, Germany).
Genome sequencing, gene prediction, and annotation.
Sequencing of the whole genome of Amycolatopsis sp. ATCC 39116, prediction of open reading frames, and automatic annotation were performed as described previously by Hiessl et al. (21). Homologues of putative VDHs were identified by using BLAST analyses, and the corresponding genes were ranked based on their homology to previously characterized VDHs.
Transfer of DNA.
Plasmids were transferred into E. coli by employing the CaCl2 method (19), whereas the transfer of plasmids and linear DNA fragments into Amycolatopsis sp. ATCC 39116 was performed by direct mycelial transformation, as described previously by Priefert et al. (22).
Plasmid and mutant construction.
The vdh gene of Amycolatopsis sp. ATCC 39116 was amplified with its own ribosomal binding site (RBS) via PCR from total chromosomal DNA by using Phusion high-fidelity polymerase and oligonucleotides vdh_for_RBS_NdeI and vdh_rev_XhoI (see Table S1 in the supplemental material). For an additional C-terminal His tag, vdh was also amplified without its native stop codon by using primer vdh_for_RBS_NdeI and primer vdh_rev_nostop_XhoI (see Table S1 in the supplemental material) as the reverse primer. Both fragments were digested with NdeI/XhoI and ligated with the NdeI/XhoI-linearized vector pET23a(+). After the transformation of E. coli Mach-1 T1, the resulting plasmids, pet23::vdh and pet23::vdhHis, were sequenced and subsequently transferred into the expression host E. coli BL21(DE3).
A precise deletion of vdh was accomplished via homologous recombination by replacing the native gene with a kanamycin resistance cassette. For this purpose, the flanking regions upstream and downstream of vdh in the genome were amplified by using the following oligonucleotides (see Table S1 in the supplemental material): vdhLF_for3 and vdhLF_rev_KmR for the upstream region and vdhRF_for_PromKmR and vdhRF_rev3 for the downstream region. Additionally, the kanamycin resistance cassette of pRLE6 (22) was amplified by using primers KmR_rev_vdhLF and PromKmR_for_vdhRF. The resulting fragments were purified and combined in a subsequent fusion PCR by using primers vdhLF_for3 and vdhRF_rev3. The resulting fragment, vdhLF_KmR_vdhRF, combined the upstream and downstream regions of vdh with the kanamycin resistance cassette and was transferred into Amycolatopsis sp. ATCC 39116, as described above. Transformants of Amycolatopsis sp. ATCC 39116 were selected on solid Caso medium containing 100 μg/ml kanamycin. Mutants were screened by colony PCR, and gene replacement was finally analyzed via diagnostic PCR with different primer combinations, showing a specific integration of the kanamycin resistance cassette into the vdh locus. Furthermore, the resulting PCR fragments were verified via digestion and sequencing.
Preparation of crude cell extracts and soluble fractions.
Cell pellets of E. coli were resuspended in 3 to 4 ml of 100 mM potassium phosphate buffer (pH 7.1). Crude extracts were obtained by three passages through a French pressure cell (120 MPa). For the preparation of soluble fractions, cell debris was removed by centrifugation for 60 min at 20,000 × g at 4°C. For subsequent enzyme assays, samples were stored on ice, while samples for polyacrylamide gel electrophoresis were stored at −20°C. His6-tagged proteins were purified by using His SpinTrap columns (GE Healthcare Europe GmbH, Freiburg, Germany), according to the manufacturer's instructions.
Polyacrylamide gel electrophoresis and Western blot analysis.
Samples from crude cell extracts and soluble fractions were analyzed for their protein contents according to the method of Bradford (23). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed by using 12.5% (wt/vol) gels, as described previously by Laemmli (24). Amounts of proteins were added as indicated. The staining of proteins was performed with Serva Blue G (25). Molecular mass reference proteins were purchased from GE Healthcare Europe GmbH (Freiburg, Germany), including low-molecular-mass SDS markers phosphorylase b, rabbit muscle (97,000 kDa); bovine serum albumin (66,000 kDa); ovalbumin, chicken egg white (45,000 kDa); carbonic anhydrase, bovine erythrocyte (30,000 kDa); trypsin inhibitor, soybean (20,100 kDa); and α-lactalbumin, bovine milk (14,400 kDa).
Proteins were blotted from SDS-polyacrylamide gels onto a Hybond-P polyvinylidene difluoride (PVDF) transfer membrane (GE Healthcare Europe GmbH, Freiburg, Germany), according to methods described previously by Towbin et al. (26), by using a PeqLab Western blotting semidry Sedec M apparatus (PeqLab GmbH, Erlangen, Germany). The transfer of proteins was accomplished for 120 min at a voltage of 14 V and a constant current of 5 mA/cm2.
The immunodetection of His6-tagged proteins was performed with a primary mouse anti-histidine tag antibody (AbD Serotec, Düsseldorf, Germany) and a secondary AP-goat anti-mouse IgG antibody (Zymed, Karlsruhe, Germany). Membranes with blotted proteins were incubated in skim milk (with 2.5% [wt/vol] skim milk in Tris-buffered saline [TBS] [2.42 g Tris-HCl {pH 7.6}, 8 g NaCl, 0.05% {wt/vol} Tween 20] in a total volume of 1 liter) at room temperature to block nonspecific binding. After washing with TBS, the membrane was incubated with the primary antibody (1:2,000 dilution in TBS-Tween [0.1%] [200 μl/cm2 membrane]) overnight at room temperature. After 3-fold washing with TBS for 10 min, the membrane was incubated with the secondary antibody (1:10,000 dilution in TBS-Tween [0.1%] [200 μl/cm2]membrane) for 2 h at room temperature. After a final 3-fold wash with TBS, as described above, the antigen-antibody complexes were visualized by using Western Blue stabilized substrate for AP (Promega GmbH, Mannheim, Germany).
VDH enzyme assay.
VDH enzyme activity was assayed according to methods described previously by Gasson et al. (27). The reaction mixture contained, in a total volume of 1 ml, 0.1 mM potassium phosphate buffer (pH 7.1), 0.5 mM NAD+, 1.2 mM pyruvate (sodium salt), lactate dehydrogenase (rabbit muscle) (1 U), 500 μg of the enzyme preparation of interest, and 0.125 mM vanillin, benzaldehyde, coniferylaldehyde, or cinnamaldehyde, respectively. The oxidation of the substrates was measured at 30°C and at a suitable wavelength: vanillin at 340 nm (ϵ = 11.492 cm2/μmol), cinnamaldehyde at 340 nm (ϵ = 0.588 cm2/μmol), benzaldehyde at 293 nm (ϵ = 0.339 cm2/μmol), and coniferylaldehyde at 410 nm (ϵ = 3.452 cm2/μmol). Enzyme activity is stated in units. One unit is defined as the amount of enzyme which converts 1 μmol vanillin per min.
