

Abstract
Streptococcus iniae causes systemic infection characterized by meningitis and sepsis. Here, we report a larval zebrafish model of S. iniae infection. Injection of wild-type S. iniae into the otic vesicle induced a lethal infection by 24 h postinfection. In contrast, an S. iniae mutant deficient in polysaccharide capsule (cpsA mutant) was not lethal, with greater than 90% survival at 24 h postinfection. Live imaging demonstrated that both neutrophils and macrophages were recruited to localized otic infection with mutant and wild-type S. iniae and were able to phagocytose bacteria. Depletion of neutrophils and macrophages impaired host survival following infection with wild-type S. iniae and the cpsA mutant, suggesting that leukocytes are critical for host survival in the presence of both the wild-type and mutant bacteria. However, zebrafish larvae with impaired neutrophil function but normal macrophage function had increased susceptibility to wild-type bacteria but not the cpsA mutant. Taking these findings together, we have developed a larval zebrafish model of S. iniae infection and have found that although neutrophils are important for controlling infection with wild-type S. iniae, neutrophils are not necessary for host defense against the cpsA mutant.
INTRODUCTION
Streptococcus iniae is a major aquatic pathogen and a potential zoonotic pathogen capable of causing systemic disease characterized by meningitis and sepsis in both fish and humans. It is both aerobic and facultatively anaerobic, Gram-positive, beta-hemolytic, and nontypeable by the Lancefield grouping system (1). First isolated from the subcutaneous abscesses of a captive Amazon freshwater dolphin (Inia geoffrensis) in the 1970s, S. iniae has a worldwide distribution and can infect over 27 species of freshwater and saltwater fish (1–3). Although it is able to colonize the surface of fish and cause skin infections, S. iniae disease usually presents as meningoencephalitits with major involvement of the central nervous system (CNS) but can also cause sepsis and invasive systemic infection involving multiple organs. Mortality rates are as high as 30 to 50% in infected fish ponds, resulting in over $100 million in global losses annually (2, 4, 5).
More recently, there have been reported human cases of S. iniae infection. The first reported human case of S. iniae infection was in Texas in 1991 (6). Since then, there have been at least 25 reported human cases of S. iniae infection, although this number is thought to be an underestimate (2). S. iniae is an opportunistic pathogen infecting mainly elderly or immunocompromised people with a recent history of fish handling and causes invasive infection with clinical pathologies similar to those caused by group A and group B streptococci. Disease in humans is usually characterized by cellulitis of the upper extremities, similar to infection with group A streptococci, but there have been cases of more systemic infection, including sepsis and meningitis, similar to infection with group B streptococci (6–10).
A better understanding of S. iniae disease pathogenesis requires an appropriate model system. The embryonic zebrafish (Danio rerio) has a number of advantages as a model that makes it an increasingly attractive vertebrate host in which to study infectious disease (11–24). The zebrafish is genetically tractable with a sequenced genome (http://www.sanger.ac.uk/Projects/D_rerio/) and is amenable to forward and reverse genetic screens. Small size and high fecundity make the zebrafish amenable to high-throughput chemical and drug screens and also allow for studies of disease progression and pathogenesis.
Zebrafish embryos and larvae are more experimentally tractable than adults. Although adaptive immunity is not functionally mature until at least 2 to 3 weeks postfertilization (25–27), zebrafish embryos have a highly conserved vertebrate innate immune system, including complement, Toll-like receptors, and neutrophils and macrophages that are capable of phagocytic activity by 28 to 30 h postfertilization (hpf) (18, 28–32). The translucency of the embryonic and larval stages makes zebrafish more optically accessible and amenable to live imaging. In addition, genetic studies relying on either the injection of synthetic mRNA for overexpression or the injection of antisense morpholino oligonucleotides (MOs) for gene knockdown (33) are available during the embryonic and larval periods.
An adult zebrafish model of S. iniae infection has previously been reported (34). Intramuscular injection of 103 CFU of S. iniae wild-type strain 9117 results in an influx of inflammatory cells at the injection site and rapid host mortality within 36 to 48 h postinfection (hpi) with evidence of systemic infection, including bacterial dissemination to the brain and other organs (34). The rapid time to death suggests that this period is too short to engage the adaptive immune system, but it is unclear how the immune system responds to S. iniae in the adult fish because the tools to study the host immune response in adult zebrafish are limited. The optical transparency and genetic tractability of the embryonic and early larval stages of the zebrafish make it an attractive model for studying the host immune response to S. iniae infection in real time in vivo.
S. iniae utilizes a number of identified virulence factors to evade host defense and establish disease (35–40). In particular, the polysaccharide capsule of S. iniae is important for virulence in adult zebrafish (41–43). A mutagenesis screen for S. iniae mutants with attenuated virulence and decreased systemic spread was done in adult zebrafish. It was found that many of the identified mutants had insertions in genes required for capsule biosynthesis, including the cpsA mutant, which has a mutation in the first gene of the putative S. iniae capsule operon (42, 43). The cpsA gene encodes a transcriptional regulator of the operon, and a polar insertion in this gene results in decreased transcription of the capsule operon and reduced production of capsule as determined by buoyant density in a Percoll gradient (42). In addition, many S. iniae capsule mutants were efficiently phagocytosed in human whole blood (43) and by cultured fish macrophages (41) compared to wild-type S. iniae.
Here, we have developed a zebrafish larva-S. iniae infection model to study host-pathogen interactions using real-time imaging. We visualized the innate immune response to S. iniae infection in real time and investigated how the innate immune response controls infection with wild-type S. iniae and the cpsA mutant. Our findings suggest that although both neutrophils and macrophages are important for surviving infection with wild-type S. iniae, neutrophils are not necessary for host defense against the cpsA mutant.
MATERIALS AND METHODS
Zebrafish maintenance.
Adult fish and embryos were maintained in accordance with the University of Wisconsin—Madison Research Animal Resources Center (Madison, WI). A light cycle of 10 h of darkness and 14 h of light was used. Wild-type fish, type AB, were used in these experiments and to generate all transgenic lines; this population of AB wild-type fish was not inbred. Embryos were obtained by natural spawning and were raised at 28.5°C in E3 medium (44). For experiments performed using embryos aged 2 days postfertilization (dpf), embryos were dechorionated with pronase. For bacterial infection and live imaging, embryos were anesthetized in E3 medium containing 0.2 mg/ml tricaine (ethyl 3-aminobenzoate; Sigma-Aldrich, St. Louis, MO). The previously published transgenic lines Tg(mpx:dendra2) (45), Tg(mpx:mCherry-2A-rac2wt), and Tg(mpx:mCherry-2A-rac2-d57n) (46) were used in these studies.
Generation of the transgenic Tg(mpeg1:dendra2) zebrafish line.
The mpeg1 promoter region was cloned from a bacterial artificial chromosome (BAC) construct (clone DKEY-67k20; Imagene, Hanover, NH) into a backbone vector containing minimal Tol2 elements (47) using previously reported primers (48) to amplify nucleotides 30437143 to 30438999 from chromosome 8. This generated a 1.86-kb sequence immediately proximal to the mpeg1 5′ untranslated region. The mpeg1 region was PCR amplified with forward primer 5′-ACGTGGATCCTTTTGCTGTCTCCTGC-3′ and reverse primer 5′-ACGTCTCGAGTGTTGGAGCACATCTG-3′ and then inserted into the Tg(mpx:dendra2) vector (45). The Tg(mpx:dendra2) vector and PCR product were cut with BamHI/XhoI and then ligated to remove the mpx promoter and replace it with the mpeg1 promoter. One-cell-stage wild-type AB embryos were injected with a 3-nl solution containing 25 ng/μl Tg(mpeg1:dendra2) DNA along with 35 ng/μl transposase mRNA and grown at 28.5°C.
Bacterial strains, media, and culture conditions.
The S. iniae wild-type strain 9117 and an S. iniae capsule mutant with a polar insertional mutation in the cpsA gene (cpsA mutant) have been previously described (42, 49). The cpsA gene is the first gene of the S. iniae capsule operon and encodes a transcriptional regulator (42). Therefore, the mutation of this gene results in decreased transcription of the capsule operon and, thus, decreased production of capsule (42). Bacteria were cultured in Todd-Hewitt medium (Sigma-Aldrich, St. Louis, MO) supplemented with 0.2% yeast extract (BD Biosciences, San Jose, CA) and 2% proteose peptone (vegetable) (Sigma-Aldrich, St. Louis, MO) (THY+P). Bacteria were cultured in sealed tubes without agitation at 37°C. Solid THY+P plates were made by supplementing liquid medium with Bacto agar (BD Biosciences, San Jose, CA) to a final concentration of 1.4%.
