

Abstract
Chlamydia trachomatis is the most common sexually transmitted bacterial pathogen and the etiologic agent of blinding trachoma. Intracellular signaling pathways leading to host cell inflammation and innate immunity to Chlamydia include those mediated by Toll-like receptors (TLRs) and nucleotide binding oligomerization domain 1 (Nod1) protein. In epithelial cells, TLR-dependent signaling contributes to local immune responses via induction of inflammatory mediators. There is evidence that TLR3, TLR4, and, particularly, TLR2 are critical for Chlamydia-mediated host cell activation and pathology. Despite the importance of TLR2, major chlamydial TLR2 antigens have not been identified so far. Numerous bacterial porins are known TLR2 agonists, i.e., porins from Neisseriae, Shigella, Salmonella, Haemophilus influenzae, and Fusobacterium nucleatum, which share structural and functional similarities with the chlamydial major outer membrane protein (MOMP), a strong antigen candidate for a potential vaccine against C. trachomatis. We describe the ability of purified, detergent-free MOMP to signal via TLR2 in vitro in TLR-overexpressing cells and TLR2-competent human reproductive tract epithelial cell lines. Using MOMP formed in pure protein micelles (proteosomes), we show the induction of TLR2-dependent interleukin-8 (IL-8) and IL-6 secretion in vitro, the involvement of TLR1 as a TLR2 coreceptor, and the activation of both NF-κB and mitogen-activated protein (MAP) kinase intracellular pathways. Interestingly, MOMP proteosomes induce cytokine secretion in endocervical epithelial cells (End/E6E7) but not in urethral epithelial cells (THUECs). A detailed understanding of the TLR2-dependent molecular mechanisms that characterize the effect of MOMP proteosomes on host cells may provide new insights for its successful development as an immunotherapeutic target against Chlamydia.
INTRODUCTION
The role of Toll-like receptors (TLRs; cell surface and intracellular receptors that recognize microbial products called pathogen-associated molecular patterns [PAMPs]) (1) in modulation of host innate and adaptive immunity by bacterial products is well established. TLR engagement and signaling induce activation of intracellular signaling pathways such as NF-κB and mitogen-activated protein (MAP) kinases that regulate secretion of inflammatory mediators by both nonhematopoietic cells and immune cells, as well as expression of costimulatory ligands and MHC molecules, ultimately enhancing the host humoral and cellular immune responses (1, 2). Among the known bacterial components with TLR agonist function, lipopolysaccharide (LPS) activates TLR4 in complex with MD2 (3), flagellin activates TLR5 (4), and CpG DNA and double-stranded RNA (dsRNA) engage TLR9 and TLR3, respectively (5, 6). A much broader repertoire of ligands characterizes TLR2, due to its heterodimerization with TLR1 or TLR6. Examples of bacterial TLR2 agonists are lipopeptides (7), peptidoglycans (8) and porins (i.e., from Neisseriae) (9, 10), Shigella (11), Salmonella (12), Haemophilus influenzae (13), and Fusobacterium nucleatum (14).
Chlamydia species are associated with a number of diseases. C. trachomatis is the most common sexually transmitted bacterial pathogen and up to 4 million to 5 million new genital C. trachomatis infections are reported annually in the United States (15). While genital infections can remain asymptomatic in women, long-term sequelae, such as infertility and ectopic pregnancy, can develop (16). C. trachomatis is also the etiologic agent of blinding trachoma, which is one of the leading causes of blindness in the world (17, 18). C. pneumoniae is a cause of community-acquired pneumonia and pharyngitis (approximately 6 to 10% of community-acquired pneumonia cases) (19) and may play a role in chronic inflammatory conditions, such as asthma, reactive arthritis, and atherosclerosis (20).
Chlamydia infection activates several innate immune pathways, leading to host cell responses, including TLR signaling (21) and the activation of the nucleotide binding oligomerization domain 1 (Nod1) protein (22), an intracellular pattern recognition receptor (23). For example, evidence of TLR3 activation by Chlamydia has been reported in murine reproductive tract epithelial cells (24), and there is strong evidence that TLR2 is critical for Chlamydia-mediated host cell activation and pathology (25, 26). However, only a few chlamydial components have been implicated in TLR-dependent cell activation, for example, the lipoprotein macrophage infectivity potentiator Mip, which signals via TLR2/TLR1/TLR6 and CD14 (27), and LPS, which signals via TLR4 (although with weaker activity than Escherichia coli LPS) (28, 29). A number of studies have also reported signaling via both TLR4 and TLR2 for chlamydial heat shock protein 60 (hsp60) in vitro and in vivo (30–32). Although the importance of TLR2 for the induction of host immune responses to Chlamydia infection and for Chlamydia virulence is recognized (21, 33–35), major chlamydial TLR2 antigens have not been identified so far. For example, expression of peptidoglycan (a TLR2 agonist) is considered to be minimal or even undetectable in Chlamydia (36, 37).
Porins, the most abundant outer membrane proteins of Gram-negative organisms, are established TLR2 ligands. Bacterial porins, including the chlamydial major outer membrane porin protein (MOMP), constitute a high percentage of the total outer membrane protein content (over 60%) (38) and share structural and functional similarities among organisms. C. trachomatis MOMP is surface exposed, has a molecular mass of ∼40 kDa in monomeric form, and is found in homotrimeric form in the bacterial outer membrane (39, 40). It is immunogenic (41) and can induce protection in the mouse and monkey models (42, 43). Since MOMP is considered a strong antigen candidate for a Chlamydia vaccine, a detailed understanding of the TLR-dependent molecular mechanisms that characterize its activity on host cells will provide new insights for its successful development as an immunotherapeutic target against Chlamydia. However, as for all purified bacterial integral membrane proteins, the addition of detergents is necessary to maintain native conformation and solubility in aqueous solutions. Due to the cytotoxic nature of detergents, it has proved difficult to establish the effects of native purified MOMP in vitro so far. To overcome this obstacle, in the present study, we have examined the ability of purified C. trachomatis MOMP formed in pure protein micelles (proteosomes) to induce inflammatory responses in vitro in a variety of cell models relevant for C. trachomatis infection. Using both a TLR2 overexpression cell model (HEK cells) and human reproductive tract epithelial cell lines, we have established that MOMP proteosomes induce cell activation via TLR2 signaling and that TLR1 is the necessary TLR2 coreceptor for its activity, while TLR4 does not play a major role in the activity of MOMP proteosomes. Furthermore, we show that interleukin-8 (IL-8) secretion is induced by MOMP proteosomes via multiple intracellular signaling pathways, namely, NF-κB nuclear translocation and the activation of MAP kinases. Interestingly, our results show that purified MOMP proteosomes induce secretion of IL-8 and IL-6 in the endocervical epithelial cell line End/E6E7 in vitro, but these proinflammatory mediators are not induced in human urethral epithelial cells.
MATERIALS AND METHODS
Preparation of native Chlamydia muridarum mouse pneumonitis (MoPn) MOMP proteosomes.
Chlamydia muridarum (strain Nigg II; previously called Chlamydia trachomatis mouse pneumonitis [ATCC]) was grown in McCoy cells (44). The extraction and purification of native MOMP were performed as described previously (40). In brief, after extraction of MOMP from bacterial lysates using 2% Anzergent 3-14 (n-tetradecyl-N,N-dimethyl-3-ammonio-1-propanesulfonate) (Z3-14), the protein suspension was subject to column chromatography on hydroxyapatite resin (Bio-Gel HTP Gel) in the presence of 0.1% Z3-14. Purified MOMP, identified by electrophoresis on 10% Tricine-SDS-PAGE and Western blotting using the monoclonal antibody MoPn40 to MOMP (40), was concentrated and resuspended in 10% d-octyl-glucoside (DOG) in 10 mM HEPES (pH 7.5), followed by extensive dialysis in phosphate-buffered saline (PBS) containing 0.02% NaN3 for the formation of detergent-free protein micelles (proteosomes) (45). The protein concentration was determined using the bicinchoninic acid (BCA) assay.