The temperature and pH optimum of VDHATCC 39116 were determined in a discontinuous assay by measuring vanillic acid formation using HPLC, since the spectrophotometric assay is additionally dependent on the activity of lactate dehydrogenase. The reaction mixture contained, in a total volume of 1 ml, 0.1 mM potassium phosphate buffer (pH 7.1), 2 mM NAD+, 500 μg of the enzyme preparation of interest, and 1.25 mM vanillin. The reactions were started by the addition of NAD+ to the mixture, and the mixture was incubated for 15 min before the reaction was stopped by heating to 85°C for 5 min. To test the temperature preference, the assay was carried out at 15°C to 65°C in steps of 5°C. After determining the optimum temperature to be between 40°C and 45°C, the temperature steps were refined to 1°C steps. The effects of different pH values (pH 5.0 to 9.5 in 0.5 steps) were observed at 30°C by using Britton-Robinson buffer consisting of 40 mM boric acid, 40 mM acetic acid, and 40 mM phosphoric acid and titrated to the desired pH with 200 mM NaOH.
Nucleotide sequence accession numbers.
The genome sequence of Amycolatopsis sp. ATCC 39116 was recently deposited in GenBank under accession no. AFWY00000000 (16). Additionally, the nucleotide sequence of vdh is provided within patent application PCT/EP2012/061600 (C. Fleige, G. Hansen, J. Kroll, J. M. Hilmer, S. Lambrecht, M. Pesaro, and A. Steinbüchel, 18 June 2012, European Union patent application PCT/EP2012/061600). Moreover, all five tested gene candidates are available in GenBank under accession no. JX292129 (vdh), JX292130, JX292131, JX292132, and JX292133.
RESULTS
In silico analyses of the genome and identification of the Amycolatopsis sp. ATCC 39116 vanillin dehydrogenase (VDHATCC 39116).
Based on the genome sequence of Amycolatopsis sp. ATCC 39116 and an automatic annotation, five genes coding for putative aldehyde dehydrogenases were identified. After the heterologous expression of these gene candidates in E. coli, only one of them led to vanillin dehydrogenase activity and might therefore be involved in the catabolism of vanillin. The gene, designated vdh, consists of 1,461 bp and encodes a protein of 486 amino acids with a theoretical molecular mass of 50.6 kDa. A comparison of the deduced amino acid sequence of VDHATCC 39116 with internet-based sequence data using BLASTP 2.2.25 (28, 29) showed homology to different (benz)aldehyde dehydrogenases from various sources. The highest percent homology (73%) was determined for an aldehyde dehydrogenase of the Nocardioidaceae bacterium Broad-1 (GenBank accession no. ZP_08197264). By using ClustalX 1.83 (30), a multiple-sequence alignment (MSA) with various representative aldehyde dehydrogenases was done for assessing their relationship to VDHATCC 39116 in a dendrogram (Fig. 1). Additionally, MSA using BioEdit Sequence Alignment Editor 7.0.5.3 (30) was performed to demonstrate the similarity at the amino acid level of VDHATCC 39116 to different enzymes with already verified vanillin dehydrogenase activity (see Fig. S1 in the supplemental material). Further in silico analysis using ScanProsite (Expert Protein Analysis System [ExPASy] [31]) revealed a characteristic pattern of aldehyde dehydrogenases within amino acids 251 to 258 including glutamic acid in the catalytic center.
Fig 1.
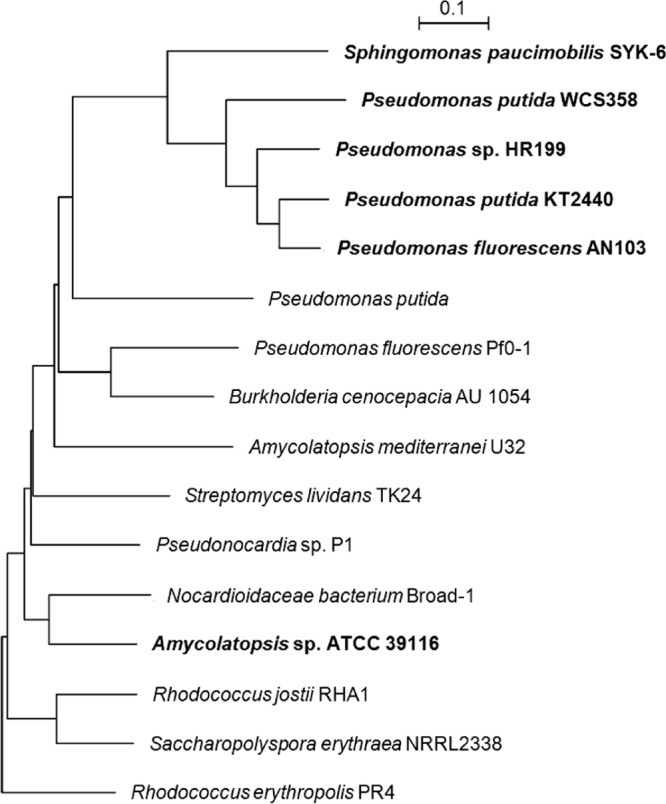
Phylogenetic tree of the putative VDH of Amycolatopsis sp. ATCC 39116 and related enzymes with aldehyde dehydrogenase or VDH activity. Multiple-sequence alignment was done by using ClustalX 1.83 based on the amino acid sequences of the following enzymes: putative VDHATCC 39116 of Amycolatopsis sp. ATCC 39116, VDH of Pseudomonas sp. strain HR199 (35), VDH of Pseudomonas fluorescens AN103 (27), VDH of P. putida WCS358 (36), VDH of P. putida KT2440 (41), VDH of Sphingomonas paucimobilis SYK-6 (42), benzaldehyde dehydrogenase (NAD+) of Nocardioidaceae bacterium Broad-1 (GenBank accession no. ZP_08197264), benzaldehyde dehydrogenase of Rhodococcus erythropolis PR4 (43), benzaldehyde dehydrogenase (NAD+) of Saccharopolyspora erythraea NRRL2338 (38), benzaldehyde dehydrogenase (NAD+) of Rhodococcus jostii RHA1 (44), aldehyde dehydrogenase of Streptomyces lividans TK24 (accession no. ZP_06526522), benzaldehyde dehydrogenase (NAD+) of Burkholderia cenocepacia AU 1054 (accession no. YP_621190), benzaldehyde dehydrogenase (NAD+) of Pseudomonas fluorescens Pf0-1 (45), benzaldehyde dehydrogenase of Pseudomonas putida (46), aldehyde dehydrogenase of Pseudonocardia sp. strain P1 (accession no. ZP_08121488), and NAD+-dependent aldehyde/benzaldehyde dehydrogenase of Amycolatopsis mediterranei U32 (47). Calculations were performed by using the neighbor-joining method (40). Enzymes with verified vanillin dehydrogenase activity are shown in boldface type.
