

Abstract
The enteropathogen Yersinia pseudotuberculosis can survive in the harsh environment of lymphoid compartments that abounds in immune cells. This capacity is dependent on the plasmid-encoded Yersinia outer proteins (Yops) that are delivered into the host cell via a mechanism involving the Yersinia type III secretion system. We show that the virulence protein YopK has a role in the mechanism by which Y. pseudotuberculosis avoids the polymorphonuclear leukocyte or neutrophil (PMN) defense. A yopK mutant, which is attenuated in the mouse infection model, where it fails to cause systemic infection, was found to colonize Peyer's patches and mesenteric lymph nodes more rapidly than the wild-type strain. Further, in mice lacking PMNs, the yopK mutant caused full disease with systemic spread and typical symptoms. Analyses of effects on PMNs revealed that both the wild-type strain and the yopK mutant inhibited internalization and reactive oxygen species production, as well as neutrophil extracellular trap formation by PMNs. However, the wild-type strain effectively avoided induction of PMN death, whereas the mutant caused a necrosis-like PMN death. Taken together, our results indicate that YopK is required for the ability of Yersinia to resist the PMN defense, which is critical for the virulence of the pathogen. We suggest a mechanism whereby YopK functions to prevent unintended Yop delivery and thereby PMN disruption, resulting in necrosis-like cell death, which would enhance the inflammatory response favoring the host.
INTRODUCTION
There are three human pathogenic species of the Gram-negative bacterium Yersiniae: Yersinia pestis, Yersinia enterocolitica, and Yersinia pseudotuberculosis (1). The enteropathogen Y. pseudotuberculosis causes the gastrointestinal disease yersiniosis (2). After entry by the oral route, it penetrates the intestinal epithelium and spreads to the lymphatic system first infecting Peyer's patches (PPs) and thereafter mesenteric lymph nodes (MLN) (3, 4). At these locations, most other bacteria are effectively destroyed and cleared by phagocytes; however, Y. pseudotuberculosis can survive and replicate in this environment by inhibiting several host immune defense mechanisms (5). In humans, Y. pseudotuberculosis infections are usually cleared at this site; however, in the mouse infection model the pathogen disseminates and causes systemic infection (2). All pathogenic Yersinia species are capable of inhibiting important host immune mechanisms in local lymph nodes. This essential virulence property is dependent on the plasmid-encoded Yersinia outer proteins (Yops): YopE, YopH, YopJ, YopM, YopT, YpkA, and YopK. Upon intimate contact with a target host cell, these Yops are delivered into the host cell via a mechanism involving the Yersinia type III secretion system (T3SS) (6). Inside the target cell, the Yop effectors interfere with several key mechanisms of the host immune defense, including phagocytosis and production of proinflammatory signaling molecules (7).
The main target cells for Y. pseudotuberculosis T3SS-mediated Yop translocation during infection are polymorphonuclear cells or neutrophils (PMNs) (8), suggesting that these innate immune cells play a key role in combating the infection. PMNs are rapidly recruited to infection sites in response to host- and pathogen-derived chemotactic factors (9). Recruited PMNs can effectively internalize bacteria by phagocytosis and then fuse antimicrobial cytoplasmic granules with the phagosome containing the pathogen. The production of reactive oxygen species (ROS) is also initiated in these cells and can activate additional enzymes and kill microbes (10). Furthermore, PMNs can kill extracellular bacteria by releasing web-like structures called neutrophil extracellular traps (NETs) (11), which are mostly comprised of DNA and associated histones, along with some granule and cytoplasmic proteins (12). NETs facilitate killing of bacteria by focusing the antimicrobial armory to the site of infection and by preventing bacterial spread from this site (13).
Previous in vitro studies have shown that Yersinia spp. can resist phagocytosis and ROS production by PMNs (14–19), enabling their survival in the lymphoid compartments. YopH and YopE are the main Yop effectors responsible for resistance against PMN-mediated phagocytic uptake (14, 15). We recently showed that the virulence protein YopK is required for precision of the targeting of antiphagocytic effectors and that this involves an interaction of YopK with the eukaryotic signaling protein receptor for activated C kinase (RACK1) that is recruited to activated integrin complexes (20). YopK has also been proposed to prevent T3SS-induced caspase-1 activation and subsequent inflammasome activation, which is suggested to enhance Yersinia survival (21). The sequence encoding YopK (YopQ in Y. enterocolitica) is present in all three human pathogenic Yersinia species (22–24), and YopK appears to be unique to Yersinia, since no similar amino acid sequence has been found in any other bacteria that harbor a T3SS. The YopK protein is delivered into the target cell, where it associates with the translocators YopB and YopD, and contributes to control of Yop effector delivery (20, 21, 25–28). In the absence of YopK, Yop delivery is uncontrolled and results in massive amounts of effectors in the target cells, whereas the opposite is seen for a strain overexpressing YopK, where the amount of translocated Yops is markedly restricted (20, 25, 26, 28). In addition to the overdelivery of Yop effectors, the absence of YopK also results in more translocators, especially YopB, in the host cell membrane (20). However, although it overdelivers Yop proteins into target cells, a yopK mutant is highly attenuated in the mouse infection model (22–24, 29), indicating that the delivery of Yop effectors must be strictly controlled to enable the establishment of infection. In the present study, we sought to further investigate the influence of YopK on Y. pseudotuberculosis pathogenesis in the mouse infection model.
