

Abstract
Capsular material of the opportunistic fungus Cryptococcus neoformans is composed mainly of a polysaccharide named glucuronoxylomannan (GXM). In this study, the effects of GXM were analyzed in an in vivo experimental system of lipopolysaccharide (LPS)-induced shock. Endotoxic shock was induced in mice by a single intraperitoneal injection of LPS from Escherichia coli. GXM treatment reduced the mortality of mice at early stages. Mice treated with LPS alone showed markedly increased plasma levels of tumor necrosis factor alpha (TNF-α), interleukin-1β (IL-1β), and IL-6, whereas mice that were also treated with GXM showed significantly lower plasma levels of these cytokines. This effect was related to a marked suppression of Akt and IκBα activation. Importantly, the inhibitory effect of GXM on proinflammatory cytokine secretion was reproduced by treatment with wortmannin, an inhibitor of the Akt transcription pathway. Our results indicate that GXM has a beneficial effect on endotoxic shock, resulting in a significant increase in the rate of survival by dampening the hyperinflammatory response.
INTRODUCTION
The fungus Cryptococcus neoformans is the causative agent of cryptococcosis and the only major fungal pathogen which possesses a polysaccharide capsule (1). Capsular polysaccharides are released into host tissues (2–5), where they exert numerous deleterious effects on the host immune function (6–8). Glucuronoxylomannan (GXM) is the principal component of the capsular material of C. neoformans, and it has been isolated from body fluids of patients with cryptococcosis (9). We previously reported that GXM inhibits proinflammatory cytokine production by macrophages (10) and induces alteration of costimulatory molecule expression (11) and apoptosis (11, 12).
These suppressive anti-inflammatory properties of GXM have been exploited to obtain beneficial effects in an in vivo experimental model of rheumatoid arthritis, a pathology in which the inflammatory reaction is the critical issue (13).
Inflammatory shock following release of lipopolysaccharide (LPS) from Gram-negative bacteria is a serious clinical concern. In humans, immune responses to LPS result in the release of many inflammatory cytokines from monocytes and macrophages, in particular tumor necrosis factor alpha (TNF-α), interleukin-1β (IL-1β), and IL-6, which can cause fever, shock, organ failure, and death (14, 15).
Toll-like receptor 4 (TLR4) is the central signaling receptor for LPS in mammals (16). LPS binds to the TLR4 receptor complex, consisting of soluble CD14 (sCD14) and MD2. This results in the recruitment of the adaptor molecule MyD88. Phosphatidylinositol 3-kinase (PI3K), through association with MyD88 via an Akt-dependent mechanism, is involved in NF-κB activation (17).
Current knowledge on the structure and function of TLR4 has opened up the possibility of developing new drug targets to fight sepsis and other diseases associated with this signaling molecule (18). Recently, direct antagonists of the LPS receptor TLR4 were developed for treating sepsis (19–21), and various molecules interfering with TLR4 expression or the TLR4-related intracellular pathway have been proposed as new therapies able to weaken the deleterious effects of an excessive host response. However, most of these have not yet been exploited, and additional studies are required to confirm their expected action (22). Furthermore, TLR4-blocking treatments (such as the TLR4 antagonist eritoran) are still under investigation (23). This study therefore aimed to examine alternative options for curing sepsis.
Given that GXM is able to reduce LPS-induced inflammatory cytokines in vitro (24) and to inhibit signal transduction triggered by TLR4 (11), the aim of this study was to evaluate the possible effect of GXM treatment on LPS-induced endotoxic shock and the related signaling that involves the MyD88/Akt/NF-κB pathway.
MATERIALS AND METHODS
Ethics statement.
All animal experiments adhered to European Union directive 2010/63. Experiments were performed according to the guidelines of the European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes (25). The protocol was approved by the Perugia University Ethics Committee. All efforts were made to minimize suffering.
Reagents and media.
RPMI 1640 with l-glutamine was obtained from Gibco BRL (Paisley, Scotland, United Kingdom). Fetal calf serum (FCS), penicillin, and Dulbecco's modified Eagle medium (DMEM) were purchased from the American Type Culture Collection (ATCC) (Manassas, VA). A purified mouse monoclonal antibody to pAkt (pS472/pS473) (clone 104A282) was purchased from BD Pharmingen (BD Biosciences, Franklin Lakes, NJ). A rabbit polyclonal antibody to Akt was purchased from Cell Signaling Technology (Beverly, MA). A goat polyclonal antibody to pIκBα (Ser-32), rabbit polyclonal antibodies to IκBα, actin (H-300), and MyD88 (HFL-296), and horseradish peroxidase (HRP)-linked rabbit polyclonal anti-goat IgG were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). A rabbit polyclonal antibody to pSHIP (Tyr pY120), a mouse monoclonal antibody to SHIP, and a goat polyclonal antibody to FcγRIIB (Ala46/Pro217) were purchased from StemCell Technologies (Vancouver, Canada), Exbio Antibodies (Czech Republic), and R&D Systems (Minneapolis, MN), respectively. HRP-linked goat polyclonal anti-rabbit IgG and HRP-linked goat polyclonal anti-mouse IgG were purchased from Bio-Rad Laboratories (Hercules, CA). Cyanine 3 (Cy3)-conjugated rabbit polyclonal anti-goat IgG was purchased from Chemicon International (Temecula, CA). Mammalian protein extraction reagent (M-PER) and Restore Western stripping buffer were obtained from Pierce (Rockford, IL). An Immun-Star HRP chemiluminescence kit was purchased from Bio-Rad Laboratories (Hercules, CA). Lipopolysaccharides from Escherichia coli O55:B5 and wortmannin from Penicillium funiculosum were purchased from Sigma-Aldrich (St. Louis, MO). All media used for cell culture were negative for endotoxin as detected by Limulus amebocyte lysate assay (Sigma-Aldrich), which had a sensitivity of approximately 0.05 to 0.1 ng of Escherichia coli lipopolysaccharide per ml.
Mice.
Eleven- to 12-week-old male C57BL/6J mice were obtained from Harlan Nossan Laboratories (Milan, Italy) and maintained under specific-pathogen-free conditions in the animal care facility of the University of Perugia (Perugia, Italy).
Cryptococcal polysaccharide.
Glucuronoxylomannan was isolated from the culture supernatant fluid of a serotype A strain (CN6) grown in liquid synthetic medium in a gyratory shaker at 30°C for 4 days, as previously described (26). GXM was isolated by differential precipitation with ethanol and hexadecyltrimethyl ammonium bromide (Sigma-Aldrich) as previously described (27).