Characterization of VDHATCC 39116.
Besides the in silico analyses, the specific vanillin dehydrogenase activity in cells of Amycolatopsis sp. ATCC 39116 was investigated after cultivating the strain with or without vanillin and structurally related compounds like ferulic acid as the carbon source, since previous investigations with Pseudomonas showed an inducing effect (27, 32). Amycolatopsis sp. ATCC 39116 was cultivated in mineral salts medium containing 0.5% (wt/vol) gluconate for 32 h at 42°C. In addition, 0.1% (wt/vol) vanillin or ferulic acid was added to one of the cultures. The cells were harvested after cultivation, soluble fractions were prepared, and the specific activity of vanillin dehydrogenase was measured photometrically, as described in Materials and Methods. The activity assays revealed specific vanillin dehydrogenase activities of 25 ± 2 U/g protein if cells were previously cultivated with vanillin as the carbon source. In cells that were cultivated with gluconate as the sole carbon source, no VDHATCC 39116 activity was detectable. The enhancing effect was also obvious during SDS-PAGE analysis of the soluble fractions, which revealed a prominent protein band at a molecular mass of approximately 54 kDa in the case of the vanillin-induced cells. A corresponding band was missing in samples of cells which were not incubated with vanillin. Comparable results were observed when cells were cultivated with ferulic acid as an inducer (data not shown).
After demonstrating the activity of a vanillin dehydrogenase in cells of Amycolatopsis sp. ATCC 39116 grown with vanillin and after identifying a putatively responsible gene, vdh was cloned and heterologously expressed in E. coli. For this, the gene was inserted into the vector pET23a(+) and expressed in E. coli BL21(DE3) cells under the control of the strong T7 promoter. In order to purify the heterologous enzyme for further investigations, vdh was also expressed without its native stop codon to append a C-terminal His6 tag provided by the vector pET23a(+). SDS-PAGE and subsequent Western blot analysis were carried out to verify the correct gene expression and purification of VDHATCC 39116 with the His6 tag (Fig. 2).
Fig 2.

Documentation of the purification of His6-tagged VDHATCC 39116 by SDS-PAGE (A) and immunodetection of the His6 tag (B). The samples were separated in two identically prepared 12.5% (wt/vol) SDS-polyacrylamide gels. The first gel was stained with Serva Blue G (A), while the proteins of the second gel were transferred onto a PVDF membrane, and the His6 tag was immunodetected as described in Materials and Methods (B). Lanes: 1 and 8, 8 μl molecular mass marker; 2, 25 μg of protein from the crude extract of E. coli BL21(DE3)/pET23a; 3, 25 μg of protein from the crude extract of E. coli BL21(DE3)/pET23a::vdhHis; 4, 25 μg of protein from the soluble fraction of E. coli BL21(DE3)/pET23a::vdhHis; 5, 25 μg of protein from the flowthrough of the purification; 6, 10 μg of protein from the first wash step; 7, 10 μg of protein from the eluate.
Protein analyses by SDS-PAGE and Western blotting revealed the presence of an appropriate protein with an apparent molecular mass of about 54 kDa. The purification of VDHHis using His SpinTrap columns yielded an almost homogenous protein (Fig. 2, lane 7). Immunodetection of the C-terminal His6 tag additionally confirmed the assumption that the occurring protein signal represents heterologously expressed VDHHis. Further analysis of the vanillin dehydrogenase activity within the eluate showed a specific activity of 72 ± 2 U/g protein, which was remarkably low due to the loss of activity during purification, which we could not overcome, although we changed the compositions of all buffers used. No enzyme activity was observed in the absence of the cofactor NAD+. Furthermore, no oxidation of vanillin was detectable when NADP+ was used as a cofactor (data not shown).
The optimal conditions for the oxidation of vanillin were determined by measuring the vanillic acid formed using cell extracts of the recombinant vdh-expressing E. coli strain. Maximum activities were found at pH 8.0 and at a temperature of 44°C. The enzyme was active at a broad pH range, from pH 5.0 to 9.0, and was able to catalyze the reaction at temperatures of between 15°C and 65°C. After storage at 4°C for 24 h, VDHATCC 39116 exhibited 86% of its former activity.
The most specific substrate was found to be vanillin, but VDHATCC 39116 also catalyzed the oxidation of coniferylaldehyde, benzaldehyde, and cinnamaldehyde, with specific activities of up to 66% (benzaldehyde) in comparison to vanillin.
In a next step, vanillin biotransformation experiments using the recombinant E. coli strain expressing VDHATCC 39116 were performed. The cells were cultivated until they reached the stationary growth phase and were then harvested and washed twice with potassium phosphate buffer. After the cells were suspended in 50 ml mineral salts medium, 14 mM vanillin was added, and the formation of vanillic acid was observed by using HPLC analysis of culture supernatants (Fig. 3). As a control experiment, E. coli BL21(DE3) cells harboring the plain vector pET23a were incubated under identical conditions.
Fig 3.