Cell culture and growth conditions.
Stably transfected HEK cells overexpressing TLR2, TLR4, TLR2/TLR1, TLR2/TLR6, or an empty vector (pcDNA) (3) were cultured at 37°C in a 5% CO2 incubator in Dulbecco's modified Eagle's medium (DMEM) containing 5% fetal bovine serum (FBS), 2 mM l-glutamine, and 10 μg/ml ciprofloxacin. HeLa cells were grown in DMEM supplemented with 10% FBS, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. The endocervical cell line End/E6E7 (46) was cultured in keratinocyte serum-free medium (KSFM) (GIBCO-Invitrogen) containing 50 μg/ml bovine pituitary extract, 0.1 ng/ml epidermal growth factor, 0.4 mM CaCl2, 100 U/ml penicillin, and 100 μg/ml streptomycin. Transduced human urethral epithelial cells (THUECs) (47) were grown in prostate epithelial growth medium (PrEGM) (Cambrex) supplemented with 5% FBS.
Cell stimulation.
A total of 105 cells/ml of HEK cells, End/E6E7 cells, HeLa cells, or THUECs were plated in 100 μl in 96-well plates and incubated with purified MOMP proteosomes (100, 10, or 1 μg/ml), Pam3CSK4 or Pam2CSK4 (0.1 μg/ml) (InvivoGen), lipoprotein-free E. coli LPS (0.1 μg/ml) (Sigma), or recombinant human tumor necrosis factor alpha (TNF-α) (0.02 μg/ml) (eBioscience) in triplicate wells for 3 h, 6 h, or 24 h. For MAP kinase signaling pathway inhibition, cells were treated with 25 μM (in 10 mM dimethyl sulfoxide [DMSO]) U0126 (MEK1/2 inhibitor, upstream kinase for ERK1/2 phosphorylation), SB203580 (inhibitor of p38 phosphorylation), or SP600125 (inhibitor of Jun N-terminal protein kinase [JNK] phosphorylation) (Sigma) or 25 μg/ml of an NF-κB inhibitory ligand (Millipore) for 1 h prior to cell stimulation.
Cytokine ELISA.
IL-8 and IL-6 secretion was measured by an enzyme-linked immunosorbent assay (ELISA) of supernatants from cell cultures stimulated as described above using OptEIA kits (BD Biosciences) per the manufacturer's protocol.
NF-κB luciferase reporter assay.
HEK cells were transiently transfected with an NF-κB luciferase reporter vector as described previously (3). Transfected cells were left to adhere overnight in 24-well cell culture plates and stimulated the next day as described above. Luciferase activity was measured using commercial reagents (Promega) per the manufacturer's protocol, and luminescence was assessed using a Wallac Victor2 luminometer. Results are expressed as relative luciferase units ± standard errors.
Statistical analysis.
Statistical analyses were calculated using GraphPad PRISM software. P values were calculated using an unpaired t test with Welch's correction, a one-sample t test, and a one-way analysis of variance (ANOVA) with a Bonferroni posttest, as described in Results.
RESULTS
Analysis of native purified C. muridarum MoPn MOMP and MOMP proteosomes.
Purified MOMP was obtained by chromatographic separation in the presence of detergent as described previously (40). Following purification, to maintain the protein in native trimeric conformation while removing the detergent to allow its use in cell-based in vitro assays, MOMP was concentrated, resuspended in 10% d-octyl-glucoside (DOG) and dialyzed extensively against PBS to allow the formation of detergent-free and lipid-free protein micelles (termed proteosomes). To assess whether inclusion of MOMP into proteosomes affected the trimeric state of the purified protein, aliquots of native purified (MoPn) MOMP and MOMP proteosomes were compared by electrophoresis on a 10% Tricine-SDS-PAGE. In Fig. 1A, purified Chlamydia MoPn MOMP (containing detergent) was heat denatured in the presence of 25 mM dithiothreitol (DTT) prior to electrophoresis and a band of approximately 39 kDa, corresponding to the monomeric form (40, 48), was observed (Fig. 1A, lane 2). When the purified MOMP was not subject to heat denaturation, a major band with an apparent molecular mass of 66 kDa, typically indicative of MOMP trimeric forms (40, 48), was observed both in the presence and in the absence of DTT (Fig. 1A, lanes 3 and 4, respectively). Furthermore, additional bands of high molecular weights were detected in the absence of DTT (Fig. 1A, lane 4).
Fig 1.
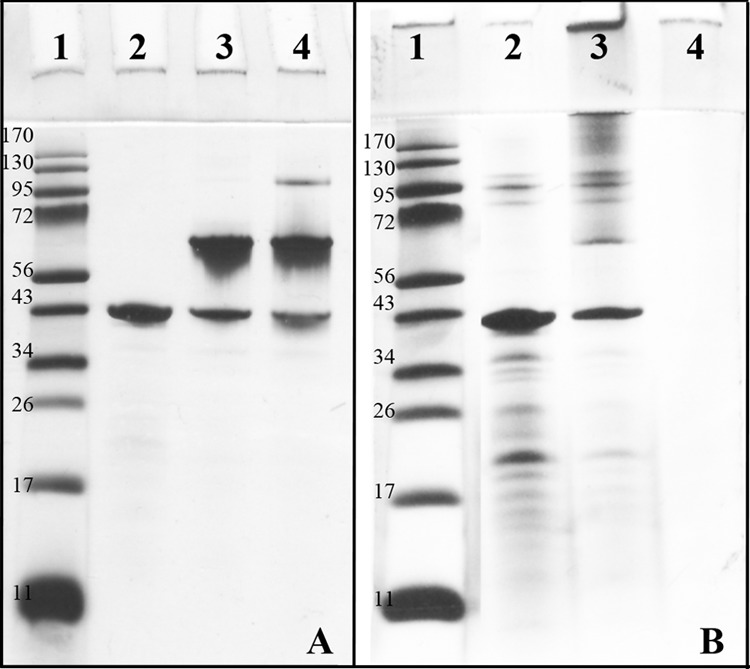
Comparison of purified Chlamydia MoPn MOMP and MOMP proteosomes. (A) Ten percent Tricine-SDS-PAGE of purified Chlamydia MoPn MOMP. Lane 1, molecular weight standard; lane 2, heat-denatured, purified MOMP containing 25 mm DTT; lane 3, not heat-denatured, purified MOMP with 25 mM DTT; and lane 4, not heat-denatured, purified MOMP in the absence of DTT. (B) Ten percent Tricine-SDS-PAGE of MOMP proteosomes. Lane 1, molecular weight standard; lane 2, heat-denatured MOMP proteosomes containing 25 mm DTT; lane 3, not heat-denatured MOMP proteosomes containing 25 mM DTT; and lane 4, not heat-denatured MOMP proteosomes in the absence of DTT.
Next, detergent-free MOMP proteosomes were examined by 10% Tricine-SDS-PAGE. In Fig. 1B, when MOMP proteosomes were heat denatured in the presence of 25 mM DTT, the monomer band (39 kDa) was mostly detected (Fig. 1B, lane 2), as were some minor bands at low molecular weights (possibly degradation products). When MOMP proteosomes were not heat denatured prior to electrophoresis in the presence of 25 mM DTT, the monomeric form was observed along with additional bands of high molecular weights, i.e., the 66-kDa band indicative of trimers, and possibly oligomeric forms (Fig. 1B, lane 3). Lastly, when the MOMP proteosome preparation was not heat denatured and no DTT was added, the sample failed to enter the gel due to the lack of denaturation (Fig. 1B, lane 4).