Endotoxic shock and GXM treatment.
Endotoxic shock was induced in 11- to 12-week-old male C57BL/6J mice by a single injection of LPS from Escherichia coli O55:B5. The GXM dosage (100 μg/mouse) was determined based on our previous experience with experimental models (28), and the number of injections was based on the results of preliminary experiments. LPS and/or GXM was given as a single dose intraperitoneally (i.p.) in 200 μl of sterile saline solution. For survival experiments, a lethal dose of LPS was used (1.5 mg/mouse) (29). Mice were randomly divided into five groups (5 mice/group): (i) control (200 μl of sterile saline solution/mouse), (ii) LPS, (iii) GXM, (iv) LPS plus GXM (GXM administration 15 min after LPS injection), and (v) GXM plus LPS (GXM administration 60 min before LPS injection). The survival rate of mice was monitored for up to 5 days.
To study GXM's effects on cytokine production and intracellular signals, a sublethal dose of LPS was used (0.6 mg/mouse) (30). Mice were randomly divided into eight groups (5 mice/group): (i) control (200 μl of sterile saline solution/mouse), (ii) LPS, (iii) GXM, (iv) LPS plus GXM (GXM administration 15 min after LPS injection), (v) GXM plus LPS (GXM administration 60 min before LPS injection), (vi) wortmannin plus LPS (i.p. injection of 0.3 mg of wortmannin per kilogram of body weight 90 min before injection of LPS), (vii) wortmannin plus GXM (i.p. injection of 0.3 mg of wortmannin per kilogram of body weight 90 min before injection of GXM), and (viii) wortmannin plus LPS plus GXM (i.p. injection of 0.3 mg of wortmannin per kilogram of body weight 90 min before injection of LPS and subsequent injection of GXM 15 min later).
TNF-α, IL-6, IL-1β, and IL-10 determinations.
Spleens and lymph nodes were recovered, homogenized, filtered by use of a Cell Strainer device (BD Biosciences), and centrifuged. Supernatant fluids were sterilized by passage through a Millipore filter (0.45-μm pore size). Serum was isolated from blood. Sera and supernatant fluids were stored at −80°C until analysis. For in vitro determination of TNF-α, RAW264.7 cells (5 × 106/ml) were treated with LPS for 30 min and then with GXM (100 μg/ml) for 1 h at 37°C with 5% CO2, and the supernatants were collected. Cytokine levels were determined by the use of commercial enzyme-linked immunoassay kits (BioLegend) according to the manufacturer's recommendations.
Western blotting for pAkt, pIκBα, and MyD88.
For Western blotting, spleens were recovered, homogenized, filtered by use of a Cell Strainer device (BD Biosciences), and centrifuged. The cell pellet was treated with hypotonic saline buffer to lyse erythrocytes, and then 30 × 106 cells of each sample were subjected to protein extraction with M-PER in the presence of protease inhibitors (Sigma-Aldrich) and phosphatase inhibitors (Sigma-Aldrich).
Protein concentrations were determined with a bicinchoninic acid (BCA) protein assay reagent kit (Pierce). The lysates (20 μg of each sample) were separated by sodium dodecyl sulfate-10% polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane (Pierce) for 1 h at 100 V in a blotting system (Bio-Rad) for Western blot analysis.
Membranes were placed in blocking buffer (3% nonfat dried milk) and incubated overnight at 4°C with a mouse monoclonal antibody to pAkt (S472/S473) (1:500), a goat polyclonal antibody to pIκBα (Ser32) (1:200), or a rabbit polyclonal antibody to MyD88 (HFL-296) (1:200). Immunoblotting with rabbit polyclonal antibodies to IκBα (1:200), Akt (1:1,000), and actin (H-300) (1:200) was used as an internal loading control to ensure equivalent amounts of protein in each lane. Detection was achieved using appropriate HRP-linked secondary antibodies followed by an Immun-Star HRP chemiluminescence kit (Bio-Rad).
Immunoreactive bands were visualized and quantified by use of Chemidoc instruments (Bio-Rad).
RAW264.7 cell line.
The murine macrophage cell line RAW264.7 was obtained from the ATCC. Cells were maintained in DMEM with l-glutamine supplemented with 10% FCS and antibiotic (100 U/ml penicillin) at 37°C and 5% CO2.
Western blotting for pSHIP and pIκBα.
RAW264.7 cells (107) were incubated in DMEM plus 10% FCS in the presence or absence of a goat polyclonal antibody to FcγRIIB (Ala46/Pro217) (0.5 μg/ml) for 30 min at 4°C for pSHIP determination and in the presence or absence of LPS (10 μg/ml) for 30 min at 37°C with 5% CO2 for pIκBα determination. The cells of each sample were washed once with phosphate-buffered saline (PBS) and incubated in the presence or absence of GXM (100 μg/ml) in DMEM plus 10% FCS for 30 min for pSHIP or 15 min for pIκBα, at 37°C with 5% CO2. The cell pellets were subjected to protein extraction with 20 μl of mammalian protein extraction reagent in the presence of protease inhibitors (Sigma-Aldrich) and phosphatase inhibitors (Sigma-Aldrich). The lysates (60 μg of each sample for pSHIP and 30 μg of each sample for pIκBα) were separated as described above, and membranes were incubated overnight at 4°C with a rabbit polyclonal antibody to pSHIP (Tyr pY120) (1:2,500). Immunoblotting with a mouse monoclonal antibody to SHIP (1:2,500) was used as an internal loading control to ensure equivalent amounts of protein in the lanes. Detection and visualization of immunoreactive bands were obtained as described above.
Flow cytometry for pIκBα.
For flow cytometry analysis, RAW264.7 cells (1 × 106/ml) were incubated in DMEM plus 10% FCS in the presence or absence of LPS (10 μg/ml). After 30 min of culture, the cells were incubated alone or with GXM (100 μg/ml) for 15 min at 37°C with 5% CO2. After incubation, cells were collected by centrifugation, fixed in 2% formalin in PBS for 10 min at room temperature, and permeabilized with ice-cold methanol (500 μl/106 cells) for 10 min at 4°C. Cells were washed twice in PBS containing 1% bovine serum albumin (BSA) and stained with a goat polyclonal antibody to pIκBα (1:50) in PBS containing 1% BSA for 30 min at room temperature. After incubation, cells were washed twice, stained with Cy3-conjugated rabbit polyclonal anti-goat IgG (1:100), and washed twice more in PBS-1% BSA, and then 5,000 events were analyzed using a FACSCalibur flow cytometer (BD Biosciences). Autofluorescence was assessed by using untreated cells. Data are expressed as mean fluorescence intensities (MFI) of labeled cells.