Preparation of streptococci.
Bacterial cultures were grown overnight at 37°C in THY+P without agitation. Overnight cultures were diluted 1:100 in fresh THY+P medium, incubated at 37°C, and harvested in the mid-logarithmic phase of growth when the optical density at 600 nm reached 0.250 (corresponding to ∼108 CFU/ml) as determined with a NanoDrop spectrophotometer. Bacteria were pelleted by centrifugation at 1,500 × g for 5 min, washed in fresh phosphate-buffered saline (PBS), repelleted, and resuspended in PBS to achieve the desired optical density at 600 nm. Phenol red tracking dye was added to bacterial aliquots prior to injection at a final concentration of 0.1%. For infections with heat-killed bacteria, an equivalent of 1,000 CFU wild-type S. iniae was heated at 95°C for 30 min. Heat killing reduced the number of viable bacteria to undetectable levels as confirmed by plating an injection volume on solid THY+P medium.
Microinjection of bacteria into zebrafish embryos.
Bacterial cells were microinjected in a 1-nl volume into the otic vesicle as previously described (50). The inoculum size was determined by injecting an equal volume of bacteria into PBS before and after zebrafish injections with each needle and plating on THY+P agar to quantify CFU. The scoring of live or dead embryos was determined based on the presence of a heartbeat and response to gentle touching with a pipette tip. After injection, zebrafish were returned to E3 medium, incubated at 28.5°C, and monitored for survival at the indicated time points or fixed for Sudan Black staining at 2 hpi.
Sudan Black staining.
Embryos were fixed at 2 hpi in 4% paraformaldehyde in PBS, and neutrophils were stained with Sudan Black as previously described (29). Neutrophil recruitment was determined by obtaining visual counts of stained neutrophils in the otic vesicle using a Nikon SMZ 745 stereomicroscope (Melville, NY).
Bacterial enumeration from infected embryos.
At the indicated times, embryos were anesthetized, rinsed in E3 medium, collected in 100 μl of 0.2% Triton X-100 in PBS, and homogenized by passing up and down 5 times through a 27 1/2-gauge needle. Serial dilutions of the homogenates were made in PBS and plated onto Columbia colistin-nalidixic acid (CNA) agar (dotScientific Inc., Burton, MI) for selective isolation of Gram-positive bacteria. CFU were enumerated after 48 h of incubation at 37°C. Log values of CFU counts were recorded.
MO injection.
All morpholino oligonucleotides (MOs) were purchased from Gene Tools, LLC (Philomath, OR), resuspended in distilled water, and stored at room temperature at a stock concentration of 1 mM. Three nanoliters of MOs was injected into the yolks of 1-cell-stage AB wild-type embryos at the indicated concentrations (Pu.1 MO, 500 μM; standard control MO, 500 μM). The translation-blocking Pu.1 MO was previously described (51). Elimination of the myeloid lineage in Pu.1 morphants was confirmed by injection into the double transgenic Tg(mpx:mcherry) × Tg(mpeg1:dendra2) line.
Microscope analysis and live imaging.
Anesthetized larvae were settled onto the bottom of a custom-made, glass-bottom dish. Time-lapse fluorescence images were acquired with a laser scanning confocal microscope (Fluoview FV1000; Olympus, Center Valley, PA) using a numerical aperture 0.75/20× objective. For confocal imaging, each fluorescent channel (488 nm and 543 nm) and differential interference contrast (DIC) images were acquired by sequential line scanning. Z-series were acquired using a 250-μm pinhole and 2- to 6-μm step sizes. Z-stacked fluorescence images were overlaid with a single DIC plane. Where indicated, bacteria were labeled with 5 μM CellTracker Red CMPTX dye (catalog number C34552; Molecular Probes, Invitrogen, Grand Island, NY) according to the manufacturer's instructions.
Statistical analyses.
All statistical analyses were performed using GraphPad Prism, versions 4 and 6. For quantification of neutrophil and macrophage recruitment, the Kruskal-Wallis test followed by Dunn's multiple-comparison posttest was used. To monitor survival, a Kaplan-Meier curve was calculated for each inoculum as well as for PBS-mock-infected larvae. Differences in survival between bacterium-infected larvae and mock-infected larvae were calculated by the log rank test.
RESULTS
Wild-type S. iniae but not a capsule mutant impairs survival of zebrafish larvae.
We tested the susceptibility of zebrafish larvae to infection with a wild-type strain of S. iniae. Larvae at ∼72 h postfertilization (hpf) were microinjected in the left otic vesicle with wild-type bacteria (Fig. 1A). The otic vesicle forms during development when the ear placodal ectoderm cavitates to form a hollow cavity of epithelium (52, 53). The otic vesicle is normally devoid of leukocytes, but in response to local bacterial infection, leukocytes are recruited to the otic cavity (50). The otic vesicle has previously been used to study localized infection (29, 46, 50, 54–56). We chose otic vesicle infection to allow for the direct observation of host neutrophil and macrophage responses to localized S. iniae infection.
Fig 1.

Zebrafish larvae are susceptible to otic vesicle infection with wild-type S. iniae but not the cpsA mutant. (A) Image of the head region of a zebrafish larva at 72 hpf. The site of microinjection in the otic vesicle is indicated. Scale bar, 200 μm. (B) Survival of 72-hpf larvae microinjected with various doses of viable wild-type S. iniae or an equivalent of 1,000 CFU wild-type bacteria heat killed at 95°C for 1 h (n = 24 per group). Compared to mock-infected larvae, each dose showed a significant difference in survival as calculated by the log rank test (P < 0.0001). The data are from 3 independent experiments, each with 24 larvae per condition. (C) Survival of 72-hpf larvae microinjected with various doses of wild-type bacteria or the cpsA mutant (n = 24 per group). Compared to larvae infected with wild-type bacteria, larvae infected with comparable doses of the cpsA mutant showed a significant difference in survival as determined by the log rank test (100 CFU cpsA versus 100 CFU wild-type S. iniae, P < 0.0001; 1,000 CFU cpsA versus 1,000 CFU wild-type S. iniae, P < 0.0001). The data are from 3 independent experiments, each with 24 larvae per condition. (D) Enumeration of viable bacteria from individual larvae at the indicated time points postinfection. The larvae were microinjected with PBS, ∼100 CFU wild-type bacteria, or ∼100 CFU cpsA bacteria, and individual larvae were euthanized, homogenized, and plated on CNA agar. Results are representative of at least 3 independent experiments. (E) Bright-field images of larvae microinjected with PBS, ∼100 CFU wild-type bacteria, or ∼100 CFU cpsA bacteria taken at 24 hpi. Scale bar, 500 μm.
Similar to infection of adult zebrafish (34), larvae were susceptible to S. iniae infection. An inoculum of ∼100 CFU resulted in the death of about 40% of larvae by 24 hpi, whereas mock infection with PBS did not result in host mortality (Fig. 1B) (P < 0.0001). Injections with increasing doses of bacteria indicated that lethality following S. iniae infection was dose dependent (Fig. 1B), with ∼1,000 CFU killing about 80% of infected larvae within 24 hpi and doses as low as ∼10 CFU killing more than 40% of infected larvae by 48 hpi. However, lethality required viable bacteria, as injection of heat-killed S. iniae did not affect host survival (Fig. 1B).
The polysaccharide capsule is an important virulence factor for S. iniae in establishing systemic disease in fish (36, 41–43, 57). We next sought to determine whether the S. iniae 9117 capsule mutant (cpsA) was virulent in zebrafish larvae. The cpsA mutant has an insertional mutation in a transcriptional regulator of the S. iniae capsule operon that results in decreased production of capsule and attenuated virulence in adult zebrafish compared to those with the wild-type strain (42). In contrast to infection with wild-type bacteria, greater than 90% of larvae injected with comparable doses of the cpsA mutant survived for at least 72 hpi (Fig. 1C). The attenuated virulence of the cpsA mutant in larvae parallels what has been shown in the adult zebrafish infection model (42). These data validate the use of zebrafish larvae in modeling streptococcal disease.