MATERIALS AND METHODS
Cells, bacterial strains, and growth conditions.
Human peripheral blood PMNs were isolated from healthy donors as described previously (30). This study was conducted using wild-type Y. pseudotuberculosis YPIII(pIB102) (31), harboring the 70-kb virulence plasmid, the plasmid-cured strain (32), and the isogenic yopK deletion mutant strain (22). For animal experiments, the bioluminescent Y. pseudotuberculosis wild-type strain [YPIII(pCD1, Xen4)] (Caliper Life Sciences) and the yopK deletion mutant [YPIII(pCD155, Xen4)] (20) of the same strain were used. Before all experiments, bacteria were grown overnight at 26°C. To induce Yop expression, the bacteria were diluted and grown at 26°C for 30 min and subsequently at 37°C for 1 to 3 h.
Mouse infection and measurement of in vivo bioluminescence.
Mouse experiments were approved (Dnr A81-08 and A104-08) and performed according to the guidelines of the Umeå University Animal Ethics Committee.
Mice were housed under standard conditions and were given food and water ad libitum. Eight-week-old female BALB/c mice (Taconic, Denmark) were orally infected with Y. pseudotuberculosis wild-type [YPIII(pCD1-Xen4)] or yopK-null [YPIII(pCD155-Xen4)] strains. The mice were deprived of food and water for 16 h before oral infection. The Yersinia strains were grown overnight in Luria-Bertani (LB) medium at 26°C and subsequently resuspended in sterile tap water supplemented with 150 mM NaCl. The infection dose was determined by viable count and drinking volume measurements. The animals were inspected daily for signs of disease and analyzed for light emission in an IVIS Spectrum (Caliper Life Sciences). Prior to imaging, the mice were anesthetized with 2.5% isoflurane (IsoFluVet; Orion Pharma Animal Health, Sweden) mixed with oxygen supplied by the XG1-8 gas system (Caliper Life Sciences). Images were acquired and analyzed using LivingImage software (Caliper Life Sciences). Total photon emission was recorded for each animal, and the light emitted from each individual mouse was quantified using the region-of-interest (ROI) tool. The data were compared by using the Student t test; differences were considered significant at a P value of <0.05 (*, P < 0.05, **, P < 0.01).
To analyze systemic spread, viable count measurement was done from infected mouse spleens. Spleens were homogenized using a Dispomix Drive (MedicTools). Homogenates were serial diluted and plated on LB agar plates supplemented with kanamycin (50 μg/ml). Colonies were counted after 48 h of incubation at 26°C.
PMN depletion.
To deplete GR1+ cells, female BALB/c mice (Taconic, Denmark) were injected intraperitoneally with 100 μg of the monoclonal anti-GR1 antibody RB6-8C5 (which recognizes PMNs and inflammatory monocytes, as well as memory CD8+ T cells, and plasmacytoid dendritic cells) (33) 1 day before and 1 and 3 days postinfection (p.i.). Mice injected with phosphate-buffered saline (PBS) served as controls. To ensure that GR1+ cells had been depleted, flow cytometric analysis of mouse blood was performed at day 1, 3, 5, and 7 p.i. Briefly, 20 μl of whole blood from the mouse tail vein were stained with anti-GR1 (BD Biosciences Pharmingen) and anti-MHC class II (BD Biosciences) for 30 min. After lysis of erythrocytes, leukocytes were collected by centrifugation and analyzed using FACSCalibur (BD Biosciences).
Visualization of NET formation.
PMNs were seeded on poly-l-lysine-coated glass coverslips in Hanks balanced salt solution (HBSS) supplemented with MgCl2 and CaCl2. PMNs were infected with Yersinia at a multiplicity of infection (MOI) of 30 and incubated at 37°C in 5% CO2 for 15 h. Thereafter, the samples were washed, fixed with 1% paraformaldehyde (PFA), and blocked with 3% cold water fish gelatin, 5% donkey serum, 1% bovine serum albumin, and 0.25% Tween 20 in PBS. The PMNs were stained using a primary antibody against CD66 (BD Biosciences, catalog no. 551354), and a rabbit anti-Yersinia primary antibody (Agrisera, Sweden) was used to stain bacteria. DAPI (4′,6′-diamidino-2-phenylindole; Sigma) was used to visualize DNA. After washing, species-specific secondary antibodies coupled to Cy2 or Cy3 (Jackson Immunoresearch) were used to detect the primary antibodies. Specimens were mounted in Mowiol (Roth) and analyzed using a DMRB microscope (Leica) equipped with a digital camera (DXM 1200; Nikon).