Statistical analysis.
Data are reported as means ± standard errors of the means (SEM) for 3 to 5 replicate experiments. Data were evaluated by one-way analysis of variance (ANOVA). Post hoc comparisons were made with Bonferroni's test. The log rank test was applied to the survival data. A P value of <0.05 was considered significant.
RESULTS
GXM improves survival of endotoxemic mice.
Given that Gram-negative infection and administration of LPS in humans and animals result in a systemic inflammatory response (28, 31) which partially mimics features of early sepsis, we tested the capacity of GXM to influence the course of LPS-induced sepsis (32). Healthy mice were treated with 100 μg of GXM 60 min before or 15 min after LPS administration. A significant (P < 0.05) increase in survival of GXM-treated mice compared to non-GXM-treated mice was observed. The survival rate rose from 20% to 60% (Fig. 1A). As evidenced in Fig. 1B, the LPS-treated mice died of endotoxic shock within 2 days. A significant increase of median survival time (MST) was observed in GXM-treated mice. Indeed, soon after LPS administration, tremor, diarrhea, crouching gait, immobility, and piloerection were observed, but the mice treated with GXM showed either no symptoms or much less severe symptoms than those of mice not treated with it. In addition, the mice sacrificed 6 h after LPS administration showed evident splenomegaly, with a concomitant increase in the total number of spleen cells (data not shown). In contrast, in the spleens of GXM-treated mice, no gross pathological abnormalities were observed, and the total number of splenocytes was not greater than that in saline-treated mice (data not shown).
Fig 1.

GXM improves survival of endotoxemic mice. (A) Mice were treated with LPS (1.5 mg/mouse) and/or GXM (100 μg/mouse) 60 min before or 15 min after LPS treatment and were monitored for death for up to 5 days, and survivors were then euthanized. Data are from three independent experiments. The percentage of survival was evaluated according to the log rank test, and the difference among experimental groups was significant. (B) Table reporting the mean survival time (MST) in days and the number of dead mice/total number of animals tested (cumulative results for all three experiments) (D/T). *, P < 0.05 (LPS-plus-GXM-treated versus LPS-treated mice).
The dose of GXM (100 μg/mouse) was extrapolated from previous in vivo experiments which demonstrated that this was the appropriate dose to inhibit proinflammatory cytokine secretion and to positively influence septic arthritis and rheumatoid arthritis, pathologies characterized by aberrant inflammatory responses (33, 34). A lower dose of GXM (10 μg/mouse) was also used, but under this experimental condition, GXM did not affect the course of LPS-induced sepsis.
GXM reduces LPS-induced IL-1β, TNF-α, and IL-6 levels and increases IL-10 levels.
LPS triggers the release of many inflammatory cytokines, and it has been implicated in lethal septic shock (14). Therefore, we analyzed IL-1β secretion by splenic macrophages from mice treated with LPS and GXM. The kinetic evaluation of IL-1β levels from supernatant fluids from spleens showed a large increase in IL-1β secretion 1 h after LPS treatment, followed by a gradual decrease. GXM treatment of mice receiving LPS produced a significant downregulation of IL-1β at 1 h and 6 h postadministration (Fig. 2A). We also evaluated the kinetics of the IL-1β response in sera from the same animals. The results showed a significant decrease in IL-1β levels in the sera of mice treated with LPS and GXM compared to the sera of LPS-treated mice throughout the period of observation (1 h, 6 h, and 24 h after LPS challenge) (Fig. 2B).
Fig 2.

GXM induces regulation of IL-1β, TNF-α, IL-6, and IL-10 production in spleens and sera of endotoxemic mice. Mice were treated with sterile saline or with LPS (0.6 mg/mouse) and/or GXM (100 μg/mouse) 15 min after LPS treatment. IL-1β (A and B), TNF-α (C and D), IL-6 (E and F), and IL-10 (G and H) levels in supernatant fluids from spleens and in sera were determined by enzyme-linked immunosorbent assay (ELISA) 1 h, 6 h, and 24 h after LPS treatment. Values represent the means and SEM for five separate experiments. *, P < 0.05 (LPS-plus-GXM-treated versus LPS-treated mice).
The kinetics of TNF-α production in spleens and sera showed that in LPS-GXM-treated mice, TNF-α subsided more quickly than it did in mice treated with LPS alone, and significant decreases (P < 0.05) of TNF-α levels were observed in the spleens and sera after 24 h (Fig. 2C and D).
IL-6 was also tested in our experimental system. The results showed significant (P < 0.05) reductions of IL-6 levels in spleens and sera (Fig. 2E and F) of LPS- and GXM-treated mice with respect to those of mice treated with LPS only. This effect was evident 1 h after LPS treatment, but no such effect was observed after 6 and 24 h.
In addition, IL-10, a prototypical anti-inflammatory cytokine, was tested. The results showed that IL-10 levels in the group treated with LPS only subsided faster than those in the LPS-GXM-treated group. This was observed in the spleens (Fig. 2G) and sera (Fig. 2H) 24 h after GXM treatment.
The ratio of proinflammatory to anti-inflammatory cytokines was analyzed, and we observed that in sera and spleens of LPS-GXM-treated mice, there were 8- and 2-fold decreases in TNF-α, respectively. Furthermore, in sera and spleens of LPS-GXM-treated mice, there were 2- and 1.5-fold increases in IL-10, respectively.
GXM produces inhibition of LPS-induced pAkt and pIκBα.
The PI3K-Akt pathway (via TRAF6) may participate in NF-κB activation induced by LPS through TLR4 stimulation (35). As a consequence, we examined the role of the PI3K-Akt pathway in our experimental system, as well as whether GXM was able to regulate LPS-induced Akt expression. We also used wortmannin, which is a pharmacologic inhibitor of the PI3K-Akt pathway. This compound has been used extensively to analyze the role of Akt in the regulation of different intracellular pathways (36, 37). Mice were treated with wortmannin (0.3 mg/kg of body weight) 90 min before LPS administration, following a previously reported procedure (23). Mice were also treated with 100 μg of GXM before (60 min) and after (15 min) LPS administration, and Akt activation was determined 24 h after LPS administration. The results (Fig. 3A) showed that both wortmannin and GXM treatment produced a strong inhibition of LPS-induced Akt activation, regardless of the time of GXM administration. The effects of wortmannin and GXM treatment on LPS-induced MyD88 recruitment were evaluated. The results (Fig. 3B) showed that both GXM and wortmannin treatment significantly (P < 0.05) reduced LPS-induced MyD88 recruitment. The effect of wortmannin on MyD88 was consistent with previous results showing that wortmannin reduced the PI3K activity in anti-MyD88 immunoprecipitates (17).