MOMP proteosomes induce IL-8 secretion in vitro via TLR2.
MOMP proteosomes were used to stimulate HEK cells overexpressing TLR2 (TLR2-HEK cells) or cells transfected with an empty vector as a negative control (pcDNA-HEK cells). After 24 h, IL-8 secretion was assessed by ELISA of culture supernatants and is expressed in pg/ml. While MOMP proteosomes (10, 1, and 0.1 μg/ml) failed to stimulate pcDNA-HEK cells (Fig. 2A, white bars), a dose-dependent increase of IL-8 production was induced in TLR2-HEK cells (Fig. 2A, black bars) (**, P = 0.001, and ****, P < 0.0001, by unpaired t test with Welch's correction). As expected, the TLR2 agonist Pam3CSK4 (0.1 μg/ml) induced high levels of IL-8 secretion in TLR2-HEK cells (Fig. 2A, black bars), which were higher than those induced by MOMP proteosomes (**, P = 0.006, and ****, P < 0.0001). In both cell types, TNF-α (0.02 μg/ml) induced equivalent IL-8 production in a TLR-independent manner (Fig. 2A, white bars and black bars). To assess whether TLR4 contributed to the effect of MOMP proteosomes, HEK cells overexpressing TLR4 were also used. In these cells, E. coli LPS (0.1 μg/ml) induced significantly higher levels of IL-8 secretion than MOMP proteosomes and Pam3CSK4 (Fig. 2B, black bars) (***, P = 0.0008, and **, P = 0.0012, respectively), while the effect of MOMP proteosomes and Pam3CSK4 was comparable to that of incubation with medium alone. An apparently high baseline of IL-8 production was observed in TLR4-HEK cells, likely due to the presence of soluble MD-2 provided by the FBS-containing cell culture medium, leading to nonspecific stimulation.
Fig 2.
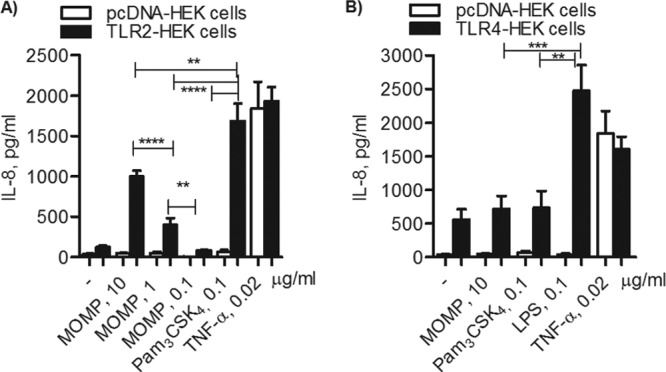
TLR2-dependent IL-8 induction by MOMP proteosomes. (A) TLR2-HEK cells (black bars) and pcDNA-HEK cells (white bars) incubated with MOMP proteosomes (10, 1, and 0.1 μg/ml), Pam3CSK4 (0.1 μg/ml), E. coli LPS (0.1 μg/ml), or TNF-α (0.02 μg/ml) for 18 h. IL-8 secretion was measured by ELISA of cell culture supernatants and expressed as pg/ml ± standard error (**, P = 0.001, **, P = 0.006, and ****, P < 0.0001, by unpaired t test with Welch's correction; n > 20). (B) TLR4-HEK cells (black bars) and pcDNA-HEK cells (white bars) were incubated as described above (***, P = 0.0008, and **, P = 0.0012; n > 10).
MOMP proteosomes induce luciferase activity in vitro via TLR2.
As an additional indicator of cell activation, induction of NF-κB-driven luciferase activity was also measured in HEK cells following stimulation with MOMP proteosomes. Luciferase activity is expressed as arbitrary relative luciferase units (RLU) normalized to nonstimulated cells ± standard errors. In TLR2-HEK cells, both MOMP proteosomes (10 μg/ml) and Pam3CSK4 (0.1 μg/ml) induced comparable levels of luciferase activity (Fig. 3A, black bars) (P > 0.05), which was predictably significantly higher than that induced by E. coli LPS (0.1 μg/ml) (**, P = 0.002). None of the stimuli used induced luciferase activity in pcDNA-HEK cells, except for TNF-α (0.02 μg/ml) (Fig. 3A and B, white bars), via a TLR-independent mechanism. Similar to IL-8, significantly high luciferase activity was induced in TLR4-HEK cells by E. coli LPS (0.1 μg/ml) compared to MOMP proteosomes and Pam3CSK4 (Fig. 3B, black bars) (**, P = 0.006 and 0.003).
Fig 3.
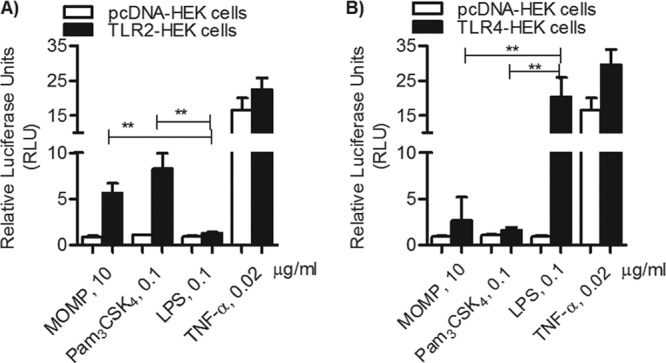
TLR2-dependent NF-κB luciferase activity. (A) TLR2-HEK cells (black bars) and pcDNA-HEK cells (white bars) incubated with MOMP proteosomes (10 μg/ml), Pam3CSK4 (0.1 μg/ml), E. coli LPS (0.1 μg/ml), or TNF-α (0.02 μg/ml) for 18 h. Luciferase activity was measured and is expressed in relative luciferase units (RLU) ± standard error normalized to nonstimulated cells (**, P = 0.002, by unpaired t test with Welch's correction; n = 6). (B) TLR4-HEK cells (black bars) and pcDNA-HEK cells (white bars) incubated as described above (**, P = 0.006 and 0.003; n = 9).
MOMP proteosomes signal via TLR2/TLR1.
TLR2 heterodimerization with TLR1 or TLR6 is required for the specific recognition of different ligands and for the induction of cell activation. To identify the TLR2 coreceptor necessary for MOMP activity in vitro, IL-8 induction was measured in HEK cells overexpressing the TLR2/TLR1 or TLR2/TLR6 receptor pair and expressed as a pg/ml fold change normalized to cells incubated with medium alone. To assess stimulation via TLR2/TLR1, Pam3CSK4 was used as a positive control, while stimulation via TLR2/TLR6 was determined using Pam2CSK4 as a positive control. In Fig. 4, TLR2/TLR1-HEK cells are indicated by the white bars. In these cells, MOMP proteosomes and Pam3CSK4 induced comparable IL-8 levels (P > 0.05 by unpaired t test with Welch's correction) and both were significantly higher than those with Pam2CSK4 (****, P < 0.0001), consistent with TLR2/TLR1-mediated cell stimulation.
Fig 4.