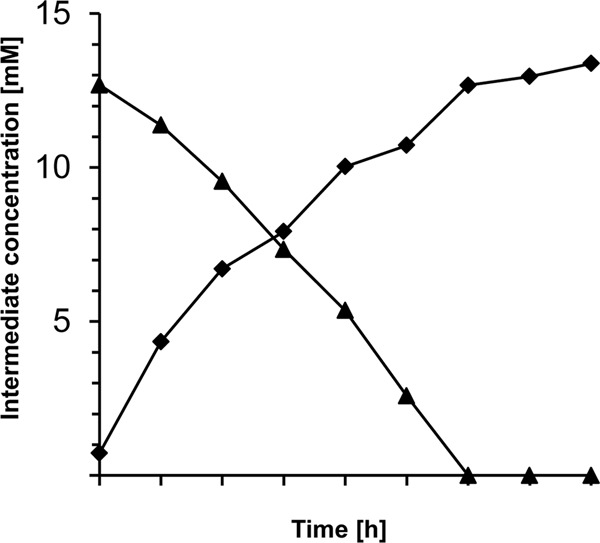
Biotransformation of vanillin by E. coli BL21(DE3)/pET23a::vdh. Cells were grown in LB with 100 μg/ml ampicillin at 30°C and harvested after 16 h in the stationary growth phase. After two wash steps with 100 mM potassium phosphate buffer (pH 7.5), cells were resuspended in 50 ml mineral salts medium containing 100 μg/ml ampicillin and 14 mM vanillin. The cultures were incubated at 30°C and at 100 rpm for 8 h, and the biotransformation of vanillin to vanillic acid was observed at the given times by using HPLC analyses, as described in Materials and Methods. ▲, vanillin; ♦, vanillic acid.
The biotransformation experiments demonstrated that the VDHATCC 39116-expressing strain is able to metabolize vanillin to vanillic acid (Fig. 3). Vanillic acid was synthesized in equimolar amounts immediately after the addition of vanillin, while transformation could not be observed in the control experiment (data not shown).
After identifying the responsible gene and allocating vanillin dehydrogenase activity to VDHATCC 39116, vdh was additionally overexpressed episomally in Amycolatopsis sp. ATCC 39116 by using a derivative of the Amycolatopsis-E. coli shuttle vector pRLE6 (22): homologous expression led to a 3.7-fold increase of the VDHATCC 39116 activity in comparison to that of the wild type.
Construction of a vdh deletion mutant of Amycolatopsis sp. ATCC 39116.
In order to investigate the impact of VDHATCC 39116 on the catabolism of vanillin in Amycolatopsis sp. ATCC 39116, a precise deletion mutant was generated by replacing the native gene with a kanamycin resistance cassette. For this, the flanking regions located upstream and downstream of vdh in the genome were amplified and combined with the resistance gene in a fusion PCR. Amycolatopsis sp. ATCC 39116 was transformed with the resulting linear fragment, and the gene replacement in the resulting Amycolatopsis sp. ATCC 39116 Δvdh::Kmr mutant was confirmed by diagnostic PCR. In a first step, the integration of the deletion construct into the genome was demonstrated by amplifying a 3.36-kb fragment (Fig. 4, lane 2). This fragment distinguishes size from the genomic organization of the wild type as a consequence of the smaller size of the resistance gene than of vdh. A second PCR with two different primer combinations verified the specific integration of the deletion fragment into the vdh locus by amplifying segments which started upstream (Fig. 4, lane 3) or downstream (Fig. 4, lane 4), respectively, of the flanking regions used. Finally, the amplified fragments were sequenced to ensure the correct replacement of vdh with the kanamycin resistance gene.
Fig 4.
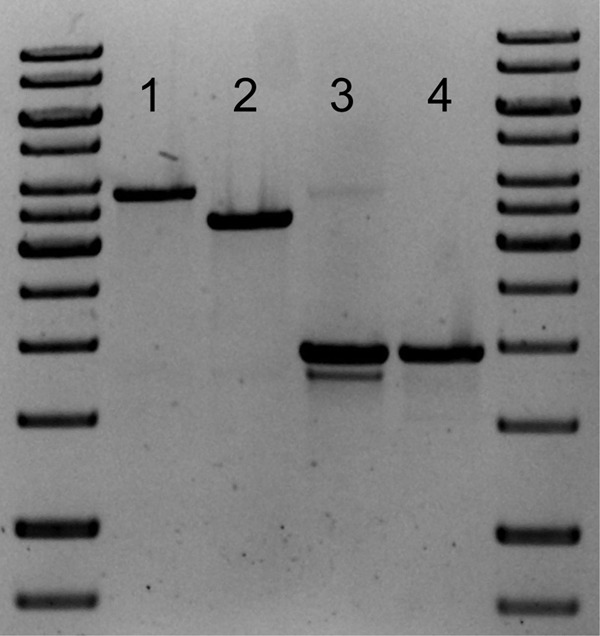
Verification of gene replacement in the Amycolatopsis sp. ATCC 39116 Δvdh::Kmr mutant. In order to verify the specific gene replacement of the vdh gene by the kanamycin resistance cassette, different diagnostic PCRs were carried out, using chromosomal DNA as the template. The resulting fragments were analyzed in a 1% agarose gel. The molecular weight marker used was a GeneRuler 1-kb DNA ladder (Fermentas GmbH, St. Leon-Rot, Germany). Lanes 1 and 2 represent the amplified product using oligonucleotides vdhLF_for_diag and vdhRF_rev2, showing the difference between wild-type Amycolatopsis sp. ATCC 39116 (lane 1) and the Amycolatopsis sp. ATCC 39116 Δvdh::Kmr mutant (lane 2). Lanes 3 and 4 present the results using primers vdhLF_for_diag and pRLE6for2 (lane 3) as well as primers KmR_rev_vdhLF and vdhRF_rev2 (lane 4) and show proof of the specific integration of the kanamycin resistance cassette into the vdh locus for the Amycolatopsis sp. ATCC 39116 Δvdh::Kmr mutant.
Characterization of the Amycolatopsis sp. ATCC 39116 Δvdh::Kmr mutant.
The vdh deletion mutant was characterized to determine the impact of the enzyme on the catabolism of vanillin in Amycolatopsis sp. ATCC 39116. For this purpose, biotransformation experiments on a 2-liter-bioreactor scale were carried out to analyze the utilization of ferulic acid. Ferulic acid is catabolized to vanillin and subsequently to vanillic acid and is therefore a potential substrate for the biotechnical production of vanillin. The difference between the vdh deletion mutant and wild-type Amycolatopsis sp. ATCC 39116 in terms of ferulic acid utilization was demonstrated for “resting cells,” independently from growth. The cells were grown in Caso medium at 42°C until they reached the stationary growth phase. Afterwards, they were harvested, washed twice with PBS, and resuspended in the same buffer (1,000 ml [pH 7.5]; 0.5 g/liter cell dry weight). Ferulic acid was added at an initial concentration of 2.0 mM (stock concentration of 210 mM NaOH [pH 8]) to the cell suspension, and the occurring intermediates were analyzed by HPLC. During the transformation process, ferulic acid was fed continuously at a rate of 0.75 mM/h. In order to promote continuing vanillin synthesis, the feeding rate was readjusted to 3.1 mM/h for 3 h, after the cells had adapted to the conversion conditions and the substrate concentration was decreasing (7 h from the start). Accordingly, feeding was stopped after 10 h, and the disappearance of ferulic acid and the occurrence of the products were observed for an additional 20 h.