Determination of bacterial uptake.
For the determination of bacterial uptake, PMNs were seeded on glass coverslips 30 min before the experiment and infected with an MOI of 10 for 30 min at 37°C in a humidified atmosphere containing 5% CO2. Intracellularly and extracellularly located bacteria were distinguished by using a previously described double-fluorescence labeling method (34, 35) using rabbit anti-Yersinia (Agrisera, Sweden) and Alexa 488-donkey anti-rabbit and Alexa 555-goat anti-rabbit (Molecular Probes/Invitrogen) antibodies. Samples were analyzed by fluorescence microscopy (Zeiss Axioskop). The numbers of extracellularly bound and total cell-associated bacteria were counted for 50 randomly selected cells per coverslip.
Determination of bacterial colonization in PPs.
For analysis of bacterial colonization, Peyer's patches (PPs) were harvested from the infected animals and frozen on isopentane prechilled on liquid nitrogen and kept in −80°C. Next, 10-μm-thick cryosections were air dried on Superfrost glass (Thermo Science), fixed with 4% PFA, and permeabilized with 0.5% Triton X-100. To block nonspecific binding, the specimens were overlaid with 0.1 M glycine, followed by 5% serum from the host of the secondary antibody. The samples were stained with rabbit anti-Yersinia (Agrisera, Sweden) primary antibody and Alexa 488-donkey anti-rabbit secondary antibody (Molecular Probes/Invitrogen). Tissue sections were analyzed in a Nikon 90i microscope. The bacterial focus size was measured using Nis-Elements AR software.
Immunohistochemistry.
Cryosections of PPs were fixed in 4% PFA, and free aldehyde groups were blocked in 0.1 M glycine for 10 min. Endogenous peroxidase was blocked in PBS with 2 mM NaN3 and 0.03% H2O2 at 37°C. Endogenous biotin was blocked with avidin-biotin treatment (Vector Laboratories). The sections were stained with rat-α-mouse Ly6G/6C (clone RB6-8C5; BD Biosciences) in 0.1% bovine serum albumin (Boehringer Mannheim). Rat IgG2A (Serotec) was used as isotype control. Biotinylated secondary antibody (biotinylated mouse-α-rat [Jackson Immunoresearch]) was used, and the signal was amplified with a Vectastain ABC enhancement kit (Vector Laboratories, Inc., Burlingame, CA) and developed with 3,3′-diaminobenzidine (Fast DAB tablet sets; Sigma). Finally, the tissue sections were stained with methyl green (Sigma) and mounted in Canada balsam (Sigma). The stained sections were examined in a Nikon 90i microscope, and images were captured using a Nikon DSFi1 camera and Nis-Elements AR software.
Determination of ROS production.
ROS production was measured by a luminal-based chemiluminescence assay. PMNs were seeded in a 96-well plate with HBSS containing 50 μM luminol and 1.2 U of horseradish peroxidase (Calbiochem)/ml. The cells were stimulated with either 100 nM phorbol myristate acetate (PMA; as a control of induction of ROS production), different strains of Y. pseudotuberculosis (MOI of 30), or left untreated. Chemiluminescence was detected using an Infinite 200 luminometer (Tecan) placed in a 5% CO2 atmosphere.
Cell death assays.
PMN cell death was determined by using the Sytox green fluorescence assay (36) and by analyses of plasma membrane binding of annexin V and incorporation of propidium iodide (PI) by flow cytometry (37). For the Sytox green assay, PMNs were seeded in a black 96-well plate in RPMI and 1 mM Sytox green (Invitrogen). The cells were stimulated using 100 nM PMA (as a control of induction of PMN cell death), different strains of Y. pseudotuberculosis (MOI of 30), left untreated, or lysed with 1% Triton X-100. Fluorescence from incorporated Sytox green in the DNA of dead cells was measured every 10 min in a Fluostar Omega fluorescence plate reader (BMG) at 37°C and 5% CO2 for a total of 16 h. Then, 5 μl of H2O was added every 60 min to balance evaporation. To calculate the percentage of cell death, the background fluorescence was subtracted from the sample values at each time point and divided by the lysis control. For flow cytometry, the cells were incubated with bacteria (MOI of 30) in HBSS supplemented with 5% heat-inactivated fetal calf serum in 5-ml round-bottom tubes (Falcon) for 3 and 6 h. Thereafter, the cells were washed, first in PBS and then in binding buffer (1 mM HEPES/NaOH [pH 7.5], 14 mM NaCl, 0.25 mM CaCl2) by centrifugation at 500 × g for 3 min in a 96-well plate with V-shaped wells (Costar). Cells were resuspended in Binding buffer and stained with fluorescein isothiocyanate (FITC)-conjugated annexin V (BD Biosciences) according to the instructions of the manufacturer, and 8 μg of PI (Sigma/ml) for 15 min in the dark at room temperature. Fluorescent signals were quantified by flow cytometry using a FACSCalibur and CellQuest software (BD Biosciences).
