

Abstract
Mycobacterium tuberculosis disease represents an enormous global health problem, with exceptionally high morbidity and mortality in HIV-seropositive (HIV+) persons. Alveolar macrophages from HIV+ persons demonstrate specific and targeted impairment of critical host cell responses, including impaired M. tuberculosis-mediated tumor necrosis factor (TNF) release and macrophage apoptosis. Vitamin D may promote anti-M. tuberculosis responses through upregulation of macrophage NO, NADPH oxidase, cathelicidin, and autophagy mechanisms, but whether vitamin D promotes anti-M. tuberculosis mechanisms in HIV+ macrophages is not known. In the current study, human macrophages exposed to M. tuberculosis demonstrated robust release of TNF, IκB degradation, and NF-κB nuclear translocation, and these responses were independent of vitamin D pretreatment. In marked contrast, HIV+ U1 human macrophages exposed to M. tuberculosis demonstrated very low TNF release and no significant IκB degradation or NF-κB nuclear translocation, whereas vitamin D pretreatment restored these critical responses. The vitamin D-mediated restored responses were dependent in part on macrophage CD14 expression. Importantly, similar response patterns were observed with clinically relevant human alveolar macrophages from healthy individuals and asymptomatic HIV+ persons at high clinical risk of M. tuberculosis infection. Taken together with the observation that local bronchoalveolar lavage fluid (BALF) levels of vitamin D are severely deficient in HIV+ persons, the data from this study demonstrate that exogenous vitamin D can selectively rescue impaired critical innate immune responses in vitro in alveolar macrophages from HIV+ persons at risk for M. tuberculosis disease, supporting a potential role for exogenous vitamin D as a therapeutic adjuvant in M. tuberculosis infection in HIV+ persons.
INTRODUCTION
Mycobacterium tuberculosis infection in HIV-seropositive (HIV+) persons represents an enormous global health problem, frequently occurs in persons in early stages of HIV disease, and is associated with exceptional morbidity and mortality, especially with multidrug-resistant (MDR) or extensively drug-resistant (XDR) tuberculosis (1, 2). However, the underlying predisposing mechanisms, particularly in HIV+ persons with relatively preserved CD4+ T-lymphocyte counts, remain incompletely understood (3–5). Alveolar macrophages (AMs) represent a critical cell type in the host defense response to M. tuberculosis (6), and alveolar macrophages from HIV+ persons demonstrate specific and targeted impairment of critical host cell responses, including impaired M. tuberculosis-mediated tumor necrosis factor (TNF) release and macrophage apoptosis (7), which may be related in part to interleukin-10 (IL-10)-mediated upregulation of BCL3 (8). Preliminary data suggest that M. tuberculosis-mediated macrophage apoptosis may be restored by exogenous TNF, suggesting that alveolar macrophages from HIV+ persons are not irreversibly impaired and may be responsive to immunomodulation (7).
Vitamin D deficiency is associated with susceptibility to M. tuberculosis disease (9–12), although the basic underlying mechanisms remain poorly understood. Early in vitro observations demonstrated that exogenous vitamin D suppressed M. tuberculosis growth in macrophages (13, 14). Vitamin D may promote anti-M. tuberculosis responses through upregulation of NO (15), NADPH oxidase (16, 17), cathelicidin (18–20), and autophagy (20) mechanisms in murine models and human macrophages. However, the effect of vitamin D on critical human alveolar macrophage host defense responses has not been investigated fully, and the influence of vitamin D on HIV+ macrophages is not known.
The purpose of this study was to examine the influence of vitamin D on human macrophage host defense responses in vitro, focusing on Toll-like receptor (TLR) signaling pathways, as TLRs represent critical innate immune host defense molecules in the recognition of pathogens, including M. tuberculosis (21–23). Furthermore, recognizing the frequent finding of vitamin D deficiency among HIV+ persons (24–26), this study also focused on HIV+ macrophages to determine whether exogenous vitamin D can rescue impaired host defense responses to M. tuberculosis, using human macrophage cell lines and clinically relevant alveolar macrophages. This study demonstrates that exogenous vitamin D can rescue impaired M. tuberculosis-mediated TNF release in HIV+ macrophages through enhanced TLR and restored IκB/NF-κB signaling; the mechanism of vitamin D-mediated rescue of restored responses was in part dependent on macrophage CD14.
MATERIALS AND METHODS
Human macrophages. (i) Human macrophage cell lines.
As a model for study of the influence of HIV infection on human macrophage function, experiments used the human monocyte U937 (American Type Culture Collection [ATCC]) and HIV-infected human monocyte U1 (subclone of U937; AIDS Research and Reference Reagent Program, Bethesda, MD) cell lines, as previously published (7, 27, 28). U1 cells contain two integrated copies of HIV-1 proviral DNA and are characterized by low levels of constitutive viral expression (29) that can be modulated with specific cytokines and phorbol myristate acetate (PMA) (30). Human U937 and U1 cells were cultured in complete RPMI 1640 medium (10% heat-inactivated fetal calf serum [FCS], 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin), except for experiments using live mycobacteria, where ceftriaxone (1 μg/ml) was substituted for streptomycin. Cells were harvested during exponential growth phase, washed, differentiated into macrophages by use of PMA (100 nM) at 37°C in 5% CO2 for 24 h, washed three times with phosphate-buffered saline (PBS), and incubated an additional 24 h before use.
(ii) Human alveolar macrophages.
For select experiments, human alveolar macrophages were used to confirm critical results observed in cell lines. Prospectively recruited healthy and asymptomatic HIV+ volunteers had no evidence of active pulmonary disease and had normal spirometry results. Healthy individuals had no known risk factors for HIV infection and were confirmed to be HIV seronegative by enzyme-linked immunosorbent assay (ELISA), which was performed according to the instructions of the manufacturer (Abbott Diagnostics). Asymptomatic HIV+ subjects had a CD4 T cell count of >200 cells/mm3 and an undetectable serum viral load (<50 HIV-1 RNA copies/ml), were on highly active antiretroviral therapy (HAART) or no therapy, and had no history of opportunistic pneumonia. Lung immune cells were obtained by bronchoalveolar lavage (BAL), using a standard technique (31). All procedures were performed on adult volunteers after informed consent, following protocols approved by the Beth Israel Deaconess Medical Center Institutional Review Board. The cells were separated from the pooled BAL fluid (BALF), and AMs were isolated by adherence for ≥72 h to plastic-bottom tissue culture plates as previously described (31). Isolation of AMs from all healthy and HIV+ persons yielded cells which were ≥98% viable, as determined by trypan blue dye exclusion, and demonstrated >95% positive nonspecific esterase staining (31).
Microbial organisms and reagents.