The biotransformation experiments revealed a significant difference between wild-type Amycolatopsis sp. ATCC 39116 and the vdh deletion mutant with regard to the occurrence of catabolism products (Fig. 5). While Amycolatopsis sp. ATCC 39116 started to accumulate vanillic acid at first, the Amycolatopsis sp. ATCC 39116 Δvdh::Kmr mutant accumulated vanillin as the first overflow product. Concerning the wild-type experiment, vanillin was not detectable in the supernatants until vanillic acid was formed at a concentration of about 1 mM. Additional vanillic acid was further synthesized from vanillin, which was meanwhile formed. The latter was completely catabolized after ferulic acid was no longer available for the cells. In contrast, only a very low concentration of vanillic acid was formed by the cells of the mutant strain, while cultures of these cells accumulated vanillin continuously up to 6.8 mM (wild type, 2.9 mM), as long as ferulic acid was present in the mixture used in the experiment. Furthermore, in contrast to the wild type, the degradation of the synthesized vanillin occurred only at a very low rate, even during the last 10 h, when ferulic acid was completely degraded. Further catabolism intermediates like guaiacol were detected only in trace amounts in cultures of the wild type.
Fig 5.

Biotransformation of ferulic acid by Amycolatopsis sp. ATCC 39116 and the Amycolatopsis sp. ATCC 39116 Δvdh::Kmr mutant. Cells were grown in 250 ml Caso medium at 42°C and harvested during the stationary growth phase. After two wash steps with phosphate-buffered saline (pH 7.5), cells were resuspended in 1,000 ml of the same buffer containing 2 mM ferulic acid. The cultures were incubated in a 2-liter bioreactor at 42°C for 30 h. The biotransformation of ferulic acid (A) to vanillin (B) and vanillic acid (C) was observed at the given times by using HPLC analyses, as described in Materials and Methods. Initially, ferulic acid was fed at a rate of 0.75 mM/h during the first 7 h. Afterwards, substrate was fed for 3 h at a rate of 3.1 mM/h, before feeding was stopped. ▲, Amycolatopsis sp. ATCC 39116; ●, Amycolatopsis sp. ATCC 39116 Δvdh::Kmr mutant.
Analyses of growth on vanillin and ferulic acid in mineral salts medium with 0.1% (wt/vol) substrates as the sole source of carbon were also carried out. While the wild type exhibited slow growth on both substrates after a long lag phase of about 30 to 40 h, the mutant did not grow at all on vanillin. During the experiment, no utilization of vanillin was detectable for the mutant strain. On ferulic acid, Amycolatopsis sp. ATCC 39116 Δvdh::Kmr cells showed a significantly reduced growth rate compared to that of the wild type, indicating that VDHATCC 39116 has a substantial impact on the metabolism of ferulic acid. However, data from the experiment suggest that ferulic acid can serve as a source of carbon and energy despite the knockout. VDH activities of both the wild-type strain and the mutant were observed subsequently. While the wild type exhibited dehydrogenase activity after growth with ferulic acid or vanillin, as previously observed, no VDH activity was detected in the mutant strain despite induction.
DISCUSSION
The actinomycete Amycolatopsis sp. ATCC 39116 is capable of synthesizing large amounts of vanillin from the natural substrate ferulic acid (12). This bacterium therefore represents the most promising platform for the biotechnical production of natural vanillin. However, significant amounts of the desired product are lost because of the inherent vanillin catabolism via vanillic acid (15). The availability of the genome sequence of Amycolatopsis sp. ATCC 39116 contributed to our understanding of vanillin catabolism in this strain. Furthermore, it offered the possibility of the identification of responsible genes as a target for the metabolic engineering of this production strain. Systematic gene deletions have been intensively used for strain development in various production processes. In order to improve industrial applications that approach the theoretical product yields, catabolic or competing pathways of the desired product were disrupted. Prominent examples are the production of amino acids, ethanol, and succinate by Corynebacterium glutamicum, Saccharomyces cerevisiae, and E. coli, respectively (33, 34).
For several vanillin-degrading bacteria, a vanillin dehydrogenase was found to catalyze the oxidation of vanillin to vanillic acid (8, 18, 27, 35, 36). The activity of a vanillin dehydrogenase could now be demonstrated for Amycolatopsis sp. ATCC 39116 as well. Moreover, the expression of the corresponding gene seems to be inducible by the enzyme's substrate and structurally related compounds, such as ferulic acid. As shown previously for Pseudomonas fluorescens AN103 (27, 37), enzyme activity could not be observed for cells cultivated in the absence of these substance.
Based on the genome sequence of Amycolatopsis sp. ATCC 39116 and its annotation, a set of putatively responsible genes showing homology to previously characterized enzymes with aldehyde dehydrogenase activity in other organisms was identified. Selected gene candidates were heterologously expressed in E. coli, and VDH activity was monitored. One of the encoded enzymes was capable of catalyzing the oxidation of vanillin to vanillic acid and thus was designated VDHATCC 39116. The enzyme exhibited homology to several aldehyde dehydrogenases also including a characteristic motif with glutamic acid as a central amino acid of the catalytic center (see Fig. S1 in the supplemental material). Interestingly, the protein does not show a remarkably high level of homology to the group of enzymes with already verified vanillin dehydrogenase activity (Fig. 1). However, all proteins characterized up to now were derived from a phylogenetically closely related group of bacteria. VDHATCC 39116 represents the first vanillin dehydrogenase that was characterized in detail for actinobacteria and might therefore represent an independently evolved protein. This is confirmed by the observation that the encoding gene vdh seems not to be part of a cluster, as previously described for other studied strains (8, 16, 35, 36, 38).