Fig 3.

GXM treatment inhibits Akt and MyD88 recruitment in endotoxemic mice. Mice were treated as described in Materials and Methods and then were sacrificed 24 h after LPS treatment. (A) Western blotting for pAkt. Optical densities of reactive bands were measured and normalized by the Akt density in the same lane. Actin was used as a loading control. (B) Western blotting for MyD88. Optical densities of reactive bands were measured and normalized by the actin density in the same lane. In both panels A and B, pAkt and MyD88 were quantified relative to the levels in saline-treated mice. Blots are representative of five independent experiments with similar results. Bars represent the means and SEM for five experiments. *, P < 0.05 (LPS-plus-GXM-treated or wortmannin [WM]-treated mice versus LPS-treated mice).
The major players involved in eliciting the functional effects of LPS are activated through the NF-κB and PI3K-Akt pathways. These pathways regulate the balance between cell viability and inflammation. Thus, 24 h after LPS administration, we analyzed the activation of NF-κB by assessment of LPS-induced pIκBα in splenic macrophages of both mice treated with and mice not treated with GXM. The results showed that in both groups, LPS-induced IκBα activation was markedly suppressed. Similar results were obtained after wortmannin administration (Fig. 4).
Fig 4.
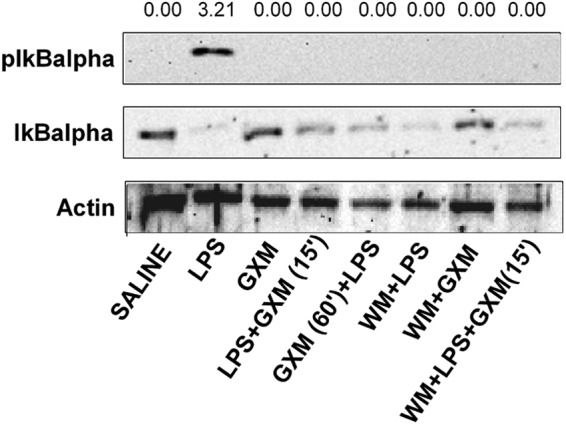
GXM treatment inhibits LPS-induced IκBα activation. Mice treated as described in Materials and Methods were sacrificed 24 h after LPS treatment, and pIκBα levels were determined by Western blotting. The optical densities of reactive bands were measured and normalized by the IκBα density in the same lane. Actin was used as a loading control. The membrane is representative of five independent experiments with similar results.
GXM dampens TNF-α production in spleens, sera, and lymph nodes of LPS-treated mice.
Finally, TNF-α, the prototype of proinflammatory cytokines, was evaluated (24 h after LPS administration) in the spleens, sera, and local lymph nodes of mice treated with LPS in the presence or absence of GXM and wortmannin. The results (Fig. 5) show that GXM treatment significantly (P < 0.05) inhibited LPS-induced TNF-α production by the spleen (Fig. 5A), serum (Fig. 5B), and lymph nodes (Fig. 5C), regardless of the time of GXM treatment. Wortmannin treatment also inhibited LPS-induced TNF-α secretion.
Fig 5.
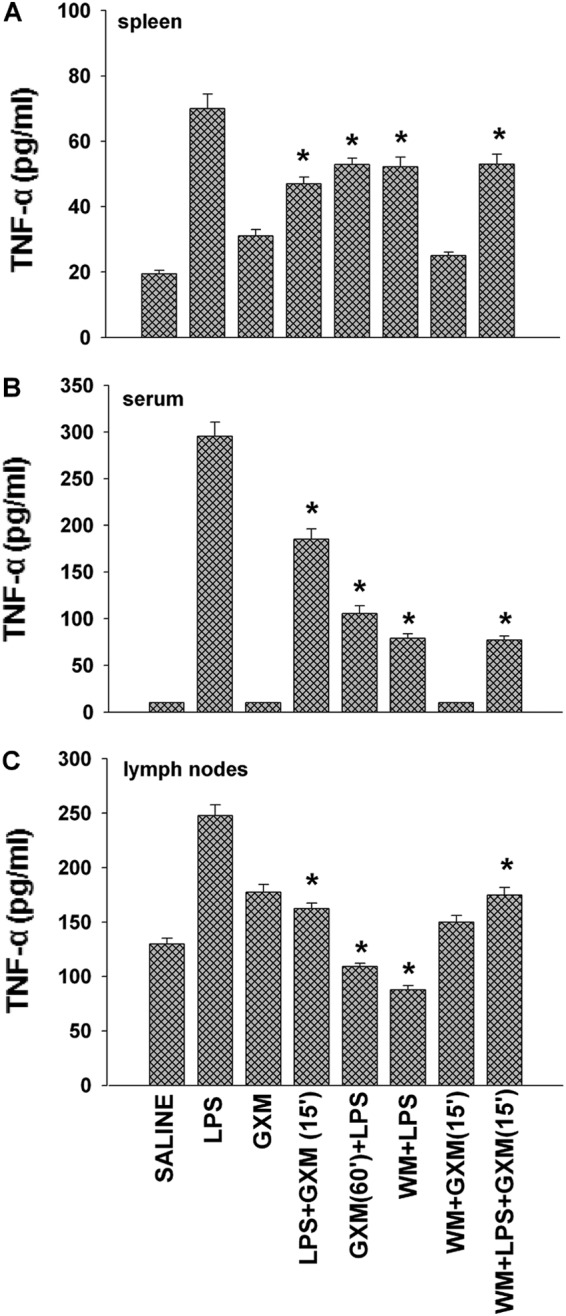
GXM dampens TNF-α production in spleens, sera, and lymph nodes of LPS-treated mice. Mice treated as described in Materials and Methods were sacrificed 24 h after LPS treatment. TNF-α levels in spleen supernatant fluids (A), sera (B), and lymph node supernatant fluids (C) were determined by ELISA. Values represent the means and SEM for five separate experiments. *, P < 0.05 (LPS-plus-GXM-treated or wortmannin-treated mice versus LPS-treated mice).
GXM treatment affects LPS-induced IκBα activation and TNF-α production in RAW264.7 cells.