Caspase-3 activity assay.
Measurements of caspase-3 activity were performed using the fluorimetric caspase-3 assay (Sigma-Aldrich) according to the manufacturer's instructions. Aliquots of 105 PMNs were left untreated or infected with bacteria (MOI of 30) for 3 and 6 h, followed by cell lysis. The relative caspase-3 activity in the lysates was determined by measuring the fluorescence of the hydrolyzed Ac-DEVD-AMC substrate cleaved by caspase-3 in a kinetic mode every 5 min for 60 min and calculation of the fluorescence intensity rate. Assays were performed in triplicate.
RESULTS
YopK is important for Y. pseudotuberculosis to spread systemically and cause full disease in mice.
Previous results from cell culture analyses have suggested different roles of YopK in bacterium-host cell interplay (20, 21). To investigate the role of YopK during infection, we here used the in vivo imaging system (IVIS) technology that allows real-time monitoring of Y. pseudotuberculosis infections in the mouse. A yopK deletion was introduced into the bioluminescent Y. pseudotuberculosis wild-type strain YPIII (pCD1 Xen4). The resulting yopK mutant strain and the wild-type strain were then used to orally infect female BALB/c mice with 108 bacteria. Infection was followed by daily visual inspections for signs of disease, and IVIS bioluminescence analysis of the mice was performed on days 1, 3, 5, and 7 postinfection (p.i.). IVIS analyses on day 1 p.i. showed higher levels of total bioluminescence emitted from mice infected with the wild-type strain compared to mice infected with the yopK mutant strain. At this early time point of infection, both strains are expected to reside in the intestine, and the observed difference in total emitted bioluminescence therefore suggested higher level of colonization for the wild-type strain (Fig. 1A and B). At day 3 p.i. the total bioluminescence signals from mice infected with the two strains reached equal values. At later time points, days 5 and 7 p.i., the signals from mice infected with the yopK mutant were slightly higher compared to those from animals infected with the wild-type strain, suggesting similar loads. There were however striking differences concerning signs of disease between the two infection groups. On days 4 to 7, mice challenged with wild-type bacteria showed clear disease symptoms, such as ruffled fur, hunchback posture, and diarrhea. In sharp contrast, mice infected with the yopK mutant strain appeared perfectly healthy throughout the experiment (Table 1), and eventually cleared the infection between days 11 and 14. A plausible explanation for the discrepancy in total bioluminescence and disease symptoms is that disseminating wild-type bacteria that reach deeper tissues and spread out actually give rise to lower levels of total bioluminescence compared to the mutant strain. The yopK strain was contained within foci and could less efficiently disseminate yet resulting in a more concentrated and thus higher bioluminescence signal.
Fig 1.

YopK is essential for Y. pseudotuberculosis to spread systemically and cause full disease in mice. Real-time in vivo imaging of Y. pseudotuberculosis infection in BALB/c mice that were orally infected with 1.1 × 108 bacteria of the wild-type [YPIII(pCD1, Xen4)] or 1.3 × 108 bacteria of the yopK [YPIII(pCD155, Xen4)] strains was performed. (A) Bioluminescent images of four mice for each bacterial strain on days 1, 3, 5, and 7 p.i. The same groups of mice are shown for the different time points indicated. The color bar presents the total number of emitted photons, with high and low bioluminescent signals indicated by red and blue, respectively. (B) Bioluminescent signals from the infected animals on the indicated days postinfection presented as photons/second. The data were compared by using the Student t test, with differences considered significant at a P of <0.05 (*, P < 0.05; **, P < 0.01). (C) Bioluminescent signals from a representative set of dissected organs for each bacterial strain on days 3, 5, and 7 p.i. Beneath each organ image, a summary presents the number of each indicated organ with signals/all dissected organs. PP, Peyer's patch; M, mesenteric lymph node (MLN); L, liver; S, spleen.
Table 1.
Signs of disease in Y. pseudotuberculosis-infected mice
| Day | Disease severitya in mice infected with various Y. pseudotuberculosis strains |
|||
|---|---|---|---|---|
| Wild type | Wild type, PMN− | yopK mutant | yopK mutant, PMN− | |
| 1 | – | – | – | – |
| 3 | + | ++ | – | + |
| 5 | +++ | ++ | – | +++ |
| 7 | +++ | +++ | – | +++ |
Disease severity was graded based on visual inspections of each mouse for symptoms such as ruffled fur, hunchback posture, and diarrhea. –, Healthy mice without any signs of disease; +, at least 50% of the mice showed signs of ruffled fur; ++, the majority of the mice had ruffled fur and diarrhea; +++, the majority of the mice had severe diarrhea and a hunchback posture.