Upon the heterologous expression of the encoding gene vdh in E. coli, a protein signal with an apparent molecular mass of 54.0 ± 2.0 kDa was obtained by SDS-PAGE analysis. This is in agreement with both the calculated molecular mass of the vdh translational product and the sizes of other enzymes exhibiting VDH activity (18, 32, 35, 36). The recombinant protein carrying a His6 tag was purified to apparent homogeneity, which was confirmed by immunodetection. VDHATCC 39116 activity was detected in the eluted protein fraction, verifying the identity of the vanillin dehydrogenase of Amycolatopsis sp. ATCC 39116. In line with this, VDH activity was detectable only in the presence of the cofactor NAD+, indicating an NAD+-dependent aldehyde dehydrogenase, as proposed previously based on the theoretical analysis of the amino acid sequence. In contrast, previous studies stated that the catabolism of vanillin in Amycolatopsis sp. ATCC 39116 is catalyzed only by an oxidase (15). NADP+ could not be used as a cofactor. A preference toward NAD+ was also reported previously for other vanillin dehydrogenases studied (18, 27). Moreover, the VDH activity of Amycolatopsis sp. ATCC 39116 could be enhanced by the supplementary episomal expression of vdh, which thereby validates the enzyme function. VDHATCC 39116 was shown to catalyze the oxidation of different aromatic aldehydes but has a preference toward vanillin, as previously shown for Sphingomonas paucimobilis SYK-6 (18). The enzyme was active under a broad range of conditions, including different pHs and temperatures, and showed its maximum activity at pH 8.0 and at a temperature of 44°C. It exhibited considerable storage stability, since 86% of its former activity could be determined after 24 h at 4°C.
Based on the results of previous studies (14, 15, 39) and in agreement with the results obtained in this study, we propose for Amycolatopsis sp. ATCC 39116 the following coenzyme A (CoA)-dependent pathway for the degradation of ferulic acid via vanillin and vanillic acid to protocatechuic acid or guaiacol with VDHATCC 39116 as a key enzyme (Fig. 6).
Fig 6.

Proposed pathway of the CoA-dependent catabolism of ferulic acid via vanillin in Amycolatopsis sp. ATCC 39116.
Following the identification of the VDHATCC 39116-encoding gene, the physiological impact of VDHATCC 39116 on the catabolism of vanillin and related compounds, such as ferulic acid, was determined. Therefore, a precise vdh deletion mutant was generated, and the catabolism of ferulic acid by the mutant was investigated and compared to that of wild-type Amycolatopsis sp. ATCC 39116. A “resting cell” approach was employed, and the occurring catabolism intermediates, including vanillin and vanillic acid, were observed by HPLC.
Both the wild type and the Amycolatopsis sp. ATCC 39116 Δvdh::Kmr mutant degrade ferulic acid in comparable manners (Fig. 5A). In contrast to data from previous studies, no negative effect regarding the catabolism of ferulic acid was observed (7, 8, 37). The authors of those studies assumed polar effects, as a consequence of the vdh deletion or disruption, to be responsible for the resulting phenotype. Since the vdh gene of Amycolatopsis sp. ATCC 39116, unlike in other studied strains, is not part of a cluster with other ferulic acid catabolism genes, a similar effect was not to be expected for the present strain (8, 16, 35, 36, 38). A remarkable difference, however, was observed for the synthesis of vanillin and vanillic acid (Fig. 5B and C). The vdh deletion mutant showed 2.3-times-enhanced vanillin accumulation, which started immediately after the exposure of the cells to the substrate. A final concentration of 6.8 mM was reached, and the vanillin excreted in the culture supernatants was degraded only very slowly, even after ferulic acid was completely metabolized. The wild type synthesized and excreted vanillin only up to 2.9 mM but only after an initial accumulation of vanillic acid up to 1 mM had occurred. This observation was also made previously for wild-type Streptomyces sp. strain ATCC 39116 by Muheim and Lerch (39). Since the accumulation of vanillin by the deletion mutant started independently from vanillic acid, a regulatory effect, which influences the expression of vdh or the activity of the enzyme in the wild type, depending on the vanillic acid concentration, could be a possible explanation. Moreover, as a result of the decreasing ferulic acid concentration, vanillin was subsequently degraded by the wild-type cells. In line with this, further catabolism intermediates like guaiacol or protocatechuic acid were formed in detectable amounts only in cultures of the wild type.
Growth analysis of the mutant strain revealed, as previously described for other knockout mutants (18, 37), that the bacteria are no longer able to use vanillin as the sole source of carbon and energy. In agreement with this, the subsequent enzyme assay showed that in contrast to the wild type, no oxidation of vanillin was detectable under the conditions used. This observation is in accordance with the fact that no other tested homologue of VDHATCC 39116 showed the desired enzyme activity. However, an alternative minor pathway of vanillin oxidation might be present, since a slow degradation of vanillin was observable in the biotransformation experiments despite the knockout.
These experiments clearly showed that VDHATCC 39116 plays a significant role in the course of ferulic acid degradation in Amycolatopsis sp. ATCC 39116. Therefore, the deletion mutant represents a promising production strain to compete with the already established vanillin production processes using wild-type strains (12, 40).
Further metabolic engineering will improve the outcome already obtained since a slow degradation of vanillin, probably due to unspecific aldehyde dehydrogenase or oxidase activities (15), occurred even for the Amycolatopsis sp. ATCC 39116 Δvdh::Kmr mutant. Similar observations were also made previously for other different vanillin production strains in which VDH-encoding genes were deleted or disrupted (6–8, 37). In addition, ferulic acid could be completely degraded to serve as source of energy through competing pathways, such as decarboxylation to 4-vinylguaiacol (2). This could be the reason why vanillin and vanillic acid were not produced in equimolar amounts in the biotransformation experiments (42% molar yield produced by the mutant and 19% by the wild type). The optimization of the fermentation process is therefore aimed at an improved vanillin synthesis rate and at a higher molar yield of vanillin with regard to ferulic acid.
Based on the present study, a strain for the highly efficient microbial production of natural vanillin from ferulic acid can now be developed. The complete prevention of the catabolism of vanillin during biotransformation and the optimization of the fermentation process will significantly improve the production of natural vanillin using the Amycolatopsis sp. ATCC 39116 Δvdh::Kmr mutant.
Supplementary Material
ACKNOWLEDGMENTS
The project was financially supported by SYMRISE AG, Holzminden, Germany, which we gratefully acknowledge.
We thank Isabelle van Hamme for technical assistance.
Footnotes
Published ahead of print 12 October 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02358-12.