Virulent (H37Rv) M. tuberculosis which had been irradiated was a generous gift from J. Belisle (Colorado State University, Fort Collins, CO) and the National Institute of Allergy and Infectious Diseases (tuberculosis research materials contract N01-AI-75320). M. bovis (BCG Pasteur) was obtained from the ATCC. Stocks were thawed, vortexed, sonicated using a bath sonicator for 15 s at 500 W, and allowed to stand for 10 min, and the upper 200 μl of solution was used for experiments (32). Lipid A (TLR4 ligand) from the Escherichia coli F583 Rd mutant and PMA were purchased from Sigma Chemical Company (St. Louis, MO). Pam3Cys-Ser-(Lys)4 hydrochloride (PamCys) (TLR3 ligand) was purchased from Calbiochem (San Diego, CA), and the 19-kDa lipoprotein from M. tuberculosis (TLR2/1 ligand) was purchased from EMC Microcollections (Tuebingen, Germany). 1-Pyrrolidinecarbodithioic acid (PDTC), an inhibitor of NF-κB activation, was purchased from Calbiochem (San Diego, CA). 1,25(OH)2Vitamin D3 (1,25D3) was purchased from Calbiochem (San Diego, CA) and used at a concentration of 100 nM unless otherwise specified.
RNA isolation and RT-PCR.
Total RNA was isolated from macrophages by use of an RNeasy kit (Qiagen, Valencia, CA), and reverse transcription-PCR (RT-PCR) was performed according to the manufacturer's protocol for the Thermoscript PCR system (Invitrogen Life Technologies). The following primers were used for amplification of the vitamin D receptor (VDR): 5′-GCC CAC CAT AAG ACC TAC GA-3′ and 5′-AGA TTG GAG AAG CTG GAC GA-3′.
Real-time PCR was performed using the following primers and probes: for TLR2, 5′-TCT GGC ATG TGC TGT GCT CT-3′ and 5′-GGA AAC GGT GGC ACA GGA C-3′, with the TaqMan probe 5′-TTC CTG CTG ATC CTG CTC ACG GG-3′; for TLR4, 5′-TGT TGT GGT GTC CCA GCA CT-3′ and 5′-CTG CCA GGT CTG AGC AAT CTC-3′, with the TaqMan probe 5′-CAT CCA GAG CCG CTG GTG TAT CTT TGA A-3′; for TNF-α, 5′-GGT GCT TGT TCC TCA GCC TC-3′ and 5′-CAG GCA GAA GAG CGT GGT G-3′, with the TaqMan probe 5′-CTC CTT CCT GAT CGT GGC AGG CG-3′; and for VDR, 5′-AAG GAC AAC CGA CGC CAC T-3′ and 5′-ATC ATG CCG ATG TCC ACA CA-3′, with the TaqMan probe 5′-CAG GCC TGC CGG CTC AAA CG-3′.
Cytokine detection in cultured supernatants by ELISA.
Isolated adherent macrophages (24-well plate; 5 × 105 cells/well) were incubated with M. tuberculosis or M. bovis BCG (multiplicity of infection [MOI] of 10:1) for 24 h in the presence or absence of 1,25D3 (10−7 M, added 24 h prior to M. tuberculosis or BCG) at 37°C in humidified 5% CO2. For select experiments, neutralizing anti-CD14 antibody or an IgG1 isotype control (R&D Systems) was added 30 min prior to M. tuberculosis. Culture supernatants were harvested and centrifuged to remove cellular debris, and aliquots were assayed immediately or stored at −80°C until assay. Specific immunoreactivity to TNF-α (R&D Systems) was measured by ELISA as described previously (28).
Flow cytometry surface receptor analysis.
TLR2, TLR4, and CD14 expression was measured via surface antibody labeling (TLR2-phycoerythrin [PE], TLR4-PE [Invivogen], and CD14-PE [MACS]) in macrophage cell suspensions with a Cytomics FC500 flow cytometer (Beckman Coulter) as previously published (28). Results were recorded as the mean relative fluorescence units (RFU) and the percentage of the population staining positive.
Western blotting.
Cell cytoplasmic protein extracts were prepared using standard ice-cold RIPA buffer with protease and phosphatase inhibitors. Western blotting was performed by utilizing a standard protocol (33) and antibodies specific to IκBα and β-actin (Cell Signaling Technology). Resolved bands were quantified by densitometry (Amersham Biosciences), and results are expressed in relative units (RU).
NF-κB ELISA.
Adherent isolated macrophages (6-well plates; 3 × 106 cells/well) were incubated with M. tuberculosis for 0 to 120 min, macrophage nuclear extracts were prepared by using an NE-PER kit (Pierce) according to the manufacturer's protocol, and an ELISA specific for p65 was performed using a Transfactor NF-κB p65 colorimetric kit according to the manufacturer's protocol (Clontech). Protein loading was standardized using the Bradford assay (Bio-Rad).
Serum and BALF vitamin D measurements.
Archived frozen clinical samples of paired BALF and serum (stored at −80°C) were available for four groups of patients who underwent bronchoscopy at the All India Institute of Medical Sciences (New Delhi, India): (i) HIV-seronegative individuals without M. tuberculosis, (ii) HIV-seronegative individuals with microbiologically confirmed active M. tuberculosis disease, (iii) HIV+ individuals without M. tuberculosis, and (iv) HIV+ individuals with microbiologically confirmed M. tuberculosis disease. Patients provided informed consent, and the study protocol was approved by the AIIMS Ethics Committee. 25(OH)Vitamin D3 and 1,25(OH)2Vitamin D3 levels were measured in paired serum and BALF samples by ELISA according to the manufacturer's protocol (IDS Ltd., Fountain Hills, AZ). Vitamin D levels were normalized with a BALF-associated dilution factor, using urea nitrogen measurements, as previously described (34, 35).
Statistical methods.
All data were analyzed using nonparametric methodology (Mann-Whitney U test), and a P value of <0.05 was considered significant. Experiments were repeated a minimum of three times.
RESULTS
Exogenous vitamin D rescues M. tuberculosis-mediated TNF release from HIV+ human macrophages.
TNF release represents a critical macrophage response to M. tuberculosis challenge (36). In the current study, unstimulated human U937 macrophages demonstrated low constitutive TNF release and a robust increase in TNF release in response to M. tuberculosis (Fig. 1A), and 1,25D3 pretreatment did not influence macrophage TNF release constitutively or in response to M. tuberculosis challenge (Fig. 1A). In HIV+ U1 macrophages, constitutive TNF release was also low, but TNF release in response to M. tuberculosis was significantly impaired compared to that for U937 cells (Fig. 1B), consistent with prior publications (7). However, in marked contrast to U937 cells, pretreatment of HIV+ U1 macrophages with 1,25D3 dramatically increased macrophage TNF release in response to M. tuberculosis, in a concentration-dependent manner, to levels comparable to those for U937 macrophages (Fig. 1B and C), whereas 1,25D3 pretreatment did not influence constitutive TNF release in HIV+ U1 macrophages. Thus, exogenous 1,25D3 selectively restored impaired M. tuberculosis-mediated TNF release in HIV+ human macrophages.
Fig 1.