To investigate whether YopK indeed was required for dissemination during infection, we applied the IVIS again, this time to analyze bioluminescence signals emitted by organs dissected from mice infected with the wild-type strain or the yopK mutant strain at days 3, 5, and 7 p.i. At day 3 p.i., the wild-type strain colonized the small intestine and the MLN and also had spread to the liver and spleen (Fig. 1C). Systemic spread of the wild-type strain to liver and spleen was also observed at days 5 and 7 p.i. (Fig. 1C). On the other hand, the yopK mutant showed a very different pattern; like the wild-type strain, the yopK mutant strain colonized the small intestine and MLN at day 3 p.i., but it did not colonize spleen or liver at day 3, 5, or 7 p.i. (Fig. 1C). Moreover, the viable bacterial count from homogenized spleens from day 7 p.i. showed the presence of the wild type (1,442,000 CFU/spleen) but not of the yopK mutant strain (0 CFU/spleen), confirming the deficient dissemination of the yopK mutant. Together, these results demonstrate that in contrast to wild-type Y. pseudotuberculosis, the yopK mutant strain is unable to spread systemically which subsequently decreases its ability to cause full disease in mice.
YopK influences early colonization in Peyer's patches.
To understand the mechanism underlying the difference in spreading capacity between the wild-type and yopK mutant strains, we next investigated the early steps of infection. To analyze initial colonization in the PPs, mice were infected as before with the bioluminescent wild-type and yopK strains. The animals were euthanized at days 1, 3, or 5 p.i., and PPs were harvested and frozen at −80°C. The frozen tissue was sectioned and subjected to immunofluorescence staining of Yersinia. The sections were analyzed by microscopy; focus sizes of <6,000 μm2 were considered small, focus sizes of 6,000 to 12,000 μm2 were considered medium, and focus sizes of >12,000 μm2 were considered large. For the wild-type strain, at day 1 p.i., no obvious bacterial foci were detected in any of the analyzed PPs, whereas after 3 days of infection, large foci were present in 83% of the analyzed PPs (Fig. 2A and B). Surprisingly, for the yopK mutant strain at day 1 p.i., foci of mostly small size were detected in all analyzed PPs (Fig. 2A). Thus, foci were detectable earlier after infection with the yopK mutant strain compared to the wild-type strain. Bacterial foci were also present in PP sections at days 3 and 5 p.i. with the yopK mutant; interestingly, these foci were smaller than the wild-type foci at days 3 (Fig. 2A and B) and 5 p.i. (data not shown). These results clearly demonstrate that the YopK-deficient strain has a different colonization pattern compared to the wild-type strain, as evidenced by earlier formation of foci, which, however, did not increase in size to the same extent as foci of wild-type bacteria. This colonization pattern might be related to impaired ability of the yopK mutant strain to spread systemically and cause full disease.
Fig 2.

The Y. pseudotuberculosis yopK mutant colonizes Peyer's patches faster than the wild-type strain. (A) Representative images of immunofluorescence staining of Y. pseudotuberculosis in sections of PP from mice infected with the indicated strains on days 1 and 3 p.i. PPs positive for bioluminescent signals were selected from IVIS analyses of dissected organs. (B) Percentages of bacterial foci of different sizes from PP of mice infected with wild-type or yopK mutant bacteria at days 1 and 3 p.i. (wild type [day 1, n = 7; day 3, n = 16] and yopK mutant [day 1, n = 10; day 3, n = 19]). Focus sizes of <6,000 μm2 were considered small, focus sizes of 6,000 to 12,000 μm2 were considered medium, and focus sizes of >12,000 μm2 were considered large. (C) Infiltration of PMNs in the PPs of mice infected with wild-type or yopK mutant bacteria at days 1 and 3 p.i. Sections of PPs were stained for bacteria with immunofluorescence and for PMNs using immunohistochemistry (upper panel, Y. pseudotuberculosis in green; lower panel, PMNs in brown). Scale bars, 200 μm.
Given that the yopK mutant strain colonized the PPs earlier than the wild-type strain, we next investigated whether this was reflected in the host's innate immune response. For this purpose, we analyzed the presence of PMNs, which are recruited to PPs upon infection as a result of the inflammatory response (38–40). Sections of PPs from mice infected with the yopK mutant or the wild-type strain were subjected to immunohistochemistry staining of PMNs. At day 1 p.i., infiltration of PMNs was detected in PPs from mice infected with the yopK mutant strain, but not in PPs from wild-type strain-infected mice (Fig. 2C). At day 3 p.i., PMN infiltration could be detected in PPs from both infection groups (Fig. 2C). Together, our data indicate that the yopK mutant strain colonizes PPs more efficiently compared to the wild-type strain and thereby initiates an earlier innate immune response involving PMN infiltration.