As a measure of disease progression, we monitored the bacterial load over time in whole animals infected with ∼100 CFU bacteria. Larvae were infected with the wild-type strain or the cpsA mutant, and at 0, 18, 24, 48, and 60 hpi, three larvae per condition were sacrificed, homogenized, and plated on colistin-nalidixic acid (CNA) agar plates to select for the growth of Gram-positive bacteria. In larvae infected with wild-type S. iniae, the amount of recoverable bacteria increased over time from 100 CFU at the time of inoculation to the greatest bacterial loads of around 104 CFU at 60 hpi (Fig. 1D). We noted that bacterial load and host susceptibility to infection corresponded with the physical appearance of infected larvae. Larvae infected with the wild-type strain developed a dark and opaque yolk sac and deteriorating necrotic body (Fig. 1E). The ability of some larvae to survive infection suggests that some individuals may be naturally more resistant to S. iniae infection than others or could reflect slight variations in the inoculum. However, it should be noted that the inoculum size as determined by enumerating bacterial loads in whole embryos sacrificed immediately following microinjection (∼100 CFU) was similar to the inoculum size determined by plating the injection volume from the needle taken before and after infections (data not shown).
In contrast to larvae infected with wild-type bacteria, the amount of viable bacteria recovered from larvae infected with the cpsA mutant remained approximately equivalent to the initial inoculum over the 60-h monitoring period (∼100 CFU) (Fig. 1D). This suggests that the cpsA mutant is deficient in its ability to proliferate in the host and establish disease. To differentiate between an inability to proliferate in the host and an overall growth defect, in vitro growth assays were performed, and the cpsA mutant was found to exhibit wild-type growth (data not shown). Larvae infected with the cpsA mutant had a slight change in opacity of the yolk sac but overall looked similar to mock (PBS)-infected animals (Fig. 1E).
Host neutrophil-S. iniae interactions during the early stages of infection.
One advantage of developing a zebrafish larval model of S. iniae infection is that the optical transparency at this developmental stage allows for real-time visualization of the host immune response to infection in vivo. Although B and T lymphocytes are not fully functional until 2 to 3 weeks postfertilization, innate immunity develops early during zebrafish development. By the age of the larvae used in this study, there are both neutrophil and macrophage populations with antimicrobial capabilities, including phagocytosis and respiratory bursts (18, 29, 58). To determine the contributions of neutrophils and macrophages to the host immune response, we used transgenic lines expressing the fluorescent protein Dendra2 specifically in neutrophils [Tg(mpx:dendra2)] (45) or macrophages [Tg(mpeg1:dendra2)]. To visualize bacteria, the wild-type strain and the cpsA mutant were labeled with the CellTracker Red CMPTX dye.
Neutrophils are an important part of the innate immune system and are typically the first responders to a site of infection. To quantify the number of neutrophils recruited to the otic vesicle during early S. iniae infection, we fixed infected larvae at 2 hpi and stained with the neutrophil-specific stain Sudan Black (29). Neutrophils were robustly recruited to the site of infection with wild-type bacteria by 2 hpi (Fig. 2A and B). The median number of neutrophils recruited to the site of infection ranged from ∼9 neutrophils following injection of ∼10 CFU wild-type bacteria to ∼20 neutrophils following injection of 1,000 CFU (Fig. 2B). Injection of increasing doses of the wild-type strain demonstrated that neutrophil recruitment was dose dependent, and neutrophils were also recruited to heat-killed bacteria (Fig. 2A and B). Moreover, neutrophils were recruited to the cpsA mutant, similar to the case for wild-type bacteria. Therefore, although the cpsA mutant produces a nonlethal infection in larvae, the initial host neutrophil responses to pathogenic wild-type S. iniae and the nonpathogenic cpsA mutant appear to be similar.
Fig 2.

Neutrophils are recruited to sites of localized S. iniae infection. (A) Sudan Black staining of neutrophils recruited to the otic vesicle in larvae infected at 72 hpf with different doses of the wild-type strain as indicated. Larvae were fixed at 2 hpi. Representative images are shown for larvae microinjected with PBS, ∼1,000 CFU wild-type bacteria, or an equivalent of 1,000 CFU heat-killed bacteria (n = 25 to 35 per group). Scale bar, 200 μm. (B) Quantification of the Sudan Black-stained larvae from panel A. Each dot represents neutrophil counts in the otic vesicle for an individual larva. (C) Sudan Black staining of neutrophils recruited to the otic vesicle in larvae infected at 72 hpf with either PBS, ∼100 CFU wild-type bacteria, or ∼100 CFU cpsA bacteria. Larvae were fixed at 2 hpi (n = 11 to 16 per group). Scale bar, 300 μm. (D) Quantification of Sudan Black-stained larvae from panel C. Each dot represents neutrophil counts for an individual larva. Results are representative of 3 independent experiments. ***, P < 0.001; **, P < 0.01; ns, not significant (as determined by the Kruskal-Wallis test with Dunn's multiple-comparison posttest).
To visualize neutrophil responses to infection with wild-type bacteria, we microinjected CellTracker Red-labeled bacteria into the left otic vesicle of the transgenic Tg(mpx:dendra2) fish at 72 hpf. The infected embryos were placed in a glass-bottom dish and immediately imaged in the left otic vesicle using a laser scanning confocal microscope. Neutrophils were recruited rapidly to the site of infection, even by the start of the movie at ∼10 min postinfection (mpi) (Fig. 3A; see Movie S1 in the supplemental material). Several green-labeled neutrophils entered the otic cavity and appeared to engulf red-labeled wild-type bacterial cells. A neutrophil could be observed extending a thin protrusion toward some bacteria (Fig. 3A, inset at 59 min; see Movie S1 in the supplemental material). As neutrophils took up bacteria, they became less motile and developed a rounded morphology (Fig. 3A, insets). After phagocytosis, the neutrophils recovered their polarized morphology and migrated to a new location.
Fig 3.

Real-time imaging of neutrophil responses to S. iniae infection. A confocal microscope was used to observe the neutrophil response to infection with the wild-type strain or the cpsA mutant using a 20× objective. Tg(mpx-dendra2) larvae were infected at 72 hpf with ∼100 CFU wild-type (A) or ∼100 CFU cpsA (B) bacteria labeled with 5 μM CellTracker Red dye. Six frames were extracted from a 96-min or a 90-min movie, respectively. The number in the lower right corner of each frame represents the time in minutes. Time zero is at ∼10 mpi. The otic vesicle is outlined in white. The insets show an enlarged picture of the cell indicated by the arrow or arrowhead colocalized with labeled bacteria. Scale bar, 40 μm. Results are representative of 3 movies from 3 independent experiments. See also Movies S1 and S2 in the supplemental material.
Tg(mpx:dendra2) larvae injected with the cpsA mutant resulted in a similar response to localized infection (Fig. 3B; see Movie S2 in the supplemental material). Green-labeled neutrophils were recruited to the site of infection by ∼10 mpi and rapidly engulfed the red-labeled cpsA mutant. The motility of these bacterium-laden neutrophils seemed to decrease, and the neutrophils developed a rounded appearance, similar to what was seen following infection with wild-type bacteria. In contrast to wild-type bacteria, the cpsA mutant was seen in several small vacuoles in some neutrophils (denoted by the arrowheads in Fig. 3B and Movie S2 in the supplemental material) and was also in large vacuoles in other neutrophils (denoted by the arrows in Fig. 3B and Movie S2 in the supplemental material).
Host macrophage-S. iniae interactions during the early stages of infection.
To characterize the macrophage response to S. iniae infection, ∼100 CFU CellTracker Red-labeled wild-type bacteria were microinjected into the left otic vesicle of Tg(mpeg1:dendra2) larvae at ∼72 hpf. Macrophage recruitment to the site of infection was quantified by fixing infected Tg(mpeg1:dendra2) larvae at 2 hpi. Macrophages were recruited following infection with either wild-type bacteria or the cpsA mutant (Fig. 4A and B), and on average between 35 and 40 macrophages were recruited to the otic vesicle following infection.
Fig 4.