Given that GXM's inhibitory effects are thought to be via SHIP activation following GXM binding to FcγRIIB, we performed experiments to evaluate whether the blockade of this interaction results in a modulation of SHIP activation. The results reported clearly show that GXM is able to induce SHIP activation and that this effect is completely inhibited by blocking the interaction of GXM with FcγRIIB (Fig. 6A). Furthermore, cells were treated with LPS in the presence or absence of GXM, and then IκBα activation and TNF-α production were tested. The results reported in Fig. 6B show that there was a significant decrease in IκBα activation in LPS-activated cells treated with GXM, and this was observed through Western blotting as well as cytofluorometric determination (MFI). This effect occurred in conjunction with the significant inhibition of TNF-α secretion by these cells (Fig. 6B).
Fig 6.

GXM induces SHIP activation by FcγRIIB binding and inhibits LPS-induced inflammatory response in RAW264.7 cells. (A) RAW264.7 cells, pretreated or not (NS) with FcγRIIB (0.5 μg/ml) antibody, were incubated with GXM (100 μg/ml), washed, and subjected to Western blotting for pSHIP determination as described in Materials and Methods. The blot is representative of three independent experiments with similar results. *, P < 0.05 (anti-FcγRIIB-plus-GXM-treated versus GXM-treated mice). (B) RAW 264.7 cells were incubated in the presence or absence (NS) of LPS (10 μg/ml), and then GXM (100 μg/ml) was added to determine IκBα activation and TNF-α production. After incubation, the cells were washed and subjected to Western blotting or flow cytometry analysis for pIκBα determination, as described in Materials and Methods. The blot is representative of three independent experiments with similar results. Bars represent the means and SEM for three separate experiments. For TNF-α evaluation, supernatants were collected and subjected to a specific ELISA. TNF-α values represent the means and SEM for five separate experiments. *, P < 0.05 (LPS-plus-GXM-treated versus LPS-treated mice).
DISCUSSION
In this study, we demonstrated that injection of the capsular polysaccharide of C. neoformans into mice with LPS-induced endotoxemia significantly improved the rate of survival. This beneficial effect was related to (i) inhibition of TNF-α secretion by splenic macrophages and a decrease of TNF-α levels in serum, (ii) downregulation of IL-6 production by splenocytes and a decrease of IL-6 levels in serum, (iii) decreased levels of IL-1β in supernatants of splenic macrophages and in serum, (iv) a decrease of Akt activation, (v) blockade of IκBα activation, and (vi) marked inhibition of MyD88 recruitment.
In vivo treatment with GXM profoundly influenced proinflammatory cytokine release induced by LPS; in particular, a singular kinetic profile for TNF-α and IL-1β was observed. Treatment with GXM resulted in a significant decrease of IL-1β secretion from splenocytes. This decrease was observed 1 h after LPS injection and was still evident after 6 h; at 24 h, the IL-1β production rapidly returned to the baseline level. The determination of IL-1β levels in serum showed that the levels of this cytokine were consistently lower than those observed in the supernatants of splenocytes, and GXM had a downregulatory effect which was still observed 24 h after LPS challenge. It is plausible that while the results obtained from ex vivo cells are indicative of a compartmentalized immune response in the spleen, the effect of GXM treatment observed in the serum accounts for the sum of events that collectively occur in vivo. In our opinion, the immunoinhibitory activity of GXM is a determinant for survival. It is conceivable to suppose that the major mechanism is through GXM binding to FcγRIIB, with consequent SHIP activation and inhibition of pAkt. This could account for the beneficial effect of GXM in the two situations, i.e., given either before or after LPS. A downregulatory role of SHIP in controlling Akt activation has in fact been reported previously (38). It has also been reported that GXM is able to bind to TLR4 (39, 40), and it is possible that it competes for LPS binding to pattern recognition receptors (PRRs). However, the inhibitory effect of GXM, observed when it was given both before and after LPS, suggests that this possible competition does not play an important role.
Previous studies have suggested that IL-6 serves as both a marker and a mediator of the severity of sepsis (41). Several reports indicate that plasma levels of IL-6 may be used as a diagnostic marker for the presence of bacteremia (42). GXM treatment decreased IL-6 levels in serum and in supernatants from splenocytes soon after LPS injection, and this effect was subsequently lost.
Conversely, GXM affected TNF-α release late in the process. Indeed, 24 h after LPS injection, there was a significant decrease in serum TNF-α levels that mirrored the decrease of TNF-α secretion by splenocytes. Indeed, the reductions of LPS-induced TNF-α observed in the spleen and serum after wortmannin and GXM treatment were substantially greater than those in untreated mice. This suggests that the observed levels of TNF-α are compatible with a therapeutic effect. This is consistent with previous reports showing that controlled production of inflammatory cytokines may be beneficial in several pathological settings (43–45) and that, conversely, aberrant secretion results in deleterious effects (46, 47).
The different profiles of proinflammatory cytokine production could imply their reciprocal regulation. In particular, there was a decrease of IL-1β and IL-6 early on. The decrease of IL-1β was prolonged, whereas the decrease of IL-6 was temporary, i.e., for 1 h after treatment.
The early decrease of IL-6 could be mediated directly by GXM ligation to the immunoinhibitory receptor FcγRIIB, but the presence of cytokines such as TNF-α may promote the late secretion of IL-6, as has been suggested before (48). However, the “cytokine storm” involved in the hyperinflammatory response during endotoxemia is very complicated, and soluble molecules other than TNF-α are thought to play a role in the cytokine profiles observed in our experimental system.
Significant inhibition of TNF-α was observed when GXM was administered before or after LPS challenge. It is possible that this drastic decrease was a consequence of GXM-induced IL-10 production. Indeed, we observed that GXM did not modulate IL-10 production 1 h or 6 h after LPS treatment, but it did induce a consistent increase of IL-10 after 24 h, and this could account for the strong decrease of TNF-α observed 24 h after LPS treatment. This is consistent with our previous results showing that GXM-mediated inhibition of TNF-α is mediated largely by induction of anti-inflammatory cytokines such as IL-10 (24).