1,25D3 rescues M. tuberculosis-mediated TNF release in HIV+ human macrophages. Differentiated U937 (A) and HIV+ U1 (B and C) macrophages were incubated with irradiated virulent M. tuberculosis (MTb) (MOI of 10:1 for 24 h) in the presence or absence of 1,25D3 (VD) pretreatment (24 h). TNF in cell culture supernatants was measured by ELISA (R&D). Figures are representative of individual experiments with similar results (n = 6). Quantitative data represent means ± standard errors of the means (SEM). US, unstimulated. *, P < 0.05.
Vitamin D promotes TNF mRNA transcripts in HIV+ human macrophages.
The main biological actions of vitamin D occur following conversion of the principle circulating 25(OH)D3 (25D3) form to 1,25D3 by the cellular enzyme 1-alpha hydroxylase (CYP27B1), with subsequent binding to intracellular VDR (37). Although the main site of CYP27B1 hydroxylase expression is the kidney, immune cells, including macrophages, express CYP27B1 hydroxylase and thus are able to independently convert 25D3 to biologically active 1,25D3 (38). In the current study, both human U937 and HIV+ U1 macrophages expressed mRNAs for VDR at comparable levels (Fig. 2A), suggesting that the observed differences in 1,25D3-mediated macrophage responses were not attributable to significant differences in levels of VDR. To determine the mechanism for 1,25D3 rescue of TNF release in HIV+ macrophages, we next examined TNF mRNA levels. Exogenous 1,25D3 pretreatment did not influence TNF mRNA levels in human U937 macrophages (Fig. 2B), whereas TNF mRNA levels were significantly increased by 1,25D3 in human HIV+ U1 macrophages in response to mycobacteria (Fig. 2B). These results suggest that increased M. tuberculosis-mediated TNF release in HIV+ U1 macrophages is associated with increased TNF mRNA.
Fig 2.
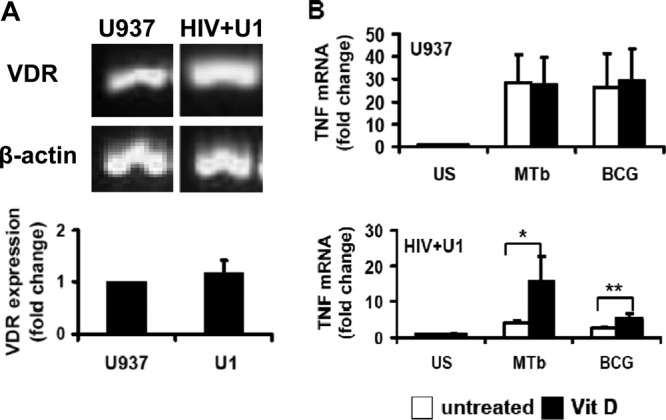
1,25D3 enhances TNF transcription in HIV+ human macrophages. (A) RT-PCR and real-time PCR for VDR were performed on total RNAs from differentiated U937 and HIV+ U1 macrophages (n = 4). (B) Differentiated U937 and HIV+ U1 cells were incubated with M. tuberculosis or BCG (M. bovis) (MOI, 10:1) for 3 h in the presence or absence of 1,25D3 pretreatment (24 h). TNF mRNA was measured by real-time PCR (n = 4). Quantitative data represent means ± SEM. *, P < 0.05; **, P < 0.01.
Vitamin D enhancement of TNF release in HIV+ human macrophages is dependent on recognition of known TLR ligands.
TLR2 and TLR4 are critical host defense signaling molecules that mediate TNF release by macrophages in response to M. tuberculosis infection (39). In the current study, human U937 macrophages released TNF in response to TLR2 and TLR4 agonists, with a significant change following 1,25D3 pretreatment for lipid A only (Fig. 3A), consistent with prior observations (40). In contrast, 1,25D3 pretreatment of human HIV+ U1 macrophages significantly increased TNF release in response to multiple TLR2 and TLR4 ligands (Fig. 3B), including the M. tuberculosis 19-kDa lipopeptide (recognized by TLR2/1). Both TLR2 and TLR4 mRNA and surface expression levels were comparable in human U937 and HIV+ U1 macrophages, and TLR2 and TLR4 levels were not significantly altered by 1,25D3 pretreatment (Fig. 3C and D). Thus, the observed rescue of M. tuberculosis-mediated TNF release following 1,25D3 pretreatment was not associated with a significant alteration in mRNA or surface expression of macrophage TLR2 or TLR4 molecules.
Fig 3.

1,25D3 increases TLR signaling but not TLR expression in HIV+ U1 macrophages. (A and B) Differentiated human U937 and HIV+ U1 macrophages were incubated with the TLR ligands lipid A (LA) (for TLR4), PamCys (for TLR2/1), and 19-kDa lipoprotein from M. tuberculosis (19 kDa MTb; 1 μg/ml) (for TLR2/1) for 24 h in the presence or absence of 1,25D3 pretreatment. Cell-free culture supernatants were assayed for TNF by ELISA (n = 3). (C) Differentiated U937 and HIV+ U1 macrophages were incubated for 24 h in the presence or absence of 1,25D3 pretreatment and then stained with PE-labeled anti-TLR antibodies or isotype control antibody. Surface expression was measured by flow cytometry. Left panels show isotype control (gray lines)- and TLR (black lines)-labeled cells; right panels show TLR-labeled cells (gray lines) and 1,25D3-treated TLR-labeled cells (black lines). Representative histograms for independent experiments with similar results (n = 3) are shown. (D) Specific TLR2 and TLR4 mRNAs were detected by real-time PCR (n = 3). Quantitative data represent means ± SEM. *, P < 0.05.
Upregulation of NF-κB signaling by vitamin D in HIV+ human macrophages.
The observation that exogenous 1,25D3 rescue of M. tuberculosis-mediated TNF release in HIV+ human macrophages was associated with increased TNF mRNA levels but not with alteration of surface expression of TLRs (major M. tuberculosis recognition signaling receptors) suggested that signaling pathways downstream of TLR may be modulated by 1,25D3. TLR signaling promotes IκB degradation and allows NF-κB nuclear translocation and subsequent host defense gene activation, including that of TNF (41, 42). In the current study, human U937 macrophages demonstrated rapid M. tuberculosis-mediated IκB degradation, with no significant change with 1,25D3 pretreatment but with expected inhibition by PDTC (an inhibitor of IκB degradation) (Fig. 4A). In marked contrast, human HIV+ U1 macrophages failed to demonstrate significant IκB degradation in response to M. tuberculosis over time (Fig. 4A), but exogenous 1,25D3 pretreatment promoted M. tuberculosis-mediated IκB degradation, although less robustly and with delayed kinetics compared to U937 cells (Fig. 4A). Consistent with these findings, human U937 macrophages in an independent assay demonstrated NF-κB nuclear translocation in response to M. tuberculosis but showed no further increase in NF-κB nuclear translocation with 1,25D3 pretreatment (Fig. 4B). In marked contrast, human HIV+ U1 macrophages demonstrated limited NF-κB nuclear translocation in response to M. tuberculosis but showed a dramatic increase in M. tuberculosis-mediated NF-κB nuclear translocation upon pretreatment with 1,25D3 (Fig. 4B). Thus, 1,25D3 selectively promoted M. tuberculosis-mediated IκB degradation and NF-κB nuclear translocation in human HIV+ U1 macrophages.