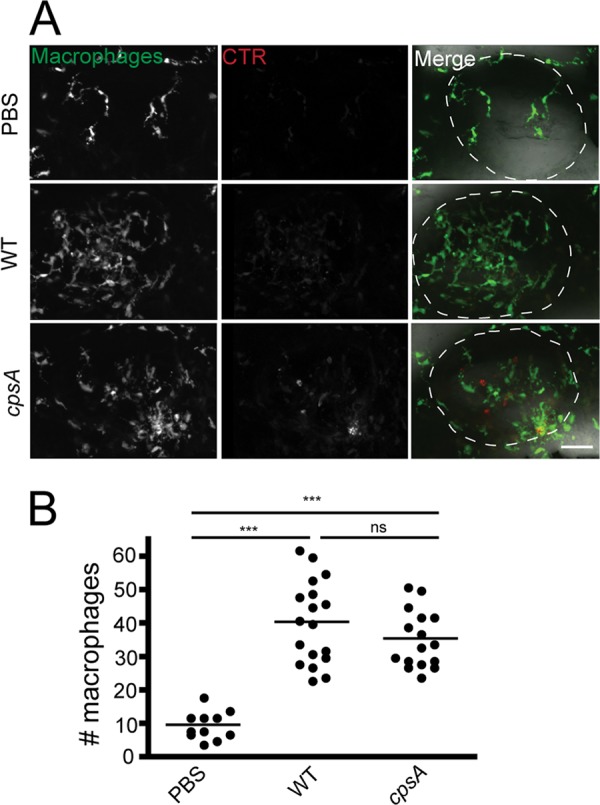
Macrophages are recruited to sites of localized S. iniae infection. (A) Fluorescence images of Tg(mpeg1:dendra2) larvae infected at 72 hpf with ∼100 CFU wild-type or ∼100 CFU cpsA bacteria. Larvae were fixed at 2 hpi and imaged using a confocal laser scanning microscope with a 20× objective. Scale bar, 40 μm. (B) Quantification of the number of macrophages recruited to the otic vesicle at 2 hpi in the larvae from panel A. Each dot represents an individual larva (n = 15 to 20 per group). Results are representative of 3 independent experiments. ***, P < 0.001; ns, not significant (as determined by the Kruskal-Wallis test followed by Dunn's multiple-comparison posttest).
Real-time imaging demonstrated that macrophages, like neutrophils, were also rapidly recruited to the site of S. iniae infection (Fig. 5A; see Movie S3 in the supplemental material). Occasionally, it was possible to observe a macrophage taking up bacteria (Fig. 5A, inset at 45 min; see Movie S3 in the supplemental material). The bacterium-carrying macrophages appeared to lose polarity, had reduced motility, and developed a rounded morphology. Additionally, there also appeared to be uninfected macrophages with large unfilled vacuoles characteristic of an activated phenotype.
Fig 5.

Real-time imaging of macrophage responses to S. iniae infection. A confocal microscope was used to observe the macrophage response to infection with wild-type bacteria or the cpsA mutant using a 20× objective. Tg(mpeg1:dendra2) larvae were infected at 72 hpf with ∼100 CFU wild-type (A) or ∼100 CFU cpsA (B) bacteria labeled with 5 μM CellTracker Red dye. Six frames were extracted from a 90-min or a 48-min movie, respectively. The number in the lower right corner of each frame represents the time in minutes. Time zero is at ∼10 mpi. The otic vesicle is outlined in white. The insets show an enlarged picture of a cell indicated by the arrow colocalized with labeled bacteria. Scale bar, 50 μm. Results are representative of 3 movies from 3 independent experiments. See also Movies S3 and S4 in the supplemental material.
Upon infection of Tg(mpeg1:dendra2) with the red-labeled cpsA mutant, macrophages were rapidly recruited to the otic vesicle at ∼10 mpi. This was similar to what was seen following infection with the wild-type strain. It was possible to observe a macrophage extend a protrusion and capture bacteria (Fig. 5B, inset at 6 min; see Movie S4 in the supplemental material). The phagocytosing macrophage lost its spindle shape and acquired a rounded appearance. The engulfed bacteria seemed to fill multiple vacuoles within the macrophage, but eventually the bacterium-filled vacuoles appeared to move together toward a central location (Fig. 5B, inset at 48 min; see Movie S4 in the supplemental material). It is also of note that it was easier to find macrophages phagocytosing the cpsA mutant than phagocytosing wild-type bacteria (data not shown). In addition, by 24 hpi, we observed both wild-type and mutant bacteria in macrophages outside the otic vesicle (data not shown), suggesting that macrophages may play a role in the dissemination of S. iniae. However, since we were unable to stably label the bacteria using fluorescent reporters, we could not observe macrophage-mediated bacterial phagocytosis and dissemination over time due to dye dilution from bacterial proliferation.
Myeloid cells are important for host survival following infection with wild-type S. iniae and the cpsA mutant.
Since both neutrophils and macrophages are rapidly recruited to the site of infection, we sought to genetically determine the contribution of host myeloid cells to defense against infection with the wild-type or mutant strain by targeted gene knockdown using morpholino oligonucleotides (33). To do this, we used a translation-blocking morpholino oligonucleotide targeting Pu.1, a transcription factor important for the differentiation of the myelo-erythroid progenitor cells into cells of the myeloid lineage, including neutrophils and macrophages (51). A Pu.1 or a mismatch control morpholino oligonucleotide was injected into double transgenic [Tg(mpx:mcherry) × Tg(mpeg1:dendra2)] embryos at the single-cell stage, and at 2 dpf, these morphants were infected with wild-type bacteria or the cpsA mutant or were mock treated with PBS. The morphants were monitored for survival. Because control larvae are highly susceptible to infection with wild-type S. iniae, we used a small inoculum (∼10 CFU) of wild-type bacteria for these studies to test whether Pu.1 morphants had increased sensitivity to infection. We found that Pu.1 morphants were significantly more susceptible to a low-dose infection with the wild-type strain than control morphants, with over 35% mortality in infected Pu.1 morphants by 24 hpi (Fig. 6A, panel ii) compared to less than 10% mortality in infected control morphants (Fig. 6A, panel i) (P < 0.0001). Thus, although wild-type S. iniae is able to establish disease and proliferate in wild-type zebrafish larvae, disease progression is more rapid in the absence of myeloid cells. This suggests a role for neutrophils and/or macrophages in controlling infection with the wild-type strain. Interestingly, Pu.1 morphants were also significantly more susceptible to infection with a higher dose (∼100 CFU) of the cpsA mutant, with almost 40% mortality in Pu.1 morphants at 24 hpi compared to less than 10% mortality in control morphants (Fig. 6A panels i and ii) (P < 0.0001). To test the sensitivity of Pu.1 morphants to infection with the cpsA mutant, we also infected morphants with a lower dose (∼10 CFU). When infected with only ∼10 CFU cpsA, there was greater than 40% mortality at 48 hpi, compared to less than 10% mortality of control morphants infected with a comparable dose of cpsA (Fig. 6A, panels i and ii) (P < 0.0001). To confirm depletion of myeloid lineage cells in Pu.1 morphants, embryos were imaged using a laser scanning confocal microscope prior to infection (Fig. 6B). Taken together, our findings show that Pu.1 morphants are more sensitive to infection with both the cpsA mutant and wild-type bacteria. Our findings also suggest that neutrophils and/or macrophages are necessary to control infection with the cpsA mutant.
Fig 6.

Myeloid cells are important for host survival following wild-type S. iniae and cpsA mutant infection. Double transgenic embryos [Tg(mpx:mCherry) × Tg(mpeg1:dendra2)] were injected at the single-cell stage with either 500 μM Pu.1 MO or 500 μM control (Ctrl) MO. (A) Survival curves of Ctrl (panel i) and Pu.1 (panel ii) morphants infected at 2 dpf with PBS, ∼10 CFU wild-type bacteria, ∼10 CFU cpsA bacteria, or ∼100 CFU cpsA bacteria (n = 24 per group). Compared to infected control morphants, Pu.1 morphants had a significant decrease in survival when infected with ∼10 CFU wild-type bacteria (P < 0.0001) or with either ∼10 CFU or ∼100 CFU of the cpsA mutant (P < 0.0001 and P < 0.0001) as determined by the log rank test. The data are from 3 independent experiments, each with 24 larvae per condition. (B) To monitor the efficiency of targeted knockdown, embryos were screened prior to infection on a laser scanning confocal microscope. Representative images are shown for Ctrl and Pu.1 morphants. Scale bar, 100 μm. (C) Enumeration of viable bacteria from morphants infected at 48 hpf with PBS, ∼10 CFU wild-type bacteria, ∼10 CFU cpsA bacteria, or ∼100 CFU cpsA bacteria. Morphants were euthanized at 24 hpi, homogenized, and plated on CNA agar. Results are representative of 3 independent experiments.