Previously, we demonstrated that GXM-mediated immunosuppression occurs via ligation to FcγRIIB and subsequent SHIP activation (24). The inflammatory process is driven by immunopathological events such as the overproduction of various proinflammatory cytokines, including TNF-α, IL-1β, IL-6, and IL-12, and inflammatory mediators (49). Production of these proinflammatory cytokines and inflammatory mediators is dependent upon the activation of PRRs, such as TLR4 and TLR2, by microbial ligands, including lipopolysaccharides (50). During activation of PRRs, a series of intracellular signaling molecules, including mitogen-activated protein kinases (MAP kinases), are activated, resulting in the upregulation of inflammatory gene expression by transcription factors such as NF-κB and activator protein 1 (AP-1) (30, 42, 43, 51).
Of the numerous signaling proteins that contribute to a large number of signals, Akt (protein kinase B [PKB]) has a major role in the regulation of metabolism, cell survival, motility, transcription, and cell cycle progress (52, 53).
Akt is involved in regulating cell survival and controlling the proliferation of many types of cancer (54). Members of the Akt family are characterized by three distinct domains that play a critical role in the PI3K-dependent activation process. This involves the generation of phosphatidylinositol (3,4,5)-trisphosphate [PI(3,4,5)P3] from phosphatidylinositol (4,5)-bisphosphate [PI(4,5)P2] and the subsequent recruitment and activation of Akt (55). LPS binding to TLR4 may stimulate PI3K through its association with the TLR4-MyD88 signaling complex (via TRAF6) (56). This transduction pathway involves Akt phosphorylation, which leads to NF-κB activation and consequently to proinflammatory cytokine gene expression (57). In our experimental system, LPS administration produced a rapid and drastic increase of Akt expression in splenocytes, whereas after GXM treatment, the intensity of phospho-Akt became even weaker than that observed in the unstimulated cells. This effect may be a consequence of GXM ligation to FcγRIIB, which, via the immunoreceptor tyrosine-based immunoinhibitory motif (ITIM), induces recruitment of SHIP, which then converts PI(3,4,5)P3 into PI(4,5)P2. Spontaneous activation of Akt in mouse splenocytes has in fact been reported (58). Thus, the inhibitory effect of GXM can occur regardless of LPS activation. Consistent with this hypothesis is the anti-inflammatory effect of GXM we previously reported for an experimental model of rheumatoid arthritis (34).
The inhibition of LPS-induced MyD88 activation strongly suggests that GXM-mediated inhibition of LPS-induced signal transduction is via the MyD88-dependent pathway and that the GXM effect occurs via upstream signal regulation (Fig. 7).
Fig 7.

Schematic representation of GXM-mediated inhibition of LPS-induced signal. GXM binds to FcγRIIB, with consequent SHIP activation and inhibition of the PI3K-Akt pathway. This results in a downregulation of LPS-induced IκBα activation, with a consequent reduction of the hyperinflammatory response.
This result is consistent with recent studies showing the molecular interactions among MyD88, PI3K, and TLR4 and suggesting that signaling is achieved upon the simultaneous interaction of multiple proteins that create a signaling platform which includes MyD88 and Akt. As a consequence, the disruption of any one interaction has a universal effect on the other protein-protein interactions involved (56).
The inhibition of PIP3 directly reflects the strong decrease of Akt activation and the suppression of IκBα activation. Indeed, IκBα is one of the most crucial signaling kinases for activation of NF-κB, a transcription factor that is crucial for inflammation, cell survival, and differentiation (30).
However, we cannot exclude the possibility that GXM-induced inhibition of IκBα is the result of multiple effects of GXM on the TLR4-mediated signaling pathway, including regulation of MAP kinases. The GXM-induced inhibition of IκBα and the inability of GXM to induce its activation are in contrast to results published by Shoham et al. (39). However, this discrepancy could be related to the different doses of GXM used: unlike Shoham et al., who used 250 μg/ml of GXM, we used 100 μg/mouse of GXM in our experiments. In addition, in our experimental system, GXM treatment was performed in vivo, while Shoham et al. used an in vitro model. These differences may explain the different results.
Because Akt has been considered a strong positive regulator of various types of cancer and autoimmune diseases (59), an effective and strong downregulation of Akt is considered a therapeutic objective for curing inflammatory diseases, particularly septic shock.
These results give evidence that GXM is a potent LPS antagonist and lacks agonistic activity in in vitro and in vivo systems, making it a potentially effective therapeutic agent for treatment of diseases caused by LPS.
ACKNOWLEDGMENTS
This work was supported by the European Commission, the FINSysB Marie Curie Initial Training 16 Network (grant FP7-214004-2), and the Fondazione Cassa di Risparmio di Perugia (grant 2010.011.0398).
No competing financial interests exist.
We thank Catherine Macpherson for editorial assistance.
Footnotes
Published ahead of print 22 October 2012
REFERENCES
- 1. Mitchell TG, Perfect JR. 1995. Cryptococcosis in the era of AIDS—100 years after the discovery of Cryptococcus neoformans. Clin. Microbiol. Rev. 8:515–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cherniak R. 1988. Soluble polysaccharides of Cryptococcus neoformans. Curr. Top. Med. Mycol. 2:40–54 [DOI] [PubMed] [Google Scholar]
- 3. Goldman DL, Lee SC, Casadevall A. 1995. Tissue localization of Cryptococcus neoformans glucuronoxylomannan in the presence and absence of specific antibody. Infect. Immun. 63:3448–3453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee SC, Casadevall A, Dickson DW. 1996. Immunohistochemical localization of capsular polysaccharide antigen in the central nervous system cells in cryptococcal meningoencephalitis. Am. J. Pathol. 148:1267–1274 [PMC free article] [PubMed] [Google Scholar]