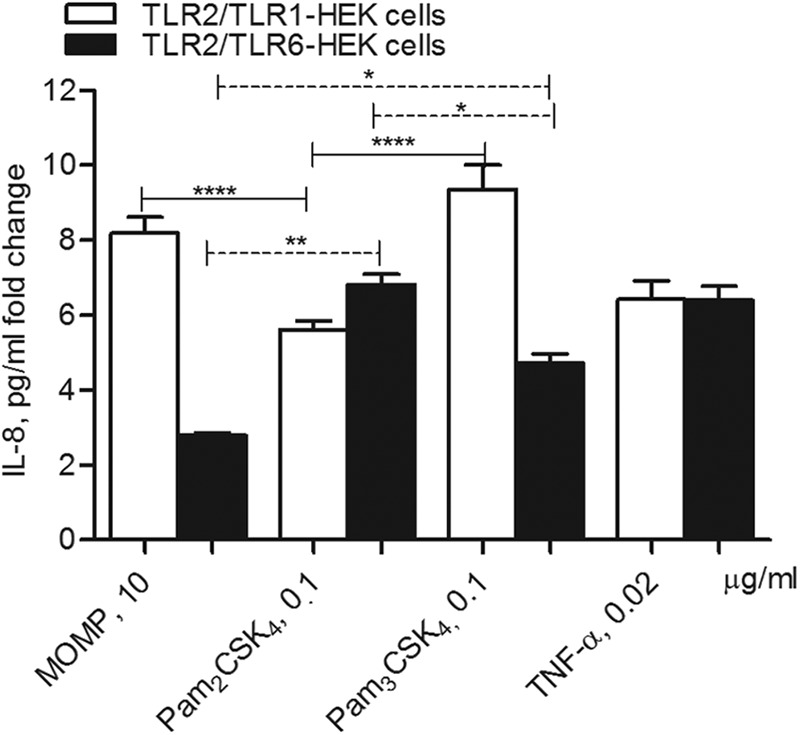
MOMP proteosome signal via TLR2/TLR1. HEK cells overexpressing TLR2/TLR1 (white bars) or TLR2/TLR6 (black bars) incubated with MOMP proteosomes (10 μg/ml), Pam2CSK4 (0.1 μg/ml), Pam3CSK4 (0.1 μg/ml), and TNF-α (0.02 μg/ml) for 18 h. IL-8 secretion was measured by ELISA of cell culture supernatants and expressed as a pg/ml fold change normalized to cells incubated with medium alone ± standard errors. For TLR2/TLR1-HEK cells, ****, P < 0.0001, by unpaired t test with Welch's correction; n = 19. For TLR2/TLR6-HEK cells, **, P = 0.005, and *, P = 0.01.
In HEK cells overexpressing TLR2 and TLR6 (Fig. 4, black bars), Pam2CSK4 induced significantly higher IL-8 levels than MOMP proteosomes (**, P = 0.005), suggesting cell activation via different receptor complexes. In these cells, as expected, Pam2CSK4 also induced higher IL-8 levels than Pam3CSK4 (*, P = 0.01). It needs to be noted that HEK cells express low endogenous levels of TLR1 and TLR6 (9), which likely accounts for the baseline level of IL-8 secretion that is consistently observed when TLR2/TLR1-HEK cells or TLR2/TLR6-HEK cells are cross-stimulated with the reciprocal agonists as well as MOMP. For example, both MOMP proteosomes and Pam3CSK4 induce some IL-8 in TLR2/TLR6-HEK cells, with Pam3CSK4 being more potent than MOMP proteosomes (*, P = 0.01), similar to that observed in TLR2/TLR1-HEK cells.
MOMP proteosome-dependent activation of intracellular signaling pathways.
IL-8 expression can be induced by the activation of the NF-κB pathway and the AP-1 pathway (via ERK1/ERK2, JNK, and p38 MAP kinases) individually or in combination, depending on the cell type and the stimulus (49). To examine the contribution of each pathway to the effect of MOMP proteosomes in vitro, specific inhibitors were used in TLR2-HEK cells. Cells were incubated with 25 μM SB203580 (p38 inhibitor), U0126 (ERK1/2 inhibitor), or SP600125 (JNK inhibitor) or with 25 μg/ml of an NF-κB inhibitory ligand for 1 h prior to stimulation with MOMP proteosomes (10 μg/ml), Pam3CSK4 (0.1 μg/ml), and TNF-α (0.02 μg/ml) for 24 h. Secretion of IL-8 was measured by ELISA, normalized to that induced in the absence of inhibitors and expressed as percentage ± standard error. When stimulation with MOMP proteosomes was carried out in the presence of each inhibitor (Fig. 5, SB203580, white bars; U0126, dotted bars; SP600125, striped bars; and NF-κB inhibitory ligand, gray bars), IL-8 secretion was reduced by approximately 50% compared to IL-8 induced in the absence of inhibitors (Fig. 5, black bar) (***, P < 0.0003, and **, P < 0.006, by a one-sample t test). Similarly, IL-8 induction by Pam3CSK4 was reduced by approximately 50% or more in the presence of SB203580, SP600125, and an NF-κB inhibitory ligand, while it was slightly less inhibited by U0126 (**, P < 0.002, and *, P = 0.02), consistent with previous results (50). Inhibition of TNF-α-induced IL-8 followed a similar trend, with a reduction of approximately 50% compared to TNF-α stimulation in the absence of inhibitors (***, P = 0.0001; **, P < 0.008; and *, P = 0.01). Thus, the ability of MOMP proteosomes to induce IL-8 secretion via TLR2 is mediated by the activation of multiple intracellular signaling pathways.
Fig 5.
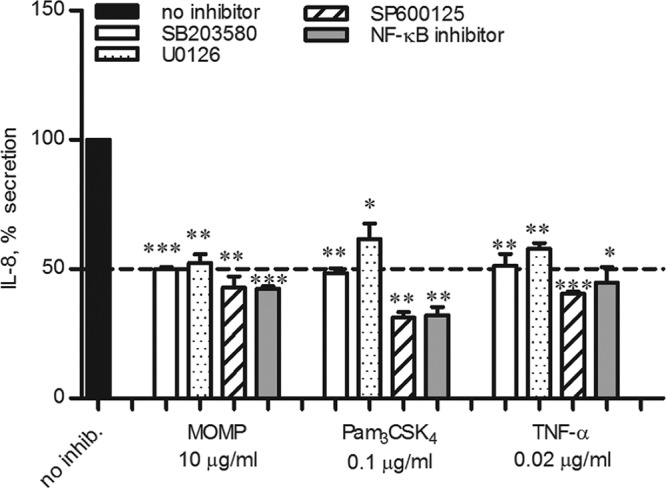
Intracellular cell signaling pathway inhibition. TLR2-HEK cells were incubated for 1 h with 25 μM p38 inhibitor SB203580 (white bars), ERK1/2 inhibitor U0126 (dotted bars), or JNK inhibitor SP600125 (striped bars) or 25 μg/ml of an NF-κB inhibitory ligand (gray bars) prior to stimulation with MOMP proteosomes (10 μg/ml), Pam3CSK4 (0.1 μg/ml), or TNF-α (0.02 μg/ml) for 24 h. Cells stimulated in the absence of inhibitors are indicated by the black bar. IL-8 secretion was measured by ELISA of cell supernatants, normalized to that induced in the absence of inhibitors and expressed as percent inhibition ± standard error. MOMP: ***, P < 0.0003, and **, P < 0.006; Pam3CSK4: **, P < 0.002, and *, P = 0.02; and TNF-α: ***, P = 0.0001, **, P < 0.008, and *, P = 0.01 (by a one-sample t test; n = 3).
TLR2-dependent induction of inflammatory mediators by MOMP proteosomes in human reproductive tract epithelial cells.