Fig 4.
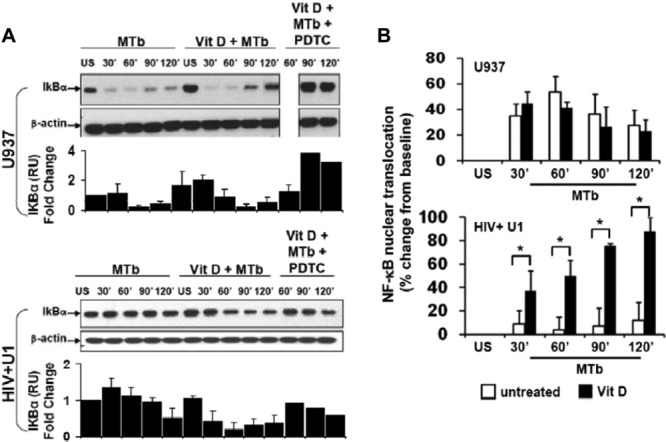
1,25D3 upregulates NF-κB signaling in HIV+ human macrophages. (A) Differentiated U937 and HIV+ U1 macrophages were incubated with M. tuberculosis (MOI, 10:1) for 0 to 120 min in the presence or absence of 1,25D3 and PDTC. Cell lysates were resolved by Western blotting using a specific antibody to IκBα. Representative blots for three independent experiments with similar results are shown. Quantitative densitometric analyses of IκBα bands are displayed directly beneath the blots. (B) NF-κB nuclear translocation in nuclear extracts was measured by ELISA (n = 3). Quantitative data represent means ± SEM. *, P < 0.05.
Vitamin D upregulates macrophage CD14 expression.
CD14 is a 55-kDa glycoprotein receptor, expressed mainly in myelomonocytic cells (including macrophages), that facilitates TLR ligand binding (43). 1,25D3 upregulates CD14 expression in human monocytes (44), and it could enhance TLR signaling. In the current study, constitutive CD14 surface expression was relatively low for both human U937 and HIV+ U1 macrophages, and 1,25D3 pretreatment significantly increased CD14 surface expression in both populations of macrophages (Fig. 5A). In human U937 macrophages pretreated with 1,25D3, M. tuberculosis-mediated TNF release was not significantly altered in the presence of anti-CD14 neutralizing antibody (Fig. 5B), whereas lipopolysaccharide (LPS)-mediated TNF release was markedly reduced by anti-CD14 antibody, as expected. However, in marked contrast, in HIV+ U1 macrophages pretreated with 1,25D3, M. tuberculosis-mediated TNF release was significantly reduced in the presence of anti-CD14 neutralizing antibody (Fig. 5C). Thus, although 1,25D3 upregulated macrophage CD14 surface expression in both U937 and HIV+ U1 cells, CD14 upregulation contributed to 1,25D3-mediated rescue of M. tuberculosis-mediated TLR signaling in HIV+ U1 macrophages, whereas TNF release was CD14 independent in U937 macrophages.
Fig 5.

1,25D3 induces CD14 expression in human macrophages. (A) Differentiated U937 and HIV+ U1 macrophages were incubated for 24 h in the presence or absence of 1,25D3 pretreatment and then stained with PE-labeled anti-CD14 antibody or isotype control antibody. Surface expression was measured by flow cytometry. Left panels show isotype control (gray lines)- and CD14 (black lines)-labeled cells; right panels show CD14-labeled cells (gray lines) and 1,25D3-treated CD14-labeled cells (black lines). Representative histograms from individual experiments with similar results (n = 3) are shown. (B and C) Differentiated U937 (B) and HIV+ U1 (C) macrophages were treated with M. tuberculosis (MOI, 10:1) or LPS (1 μg/ml) in the presence or absence of vitamin D pretreatment (24 h) and the indicated antibodies. TNF in cell culture supernatants was measured by ELISA (R&D). The data are representative of three individual experiments with similar results. Quantitative data represent means ± SEM. *, P < 0.05.
Vitamin D rescues M. tuberculosis-mediated TNF release in human alveolar macrophages.
To validate the above findings, select experiments were next performed using clinically relevant human alveolar macrophages. Consistent with the results obtained using human macrophage cell lines, human alveolar macrophages from healthy individuals demonstrated significant release of TNF in response to M. tuberculosis or BCG, but without a significant influence following 1,25D3 pretreatment (Fig. 6A), whereas 1,25D3 pretreatment significantly increased M. tuberculosis-mediated TNF release in alveolar macrophages from asymptomatic HIV+ persons (Fig. 6A), even in immune-reconstituted subjects on HAART with preserved baseline TNF responses. Similar to the case with the human macrophage cell lines, TLR2 and TLR4 mRNA (Fig. 6B) and surface (Fig. 6C) expression levels were comparable in human alveolar macrophages from healthy individuals and asymptomatic HIV+ persons, and TLR expression was not influenced by 1,25D3 (Fig. 6B and C). Although 1,25D3 upregulated CD14 expression in alveolar macrophages from both healthy and HIV+ persons (Fig. 7A), in alveolar macrophages from asymptomatic HIV+ persons pretreated with 1,25D3, neutralizing anti-CD14 antibody significantly reduced M. tuberculosis-mediated TNF release (Fig. 7B), whereas neutralizing anti-CD14 antibody had no effect on alveolar macrophages from healthy persons (data not shown). Collectively, these experiments validate the results observed with human U937 and HIV+ U1 macrophages, and they suggest that 1,25D3 may selectively rescue M. tuberculosis-mediated TNF release in alveolar macrophages from HIV+ persons, in part through a CD14-dependent mechanism.
Fig 6.

1,25D3 rescues M. tuberculosis-mediated TNF release in human alveolar macrophages from HIV+ persons. (A) AMs from healthy (n = 6) and HIV+ (n = 2) persons were incubated with M. tuberculosis or BCG (MOI of 10:1 for 24 h) in the presence or absence of 1,25D3 pretreatment (24 h). TNF in cell culture supernatants was measured by ELISA (R&D). (B) Specific TLR2 and TLR4 mRNAs were detected by real-time PCR (n = 3). (C) AMs were incubated for 24 h in the presence or absence of 1,25D3 pretreatment and then stained with PE-labeled anti-TLR or isotype control antibody. Surface expression was measured by flow cytometry. Left panels show isotype control (gray lines)- and receptor (black lines)-labeled cells; right panels show receptor-labeled cells (gray lines) and 1,25D3-treated receptor-labeled cells (black lines). Representative histograms for individual experiments with similar results (n = 3 for healthy individuals and 2 for HIV+ individuals) are shown. Quantitative data represent means ± SEM. *, P < 0.05; **, P < 0.01.