- 5. Cherniak R, Sundstrom JB. 1994. Polysaccharide antigens of the capsule of Cryptococcus neoformans. Infect. Immun. 62:1507–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vecchiarelli A. 2007. Fungal capsular polysaccharide and T-cell suppression: the hidden nature of poor immunogenicity. Crit. Rev. Immunol. 27:547–557 [DOI] [PubMed] [Google Scholar]
- 7. Yauch LE, Lam JS, Levitz SM. 2006. Direct inhibition of T-cell responses by the Cryptococcus capsular polysaccharide glucuronoxylomannan. PLoS Pathog. 2:e120 doi:10.1371/journal.ppat.0020120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zaragoza O, Rodrigues ML, De Jesus M, Frases S, Dadachova E, Casadevall A. 2009. The capsule of the fungal pathogen Cryptococcus neoformans. Adv. Appl. Microbiol. 68:133–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bennett JE, Hasenclever HF, Tynes BS. 1964. Detection of cryptococcal polysaccharide in serum and spinal fluid: value in diagnosis and prognosis. Trans. Assoc. Am. Physicians 77:145–150 [PubMed] [Google Scholar]
- 10. Vecchiarelli A, Retini C, Pietrella D, Monari C, Tascini C, Beccari T, Kozel TR. 1995. Downregulation by cryptococcal polysaccharide of tumor necrosis factor alpha and interleukin-1 beta secretion from human monocytes. Infect. Immun. 63:2919–2923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Monari C, Bistoni F, Casadevall A, Pericolini E, Pietrella D, Kozel TR, Vecchiarelli A. 2005. Glucuronoxylomannan, a microbial compound, regulates expression of costimulatory molecules and production of cytokines in macrophages. J. Infect. Dis. 191:127–137 [DOI] [PubMed] [Google Scholar]
- 12. Monari C, Paganelli F, Bistoni F, Kozel TR, Vecchiarelli A. 2008. Capsular polysaccharide induction of apoptosis by intrinsic and extrinsic mechanisms. Cell. Microbiol. 10:2129–2137 [DOI] [PubMed] [Google Scholar]
- 13. McInnes IB, Schett G. 2007. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 7:429–442 [DOI] [PubMed] [Google Scholar]
- 14. Parrillo JE, Parker MM, Natanson C, Suffredini AF, Danner RL, Cunnion RE, Ognibene FP. 1990. Septic shock in humans. Advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Ann. Intern. Med. 113:227–242 [DOI] [PubMed] [Google Scholar]
- 15. Guha M, Mackman N. 2001. LPS induction of gene expression in human monocytes. Cell. Signal. 13:85–94 [DOI] [PubMed] [Google Scholar]
- 16. Lu YC, Yeh WC, Ohashi PS. 2008. LPS/TLR4 signal transduction pathway. Cytokine 42:145–151 [DOI] [PubMed] [Google Scholar]
- 17. Ojaniemi M, Glumoff V, Harju K, Liljeroos M, Vuori K, Hallman M. 2003. Phosphatidylinositol 3-kinase is involved in Toll-like receptor 4-mediated cytokine expression in mouse macrophages. Eur. J. Immunol. 33:97–605 [DOI] [PubMed] [Google Scholar]
- 18. Stoll LL, Denning GM, Weintraub NL. 2006. Endotoxin, TLR4 signaling and vascular inflammation: potential therapeutic targets in cardiovascular disease. Curr. Pharm. Des. 12:4229–4245 [DOI] [PubMed] [Google Scholar]
- 19. Johnson DA, Sowell CG, Johnson CL, Livesay MT, Keegan DS, Rhodes MJ, Ulrich JT, Ward JR, Cantrell JL, Brookshire VG. 1999. Synthesis and biological evaluation of a new class of vaccine adjuvants: aminoalkyl glucosaminide 4-phosphates (AGPs). Bioorg. Med. Chem. Lett. 9:2273–2278 [DOI] [PubMed] [Google Scholar]
- 20. Christ WJ, Asano O, Robidoux ALC, Perez M, Wang Y, Dubuc GR, Gavin WE, Hawkins LD, McGuinness PD, Mullarkey MA, Lewis MD, Kishi Y, Kawata T, Bristol JR, Rose JR, Rossignol DP, Kobayashi S, Hishinuma L, Kimura A, Asakawa N, Katayama K, Yamatsu I. 1995. E5531, a pure endotoxin antagonist of high potency. Science 268:80–83 [DOI] [PubMed] [Google Scholar]
- 21. Kitazawa T, Tsujimoto T, Kawaratani H, Fukui H. 2010. Salvage effect of E5564, Toll-like receptor 4 antagonist on d-galactosamine and lipopolysaccharide-induced acute liver failure in rats. J. Gastroenterol. Hepatol. 25:1009–1012 [DOI] [PubMed] [Google Scholar]
- 22. Wittebole X, Castanares-Zapatero D, Laterre PF. 2010. Toll-like receptor 4 modulation as a strategy to treat sepsis. Mediators Inflamm. 2010:568396 doi:10.1155/2010/568396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barochia A, Solomon S, Cui X, Natanson C, Eichacker PQ. 2011. Eritoran tetrasodium (E5564) treatment for sepsis: review of preclinical and clinical studies. Expert Opin. Drug Metab. Toxicol. 7:479–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Monari C, Kozel TR, Paganelli F, Pericolini E, Perito S, Bistoni F, Casadevall A, Vecchiarelli A. 2006. Microbial immune suppression mediated by direct engagement of inhibitory Fc receptor. J. Immunol. 177:6842–6851 [DOI] [PubMed] [Google Scholar]
- 25. Council of Europe 2010. European convention for the protection of vertebrate animals used for experimental and other scientific purposes. Directive 2010/63/EU. Council of Europe, Strasbourg, Brussels, Belgium [Google Scholar]
- 26. Cherniak R, Reiss E, Slodki ME, Plattner RD, Blumer SO. 1980. Structure and antigenic activity of the capsular polysaccharide of Cryptococcus neoformans serotype A. Mol. Immunol. 17:1025–1032 [DOI] [PubMed] [Google Scholar]
- 27. Houpt DC, Pfrommer GS, Young BJ, Larson TA, Kozel TR. 1994. Occurrences, immunoglobulin classes, and biological activities of antibodies in normal human serum that are reactive with Cryptococcus neoformans glucuronoxylomannan. Infect. Immun. 62:2857–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Andreasen AS, Krabbe KS, Krogh-Madsen R, Taudorf S, Pedersen BK, Møller K. 2008. Human endotoxemia as a model of systemic inflammation. Curr. Med. Chem. 15:1697–1705 [DOI] [PubMed] [Google Scholar]