Evidence of TLR2-dependent signaling in the induction of inflammation in the female mouse model of genital tract Chlamydia infection has been shown, and a role of TLR2 in site-specific mucosal inflammation has been hypothesized (35). To evaluate the effect of MOMP proteosomes in a naturally TLR2-competent cell model representative of the human female reproductive tract, the endocervical cell line End/E6E7 was used and secretion of inflammatory mediators IL-8 and IL-6 was examined. Due to high baseline cytokine secretion in growth factor-containing KSFM culture medium, IL-8 and IL-6 are expressed as a pg/ml fold change relative to cells incubated with medium alone. End/E6E7 cells, which lack expression of TLR4 and MD-2 (51), did not secrete IL-8 or IL-6 above the baseline level in response to stimulation with E. coli LPS (0.1 μg/ml) for 24 h (Fig. 6A and B). MOMP proteosomes and Pam3CSK4 induced a similar fold increase in IL-8 secretion (Fig. 6A) (*, P < 0.05, by a one-sample t test). In these cells, MOMP proteosomes also induced a statistically significant increase in IL-6 secretion, although the effect of Pam3CSK4 was more pronounced (Fig. 6B). As expected, TNF-α induced TLR2-independent IL-8 and IL-6 secretion (Fig. 6A and B).
Fig 6.
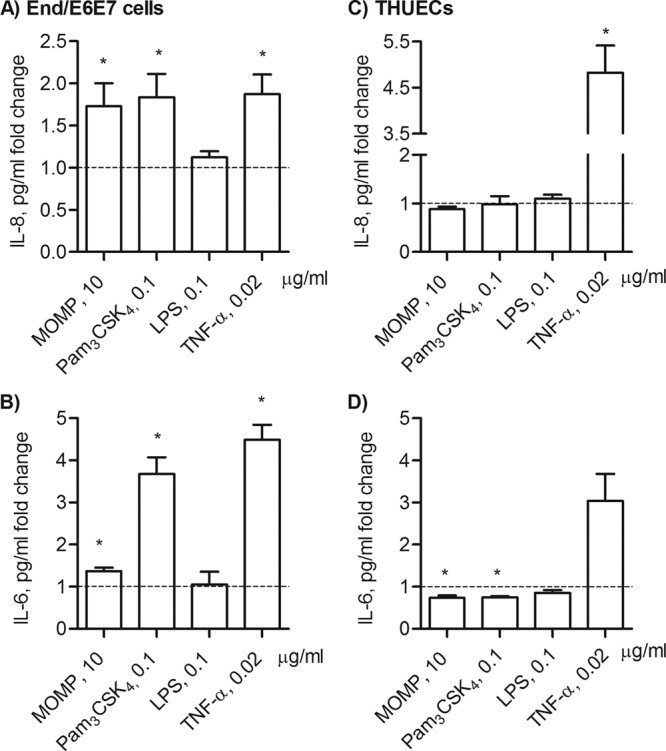
Inflammatory responses of human reproductive tract epithelial cells induced by MOMP proteosomes. IL-8 secretion (A) and IL-6 secretion (B) by End/E6E7 cells incubated with MOMP proteosomes (10 μg/ml), Pam3CSK4 (0.1 μg/ml), E. coli LPS (0.1 μg/ml), or TNF-α (0.02 μg/ml) for 24 h. Cytokines were measured by ELISA of cell supernatants and expressed as a pg/ml fold change relative to cells incubated with medium alone ± standard error (*, P < 0.05, by a one-sample t test; n = 9). IL-8 (C) and IL-6 (D) induced in THUECs incubated as described above (*, P < 0.05; n = 3).
The effect of MOMP proteosomes on HeLa cells, previously used as a cell model to study Chlamydia infection (52, 53), was also examined. However, since HeLa cells lack TLR1 expression and are nonresponsive to TLR2/TLR1 ligands (54, 55), no induction of IL-8 or IL-6 by MOMP proteosomes or by Pam3CSK4 was detected (not shown). In contrast to End/E6E7 cells, HeLa cells expressed TLR4 and were stimulated by E. coli LPS as well as by TNF-α in a TLR-independent manner (not shown). The responses of male urethral epithelial cells, THUECs (47, 52), were assessed. Similar to HeLa cells, no IL-8 secretion was induced in THUECs by MOMP proteosomes, Pam3CSK4, or even E. coli LPS incubation for 24 h (Fig. 6C). Furthermore, a below-baseline trend of IL-6 production was observed following incubation with MOMP proteosomes or Pam3CSK4 while TNF-α successfully induced IL-8 and IL-6 secretion (Fig. 6D). To determine whether inflammatory responses were elicited in End/E6E7 and THUECs by MOMP proteosomes with different kinetics, time-dependent IL-8 and IL-6 secretion was examined. In End/E6E7 cells, Pam3CSK4 and TNF-α induced high IL-8 secretion as early as 3 h and this remained sustained up to 24 h, while MOMP proteosomes only induced elevated IL-8 secretion at 24 h (Fig. 7A) (*, P < 0.05, by one-way ANOVA with a Bonferroni posttest). Stimulation of End/E6E7 cells with Pam3CSK4 for 3 h, 6 h, and 24 h showed a trend of increased IL-6 production over time (Fig. 7B), while an opposite trend was observed for TNF-α, where IL-6 secretion appeared reduced at 24 h compared to 3 h and 6 h (Fig. 7B). Similar to Pam3CSK4, MOMP proteosomes induced an apparent 2-fold increase in IL-6 secretion at 3 h and 6 h, but this remained unchanged (or possibly even slightly decreased) at 24 h (Fig. 7B). Conversely, in THUECs, sustained IL-8 and IL-6 secretion over time was only induced by TNF-α stimulation (Fig. 7C and D).
Fig 7.

Time course of IL-8 and IL-6 secretion in human reproductive tract epithelial cells. Kinetics of IL-8 (A) and IL-6 (B) secretion in End/E6E stimulated with MOMP proteosomes (10 μg/ml), Pam3CSK4 (0.1 μg/ml), E. coli LPS (0.1 μg/ml), or TNF-α (0.02 μg/ml) for 3 h (white bars), 6 h (striped bars), and 24 h (black bars) (*, P < 0.05, by one-way ANOVA with a Bonferroni posttest; n = 3). IL-8 (C) and IL-6 (D) production by THUECs incubated as described above.
DISCUSSION
TLRs and other pattern recognition receptors (PRRs) recognize microbial products and modulate acute inflammatory responses, host innate and adaptive immune responses, and site-specific defense mechanisms. In the past decade, much progress has been made in defining how bacterial outer membrane components induce such responses via TLR signaling. For example, bacterial porins are well-established TLR2 agonists in combination with the coreceptors TLR1 and TLR6. The effect of numerous bacterial porins has been examined in vitro and in vivo (9–11, 13, 14), thus providing relevant information on how these products may influence initial mucosal host responses in specific cell niches by regulating TLR-dependent cell signaling.
A number of pattern recognition receptors, including TLRs and Nods (21, 22), are involved in host responses to Chlamydia infection and to purified Chlamydia components, but the role of the chlamydial MOMP had not been examined so far. Since MOMP is an integral outer membrane protein, the addition of detergents is required to preserve its native structure and to ensure solubility when it is purified from Chlamydia organisms. Detergent-based MOMP preparations have been used in animal immunization studies, but these cannot be successfully used in vitro due to the cytotoxic nature of detergents, which is amplified in the confined tissue culture well microenvironment. Thus, native purified MOMP was incorporated into detergent-free protein micelles, termed proteosomes (45), a strategy that has been successfully applied to investigate the in vitro and in vivo effects of bacterial porins. In all the cell types used in this work, no indication of cell toxicity was observed for MOMP proteosomes (not shown). In addition, the trimeric nature of MOMP was preserved, as observed by the presence of high-molecular-weight forms corresponding to trimers (and oligomers) by electrophoretic analysis (48).