Fig 7.

1,25D3 rescue of M. tuberculosis-mediated TNF release in human alveolar macrophages from HIV+ persons is dependent on CD14. (A) AMs were incubated for 24 h in the presence or absence of 1,25D3 pretreatment and then stained with PE-labeled anti-CD14 or isotype control antibody. Surface expression was measured by flow cytometry. Left panels show isotype control (gray lines)- and receptor (black lines)-labeled cells; right panels show receptor-labeled cells (gray lines) and 1,25D3-treated receptor-labeled cells (black lines) (n = 3). (B) HIV+ AMs were treated with M. tuberculosis (MOI of 0.25:1), BCG (MOI of 10:1), or LPS (1 μg/ml) in the presence or absence of 1,25D3 pretreatment (24 h) and the indicated antibodies. TNF in cell culture supernatants was measured by ELISA (n = 2). (C) 25D3 levels were measured in the cell-free BALF of healthy and HIV-infected Indian patients with and without active M. tuberculosis infection (for HIV− M. tuberculosis− individuals, n = 38; for HIV− M. tuberculosis+ individuals, n = 35; for HIV+ M. tuberculosis− individuals, n = 12; and for HIV+ M. tuberculosis+ individuals, n = 17) by ELISA. Quantitative data represent means ± SEM. *, P < 0.05.
Reduced BALF vitamin D levels in HIV+ patients with active tuberculosis.
Serum levels of 25D3 are reduced in persons with active tuberculosis (10, 12), and HIV infection is associated with reduced serum levels of 25D3 (24–26). However, vitamin D levels in the lungs of persons with HIV or HIV-M. tuberculosis coinfection have not been reported. In the current study, biologically active 1,25D3 was not detected in any cell-free BALF specimen (data not shown). In contrast, 25D3 levels were readily detected in the BALF of all persons but were lowest in persons with HIV infection, especially in HIV-infected persons with active M. tuberculosis disease (Fig. 7C). These data suggest that HIV infection is associated with a local vitamin D deficiency in the alveolar airspace, especially in HIV+ persons coinfected with M. tuberculosis.
DISCUSSION
This study shows that exogenous 1,25D3 rescues M. tuberculosis-mediated TNF release in HIV+ human macrophages. In the absence of HIV infection, human macrophages exposed to M. tuberculosis demonstrated a robust release of TNF, IκB degradation, and NF-κB nuclear translocation, and these responses were independent of 1,25D3 pretreatment. In marked contrast, HIV+ U1 human macrophages exposed to M. tuberculosis demonstrated very little TNF release and no significant IκB degradation or NF-κB nuclear translocation, but there was a significant rescue of these responses with 1,25D3 pretreatment. Furthermore, the 1,25D3-mediated rescue of macrophage function in response to M. tuberculosis was dependent in part on CD14 expression. Importantly, similar response patterns were observed with clinically relevant human alveolar macrophages from healthy individuals and asymptomatic HIV+ persons at high clinical risk of M. tuberculosis infection. Taken together, these data support the concept that 1,25D3 pretreatment rescues impaired M. tuberculosis-mediated TNF release in HIV+ macrophages through restored IκB/NF-κB signaling that is in part CD14 dependent.
This is the first study, to our knowledge, to examine the immunomodulatory effects of exogenous vitamin D on the response of HIV+ macrophages to M. tuberculosis. The clinical implications of the current investigation are of particular importance given that the global M. tuberculosis epidemic disproportionately affects HIV+ persons. Epidemiologic data show that unlike the case for other opportunistic infections, the risk of M. tuberculosis disease rises soon after HIV seroconversion, despite relatively preserved CD4 counts, and is not completely reversed by HAART (4, 45). Previous studies from our laboratory and other investigators have demonstrated that HIV is associated with specific and targeted defects in alveolar macrophage innate host defense responses to M. tuberculosis, including intracellular signaling, chemokine production, TNF-α and other proinflammatory cytokine release, and macrophage apoptosis (7, 32), which may in part contribute to the elevated risk of M. tuberculosis disease in the absence of significantly reduced circulating CD4 T-lymphocyte counts. In the current study, macrophage innate immune function was restored by exogenous 1,25D3. Specifically, in HIV+ macrophages, exogenous 1,25D3 restored TNF release, upregulated TNF mRNA, enhanced TLR2 and TLR4 responses, and rescued IκB degradation and NF-κB nuclear translocation. Furthermore, these 1,25D3-restored host defense responses were dependent on CD14 expression in HIV+ macrophages. Taken together, these findings support the concept that vitamin D may selectively restore TLR signaling, a critical recognition signaling pathway in the host cell response to M. tuberculosis challenge.
The mechanism for vitamin D rescue of macrophage innate function in HIV+ macrophages is through TLR signaling. The differences in influence of 1,25D3 on U937 and HIV+ U1 macrophages were not explained by obvious differences in the levels of the principal receptor for vitamin D, VDR, which were similar in the U937 and HIV+ U1 macrophages and in human alveolar macrophages. Furthermore, the findings that TLR2 and TLR4 ligand-mediated TNF release was enhanced by vitamin D (including the TLR2 ligand 19-kDa M. tuberculosis lipoprotein-mediated TNF release) and that the IκB/NF-κB pathway was restored in HIV+ U1 macrophages, while constitutive surface expression levels of TLR2 and TLR4 were similar and without significant alterations in response to 1,25D3, suggest that 1,25D3 stimulates other components of the TLR signaling pathway in HIV+ macrophages. Finally, the findings that 1,25D3 upregulated the TLR coreceptor CD14 and that neutralizing CD14 in HIV+ macrophages pretreated with 1,25D3 reduced M. tuberculosis-mediated TNF release suggest that TLR signaling may be enhanced through modulation of the TLR coreceptor CD14, whereas in the absence of HIV infection, M. tuberculosis-mediated TNF release is mediated through IκB/NF-κB signaling but is CD14 independent. The CD14 independence of M. tuberculosis-mediated TNF release in healthy cells may be due to activation of alternate pathways or to expression of alternate costimulatory molecules that may be suppressed in HIV-infected cells, or perhaps to other mechanisms. Determining the specific pathways involved in the macrophage response to M. tuberculosis represents an area of active investigation.