To monitor disease progression in the Pu.1 morphants, the bacterial loads were measured at 24 hpi. We found that the bacterial burden at this time point appeared to predict host mortality, whereas at later time points, it was difficult to measure bacterial load because the majority of the Pu.1 morphants had already succumbed to infection. The overall trend was that Pu.1 morphants had higher bacterial loads than control morphants following infection with either wild-type bacteria or the cpsA mutant. By 24 hpi, bacterial loads in Pu.1 morphants increased to ∼103 to 104 CFU following infection with ∼10 CFU wild-type bacteria, while in control morphants bacterial loads were about 10-fold lower (Fig. 6C). The increased susceptibility to infection with the cpsA mutant was also reflected in relative bacterial burdens. Pu.1 embryos infected with ∼10 CFU of the cpsA mutant had bacterial loads of ∼102.5 CFU by 24 hpi, and those infected with ∼100 CFU of the cpsA mutant had bacterial loads of 103 to 104 CFU. In contrast, in control morphants, there was not much increase in cpsA mutant levels above the initial inocula (Fig. 6C). This suggests that the ability of the bacteria to survive and proliferate in a host corresponds with increased mortality, and in the absence of myeloid cells, zebrafish larvae are susceptible to both a pathogenic strain and a nonpathogenic strain of S. iniae.
Neutrophils are important for controlling infection with wild-type S. iniae but not the cpsA mutant.
To determine the role of neutrophils in controlling S. iniae infection, we monitored the infection process in larvae with impaired neutrophil function. We used a previously generated zebrafish model of leukocyte adhesion deficiency in which the human dominant inhibitory mutation in Rac2, D57N, is expressed specifically in neutrophils (46). Neutrophils expressing the inhibitory Rac2(D57N) mutation are unable to respond to inflammatory stimuli, including otic infection with Pseudomonas aeruginosa (46). When larvae with the inhibitory Rac2(D57N) mutation expressed specifically in neutrophils were infected with the wild-type strain or the cpsA mutant, we observed that neutrophils were not recruited to the otic vesicle even though they were recruited in larvae ectopically expressing the wild-type Rac2 gene [Rac2(WT)] as shown by Sudan Black staining (Fig. 7A and B). This corresponds to what was previously shown regarding the inability of neutrophils expressing the mutated Rac2(D57N) to respond to local bacterial infection (46). However, the Rac2(D57N) dominant inhibitory mutation expressed in neutrophils did not affect macrophage recruitment to the otic vesicle following infection with either wild-type bacteria or the cpsA mutant (Fig. 7C and D). The Rac2(D57N) larvae were more susceptible to infection with a low-dose inoculum (∼10 CFU) of wild-type bacteria than larvae with the wild-type Rac2 gene [Rac2(WT)], with over 70% mortality of Rac2(D57N) fish compared to only 40% mortality of Rac2(WT) fish by 48 hpi (Fig. 7E) (P = 0.0004). This supports a role for neutrophils in controlling infection with wild-type bacteria. These data, when considered in combination with the Pu.1 morphants, indicate that both neutrophils and macrophages contribute to host defense against wild-type S. iniae infection. Interestingly, and in contrast to the response to wild-type bacteria, the Rac2(D57N) fish were not susceptible to infection with even ∼100 CFU of the cpsA mutant (Fig. 7E). These findings suggest that neutrophils are not important for host defense against the cpsA mutant, suggesting that macrophages are likely to play a key role in controlling infection with the cpsA mutant. Therefore, both neutrophils and macrophages are important for controlling infection with wild-type S. iniae, but neutrophils are not necessary to control infection with the cpsA mutant.
Fig 7.

Neutrophils are important for controlling infection with wild-type S. iniae but not the cpsA mutant. Tg(mpx:mCherry-2A-Rac2) [Rac2(WT)] or Tg(mpx:mCherry-2A-Rac2D57N) [Rac2(D57N)] larvae were infected at 48 or 72 hpf with PBS, wild-type S. iniae, or the cpsA mutant. (A) Larvae infected at 72 hpf were fixed at 2 hpi and stained with Sudan Black. Representative images are shown for larvae microinjected with PBS, ∼100 CFU wild-type bacteria, or ∼100 CFU cpsA bacteria. Scale bar, 300 μm. (B) Quantification of neutrophil recruitment to the otic vesicle shown in panel A (n = 8 to 15 per group). Medians with common letters indicate a P value of >0.05 by the Kruskal-Wallis test with Dunn's multiple-comparison posttest. Results are representative of 3 independent experiments. (C) Both the Rac2(WT) and Rac2(D57N) transgenic lines were crossed to Tg(mpeg1:dendra2), and the resulting double transgenic lines were infected at 72 hpf and fixed at 2 hpi. Representative images are shown for larvae microinjected with PBS, ∼100 CFU wild-type bacteria, or ∼100 CFU cpsA bacteria. Scale bar, 20 μm. (D) Quantification of macrophage recruitment to the otic vesicle shown in panel C (n = 20 per group). Medians with common letters indicate a P value of >0.05 by the Kruskal-Wallis test with Dunn's multiple-comparison posttest. Results are representative of 3 independent experiments. (E) Survival curves of larvae infected at 48 hpf with PBS, ∼10 CFU wild-type bacteria, or ∼100 CFU cpsA bacteria (n = 24 per group). Compared to infected Rac2(WT) larvae, Rac2(D57N) larvae had a significant difference in mortality when infected with wild-type bacteria (P = 0.0004) but no significant difference when infected with the cpsA mutant as determined by the log rank test. The data are from 3 independent experiments, each with 24 larvae per condition.
DISCUSSION
Here, we report a new model for the real-time observation of the innate immune response to S. iniae disease using zebrafish larvae. Injection of wild-type S. iniae into the otic vesicles of zebrafish larvae resulted in a dose-dependent host mortality accompanied by an increase in bacterial burden (Fig. 1). Zebrafish larvae were highly sensitive to wild-type S. iniae infection, with as few as ∼10 CFU resulting in lethal infection (Fig. 1B) and increasing bacterial burdens during the first 60 hpi (Fig. 1D). This infection model provided a powerful tool to differentiate host response to and outcome of infection with an S. iniae capsule mutant with an insertion in the cpsA gene. The S. iniae cpsA mutant, which had attenuated virulence in adult zebrafish, was also attenuated in zebrafish larvae, further validating the use of this model (42) (Fig. 1). Unlike infection with wild-type bacteria, infection of zebrafish larvae with the cpsA mutant at doses as large as 1,000 CFU was not lethal (Fig. 1C), and there was no increase in relative bacterial burden in the first 60 hpi (Fig. 1D), indicating that the host was able to control infection.
Although the majority of larvae infected with wild-type S. iniae did not survive infection, some larvae survived past 96 hpi. This variability may be due to individual variation in the ability of the host to control S. iniae disease because zebrafish lines cannot be highly inbred. In support of this idea, some of the surviving embryos had nonlethal carriage of dye-labeled wild-type and mutant S. iniae up to 72 hpi (data not shown). It is also possible that the variability in survival was due to differences in the dose of inoculum or the site of injection, since we cannot exclude the possibility that when the microinjection needle is retracted, some bacteria may be deposited outside the otic vesicle, as was previously reported in a study by Colucci-Guyon et al. (55). An additional consideration is that our method of measuring bacterial burden was not a specific measure of S. iniae burden but, rather, was a measure of the number of culturable Gram-positive organisms. Indeed, there were bacteria recovered from the PBS-mock-infected larvae that are likely to be part of the normal zebrafish microbiota. According to a study done by Roeselers et al., some potential members of the zebrafish gut microbiota are Gram-positive bacteria, including members from the phyla Actinobacteria and Firmicutes (59). Despite these caveats, there was an overall trend where wild-type-infected larvae had an increase in bacterial burden over time while cpsA-infected larvae had relatively constant bacterial burdens. However, we cannot rule out the possibility that the increase in bacterial burden in the wild-type-infected larvae may have resulted from infection-associated changes in the host that altered the composition of the Gram-positive members of the microbiota.