Y. pseudotuberculosis requires YopK for protection against PMNs during infection.
Our finding that the colonization pattern of YopK-deficient bacteria is distinct from that of wild-type bacteria during the initial phase of PP infection, together with the inability of this mutant to spread systemically, suggest a role for this virulence protein in the interplay of the bacteria with the early innate immune defense. PMNs play a critical role in immediate clearance of invading bacteria and are considered as potential putative key targets for pathogens that manage to establish and maintain infection in host tissues (10). To investigate the role of these immune cells in the mechanism behind the inability of the yopK mutant to cause full disease, we examined experimental yersiniosis in PMN-depleted and nondepleted mice. Intraperitoneal administration of the anti-GR1 monoclonal antibody RB6-8C5 was used to deplete GR1+ cells. The administration of this antibody eliminated 91% of circulating GR1+ cells, as confirmed by flow cytometric analysis (data not shown).
PMN-depleted and nondepleted mice were infected with the bioluminescent Y. pseudotuberculosis strains, visually inspected, and analyzed by IVIS, as in previous experiments. PMN-depleted mice infected with the yopK mutant showed typical signs of disease that were indistinguishable from control mice infected with wild-type bacteria (Table 1). IVIS analysis of whole animals indicated similar overall infection levels in PMN-depleted mice infected with wild-type and yopK mutant bacteria at day 1 p.i. (Fig. 3A); however, at day 3 p.i., there were significantly higher total bioluminescence signals emitted from mice infected with the yopK mutant strain (Fig. 3A). Interestingly, comparing control and neutropenic mice infected with the wild-type strain, there were higher total bioluminescence signals emitted from the former, which suggest higher colonization in the presence of PMNs (Fig. 3A). Such difference was not seen in mice infected with the yopK mutant strain where the levels of total emitted bioluminescence were similar in control and neutropenic mice (Fig. 3A).
Fig 3.

Depletion of PMNs allows the yopK mutant to spread systemically. Real-time monitoring of Y. pseudotuberculosis infection using in vivo imaging. PMN-depleted mice treated intraperitoneally with α-Ly6G/6C and control mice receiving PBS were orally infected with 1.2 × 108 wild-type bacteria [YPIII(pCD1, Xen4)] or 1.3 × 108 yopK mutant [YPIII(pCD155, Xen4)] bacterial strains. (A) Bioluminescent signals from the infected animals on the indicated days postinfection are presented as photons/second. The data were compared by using the Student t test; differences were considered significant at a P value of <0.05 (*, P < 0.05; **, P < 0.01). (B) Bioluminescent signals from a representative set of dissected organs for each bacterial strain on days 1, 3, 5, and 7 p.i. Beneath each organ image is a summary presenting the number of each indicated organ with signals/all dissected organs. PP, Peyer's patches; M, mesenteric lymph node (MLN); L, liver; S, spleen.
Next, we analyzed dissected organs from the infected mice. Interestingly, the yopK mutant strain, but not the wild-type strain, had already colonized MLN at day 1 p.i. in both control and PMN-depleted mice (Fig. 3B). At day 3 p.i., the yopK mutant had spread to the liver in PMN-depleted mice (Fig. 3B), whereas at this time point, in this experiment, systemic spread of the wild-type strain was not observed in either nondepleted or PMN-depleted mice (Fig. 3B). Hence, in the absence of PMNs, the yopK mutant strain colonized the intestinal organs and likely also spleen and liver more efficiently compared to the wild-type strain. At days 5 and 7 p.i., both strains had spread to the spleen and liver in PMN-depleted mice, whereas, as previously shown (Fig. 1) (22, 29), only the wild-type strain had spread systemically in nondepleted mice (Fig. 3B). As expected, no systemic spread of the yopK mutant was observed in nondepleted mice throughout the experiment (Fig. 3B). Thus, wild-type Y. pseudotuberculosis effectively avoids the PMN defense and establishes infection in PPs and MLN, after which it spreads systemically, totally unaffected by attacking PMNs. In fact, our data suggest that the presence of PMNs might benefit establishment of infection. Moreover, we demonstrate a requirement for YopK for Yersinia resistance against PMN attack, since the yopK mutant caused full disease only in PMN-depleted mice.
YopK is required for avoiding PMN death.
To further investigate the influence of YopK on PMNs, we next performed in vitro infection analyses with Y. pseudotuberculosis strains and PMNs purified from human blood. Analyses of the inhibition of phagocytosis showed that the yopK mutant retained the ability to inhibit phagocytosis by PMNs (Fig. 4A). In analogy to that previously shown in experiments with HeLa cells (20), inhibition of phagocytosis was more pronounced for the mutant than for the wild-type strain, which reflects the hypertranslocation phenotype of the mutant (26, 28). PMNs attack pathogens by different mechanisms, including the generation of highly microbicidal ROS, known as the respiratory burst. Upon PMN activation, the large NADP (NADPH) oxidase assembles at the phagosomal membrane, where it transfers electrons to molecular oxygen to generate superoxide anions which are delivered into the phagocytic vacuole, as well as to the extracellular environment (41). Therefore, Yersinia, which mainly remains extracellular in the PPs, presumably encounters high levels of antimicrobial oxidants. Thus, we analyzed the effects of Yersinia and YopK on ROS production by the PMNs. In analogy with results from previous studies (15–17), the wild-type Y. pseudotuberculosis strain inhibited ROS production (Fig. 4B). The yopK mutant strain inhibited ROS production to a similar extent as the wild-type strain (Fig. 4B), suggesting that YopK does not contribute to Yersinia-mediated blocking of PMN ROS production.