In the current study, the mechanism of rescued M. tuberculosis-mediated TNF release in HIV+ macrophages was attributed in part to CD14 expression or signaling. However, other host defense receptors and signaling pathways may also contribute but were not specifically investigated. Although 1,25D3 rescued M. tuberculosis-mediated human HIV+ macrophage TNF release, its influences on other cytokines and other macrophage host defense functions were not investigated. Other limitations of the current study include the experimental design, which examined 1,25D3 pretreatment but did not examine the influence of 1,25D3 on macrophages previously (or simultaneously) infected with M. tuberculosis. Although 25D3 levels were very low in BALF from HIV+ persons, especially from persons coinfected with M. tuberculosis, detailed clinical characteristics, a specific correlation with serum 1,25D3 levels, and a correlation with macrophage function for individuals were not available. The use of human macrophage cell lines may not reflect the behavior of primary human macrophages, although the consistent finding of similar response patterns in human alveolar macrophages in both the current study (although the number of subjects was limited) and previous studies (7, 27, 46) validates these observations and supports the use of these human macrophage cell lines as an experimental model. Differences in the magnitude of observed biological responses in comparing HIV+ U1 macrophage cell lines and alveolar macrophages from HIV+ persons may in part reflect differences in the level of HIV infection (as 100% of U1 macrophages contain the HIV genome, whereas <10% of human alveolar macrophages contain the HIV genome) (7, 28, 33, 34). The use of irradiated virulent M. tuberculosis may not accurately predict the influence of live M. tuberculosis on human macrophage function, although we previously observed similar human macrophage TNF responses in comparing irradiated to live M. tuberculosis H37Rv (7, 8). The use of irradiated M. tuberculosis did not allow determination of the influence of 1,25D3 on M. tuberculosis growth. Finally, in vitro experiments may not accurately reflect in vivo behavior, although the inclusion of clinically relevant primary human alveolar macrophages may allow for more direct translation of these findings to human disease. Our data provide the rationale for further study, including further validation using alveolar macrophages from a larger number of HIV+ persons.
Our results are consistent with several earlier studies that showed a stimulatory effect of 1,25D3 on monocyte-macrophage responses to M. tuberculosis, including respiratory burst, autophagy, and antimicrobial protein production (17, 19, 20). Our finding of a select benefit of exogenous 1,25D3 on HIV+ human macrophages (but not healthy macrophages) is consistent with one previous study which showed that 1,25D3 suppressed replication of Mycobacterium avium in macrophages from HIV+ subjects but had no effect on macrophages from healthy individuals (47). These observations suggest that the innate immune modulatory effects of exogenous 1,25D3 are further modulated in the setting of HIV infection. HIV does not appear to grossly alter macrophage VDR expression. Other possible explanations for differences in measured responses of human HIV+ macrophages to exogenous 1,25D3 include differences in host defense gene expression induced by HIV infection, the requirement of TLR or other receptor expression to critical or threshold levels to activate signaling pathways, or differences in activation states of HIV+ macrophages compared to macrophages from healthy persons (48), although these were not specifically investigated in the current study.
The potential benefit of vitamin D supplementation in the treatment of M. tuberculosis disease in HIV+ persons has not yet been established. To date, two clinical trials have investigated the effect of vitamin D supplementation on M. tuberculosis disease, and neither demonstrated a significant benefit. However, neither trial included significant numbers of HIV+ patients. Furthermore, both trials investigated vitamin D as an adjunctive therapy to antimicrobials in the treatment of established active M. tuberculosis disease (49, 50). Our central observation is that vitamin D pretreatment can rescue defective M. tuberculosis-mediated TNF release in HIV+ human macrophages. Clinically, TNF is crucial to maintaining latency in M. tuberculosis-infected individuals, as evidenced by the high incidence of reactivation of M. tuberculosis in patients treated with anti-TNF strategies (51). Our observation that vitamin D augments the M. tuberculosis-mediated TNF response suggests that vitamin D supplementation may be more effective in preventing M. tuberculosis disease in HIV+ individuals than as a primary treatment for active M. tuberculosis infection, although this hypothesis has yet to be investigated clinically.
In conclusion, exogenous vitamin D rescues M. tuberculosis-mediated TNF release in HIV+ macrophages by restoring TLR-mediated NF-κB signaling, in part through a CD14-dependent mechanism, whereas vitamin D does not influence M. tuberculosis-mediated TNF release in healthy macrophages. These data further support the important concept that alveolar macrophages from HIV+ persons prescribed HAART and with clinically controlled HIV infection (as determined by CD4 T-lymphocyte counts of >200 and an undetectable viral load) continue to exhibit evidence of intrinsic macrophage dysfunction, suggesting that HAART is not sufficient to restore macrophage innate function. Furthermore, this study supports the concept that macrophages from HIV+ persons that demonstrate impaired innate immune function can be immunomodulated to rescue or restore function in vitro. Taken together with the observation that local BALF levels of vitamin D are severely deficient in HIV+ persons, the current finding that exogenous 1,25D3 partially rescues the impaired innate macrophage host defense response in vitro suggests a potential therapeutic role for 1,25D3 supplementation for HIV+ persons at risk for M. tuberculosis disease. This study provides the rationale to pursue additional in vitro investigations to allow the design of appropriate clinical trials to define the role of exogenous vitamin D as a preventive or therapeutic adjuvant for M. tuberculosis infection, particularly in highly susceptible HIV+ persons.
ACKNOWLEDGMENTS
We thank all volunteers who consented to research bronchoscopy. We thank Elizabeth Vassar-Sternburg, Kristin Linnell, Ann Hougland, Xiomarra Guerra, Johanna Leary, Cynthia Peguero, Jose Munguia, and the BIDMC West Procedure Center staff for technical assistance with research bronchoscopies.
This work was supported by NIH grants T32-HL007118-33, R01 HL063655 (H.K.), R01 HL092811 (S.D.T.), and K08AI064014 (N.R.P.) and by an ALA biomedical research grant (N.R.P.).