Although the wild-type strain and the cpsA mutant differed in their ability to establish lethal infection and proliferate in the larval host, both strains were similar in their ability to attract neutrophils and macrophages to localized sites of infection (Fig. 2 to 5). The mechanism of neutrophil and macrophage recruitment into the otic vesicle is not clear, but it likely involves transmigration from blood vessels and movement through interstitial spaces in response to chemoattractive signals. It is possible that only a subset of host phagocytes have the surface receptor expression required to sense the signals that recruit the cells into the otic vesicle. Neutrophils are among the first cells recruited to the site of infection and were able to phagocytose both wild-type bacteria and the cpsA mutant (Fig. 3; see Movies S1 and S2 in the supplemental material). Neutrophils that phagocytosed bacteria developed a rounded appearance that was accompanied by decreased motility. This motility was quickly recovered in the case of infection with the wild-type strain (see Movie S1 in the supplemental material), but neutrophils phagocytosing the cpsA mutant became laden with bacteria and did not quickly regain their motility (see Movie S2 in the supplemental material). It is possible that neutrophils are more efficient at phagocytosing the cpsA mutant and that their apparent inability to immediately regain motility was due to larger amounts of internalized bacteria than in infection with wild-type bacteria.
Macrophages were also able to phagocytose both wild-type bacteria and the cpsA mutant. In the case of wild-type infection, it was possible to find many uninfected macrophages that had an activated morphology (Fig. 5A; see Movie S3 in the supplemental material), suggesting that even in the absence of phagocytosis, the macrophages still became activated. It was more common to see macrophages that had phagocytosed the cpsA mutant than wild-type bacteria. In addition, the cpsA mutant was contained in multiple separate vacuoles in the macrophages, as well as in single phagosomes (Fig. 5B; see Movie S4 in the supplemental material). Although our findings suggested that macrophages were more efficient at phagocytosing the cpsA mutant than wild-type bacteria, we were not able to quantify the efficiency of phagocytosis by neutrophils and macrophages because of the inability to reliably track S. iniae using stably fluorescent bacteria.
Wild-type S. iniae and the cpsA mutant differ in terms of polysaccharide capsule production. The capsule of S. iniae is closely related to the capsule of Streptococcus agalactiae (group B Streptococcus), which is known to have antiphagocytic properties. The polysaccharide capsule of group B Streptococcus interferes with the deposition of the complement molecule C3b, preventing activation of the alternative complement pathway and also disrupting the opsonophagocytic abilities of host phagocytes (60). The capsule of S. iniae is thought to have similar antiphagocytic properties. In vitro studies have demonstrated that S. iniae mutants with decreased capsule production are more prone to whole-blood killing in either human (43) or fish (41) blood and are more susceptible to phagocytosis by cultured fish macrophages (41). Accordingly, we found that the cpsA mutant seemed more susceptible to phagocytosis, since we observed more host phagocytes with internalized cpsA mutant bacteria than with wild-type bacteria. Although initial recruitment of host phagocytes to the site of infection is similar for both wild-type bacteria and the cpsA mutant, subsequent bacterium-phagocyte interactions seem to be different and likely contribute to the wild-type strain being pathogenic and the cpsA mutant nonpathogenic.
Phagocytosis of bacteria is important for controlling infection but may also play a role in bacterial dissemination. Intracellular residence in macrophages has been suggested to provide S. iniae with an intracellular niche for dissemination throughout the host (61). It has been shown that S. iniae can survive inside cultured mouse RAW 267.4 macrophages (62) and salmon macrophages (61) for at least 24 h. A challenge for future investigation will be to develop the tools to determine if host phagocytes contribute to bacterial dissemination with S. iniae infection in zebrafish larvae.
As a first step, we were interested in determining if host phagocytes control disease in S. iniae infection of zebrafish. To examine the importance of neutrophils and macrophages in host defense, we depleted myeloid cells using a Pu.1 morpholino oligonucleotide. It has been previously shown that myeloid cells are important for controlling infection with P. aeruginosa (11, 14), Mycobacterium marinum (15), Streptococcus pneumoniae (22), and Staphylococcus aureus (21). However, clearance of bacterial pathogens is not always dependent on the presence of myeloid cells, as Pu.1 morphants are able to clear infection with nonpathogenic Escherichia coli (15). Using the Pu.1 morphants, we show that myeloid cells are important for host defense in S. iniae infection, since the Pu.1 morphants had increased lethality following localized infection with both wild-type bacteria and the cpsA mutant (Fig. 6A). In the case of the Pu.1 morphants, deposition of even a few bacteria outside the otic vesicle could explain why the morphants develop a lethal infection when there are no host phagocytes to disseminate bacteria. However, the lethality in the Pu.1 morphants may also occur because without host phagocytes, the bacterial burden can rapidly reach lethal numbers.
Our findings also demonstrate that neutrophils are necessary for controlling infection with wild-type S. iniae but not the cpsA mutant (Fig. 7E). Rac2(D57N) larvae were more susceptible than control larvae to a low-dose inoculum of wild-type bacteria but not to the cpsA mutant (Fig. 7E). These results suggest that although neutrophils play a role in controlling infection with wild-type S. iniae, they are dispensable for controlling infection with the nonpathogenic cpsA mutant. These findings raise the intriguing idea that macrophages, but not neutrophils, are critical for controlling infection with the cpsA mutant. The development of transgenic zebrafish lines with conditional tissue-specific ablation will help further elucidate the individual contribution of macrophages to host defense (63, 64). A further challenge will be to determine if macrophages are able to serve as “Trojan horses” which facilitate systemic spread of wild-type S. iniae under some conditions (61).
The ability of neutrophils and macrophages to respond and phagocytose bacteria has been shown to be pathogen specific in various zebrafish models of microbial infection. Both primitive neutrophils and macrophages phagocytose P. aeruginosa (11, 14), but neutrophils, and not macrophages, are necessary to control P. aeruginosa infection (46). Although both zebrafish neutrophils and macrophages are recruited to sites of infection with E. coli (15, 18, 29, 65), Staphylococcus aureus (21), Listeria monocytogenes (19), Burkholderia cenocepacia (24), and Salmonella enterica serovar Typhimurium (32, 65), primitive macrophages play a more critical role in the phagocytosis of these microorganisms, with only minimal phagocytosis by neutrophils. Additionally, macrophages, but not neutrophils, are recruited and are important for the phagocytosis of Mycobacterium marinum (15). Host phagocytes are also important for defense against S. pneumoniae infection (22), since Pu.1 morphants are more susceptible to disease. Here, we have shown that both neutrophils and macrophages are crucial for the control of wild-type S. iniae infection but that in the absence of functional neutrophils, other host leukocytes such as macrophages control infection with the nonpathogenic S. iniae cpsA mutant. Interestingly, the observation that neutrophils phagocytose wild-type S. iniae and the cpsA mutant following infection in the otic vesicle is in contrast with a recent report from Colucci-Guyon et al. (55). This report showed that unlike macrophages, neutrophils were ineffective at phagocytosing E. coli injected into closed fluid-filled body cavities such as the otic vesicle and were highly phagocytic only when bacteria were attached to a surface (55). This suggests that the ability of neutrophils to efficiently phagocytose bacteria in fluid-filled body cavities may be dependent on the particular type of invading microbe.
In summary, we have developed a zebrafish larval infection model with the fish pathogen S. iniae. The optical accessibility of this model allowed for the real-time observation of host phagocyte-S. iniae interactions in vivo. The genetic tractability of the zebrafish allowed for components of the host immune system, such as neutrophils or macrophages, to be altered to investigate how defects in innate immunity affect S. iniae disease outcome. We found that while both neutrophils and macrophages provide protection against infection with wild-type S. iniae, neutrophils are not necessary for host survival following infection with the S. iniae cpsA capsule mutant. Moreover, this model was able to differentiate between S. iniae strains with altered virulence, allowing for the real-time observation of host-pathogen interactions when both host and pathogen components are altered.
Supplementary Material
ACKNOWLEDGMENTS
We thank P. Y. Lam and D. C. LeBert for critical reading of the manuscript and lab members for zebrafish maintenance.
This work was supported by National Institutes of Health National Research Service Award AI55397 to E. A. Harvie and by a Burroughs Wellcome grant to A. Huttenlocher.