Fig 4.

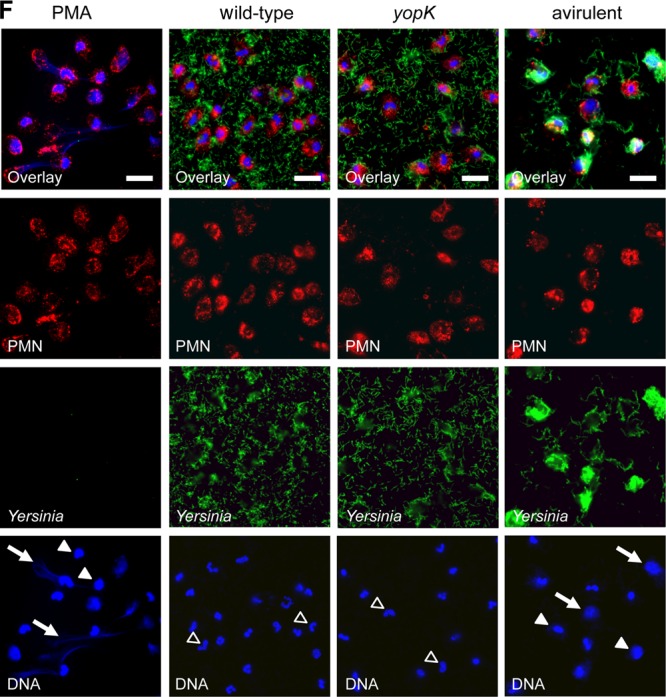
YopK is required for Y. pseudotuberculosis-mediated silencing of bacterium-induced PMN cell death. (A) Internalization of the indicated Y. pseudotuberculosis strains by PMNs. The number of bacteria internalized is presented as the percentage of the total number associated with PMNs. The illustrated data represent the means and standard errors of the mean (n = 3). (B) ROS production by PMNs infected with the indicated Y. pseudotuberculosis strains measured by a luminometric assay. The y axis shows the relative light units (RLU). The illustrated data represent means and standard deviations (n = 3). (C) Cell death of PMNs infected with the indicated Y. pseudotuberculosis strains for a period of 960 min. Cell death was measured in 10-min intervals as the fluorescence from incorporated Sytox green in the DNA of dead cells. One representative graph is displayed, and similar results were obtained from at least four independent experiments. (D) Annexin V and PI staining of uninfected or PMNs infected with the indicated Y. pseudotuberculosis strains for 6 h. Error bars show the standard errors of the mean (n = 4). A comparison of groups was performed using one-way analysis of variance (ANOVA), followed by the Bonferroni post test (*, P < 0.05; **, P < 0.01; ***, P < 0.001). For caspase-1 inhibition in panels C and D, the cells were pretreated for 60 min with 100 μM caspase-1 inhibitor II (Calbiochem, Darmstadt, Germany) dissolved in dimethyl sulfoxide. (E) Caspase-3 activity in PMNs that were left untreated or infected with Y. pseudotuberculosis strains indicated for 3 h (gray) or 6 h (black). The fold difference in caspase-3 activity, relative to the 3-h untreated sample, is indicated. Staurosporine (2 μM) was used as positive control for the induction of caspase-3 activity (35-fold increase after 6 h). The data are presented as means with the standard errors of the mean (n = 3). Comparison of groups was performed using one-way ANOVA, followed by the Bonferroni post test (***, P < 0.001). (F) Immunofluorescence staining of human PMNs infected with the indicated Y. pseudotuberculosis strains or stimulated with PMA as a control for NET formation. Specimens were fixed after 15 h and stained for immunofluorescence analysis, with DNA stain in blue (DAPI), anti-PMN stain in red (anti-CD66), and Yersinia in green (anti-Yersinia). The superimposition of all signals is shown in the first row. The images are representative of at least three independent experiments. Scale bars, 10 μm. Arrows indicate NETs, arrowheads indicate a decondensed nucleus, and open arrowheads indicate an intact nucleus.