Footnotes
Published ahead of print 15 October 2012
REFERENCES
- 1. Anandaiah A, Dheda K, Keane J, Koziel H, Moore DA, Patel NR. 2011. Novel developments in the epidemic of human immunodeficiency virus and tuberculosis coinfection. Am. J. Respir. Crit. Care Med. 183:987–997 doi:10.1164/rccm.201008-1246CI [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. WHO 2010. Global tuberculosis control: WHO report 2010. WHO, Geneva, Switzerland [Google Scholar]
- 3. Moreno S, Baraia-Etxaburu J, Bouza E, Parras F, Perez-Tascon M, Miralles P, Vicente T, Alberdi JC, Cosin J, Lopez-Gay D. 1993. Risk for developing tuberculosis among anergic patients infected with HIV. Ann. Intern. Med. 119:194–198 [DOI] [PubMed] [Google Scholar]
- 4. Sonnenberg P, Glynn JR, Fielding K, Murray J, Godfrey-Faussett P, Shearer S. 2005. How soon after infection with HIV does the risk of tuberculosis start to increase? A retrospective cohort study in South African gold miners. J. Infect. Dis. 191:150–158 [DOI] [PubMed] [Google Scholar]
- 5. Williams BG, Dye C. 2003. Antiretroviral drugs for tuberculosis control in the era of HIV/AIDS. Science 301:1535–1537 [DOI] [PubMed] [Google Scholar]
- 6. Kaufmann SH. 2001. How can immunology contribute to the control of tuberculosis? Nat. Rev. Immunol. 1:20–30 [DOI] [PubMed] [Google Scholar]
- 7. Patel NR, Zhu J, Tachado SD, Zhang J, Wan Z, Saukkonen J, Koziel H. 2007. HIV impairs TNF-alpha mediated macrophage apoptotic response to Mycobacterium tuberculosis. J. Immunol. 179:6973–6980 [DOI] [PubMed] [Google Scholar]
- 8. Patel NR, Swan K, Li X, Tachado SD, Koziel H. 2009. Impaired M. tuberculosis-mediated apoptosis in alveolar macrophages from HIV+ persons: potential role of IL-10 and BCL-3. J. Leukoc. Biol. 86:53–60 doi:10.1189/jlb.0908574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Davies PD. 1985. A possible link between vitamin D deficiency and impaired host defence to Mycobacterium tuberculosis. Tubercle 66:301–306 [DOI] [PubMed] [Google Scholar]
- 10. Davies PD, Brown RC, Woodhead JS. 1985. Serum concentrations of vitamin D metabolites in untreated tuberculosis. Thorax 40:187–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Martineau AR, Nhamoyebonde S, Oni T, Rangaka MX, Marais S, Bangani N, Tsekela R, Bashe L, de Azevedo V, Caldwell J, Venton TR, Timms PM, Wilkinson KA, Wilkinson RJ. 2011. Reciprocal seasonal variation in vitamin D status and tuberculosis notifications in Cape Town, South Africa. Proc. Natl. Acad. Sci. U. S. A. 108:19013–19017 doi:10.1073/pnas.1111825108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wilkinson RJ, Llewelyn M, Toossi Z, Patel P, Pasvol G, Lalvani A, Wright D, Latif M, Davidson RN. 2000. Influence of vitamin D deficiency and vitamin D receptor polymorphisms on tuberculosis among Gujarati Asians in west London: a case-control study. Lancet 355:618–621 doi:10.1016/S0140-6736(99)02301-6 [DOI] [PubMed] [Google Scholar]
- 13. Crowle AJ, Ross EJ, May MH. 1987. Inhibition by 1,25(OH)2-vitamin D3 of the multiplication of virulent tubercle bacilli in cultured human macrophages. Infect. Immun. 55:2945–2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rook GA, Steele J, Fraher L, Barker S, Karmali R, O'Riordan J, Stanford J. 1986. Vitamin D3, gamma interferon, and control of proliferation of Mycobacterium tuberculosis by human monocytes. Immunology 57:159–163 [PMC free article] [PubMed] [Google Scholar]
- 15. Rockett KA, Brookes R, Udalova I, Vidal V, Hill AV, Kwiatkowski D. 1998. 1,25-Dihydroxyvitamin D3 induces nitric oxide synthase and suppresses growth of Mycobacterium tuberculosis in a human macrophage-like cell line. Infect. Immun. 66:5314–5321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sly LM, Hingley-Wilson SM, Reiner NE, McMaster WR. 2003. Survival of Mycobacterium tuberculosis in host macrophages involves resistance to apoptosis dependent upon induction of antiapoptotic Bcl-2 family member Mcl-1. J. Immunol. 170:430–437 [DOI] [PubMed] [Google Scholar]
- 17. Sly LM, Lopez M, Nauseef WM, Reiner NE. 2001. 1α,25-Dihydroxyvitamin D3-induced monocyte antimycobacterial activity is regulated by phosphatidylinositol 3-kinase and mediated by the NADPH-dependent phagocyte oxidase. J. Biol. Chem. 276:35482–35493 doi:10.1074/jbc.M102876200 [DOI] [PubMed] [Google Scholar]
- 18. Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, Ochoa MT, Schauber J, Wu K, Meinken C, Kamen DL, Wagner M, Bals R, Steinmeyer A, Zugel U, Gallo RL, Eisenberg D, Hewison M, Hollis BW, Adams JS, Bloom BR, Modlin RL. 2006. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 311:1770–1773 doi:10.1126/science.1123933 [DOI] [PubMed] [Google Scholar]
- 19. Liu PT, Stenger S, Tang DH, Modlin RL. 2007. Cutting edge: vitamin D-mediated human antimicrobial activity against Mycobacterium tuberculosis is dependent on the induction of cathelicidin. J. Immunol. 179:2060–2063 [DOI] [PubMed] [Google Scholar]
- 20. Yuk JM, Shin DM, Lee HM, Yang CS, Jin HS, Kim KK, Lee ZW, Lee SH, Kim JM, Jo EK. 2009. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe 6:231–243 doi:10.1016/j.chom.2009.08.004 [DOI] [PubMed] [Google Scholar]
- 21. Heldwein KA, Fenton MJ. 2002. The role of Toll-like receptors in immunity against mycobacterial infection. Microbes Infect. 4:937–944 [DOI] [PubMed] [Google Scholar]
- 22. Means TK, Wang S, Lien E, Yoshimura A, Golenbock DT, Fenton MJ. 1999. Human Toll-like receptors mediate cellular activation by Mycobacterium tuberculosis. J. Immunol. 163:3920–3927 [PubMed] [Google Scholar]
- 23. Underhill DM, Ozinsky A, Smith KD, Aderem A. 1999. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc. Natl. Acad. Sci. U. S. A. 96:14459–14463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Haug C, Muller F, Aukrust P, Froland SS. 1994. Subnormal serum concentration of 1,25-vitamin D in human immunodeficiency virus infection: correlation with degree of immune deficiency and survival. J. Infect. Dis. 169:889–893 [DOI] [PubMed] [Google Scholar]
- 25. Mueller NJ, Fux CA, Ledergerber B, Elzi L, Schmid P, Dang T, Magenta L, Calmy A, Vergopoulos A, Bischoff-Ferrari HA, Swiss HIV Cohort Study 2010. High prevalence of severe vitamin D deficiency in combined antiretroviral therapy-naive and successfully treated Swiss HIV patients. AIDS 24:1127–1134 doi:10.1097/QAD.0b013e328337b161 [DOI] [PubMed] [Google Scholar]
- 26. Wasserman P, Rubin DS. 2010. Highly prevalent vitamin D deficiency and insufficiency in an urban cohort of HIV-infected men under care. AIDS Patient Care STDS 24:223–227 doi:10.1089/apc.2009.0241 [DOI] [PubMed] [Google Scholar]