Footnotes
Published ahead of print 22 October 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00642-12.
REFERENCES
- 1. Pier GB, Madin SH. 1976. Streptococcus iniae sp. nov., a beta-hemolytic Streptococcus isolated from an Amazon freshwater dolphin, Inia geoffrensis. Int. J. Syst. Bacteriol. 26:545–553 [Google Scholar]
- 2. Agnew W, Barnes A. 2007. Streptococcus iniae: An aquatic pathogen of global veterinary significance and a challenging candidate for reliable vaccination. Vet. Microbiol. 122:1–15 [DOI] [PubMed] [Google Scholar]
- 3. Pier GB, Madin SH, Al-Nakeeb S. 1978. Isolation and characterization of a second isolate of Streptococcus iniae. Int. J. Syst. Bacteriol. 28:311–314 [Google Scholar]
- 4. Eldar A, Bejerano Y, Bercovier H. 1995. Experimental streptococcal meningo-encephalitis in cultured fish. Vet. Microbiol. 43:33–40 [DOI] [PubMed] [Google Scholar]
- 5. Shoemaker CA, Klesius PH, Evans JJ. 2001. Prevalence of Streptococcus iniae in tilapia, hybrid striped bass, and channel catfish on commercial fish farms in the United States. Am. J. Vet. Res. 62:174–177 [DOI] [PubMed] [Google Scholar]
- 6. Weinstein MR, Litt M, Kertesz DA, Wyper P, Rose D, Coulter M, McGeer A, Facklam R, Ostach C, Willey BM, Borczyk A, Low DE. 1997. Invasive infections due to a fish pathogen, Streptococcus iniae. N. Engl. J. Med. 337:589–594 [DOI] [PubMed] [Google Scholar]
- 7. Facklam R, Elliott J, Shewmaker L, Reingold A. 2005. Identification and characterization of sporadic isolates of Streptococcus iniae isolated from humans. J. Clin. Microbiol. 43:933–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koh TH, Kurop A, Chen J. 2004. Streptococcus iniae discitis in Singapore. Emerg. Infect. Dis. 10:1694–31696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lau SKP, Woo PCY, Luk W-K, Fung AMY, Hui W-T, Fong AHC, Chow C-W, Wong SSY, Yuen K-Y. 2006. Clinical isolates of Streptococcus iniae from Asia are more mucoid and β-hemolytic than those from North America. Diagn. Microbiol. Infect. Dis. 54:177–181 [DOI] [PubMed] [Google Scholar]
- 10. Lau SKP, Woo PCY, Tse H, Leung K-W, Wong SSY, Yuen K-Y. 2003. Invasive Streptococcus iniae infections outside North America. J. Clin. Microbiol. 41:1004–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brannon MK, Davis JM, Mathias JR, Hall CJ, Emerson JC, Crosier PS, Huttenlocher A, Ramakrishnan L, Moskowitz SM. 2009. Pseudomonas aeruginosa type III secretion system interacts with phagocytes to modulate systemic infection of zebrafish embryos. Cell. Microbiol. 11:755–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brothers KM, Newman ZR, Wheeler RT. 2011. Live imaging of disseminated candidiasis in zebrafish reveals role of phagocyte oxidase in limiting filamentous growth. Eukaryot. Cell 10:932–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chao CC, Hsu PC, Jen CF, Chen IH, Wang CH, Chan HC, Tsai PW, Tung KC, Wang CH, Lan CY, Chuang YJ. 2010. Zebrafish as a model host for Candida albicans infection. Infect. Immun. 78:2512–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clatworthy AE, Lee JSW, Leibman M, Kostun Z, Davidson AJ, Hung DT. 2009. Pseudomonas aeruginosa infection of zebrafish involves both host and pathogen determinants. Infect. Immun. 77:1293–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Clay H, Davis JM, Beery D, Huttenlocher A, Lyons SE, Ramakrishnan L. 2007. Dichotomous role of the macrophage in early Mycobacterium marinum infection of the zebrafish. Cell Host Microbe 2:29–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Davis JM, Clay H, Lewis JL, Ghori N, Herbomel P, Ramakrishnan L. 2002. Real-time visualization of mycobacterium-macrophage interactions leading to initiation of granuloma formation in zebrafish embryos. Immunity 17:693–702 [DOI] [PubMed] [Google Scholar]
- 17. Davis JM, Haake DA, Ramakrishnan L. 2009. Leptospira interrogans stably infects zebrafish embryos, altering phagocyte behavior and homing to specific tissues. PLoS Negl Trop. Dis. 3:e463 doi:10.1371/journal.pntd.0000463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Herbomel P, Thisse B, Thisse C. 1999. Ontogeny and behaviour of early macrophages in the zebrafish embryo. Development. 126:3735–3745 [DOI] [PubMed] [Google Scholar]
- 19. Levraud J-P, Disson O, Kissa K, Bonne I, Cossart P, Herbomel P, Lecuit M. 2009. Real-time observation of Listeria monocytogenes-phagocyte interactions in living zebrafish larvae. Infect. Immun. 77:3651–3660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lin A, Loughman JA, Zinselmeyer BH, Miller MJ, Caparon MG. 2009. Streptolysin S inhibits neutrophil recruitment during the early stages of Streptococcus pyogenes infection. Infect. Immun. 77:5190–5201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Prajsnar TK, Cunliffe VT, Foster SJ, Renshaw SA. 2008. A novel vertebrate model of Staphylococcus aureus infection reveals phagocyte-dependent resistance of zebrafish to non-host specialized pathogens. Cell. Microbiol. 10:2312–2325 [DOI] [PubMed] [Google Scholar]
- 22. Rounioja S, Saralahti A, Rantala L, Parikka M, Henriques-Normark B, Silvennoinen O, Rämet M. 2012. Defense of zebrafish embryos against Streptococcus pneumoniae infection is dependent on the phagocytic activity of leukocytes. Dev. Comp. Immunol. 36:342–348 [DOI] [PubMed] [Google Scholar]
- 23. van Soest JJ, Stockhammer OW, Ordas A, Bloemberg GV, Spaink HP, Meijer AH. 2011. Comparison of static immersion and intravenous injection systems for exposure of zebrafish embryos to the natural pathogen Edwardsiella tarda. BMC Immunol. 12:58 doi:10.1186/1471-2172-12-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vergunst AC, Meijer AH, Renshaw SA, O'Callaghan D. 2010. Burkholderia cenocepacia creates an intramacrophage replication niche in zebrafish embryos, followed by bacterial dissemination and establishment of systemic infection. Infect. Immun. 78:1495–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Danilova N, Steiner LA. 2002. B cells develop in the zebrafish pancreas. Proc. Natl. Acad. Sci. U. S. A. 99:13711–13716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lam SH, Chua HL, Gong Z, Lam TJ, Sin YM. 2004. Development and maturation of the immune system in zebrafish, Danio rerio: a gene expression profiling, in situ hybridization and immunological study. Dev. Comp. Immunol. 28:9–28 [DOI] [PubMed] [Google Scholar]
- 27. Willett CE, Cortes A, Zuasti A, Zapata A. 1999. Early hematopoiesis and developing lymphoid organs in the zebrafish. Dev. Dyn. 214:323–336 [DOI] [PubMed] [Google Scholar]
- 28. Jault C, Pichon L, Chluba J. 2004. Toll-like receptor gene family and TIR-domain adapters in Danio rerio. Mol. Immunol. 40:759–771 [DOI] [PubMed] [Google Scholar]
- 29. Le Guyader D, Redd MJ, Colucci-Guyon E, Murayama E, Kissa K, Briolat V, Mordelet E, Zapata A, Shinomiya H, Herbomel P. 2008. Origins and unconventional behavior of neutrophils in developing zebrafish. Blood 111:132–141 [DOI] [PubMed] [Google Scholar]
- 30. Meijer AH, Gabby Krens SF, Medina Rodriguez IA, He S, Bitter W, Ewa Snaar-Jagalska B, Spaink HP. 2004. Expression analysis of the Toll-like receptor and TIR domain adaptor families of zebrafish. Mol. Immunol. 40:773–783 [DOI] [PubMed] [Google Scholar]
- 31. Seeger A, Mayer WE, Klein J. 1996. A complement factor B-like cDNA clone from the zebrafish (Brachydanio rerio). Mol. Immunol. 33:511–520 [DOI] [PubMed] [Google Scholar]
- 32. van der Sar AM, Musters RJP, van Eeden FJM, Appelmelk BJ, Vandenbroucke-Grauls CMJE, Bitter W. 2003. Zebrafish embryos as a model host for the real time analysis of Salmonella typhimurium infections. Cell. Microbiol. 5:601–611 [DOI] [PubMed] [Google Scholar]