REFERENCES
- 1. Converti A, Aliakbarian B, Domínguez JM, Vázquez GB, Perego P. 2010. Microbial production of biovanillin. Braz. J. Microbiol. 41:519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Priefert H, Rabenhorst J, Steinbüchel A. 2001. Biotechnological production of vanillin. Appl. Microbiol. Biotechnol. 56:296–314 [DOI] [PubMed] [Google Scholar]
- 3. European Commission 2008. European Commission regulation no 1334/2008 of the European Parliament and of the Council of 16 December 2008 on flavourings and certain food ingredients with flavouring properties for use in and on foods and amending regulation (EC) no 1601/91 of the Council, regulations (EC) no 2232/96 and (EC) no 110/2008 and directive 2000/13/EC. [Google Scholar]
- 4. Hansen EH, Møller BL, Kock GR, Bünner CM, Kristensen C, Jensen OR, Okkels FT, Olsen CE, Motawia MS, Hansen J. 2009. De novo biosynthesis of vanillin in fission yeast (Schizosaccharomyces pombe) and baker's yeast (Saccharomyces cerevisiae). Appl. Environ. Microbiol. 75:2765–2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Narbad A, Gasson MJ. 1998. Metabolism of ferulic acid via vanillin using a novel CoA-dependent pathway in a newly-isolated strain of Pseudomonas fluorescens. Microbiology 144:1397–1405 [DOI] [PubMed] [Google Scholar]
- 6. Overhage J, Priefert H, Steinbüchel A. 1999. Biochemical and genetic analyses of ferulic acid catabolism in Pseudomonas sp. strain HR199. Appl. Environ. Microbiol. 65:4837–4847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Plaggenborg R, Overhage J, Loos A, Archer JAC, Lessard P, Sinskey AJ, Steinbüchel A, Priefert H. 2006. Potential of Rhodococcus strains for biotechnological vanillin production from ferulic acid and eugenol. Appl. Microbiol. Biotechnol. 72:745–755 [DOI] [PubMed] [Google Scholar]
- 8. Plaggenborg R, Overhage J, Steinbüchel A, Priefert H. 2003. Functional analyses of genes involved in the metabolism of ferulic acid in Pseudomonas putida KT2440. Appl. Microbiol. Biotechnol. 61:528–535 [DOI] [PubMed] [Google Scholar]
- 9. Tilay A, Bule M, Annapure U. 2010. Production of biovanillin by one-step biotransformation using fungus Pycnoporous cinnabarinus. J. Agric. Food Chem. 58:4401–4405 [DOI] [PubMed] [Google Scholar]
- 10. Barghini P, Di Gioia D, Fava F, Ruzzi M. 2007. Vanillin production using metabolically engineered Escherichia coli under non-growing conditions. Microb. Cell Fact. 6:13 doi:10.1186/1475-2859-6-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Overhage J, Steinbüchel A, Priefert H. 2003. Highly efficient biotransformation of eugenol to ferulic acid and further conversion to vanillin in recombinant strains of Escherichia coli. Appl. Environ. Microbiol. 69:6569–6576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Muheim A, Müller B, Münch Wetli TM. February 2006. Process for the production of vanillin. European Union patent EP0885968
- 13. Rabenhorst J, Hopp R. November 2002. Process for the preparation of vanillin and suitable microorganisms. European Union patent EP0761817
- 14. Achterholt S, Priefert H, Steinbüchel A. 2000. Identification of Amycolatopsis sp. strain HR167 genes, involved in the bioconversion of ferulic acid to vanillin. Appl. Microbiol. Biotechnol. 54:799–807 [DOI] [PubMed] [Google Scholar]
- 15. Sutherland JB, Crawford DL, Pometto AL. 1983. Metabolism of cinnamic, p-coumaric, and ferulic acids by Streptomyces setonii. Can. J. Microbiol. 29:1253–1257 [DOI] [PubMed] [Google Scholar]
- 16. Davis JR, Goodwin LA, Woyke T, Teshima H, Bruce D, Detter C, Tapia R, Han S, Han J, Pitluck S, Nolan M, Mikhailova N, Land ML, Sello JK. 2012. Genome sequence of Amycolatopsis sp. strain ATCC 39116, a plant biomass-degrading actinomycete. J. Bacteriol. 194:2396–2397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Di Gioia D, Luziatelli F, Negroni A, Ficca AG, Fava F, Ruzzi M. 2010. Metabolic engineering of Pseudomonas fluorescens for the production of vanillin from ferulic acid. J. Biotechnol. 156:309–316 [DOI] [PubMed] [Google Scholar]
- 18. Masai E, Yamamoto Y, Inoue T, Takamura K, Hara H, Kasai D, Katayama Y, Fukuda M. 2007. Characterization of ligV essential for catabolism of vanillin by Sphingomonas paucimobilis SYK-6. Biosci. Biotechnol. Biochem. 71:2487–2492 [DOI] [PubMed] [Google Scholar]
- 19. Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 20. Kroll J, Klinter S, Steinbüchel A. 2011. A novel plasmid addiction system for large-scale production of cyanophycin in Escherichia coli using mineral salts medium. Appl. Microbiol. Biotechnol. 89:593–604 [DOI] [PubMed] [Google Scholar]
- 21. Hiessl S, Schuldes J, Thürmer A, Halbsguth T, Bröker D, Angelov A, Liebl W, Daniel R, Steinbüchel A. 2012. Involvement of two latex-clearing proteins during rubber degradation and insights into the subsequent degradation pathway revealed by the genome sequence of Gordonia polyisoprenivorans strain VH2. Appl. Environ. Microbiol. 78:2874–2887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Priefert H, Achterholt S, Steinbüchel A. 2002. Transformation of the Pseudonocardiaceae Amycolatopsis sp. strain HR167 is highly dependent on the physiological state of the cells. Appl. Microbiol. Biotechnol. 58:454–460 [DOI] [PubMed] [Google Scholar]
- 23. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 24. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 25. Weber K, Osborn M. 1969. The reliability of molecular weight determinations by dodecyl sulfate-polyacrylamide gel electrophoresis. J. Biol. Chem. 244:4406–4412 [PubMed] [Google Scholar]
- 26. Towbin H, Staehelin T, Gordon J. 1992. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. 1979. Biotechnology 24:145–149 [PubMed] [Google Scholar]