- 27. Tachado SD, Li X, Bole M, Swan K, Anandaiah A, Patel NR, Koziel H. 2010. MyD88-dependent TLR4 signaling is selectively impaired in alveolar macrophages from asymptomatic HIV+ persons. Blood 115:3606–3615 doi:10.1182/blood-2009-10-250787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tachado SD, Zhang J, Zhu J, Patel N, Koziel H. 2005. HIV impairs TNF-alpha release in response to Toll-like receptor 4 stimulation in human macrophages in vitro. Am. J. Respir. Cell Mol. Biol. 33:610–621 [DOI] [PubMed] [Google Scholar]
- 29. Folks TM, Justement J, Kinter A, Dinarello CA, Fauci AS. 1987. Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cell line. Science 238:800–802 [DOI] [PubMed] [Google Scholar]
- 30. Folks TM, Justement J, Kinter A, Schnittman S, Orenstein J, Poli G, Fauci AS. 1988. Characterization of a promonocyte clone chronically infected with HIV and inducible by 13-phorbol-12-myristate acetate. J. Immunol. 140:1117–1122 [PubMed] [Google Scholar]
- 31. Koziel H, Eichbaum Q, Kruskal BA, Pinkston P, Rogers RA, Armstrong MY, Richards FF, Rose RM, Ezekowitz RA. 1998. Reduced binding and phagocytosis of Pneumocystis carinii by alveolar macrophages from persons infected with HIV-1 correlates with mannose receptor downregulation. J. Clin. Invest. 102:1332–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saukkonen JJ, Bazydlo B, Thomas M, Strieter RM, Keane J, Kornfeld H. 2002. Beta-chemokines are induced by Mycobacterium tuberculosis and inhibit its growth. Infect. Immun. 70:1684–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang J, Zhu J, Imrich A, Cushion M, Kinane TB, Koziel H. 2004. Pneumocystis activates human alveolar macrophage NF-kappaB signaling through mannose receptors. Infect. Immun. 72:3147–3160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koziel H, Kim S, Reardon C, Li X, Garland R, Pinkston P, Kornfeld H. 1999. Enhanced in vivo human immunodeficiency virus-1 replication in the lungs of human immunodeficiency virus-infected persons with Pneumocystis carinii pneumonia. Am. J. Respir. Crit. Care Med. 160:2048–2055 [DOI] [PubMed] [Google Scholar]
- 35. Rennard SI, Basset G, Lecossier D, O'Donnell KM, Pinkston P, Martin PG, Crystal RG. 1986. Estimation of volume of epithelial lining fluid recovered by lavage using urea as marker of dilution. J. Appl. Physiol. 60:532–538 [DOI] [PubMed] [Google Scholar]
- 36. Harris J, Hope JC, Keane J. 2008. Tumor necrosis factor blockers influence macrophage responses to Mycobacterium tuberculosis. J. Infect. Dis. 198:1842–1850 doi:10.1086/593174 [DOI] [PubMed] [Google Scholar]
- 37. White JH. 2008. Vitamin D signaling, infectious diseases, and regulation of innate immunity. Infect. Immun. 76:3837–3843 doi:10.1128/IAI.00353-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mora JR, Iwata M, von Andrian UH. 2008. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat. Rev. Immunol. 8:685–698 doi:10.1038/nri2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Quesniaux V, Fremond C, Jacobs M, Parida S, Nicolle D, Yeremeev V, Bihl F, Erard F, Botha T, Drennan M, Soler MN, Le Bert M, Schnyder B, Ryffel B. 2004. Toll-like receptor pathways in the immune responses to mycobacteria. Microbes Infect. 6:946–959 doi:10.1016/j.micinf.2004.04.016 [DOI] [PubMed] [Google Scholar]
- 40. Prehn JL, Fagan DL, Jordan SC, Adams JS. 1992. Potentiation of lipopolysaccharide-induced tumor necrosis factor-alpha expression by 1,25-dihydroxyvitamin D3. Blood 80:2811–2816 [PubMed] [Google Scholar]
- 41. Baldwin AS., Jr 1996. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu. Rev. Immunol. 14:649–683 [DOI] [PubMed] [Google Scholar]
- 42. Muzio M, Natoli G, Saccani S, Levrero M, Mantovani A. 1998. The human Toll signaling pathway: divergence of nuclear factor kappaB and JNK/SAPK activation upstream of tumor necrosis factor receptor-associated factor 6 (TRAF6). J. Exp. Med. 187:2097–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ostuni R, Zanoni I, Granucci F. 2010. Deciphering the complexity of Toll-like receptor signaling. Cell. Mol. Life Sci. 67:4109–4134 doi:10.1007/s00018-010-0464-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu HZ, Gong JP, Wu CX, Peng Y, Li XH, You HB. 2005. The U937 cell line induced to express CD14 protein by 1,25-dihydroxyvitamin D3 and be sensitive to endotoxin stimulation. Hepatobiliary Pancreat. Dis. Int. 4:84–89 [PubMed] [Google Scholar]
- 45. Lawn SD, Bekker LG, Wood R. 2005. How effectively does HAART restore immune responses to Mycobacterium tuberculosis? Implications for tuberculosis control. AIDS 19:1113–1124 [DOI] [PubMed] [Google Scholar]
- 46. Li X, Han X, Llano J, Bole M, Zhou X, Swan K, Anandaiah A, Nelson B, Patel NR, Reinach PS, Koziel H, Tachado SD. 2011. Mammalian target of rapamycin inhibition in macrophages of asymptomatic HIV+ persons reverses the decrease in TLR-4-mediated TNF-alpha release through prolongation of MAPK pathway activation. J. Immunol. 187:6052–6058 doi:10.4049/jimmunol.1101532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Haug CJ, Muller F, Aukrust P, Froland SS. 1998. Different effect of 1,25-dihydroxyvitamin D3 on replication of Mycobacterium avium in monocyte-derived macrophages from human immunodeficiency virus-infected subjects and healthy controls. Immunol. Lett. 63:107–112 [DOI] [PubMed] [Google Scholar]
- 48. Buhl R, Jaffe HA, Holroyd KJ, Borok Z, Roum JH, Mastrangeli A, Wells FB, Kirby M, Saltini C, Crystal RG. 1993. Activation of alveolar macrophages in asymptomatic HIV-infected individuals. J. Immunol. 150:1019–1028 [PubMed] [Google Scholar]
- 49. Martineau AR, Timms PM, Bothamley GH, Hanifa Y, Islam K, Claxton AP, Packe GE, Moore-Gillon JC, Darmalingam M, Davidson RN, Milburn HJ, Baker LV, Barker RD, Woodward NJ, Venton TR, Barnes KE, Mullett CJ, Coussens AK, Rutterford CM, Mein CA, Davies GR, Wilkinson RJ, Nikolayevskyy V, Drobniewski FA, Eldridge SM, Griffiths CJ. 2011. High-dose vitamin D(3) during intensive-phase antimicrobial treatment of pulmonary tuberculosis: a double-blind randomised controlled trial. Lancet 377:242–250 doi:10.1016/S0140-6736(10)61889-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wejse C, Gomes VF, Rabna P, Gustafson P, Aaby P, Lisse IM, Andersen PL, Glerup H, Sodemann M. 2009. Vitamin D as supplementary treatment for tuberculosis: a double-blind, randomized, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 179:843–850 doi:10.1164/rccm.200804-567OC [DOI] [PubMed] [Google Scholar]
- 51. Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, Siegel JN, Braun MM. 2001. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N. Engl. J. Med. 345:1098–1104 [DOI] [PubMed] [Google Scholar]