- 33. Nasevicius A, Ekker SC. 2000. Effective targeted gene “knockdown” in zebrafish. Nat. Genet. 26:216–220 [DOI] [PubMed] [Google Scholar]
- 34. Neely MN, Pfeifer JD, Caparon MG. 2002. Streptococcus-zebrafish model of bacterial pathogenesis. Infect. Immun. 70:3904–3914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baiano JC, Tumbol RA, Umapathy A, Barnes AC. 2008. Identification and molecular characterisation of a fibrinogen binding protein from Streptococcus iniae. BMC Microbiol. 8:67 doi:10.1186/1471-2180-8-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Buchanan JT, Stannard JA, Lauth X, Ostland VE, Powell HC, Westerman ME, Nizet V. 2005. Streptococcus iniae phosphoglucomutase is a virulence factor and a target for vaccine development. Infect. Immun. 73:6935–6944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eyngor M, Lublin A, Shapira R, Hurvitz A, Zlotkin A, Tekoah Y, Eldar A. 2010. A pivotal role for the Streptococcus iniae extracellular polysaccharide in triggering proinflammatory cytokines transcription and inducing death in rainbow trout. FEMS Microbiol. Lett. 305:109–120 [DOI] [PubMed] [Google Scholar]
- 38. Fuller J, Camus A, Duncan C, Nizet V, Bast D, Thune R, Low DE, DE Azavedo JCS. 2002. Identification of a Streptolysin S-associated gene cluster and its role in the pathogenesis of Streptococcus iniae disease. Infect. Immun. 70:5730–5739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Locke JB, Aziz RK, Vicknair MR, Nizet V, Buchanan JT. 2008. Streptococcus iniae M-Like protein contributes to virulence in fish and is a target for live attenuated vaccine development. PLoS One 3:e2824 doi:10.1371/journal.pone.0002824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Milani CJE, Aziz RK, Locke JB, Dahesh S, Nizet V, Buchanan JT. 2010. The novel polysaccharide deacetylase homologue Pdi contributes to virulence of the aquatic pathogen Streptococcus iniae. Microbiology 156:543–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Locke JB, Colvin KM, Datta AK, Patel SK, Naidu NN, Neely MN, Nizet V, Buchanan JT. 2007. Streptococcus iniae capsule impairs phagocytic clearance and contributes to virulence in fish. J. Bacteriol. 189:1279–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lowe BA, Miller JD, Neely MN. 2007. Analysis of the polysaccharide capsule of the systemic pathogen Streptococcus iniae and its implications in virulence. Infect. Immun. 75:1255–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miller JD, Neely MN. 2005. Large-scale screen highlights the importance of capsule for virulence in the zoonotic pathogen Streptococcus iniae. Infect. Immun. 73:921–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nüsslein-Volhard C, Dahm R. 2002. Zebrafish: a practical approach. Oxford University Press, New York, NY [Google Scholar]
- 45. Yoo SK, Huttenlocher A. 2011. Spatiotemporal photolabeling of neutrophil trafficking during inflammation in live zebrafish. J. Leukoc. Biol. 89:661–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Deng Q, Yoo SK, Cavnar PJ, Green JM, Huttenlocher A. 2011. Dual roles for Rac2 in neutrophil motility and active retention in zebrafish hematopoietic tissue. Dev. Cell 21:735–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Urasaki A, Morvan G, Kawakami K. 2006. Functional dissection of the Tol2 transposable element identified the minimal cis-sequence and a highly repetitive sequence in the subterminal region essential for transposition. Genetics 174:639–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ellett F, Pase L, Hayman JW, Andrianopoulos A, Lieschke GJ. 2011. mpeg1 promoter transgenes direct macrophage-lineage expression in zebrafish. Blood 117:e49–e56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fuller J, Bast D, Nizet V, Low D. 2001. Streptococcus iniae virulence is associated with a distinct genetic profile. Infect. Immun. 69:1994–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Levraud J-P, Colucci-Guyon E, Redd MJ, Lutfalla G, Herbomel P. 2008. In vivo analysis of zebrafish innate immunity. Methods Mol. Biol. 415:337–363 [DOI] [PubMed] [Google Scholar]
- 51. Rhodes J, Hagen A, Hsu K, Deng M, Liu TX, Look AT, Kanki JP. 2005. Interplay of pu. 1 and gata1 determines myelo-erythroid progenitor cell fate in zebrafish. Dev. Cell 8:97–108 [DOI] [PubMed] [Google Scholar]
- 52. Haddon C, Lewis J. 1996. Early ear development in the embryo of the zebrafish, Danio rerio. J. Comp. Neurol. 365:113–128 [DOI] [PubMed] [Google Scholar]
- 53. Whitfield TT, Riley BB, Chiang M-Y, Phillips B. 2002. Development of the zebrafish inner ear. Dev. Dyn. 223:427–458 [DOI] [PubMed] [Google Scholar]
- 54. Benard EL, van der Sar AM, Ellett F, Lieschke GJ, Spaink HP, Meijer AH. 2012. Infection of zebrafish embryos with intracellular bacterial pathogens. J. Vis Exp. 15:3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Colucci-Guyon E, Tinevez JY, Renshaw SA, Herbomel P. 2011. Strategies of professional phagocytes in vivo: unlike macrophages, neutrophils engulf only surface-associated microbes. J. Cell Sci. 124:3053–3059 [DOI] [PubMed] [Google Scholar]
- 56. Deng Q, Harvie EA, Huttenlocher A. 2011. Distinct signaling mechanisms mediate neutrophil attraction to bacterial infection and tissue injury. Cell. Microbiol. 14:517–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bolotin S, Fuller JD, Bast DJ, Beveridge TJ, de Azavedo JCS. 2007. Capsule expression regulated by a two-component signal transduction system in Streptococcus iniae. FEMS Immunol. Med. Microbiol. 50:366–374 [DOI] [PubMed] [Google Scholar]
- 58. Hermann AC, Millard PJ, Blake SL, Kim CH. 2004. Development of a respiratory burst assay using zebrafish kidneys and embryos. J. Immunol. Methods 292:119–129 [DOI] [PubMed] [Google Scholar]
- 59. Roeselers G, Mittge EK, Stephens WZ, Parichy DM, Cavanaugh CM, Guillemin K, Rawls JF. 2011. Evidence for a core gut microbiota in the zebrafish. ISME J. 5:1595–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Marques MB, Kasper DL, Pangburn MK, Wessels MR. 1992. Prevention of C3 deposition by capsular polysaccharide is a virulence mechanism of type III group B streptococci. Infect. Immun. 60:3986–3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zlotkin A, Chilmonczyk S, Eyngor M, Hurvitz A, Ghittino C, Eldar A. 2003. Trojan horse effect: phagocyte-mediated Streptococcus iniae infection of fish. Infect. Immun. 71:2318–2325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Miller JD, Neely MN. 2004. Zebrafish as a model host for streptococcal pathogenesis. Acta Trop. 91:53–68 [DOI] [PubMed] [Google Scholar]
- 63. Curado S, Anderson RM, Jungblut B, Mumm J, Schroeter E, Stainier DYR. 2007. Conditional targeted cell ablation in zebrafish: a new tool for regeneration studies. Dev. Dyn. 236:1025–1035 [DOI] [PubMed] [Google Scholar]
- 64. Curado S, Stainier DYR, Anderson RM. 2008. Nitroreductase-mediated cell/tissue ablation in zebrafish: a spatially and temporally controlled ablation method with applications in developmental and regeneration studies. Nat. Protoc. 3:948–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Meijer AH, van der Sar AM, Cunha C, Lamers GEM, Laplante MA, Kikuta H, Bitter W, Becker TS, Spaink HP. 2008. Identification and real-time imaging of a myc-expressing neutrophil population involved in inflammation and mycobacterial granuloma formation in zebrafish. Dev. Comp. Immunol. 32:36–49 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.