- 29. Zhang WJ, Wei H, Hagen T, Frei B. 2007. Alpha-lipoic acid attenuates LPS-induced inflammatory responses by activating the phosphoinositide 3-kinase/Akt signaling pathway. Proc. Natl. Acad. Sci. U. S. A. 104:4077–4082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lawrence T. 2009. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 1:a001651 doi:10.1101/cshperspect.a001651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lepper PM, Held TK, Schneider EM, Bölke E, Gerlach H, Trautmann M. 2002. Clinical implications of antibiotic-induced endotoxin release in septic shock. Intensive Care Med. 28:824–833 [DOI] [PubMed] [Google Scholar]
- 32. Poli-de-Figueiredo LF, Garrido AG, Nakagawa N, Sannomiya P. 2008. Experimental models of sepsis and their clinical relevance. Shock 30(Suppl 1):53–59 [DOI] [PubMed] [Google Scholar]
- 33. Tissi L, Puliti M, Bistoni F, Mosci P, Kozel TR, Vecchiarelli A. 2004. Glucuronoxylomannan, the major capsular polysaccharide of Cryptococcus neoformans, inhibits the progression of group B streptococcal arthritis. Infect. Immun. 72:6367–6372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Monari C, Bevilacqua S, Piccioni M, Pericolini E, Perito S, Calvitti M, Bistoni F, Kozel TR, Vecchiarelli A. 2009. A microbial polysaccharide reduces the severity of rheumatoid arthritis by influencing Th17 differentiation and proinflammatory cytokines production. J. Immunol. 183:191–200 [DOI] [PubMed] [Google Scholar]
- 35. Dauphinee SM, Karsan A. 2006. Lipopolysaccharide signaling in endothelial cells. Lab. Invest. 86:9–22 [DOI] [PubMed] [Google Scholar]
- 36. Ward SG, Finan P. 2003. Isoform-specific phosphoinositide 3-kinase inhibitors as therapeutic agents. Curr. Opin. Pharmacol. 3:426–434 [DOI] [PubMed] [Google Scholar]
- 37. Gharbi SI, Zvelebil MJ, Shuttleworth SJ, Hancox T, Saghir N, Timms JF, Waterfield MD. 2007. Exploring the specificity of the PI3K family inhibitor LY294002. Biochem. J. 404:15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Carver DJ, Aman MJ, Ravichandran KS. 2000. SHIP inhibits Akt activation in B cells through regulation of Akt membrane localization. Blood 96:1449–1456 [PubMed] [Google Scholar]
- 39. Yauch LE, Mansour MK, Shoham S, Rottman JB, Levitz SM. 2004. Involvement of CD14, Toll-like receptors 2 and 4, and MyD88 in the host response to the fungal pathogen Cryptococcus neoformans in vivo. Infect. Immun. 72:5373–5382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shoham S, Huang C, Chen JM, Golenbock DT, Levitz SM. 2001. Toll-like receptor 4 mediates intracellular signaling without TNF-alpha release in response to Cryptococcus neoformans polysaccharide capsule. J. Immunol. 166:4620–4626 [DOI] [PubMed] [Google Scholar]
- 41. Cavaillon JM, Annane D. 2006. Compartmentalization of the inflammatory response in sepsis and SIRS. J. Endotoxin Res. 12:151–170 [DOI] [PubMed] [Google Scholar]
- 42. Martin H, Olander B, Norman M. 2001. Reactive hyperemia and interleukin 6, interleukin 8, and tumor necrosis factor-alpha in the diagnosis of early-onset neonatal sepsis. Pediatrics 108:E61. [DOI] [PubMed] [Google Scholar]
- 43. Koninckx PR, Craessaerts M, Timmerman D, Cornillie F, Kennedy S. 2008. Anti-TNF-alpha treatment for deep endometriosis-associated pain: a randomized placebo-controlled trial. Hum. Reprod. 23:2017–2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Choi YH, Park HY. 2012. Anti-inflammatory effects of spermidine in lipopolysaccharide-stimulated BV2 microglial cells. J. Biomed. Sci. 19:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zeytun A, Chaudhary A, Pardington P, Cary R, Gupta G. 2010. Induction of cytokines and chemokines by Toll-like receptor signaling: strategies for control of inflammation. Crit. Rev. Immunol. 30:53–67 [DOI] [PubMed] [Google Scholar]
- 46. Hansson GK, Robertson AK, Soderberg-Naucler C. 2006. Inflammation and atherosclerosis. Annu. Rev. Pathol. 1:297–329 [DOI] [PubMed] [Google Scholar]
- 47. O'Shea JJ, Ma A, Lipsky P. 2002. Cytokines and autoimmunity. Nat. Rev. Immunol. 2:37–45 [DOI] [PubMed] [Google Scholar]
- 48. Williams LM, Lali F, Willetts K, Balague C, Godessart N, Brennan F, Feldmann M, Foxwell BM. 2008. Rac mediates TNF-induced cytokine production via modulation of NF-kappaB. Mol. Immunol. 45:2446–2454 [DOI] [PubMed] [Google Scholar]
- 49. Lin WW, Karin M. 2007. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Invest. 117:117511–117583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kumar H, Kawai T, Akira S. 2009. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 388:621–625 [DOI] [PubMed] [Google Scholar]
- 51. Turjanski AG, Vaque JP, Gutkind JS. 2007. MAP kinases and the control of nuclear events. Oncogene 26:3240–3253 [DOI] [PubMed] [Google Scholar]
- 52. Yang ZZ, Tschopp O, Baudry A, Dümmler B, Hynx D, Hemmings BA. 2004. Physiological functions of protein kinase B/Akt. Biochem. Soc. Trans. 32:350–354 [DOI] [PubMed] [Google Scholar]
- 53. Dummler B, Hemmings BA. 2007. Physiological roles of PKB/Akt isoforms in development and disease. Biochem. Soc. Trans. 35:231–235 [DOI] [PubMed] [Google Scholar]
- 54. Song G, Ouyang G, Bao S. 2005. The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med. 9:59–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liao Y, Hung MC. 2010. Physiological regulation of Akt activity and stability. Am. J. Transl. Res. 2:19–42 [PMC free article] [PubMed] [Google Scholar]
- 56. Laird MH, Rhee SH, Perkins DJ, Medvedev AE, Piao W, Fenton MJ, Vogel SN. 2009. TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J. Leukoc. Biol. 85:966–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kane LP, Shapiro VS, Stokoe D, Weiss A. 1999. Induction of NF-kappaB by the Akt/PKB kinase. Curr. Biol. 9:601–604 [DOI] [PubMed] [Google Scholar]
- 58. Wang ZL, Wu XH, Song LF, Wang YS, Hu XH, Luo YF, Chen ZZ, Ke J, Peng XD, He CM, Zhang W, Chen LJ, Wei YK. 2009. Phosphoinositide 3-kinase gamma inhibitor ameliorates concanavalin A-induced hepatic injury in mice. Biochem. Biophys. Res. Commun. 386:569–574 [DOI] [PubMed] [Google Scholar]
- 59. Kim D, Dan HC, Park S, Yang L, Liu Q, Kaneko F, Ning J, He L, Yang H, Sun M, Nicosia SV, Cheng JQ. 2005. AKT/PKB signaling mechanisms in cancer and chemoresistance. Front. Biosci. 10:975–987 [DOI] [PubMed] [Google Scholar]