Using a human nonhematopoietic cell model of TLR expression (HEK cells), we demonstrate that MOMP proteosomes induce TLR2-dependent IL-8 secretion and the induction of NF-κB-dependent luciferase activity, consistent with the known TLR2-dependent activity of bacterial porins. Furthermore, TLR4 did not appear to play a role in the activity of MOMP proteosomes in vitro.
TLR2 signaling is dependent on the expression of coreceptors. A TLR2/TLR1 complex drives TLR2-dependent cell activation by Neisseria PorB, while TLR2/TLR6 mediates the activity of Shigella porins. Using HEK cells that overexpress the TLR2/TLR1 or TLR2/TLR6 receptor pair, we show that MOMP proteosomes induce high levels of IL-8 secretion when TLR2 and TLR1 are present on the cell surface, similar to the effect of the synthetic triacylated lipopeptide Pam3CSK4. In contrast, in cells that overexpress TLR2 and TLR6, MOMP proteosomes (and Pam3CSK4) induced low IL-8 secretion, while the diacylated lipopeptide Pam2CSK4 induced high levels of IL-8, consistent with TLR2/TLR6 signaling. As previously mentioned, HEK cells express low endogenous TLR1 and TLR6 levels, which can explain the background level of cell activation in response to each of the stimuli used.
Maximal expression of IL-8 is induced via combined activation of both NF-κB and AP-1 signaling pathways (via ERK1/ERK2, JNK, and p38 MAP kinases), particularly in nonhematopoietic cells (49). To dissect the intracellular signaling cascades induced by MOMP proteosomes in vitro, specific inhibitors of NF-κB and MAP kinase phosphorylation and activation were used. Inhibition of p38, ERK1/2, and JNK signaling, as well as NF-κB, prevented TLR2-dependent IL-8 induction by MOMP proteosomes and Pam3CSK4, as well as TLR2-independent cell activation by TNF-α, in agreement with multiple signaling pathways involved in TLR2-dependent (and -independent) induction of IL-8.
The use of HEK cells overexpressing TLRs is a well-established tool for the characterization of the activity of various TLR agonists. However, the overabundance of TLR expression may, in part, supersede variations of activity of different agonists. For this reason, naturally TLR2-competent cells were also used to test the activity of MOMP proteosomes. Because Chlamydia infection is a significant public health problem, it is important to understand the mechanism(s) by which chlamydial products may activate host cell inflammatory responses at the site of colonization and infection. In the reproductive tract epithelium, as well as in other nonsterile epithelia, TLRs are selectively expressed for a tight regulation of inflammatory responses following bacterial colonization or host cell infection (56–58).
Evidence exists that TLR2 signaling may contribute to regulation of site-specific mucosal inflammation following Chlamydia infection, for example, more severe respiratory pathology and inflammation have been observed in TLR2 knockout mice infected with C. muridarum compared to wild-type mice (35). The human female epithelial reproductive tract cell line EndE6/E7 is a relevant cell model for studying the outcomes of cell stimulation with MOMP proteosomes, since Chlamydia infects the human reproductive tract epithelium. An additional advantage of these cells is their unresponsiveness to TLR4 agonists, due to the lack of TLR4 and MD-2 expression required for LPS signaling (51). In these cells, MOMP proteosomes induced secretion of the proinflammatory mediators IL-8 and IL-6, similar to the effect of Pam3CSK4 (although Pam3CSK4 was more potent in inducing IL-6 than MOMP proteosomes). In HeLa cells, MOMP proteosomes failed to induce both IL-8 and IL-6 secretion, possibly due to the lack of expression of TLR1 by these cells (54), which also explains the lack of responsiveness to Pam3CSK4 (55). Since Chlamydia also infects the male urethral and bladder epithelia, epithelial cells from both the female and male reproductive tracts have been used as models for Chlamydia infection (as well as for Neisseria gonorrhoeae) (47, 51). Studies in both human and rat prostate epithelial cell lines have shown that these cells express TLR4 and CD14 mRNA as well as TLR2 and TLR9 (57). It is known that, after prolonged exposure of THUECs to live Chlamydia organisms, IL-6, IL-1β, and, to a lesser extent, IL-8 are induced in the cell supernatant (52). However, stimulation of THUECs with MOMP proteosomes, Pam3CSK4, or even E. coli LPS for 24 h did not induce IL-8 or IL-6 secretion, while only TNF-α stimulation led to high levels of inflammatory cytokine production.
To determine whether IL-8 and IL-6 secretion may follow different kinetics in female and male reproductive tract epithelial cells, early time points were also examined, but a time-dependent, increased IL-8 induction by MOMP proteosomes was only detected in End/E6E7. Furthermore, in End/E6E7, a reduced IL-6 secretion trend was apparent after 24 h of incubation with MOMP proteosomes and with TNF-α. From these initial observations, the speculation that MOMP could contribute to suppression of cellular inflammatory responses in TLR2-competent female reproductive tract epithelial cells during host exposure to chlamydial products may arise. It is possible that a longer stimulation with MOMP may be required to mimic the consequences of prolonged cell exposure to Chlamydia that would recapitulate a chronic infection. Nevertheless, our comparison of female and male reproductive tract epithelial cells may, in part, contribute to explain the differences in the inflammatory responses in vivo following Chlamydia infection, although the limitations of these in vitro models need to be carefully considered.
A better understanding of both the host immune responses and the bacterial immune effectors is essential for guiding new and improved approaches to control and prevent chlamydial pathogenesis. Crucial studies have suggested that TLR2-dependent host responses and murine genital tract and respiratory infections by Chlamydia are influenced by the presence of a cryptic plasmid in strains of C. muridarum and C. trachomatis (26), while plasmid-cured Chlamydophila caviae retains the ability to signal and induce pathogenesis in guinea pigs (25). However, there is no evidence that plasmid-encoded proteins are directly responsible for TLR2-mediated signaling by Chlamydia. In addition, since the plasmid-cured strains replicate less efficiently than the wild-type strains, a potential overall smaller amount of antigens, including MOMP, may be present in the coculture experiments in vitro. Whether MOMP purified from these plasmid-cured strains induces TLR2-dependent cell activation in vitro has not been examined. However, since the DNA sequence of the MOMP in these isolates is identical to that of the original strains, one could predict that there will be no differences in activity.
In conclusion, our results provide evidence of TLR2-dependent activity for purified MOMP proteosomes and lay the basis for investigating its potential role, as a TLR2-dependent agonist, in the induction of the inflammatory response during a chlamydial infection. Furthermore, as a vaccine candidate, it will be important to further evaluate the potential adjuvant activity of MOMP proteosomes in vitro and in vivo.
ACKNOWLEDGMENTS
We thank Ryan McClure for help with the End/E6E7 cell and THUEC cultures. We also thank Ryan McClure and Robin Ingalls for critical readings of the manuscript.
This work was supported by Public Health Service grants AI067888 and AI092129 from the National Institute of Allergy and Infectious Diseases.