- 27. Gasson MJ, Kitamura Y, McLauchlan WR, Narbad A, Parr AJ, Parsons ELH, Payne J, Rhodes MJC, Walton NJ. 1998. Metabolism of ferulic acid to vanillin: a bacterial gene of the enoyl-SCoA hydratase/isomerase superfamily encodes an enzyme for the hydration and cleavage of a hydroxycinnamic acid SCoA thioester. J. Biol. Chem. 273:4163–4170 [DOI] [PubMed] [Google Scholar]
- 28. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Altschul SF, Wootton JC, Gertz EM, Agarwala R, Morgulis A, Schaffer AA, Yu YK. 2005. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 272:5101–5109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41:95–98 [Google Scholar]
- 31. Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. 2003. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31:3784–3788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bare G, Swiatkowski T, Moukil A, Gerday C, Thonart P. 2002. Purification and characterization of a microbial dehydrogenase: a vanillin:NAD(P)+ oxidoreductase. Appl. Biochem. Biotechnol. 98–100:415–428 [DOI] [PubMed] [Google Scholar]
- 33. Becker J, Wittmann C. 13 January 2012. Systems and synthetic metabolic engineering for amino acid production—the heartbeat of industrial strain development. Curr. Opin. Biotechnol. doi:10.1016/j.copbio.2011.12.025 [DOI] [PubMed] [Google Scholar]
- 34. Wendisch VF, Bott M, Eikmanns BJ. 2006. Metabolic engineering of Escherichia coli and Corynebacterium glutamicum for biotechnological production of organic acids and amino acids. Curr. Opin. Microbiol. 9:268–274 [DOI] [PubMed] [Google Scholar]
- 35. Priefert H, Rabenhorst J, Steinbüchel A. 1997. Molecular characterization of genes of Pseudomonas sp. strain HR199 involved in bioconversion of vanillin to protocatechuate. J. Bacteriol. 179:2595–2607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Venturi V, Zennaro F, Degrassi G, Okeke BC, Bruschi CV. 1998. Genetics of ferulic acid bioconversion to protocatechuic acid in plant-growth-promoting Pseudomonas putida WCS358. Microbiology 144:965–973 [DOI] [PubMed] [Google Scholar]
- 37. Martínez-Cuesta MDC, Payne J, Hanniffy SB, Gasson MJ, Narbad A. 2005. Functional analysis of the vanillin pathway in a vdh-negative mutant strain of Pseudomonas fluorescens AN103. Enzyme Microb. Technol. 37:131–138 [Google Scholar]
- 38. Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N, Dickens S, Haydock SF, Leadlay PF. 2007. Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat. Biotechnol. 25:447–453 [DOI] [PubMed] [Google Scholar]
- 39. Muheim A, Lerch K. 1999. Towards a high-yield bioconversion of ferulic acid to vanillin. Appl. Microbiol. Biotechnol. 51:456–461 [Google Scholar]
- 40. Saitou N, Nei M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4:406–425 [DOI] [PubMed] [Google Scholar]
- 41. Nelson KE, Weinel C, Paulsen IT, Dodson RJ, Hilbert H, Martins dos Santos VAP, Fouts DE, Gill SR, Pop M, Holmes M, Brinkac L, Beanan M, DeBoy RT, Daugherty S, Kolonay J, Madupu R, Nelson W, White O, Peterson J, Khouri H, Hance I, Lee PC, Holtzapple E, Scanlan D, Tran K, Moazzez A, Utterback T, Rizzo M, Lee K, Kosack D, Moestl D, Wedler H, Lauber J, Stjepandic D, Hoheisel J, Straetz M, Heim S, Kiewitz C, Eisen J, Timmis KN, Düsterhöft A, Tümmler B, Fraser CM. 2002. Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ. Microbiol. 4:799–808 [DOI] [PubMed] [Google Scholar]
- 42. Masai E, Harada K, Peng X, Kitayama H, Katayama Y, Fukuda M. 2002. Cloning and characterization of the ferulic acid catabolic genes of Sphingomonas paucimobilis SYK-6. Appl. Environ. Microbiol. 68:4416–4424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sekine M, Tanikawa S, Omata S, Saito M, Fujisawa T, Tsukatani N, Tajima T, Sekigawa T, Kosugi H, Matsuo Y, Nishiko R, Imamura K, Ito M, Narita H, Tago S, Fujita N, Harayama S. 2006. Sequence analysis of three plasmids harboured in Rhodococcus erythropolis strain PR4. Environ. Microbiol. 8:334–346 [DOI] [PubMed] [Google Scholar]
- 44. McLeod MP, Warren RL, Hsiao WW, Araki N, Myhre M, Fernandes C, Miyazawa D, Wong W, Lillquist AL, Wang D, Dosanjh M, Hara H, Petrescu A, Morin RD, Yang G, Stott JM, Schein JE, Shin H, Smailus D, Siddiqui AS, Marra MA, Jones SJ, Holt R, Brinkman FS, Miyauchi K, Fukuda M, Davies JE, Mohn WW, Eltis LD. 2006. The complete genome of Rhodococcus sp. RHA1 provides insights into a catabolic powerhouse. Proc. Natl. Acad. Sci. U. S. A. 103:15582–15587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Silby MW, Cerdeno-Tarraga AM, Vernikos GS, Giddens SR, Jackson RW, Preston GM, Zhang XX, Moon CD, Gehrig SM, Godfrey SA, Knight CG, Malone JG, Robinson Z, Spiers AJ, Harris S, Challis GL, Yaxley AM, Harris D, Seeger K, Murphy L, Rutter S, Squares R, Quail MA, Saunders E, Mavromatis K, Brettin TS, Bentley SD, Hothersall J, Stephens E, Thomas CM, Parkhill J, Levy SB, Rainey PB, Thomson NR. 2009. Genomic and genetic analyses of diversity and plant interactions of Pseudomonas fluorescens. Genome Biol. 10:R51 doi:10.1186/gb-2009-10-5-r51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Greated A, Lambertsen L, Williams PA, Thomas CM. 2002. Complete sequence of the IncP-9 TOL plasmid pWW0 from Pseudomonas putida. Environ. Microbiol. 4:856–871 [DOI] [PubMed] [Google Scholar]
- 47. Zhao W, Zhong Y, Yuan H, Wang J, Zheng H, Wang Y, Cen X, Xu F, Bai J, Han X, Lu G, Zhu Y, Shao Z, Yan H, Li C, Peng N, Zhang Z, Zhang Y, Lin W, Fan Y, Qin Z, Hu Y, Zhu B, Wang S, Ding X, Zhao GP. 2010. Complete genome sequence of the rifamycin SV-producing Amycolatopsis mediterranei U32 revealed its genetic characteristics in phylogeny and metabolism. Cell Res. 20:1096–1108 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.