Footnotes
Published ahead of print 6 November 2012
REFERENCES
- 1. Medzhitov R, Janeway C., Jr 2000. The Toll receptor family and microbial recognition. Trends Microbiol. 8:452–456 [DOI] [PubMed] [Google Scholar]
- 2. Akira S, Takeda K, Kaisho T. 2001. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2:675–680 [DOI] [PubMed] [Google Scholar]
- 3. Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. 1999. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J. Biol. Chem. 274:10689–10692 [DOI] [PubMed] [Google Scholar]
- 4. Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. 2001. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410:1099–1103 [DOI] [PubMed] [Google Scholar]
- 5. Bauer S, Kirschning CJ, Hacker H, Redecke V, Hausmann S, Akira S, Wagner H, Lipford GB. 2001. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc. Natl. Acad. Sci. U. S. A. 98:9237–9242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413:732–738 [DOI] [PubMed] [Google Scholar]
- 7. Kang JY, Nan X, Jin MS, Youn SJ, Ryu YH, Mah S, Han SH, Lee H, Paik SG, Lee JO. 2009. Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 31:873–884 [DOI] [PubMed] [Google Scholar]
- 8. Asong J, Wolfert MA, Maiti KK, Miller D, Boons GJ. 2009. Binding and cellular activation studies reveal that Toll-like receptor 2 can differentially recognize peptidoglycan from Gram-positive and Gram-negative bacteria. J. Biol. Chem. 284:8643–8653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Massari P, Visintin A, Gunawardana J, Halmen KA, King CA, Golenbock DT, Wetzler LM. 2006. Meningococcal porin PorB binds to TLR2 and requires TLR1 for signaling. J. Immunol. 176:2373–2380 [DOI] [PubMed] [Google Scholar]
- 10. Liu X, Wetzler LM, Nascimento LO, Massari P. 2010. Human airway epithelial cell responses to Neisseria lactamica and purified porin via Toll-like receptor 2-dependent signaling. Infect. Immun. 78:5314–5323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Biswas A, Banerjee P, Mukherjee G, Biswas T. 2007. Porin of Shigella dysenteriae activates mouse peritoneal macrophage through Toll-like receptors 2 and 6 to induce polarized type I response. Mol. Immunol. 44:812–820 [DOI] [PubMed] [Google Scholar]
- 12. Cervantes-Barragan L, Gil-Cruz C, Pastelin-Palacios R, Lang KS, Isibasi A, Ludewig B, Lopez-Macias C. 2009. TLR2 and TLR4 signaling shapes specific antibody responses to Salmonella Typhi antigens. Eur. J. Immunol. 39:126–135 [DOI] [PubMed] [Google Scholar]
- 13. Galdiero M, Galdiero M, Finamore E, Rossano F, Gambuzza M, Catania MR, Teti G, Midiri A, Mancuso G. 2004. Haemophilus influenzae porin induces Toll-like receptor 2-mediated cytokine production in human monocytes and mouse macrophages. Infect. Immun. 72:1204–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Toussi DN, Liu X, Massari P. 2012. The FomA porin from Fusobacterium nucleatum is a Toll-like receptor 2 agonist with immune adjuvant activity. Clin. Vaccine Immunol. 19:1093–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Centers for Disease Control and Prevention 2009. Chlamydia screening among sexually active young female enrollees of health plans—United States, 2000–2007. MMWR Morb. Mortal. Wkly. Rep. 58:362–365 [PubMed] [Google Scholar]
- 16. Westrom LV. 1996. Chlamydia and its effect on reproduction. J. Br. Fer. Soc. 1:23–30 [PubMed] [Google Scholar]
- 17. Grayston JT, Wang S. 1975. New knowledge of chlamydiae and the diseases they cause. J. Infect. Dis. 132:87–105 [DOI] [PubMed] [Google Scholar]
- 18. Schachter J, Dawson CR. 2002. Elimination of blinding trachoma. Curr. Opin. Infect. Dis. 15:491–495 [DOI] [PubMed] [Google Scholar]
- 19. Kumar S, Hammerschlag MR. 2007. Acute respiratory infection due to Chlamydia pneumoniae: current status of diagnostic methods. Clin. Infect. Dis. 44:568–576 [DOI] [PubMed] [Google Scholar]
- 20. Jackson LA, Campbell LA, Schmidt RA, Kuo C, Cappuccio AL, Lee MJ, Grayston JT. 2000. Specificity of detection of Chlamydia pneumoniae in cardiovascular atheroma. J. Infect. Dis. 181(Suppl 3):S447–S448 [DOI] [PubMed] [Google Scholar]
- 21. Joyee AG, Yang X. 2008. Role of toll-like receptors in immune responses to chlamydial infections. Curr. Pharm. Des. 14:593–600 [DOI] [PubMed] [Google Scholar]
- 22. Welter-Stahl L, Ojcius DM, Viala J, Girardin S, Liu W, Delarbre C, Philpott D, Kelly KA, Darville T. 2006. Stimulation of the cytosolic receptor for peptidoglycan, Nod1, by infection with Chlamydia trachomatis or Chlamydia muridarum. Cell. Microbiol. 8:1047–1057 [DOI] [PubMed] [Google Scholar]
- 23. Chamaillard M, Girardin SE, Viala J, Philpott DJ. 2003. Nods, Nalps and Naip: intracellular regulators of bacterial-induced inflammation. Cell. Microbiol. 5:581–592 [DOI] [PubMed] [Google Scholar]
- 24. Derbigny WA, Shobe LR, Kamran JC, Toomey KS, Ofner S. 2012. Identifying a role for Toll-like receptor 3 in the innate immune response to Chlamydia muridarum infection in murine oviduct epithelial cells. Infect. Immun. 80:254–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Frazer LC, Darville T, Chandra-Kuntal K, Andrews CW, Jr, Zurenski M, Mintus M, AbdelRahman YM, Belland RJ, Ingalls RR, O'Connell CM. 2012. Plasmid-cured Chlamydia caviae activates TLR2-dependent signaling and retains virulence in the guinea pig model of genital tract infection. PLoS One 7:e30747 doi:10.1371/journal.pone.0030747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. O'Connell CM, AbdelRahman YM, Green E, Darville HK, Saira K, Smith B, Darville T, Scurlock AM, Meyer CR, Belland RJ. 2011. Toll-like receptor 2 activation by Chlamydia trachomatis is plasmid dependent, and plasmid-responsive chromosomal loci are coordinately regulated in response to glucose limitation by C. trachomatis but not by C. muridarum. Infect. Immun. 79:1044–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bas S, Neff L, Vuillet M, Spenato U, Seya T, Matsumoto M, Gabay C. 2008. The proinflammatory cytokine response to Chlamydia trachomatis elementary bodies in human macrophages is partly mediated by a lipoprotein, the macrophage infectivity potentiator, through TLR2/TLR1/TLR6 and CD14. J. Immunol. 180:1158–1168 [DOI] [PubMed] [Google Scholar]
- 28. Ingalls RR, Rice PA, Qureshi N, Takayama K, Lin JS, Golenbock DT. 1995. The inflammatory cytokine response to Chlamydia trachomatis infection is endotoxin mediated. Infect. Immun. 63:3125–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Heine H, Lien E. 2003. Toll-like receptors and their function in innate and adaptive immunity. Int. Arch. Allergy Immunol. 130:180–192 [DOI] [PubMed] [Google Scholar]
- 30. Bulut Y, Faure E, Thomas L, Karahashi H, Michelsen KS, Equils O, Morrison SG, Morrison RP, Arditi M. 2002. Chlamydial heat shock protein 60 activates macrophages and endothelial cells through Toll-like receptor 4 and MD2 in a MyD88-dependent pathway. J. Immunol. 168:1435–1440 [DOI] [PubMed] [Google Scholar]
- 31. Vabulas RM, Ahmad-Nejad P, da Costa C, Miethke T, Kirschning CJ, Hacker H, Wagner H. 2001. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J. Biol. Chem. 276:31332–31339 [DOI] [PubMed] [Google Scholar]
- 32. Da Costa CU, Wantia N, Kirschning CJ, Busch DH, Rodriguez N, Wagner H, Miethke T. 2004. Heat shock protein 60 from Chlamydia pneumoniae elicits an unusual set of inflammatory responses via Toll-like receptor 2 and 4 in vivo. Eur. J. Immunol. 34:2874–2884 [DOI] [PubMed] [Google Scholar]
- 33. Beckett EL, Phipps S, Starkey MR, Horvat JC, Beagley KW, Foster PS, Hansbro PM. 2012. TLR2, but not TLR4, is required for effective host defence against chlamydia respiratory tract infection in early life. PLoS One 7:e39460 doi:10.1371/journal.pone.0039460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. O'Connell CM, Ionova IA, Quayle AJ, Visintin A, Ingalls RR. 2006. Localization of TLR2 and MyD88 to Chlamydia trachomatis inclusions. Evidence for signaling by intracellular TLR2 during infection with an obligate intracellular pathogen. J. Biol. Chem. 281:1652–1659 [DOI] [PubMed] [Google Scholar]
- 35. He X, Nair A, Mekasha S, Alroy J, O'Connell CM, Ingalls RR. 2011. Enhanced virulence of Chlamydia muridarum respiratory infections in the absence of TLR2 activation. PLoS One 6:e20846 doi:10.1371/journal.pone.0020846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McCoy AJ, Maurelli AT. 2005. Characterization of Chlamydia MurC-Ddl, a fusion protein exhibiting D-alanyl-D-alanine ligase activity involved in peptidoglycan synthesis and D-cycloserine sensitivity. Mol. Microbiol. 57:41–52 [DOI] [PubMed] [Google Scholar]
- 37. Patin D, Bostock J, Chopra I, Mengin-Lecreulx D, Blanot D. 2012. Biochemical characterisation of the chlamydial MurF ligase, and possible sequence of the chlamydial peptidoglycan pentapeptide stem. Arch. Microbiol. 194:505–512 [DOI] [PubMed] [Google Scholar]
- 38. Bavoil P, Ohlin A, Schachter J. 1984. Role of disulfide bonding in outer membrane structure and permeability in Chlamydia trachomatis. Infect. Immun. 44:479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rodriguez-Maranon MJ, Bush RM, Peterson EM, Schirmer T, de la Maza LM. 2002. Prediction of the membrane-spanning beta-strands of the major outer membrane protein of Chlamydia. Protein Sci. 11:1854–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sun G, Pal S, Sarcon AK, Kim S, Sugawara E, Nikaido H, Cocco MJ, Peterson EM, de la Maza LM. 2007. Structural and functional analyses of the major outer membrane protein of Chlamydia trachomatis. J. Bacteriol. 189:6222–6235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kawa DE, Schachter J, Stephens RS. 2004. Immune response to the Chlamydia trachomatis outer membrane protein PorB. Vaccine 22:4282–4286 [DOI] [PubMed] [Google Scholar]
- 42. Kari L, Whitmire WM, Crane DD, Reveneau N, Carlson JH, Goheen MM, Peterson EM, Pal S, de la Maza LM, Caldwell HD. 2009. Chlamydia trachomatis native major outer membrane protein induces partial protection in nonhuman primates: implication for a trachoma transmission-blocking vaccine. J. Immunol. 182:8063–8070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cheng C, Pal S, Bettahi I, Oxford KL, Barry PA, de la Maza LM. 2011. Immunogenicity of a vaccine formulated with the Chlamydia trachomatis serovar F, native major outer membrane protein in a nonhuman primate model. Vaccine 29:3456–3464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sabet SF, Simmons J, Caldwell HD. 1984. Enhancement of Chlamydia trachomatis infectious progeny by cultivation of HeLa 229 cells treated with DEAE-dextran and cycloheximide. J. Clin. Microbiol. 20:217–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Massari P, King CA, Macleod H, Wetzler LM. 2005. Improved purification of native meningococcal porin PorB and studies on its structure/function. Protein Expr. Purif. 44:136–146 [DOI] [PubMed] [Google Scholar]
- 46. Fichorova RN, Rheinwald JG, Anderson DJ. 1997. Generation of papillomavirus-immortalized cell lines from normal human ectocervical, endocervical, and vaginal epithelium that maintain expression of tissue-specific differentiation proteins. Biol. Reprod. 57:847–855 [DOI] [PubMed] [Google Scholar]
- 47. Harvey HA, Post DM, Apicella MA. 2002. Immortalization of human urethral epithelial cells: a model for the study of the pathogenesis of and the inflammatory cytokine response to Neisseria gonorrhoeae infection. Infect. Immun. 70:5808–5815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yen TY, Pal S, de la Maza LM. 2005. Characterization of the disulfide bonds and free cysteine residues of the Chlamydia trachomatis mouse pneumonitis major outer membrane protein. Biochemistry 44:6250–6256 [DOI] [PubMed] [Google Scholar]
- 49. Roebuck KA, Carpenter LR, Lakshminarayanan V, Page SM, Moy JN, Thomas LL. 1999. Stimulus-specific regulation of chemokine expression involves differential activation of the redox-responsive transcription factors AP-1 and NF-kappaB. J. Leukoc. Biol. 65:291–298 [DOI] [PubMed] [Google Scholar]
- 50. Toussi DN, Carraway M, Wetzler LM, Lewis LA, Liu X, Massari P. 2012. The amino acid sequence of Neisseria lactamica PorB surface-exposed loops influences Toll-like receptor 2-dependent cell activation. Infect. Immun. 80:3417–3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fichorova RN, Cronin AO, Lien E, Anderson DJ, Ingalls RR. 2002. Response to Neisseria gonorrhoeae by cervicovaginal epithelial cells occurs in the absence of toll-like receptor 4-mediated signaling. J. Immunol. 168:2424–2432 [DOI] [PubMed] [Google Scholar]
- 52. Al-Mously N, Eley A. 2007. Interaction of Chlamydia trachomatis serovar E with male genital tract epithelium results in secretion of proinflammatory cytokines. J. Med. Microbiol. 56:1025–1032 [DOI] [PubMed] [Google Scholar]
- 53. Rasmussen SJ, Eckmann L, Quayle AJ, Shen L, Zhang YX, Anderson DJ, Fierer J, Stephens RS, Kagnoff MF. 1997. Secretion of proinflammatory cytokines by epithelial cells in response to Chlamydia infection suggests a central role for epithelial cells in chlamydial pathogenesis. J. Clin. Invest. 99:77–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wyllie DH, Kiss-Toth E, Visintin A, Smith SC, Boussouf S, Segal DM, Duff GW, Dower SK. 2000. Evidence for an accessory protein function for Toll-like receptor 1 in anti-bacterial responses. J. Immunol. 165:7125–7132 [DOI] [PubMed] [Google Scholar]
- 55. Massari P, Gunawardana J, Liu X, Wetzler LM. 2010. Meningococcal porin PorB prevents cellular apoptosis in a toll-like receptor 2- and NF-kappaB-independent manner. Infect. Immun. 78:994–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fazeli A, Bruce C, Anumba DO. 2005. Characterization of Toll-like receptors in the female reproductive tract in humans. Hum. Reprod. 20:1372–1378 [DOI] [PubMed] [Google Scholar]
- 57. Mackern-Oberti JP, Maccioni M, Cuffini C, Gatti G, Rivero VE. 2006. Susceptibility of prostate epithelial cells to Chlamydia muridarum infection and their role in innate immunity by recruitment of intracellular Toll-like receptors 4 and 2 and MyD88 to the inclusion. Infect. Immun. 74:6973–6981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gribar SC, Richardson WM, Sodhi CP, Hackam DJ. 2008. No longer an innocent bystander: epithelial toll-like receptor signaling in the development of mucosal inflammation. Mol. Med. 14:645–659 [DOI] [PMC free article] [PubMed] [Google Scholar]