ROS production is essential for microbe-induced NET formation, a newly discovered antimicrobial defense of PMNs (30). NET release is accompanied by the death of PMNs associated with exposure of DNA to the extracellular environment. To investigate eventual interference with NET formation, we used a fluorescence assay with Sytox green, a membrane-impermeable DNA-binding dye, to determine PMN cell death after infection with the wild-type, plasmid-cured, and yopK mutant strains. The wild-type and the plasmid-cured strain had distinct effects on the PMNs, with the level of cell death induced by the wild-type strain being considerably lower than that caused by the plasmid-cured strain. This suggests that the virulent wild-type strain can avoid the bacterium-induced PMN cell death, which indeed is in agreement with the previously observed inhibition of phagocytosis and ROS production. Interestingly, the yopK mutant induced more PMN cell death in a shorter time compared to the plasmid-cured strain (Fig. 4C). This effect appeared to be specific for YopK, since infection with yopEH, yopJ, and yopM mutant strains did not result in PMN death (data not shown). Further, since YopK recently was suggested to act by protecting against inflammasome activation (21), we sought to elucidate whether PMN cell death induced by YopK-deficient bacteria may be related to the inflammasome. Therefore, we repeated cell death experiments in the presence or absence of a caspase-1 inhibitor. However, caspase-1 inhibition did not affect the levels of PMN death induced by the yopK mutant strain (Fig. 4C), indicating that this cell death is not related to the inflammasome. As expected from this finding, minor levels of wild-type-induced cell death also remained unaffected by the caspase-1 inhibitor treatment (Fig. 4C).
In order to further investigate the nature behind the observed PMN death, we next analyzed whether the death occurred in an apoptotic or necrosis-like manner. For this purpose, we first determined binding of FITC-annexin V and exclusion of PI as a measurement of apoptosis. The data obtained indicated that neither the wild-type strain nor the yopK mutant strain induced apoptosis of PMNs. For cells infected with these strains for 6 h, the levels of annexin V single-positive cells were somewhat lower than that those observed for untreated cells (Fig. 4D). Measurements were also performed at 3 h p.i., but also at this time point no increased levels of single-positive cells could be detected (data not shown), suggesting that none of these strains induced apoptosis in PMNs. For cells infected with the avirulent plasmid-cured strain, a slight increase in annexin V single-positive cells was observed. However, when we compared the levels of the annexin V/PI double-positive cells, a clearly distinct effect was observed for cells infected with the yopK mutant strain, with a significantly higher amount of double-positive cells after 6 h of infection (Fig. 4D). This was not seen for PMNs infected with the wild-type or the plasmid- cured strain. Hence, since there were no signs of early apoptosis, the accumulation annexin V/PI double-positive cells indicates that YopK-deficient bacteria induce a nonapoptotic cell death associated with disrupted membrane integrity. In analogy with the results from the Sytox assay, the presence of the caspase-1 inhibitor did not affect the level of cell death (Fig. 4D). To further confirm the absence of an apoptosis-connected mechanism, we also measured the activation of the apoptotic mediator caspase-3. For this, the cells were left untreated or infected with the wild-type, yopK mutant, and plasmid-cured strains of Y. pseudotuberculosis for 3 and 6 h, and the caspase-3 activity was determined. These time points were chosen since 3 h after infection was just prior to a detectable induction of cell death, whereas after 6 h cell death was appropriately induced by the yopK mutant strain. However, no difference in caspase-3 activation could be detected for the different strains, which supported the result from the annexin V binding analyses (Fig. 4E). Notable, however, was the fact that all of the infections appeared to have a dampening effect on the spontaneous activation of caspase-3 that occurred in uninfected cells (Fig. 4E).
Immunofluorescence staining of DNA was used to visualize NET formation in cells infected with the different strains. For microscopic analyses, we used later time points (15 h) to ensure that the eventually observed differences between mutant and wild-type strains are not solely due to a delayed induction. Only the plasmid-cured avirulent strain, and not the wild-type strain or the yopK mutant strain, triggered NET release (Fig. 4F). Signs that are typical for NET formation, such as decondensation of nuclei and web-like effuse areas of DNA (30), were observed in PMNs infected with the plasmid-cured strain. These web-like structures were also very similar to the NETs in the positive control, where we induced NET formation with the phorbol ester PMA (Fig. 4F). In contrast, a large proportion of the nuclei of wild type- and yopK mutant-infected PMNs were still lobulated even after 15 h of infection, indicating that no NET formation had occurred. In addition, this appearance of nuclei and absence of web-like structures was already evident with these strains after 4 h of infection (data not shown).
Taken together, our data indicate that the cell death induced by the yopK mutant observed by both Sytox assay and PI incorporation is distinct from both apoptosis and NET-associated apoptosis, so called NETosis. We therefore suggest that the observed cell death is due to a necrosis-like effect, but the more detailed mechanism behind remains to be elucidated.
DISCUSSION
Pathogenic yersiniae effectively evade the innate host response and can survive in host lymphoid organs that are designated to recognize and destroy invading microorganisms. PMN recruitment is one critical early event during bacterial infection, which most often clears invading bacteria at these sites. Upon oral infection, enteropathogenic Yersinia initially colonize the PPs of the small intestine, resulting in high numbers of both bacteria and immune cells containing Yop effectors in PPs. PMNs are the cells that are most targeted concerning the delivery of Yop effectors (8), and in the present study we report a role for the virulence protein YopK in Y. pseudotuberculosis resistance to these immune cells.
Analysis of mice infected with Y. pseudotuberculosis lacking YopK showed overall colonization of the intestinal organs; however, infection with this mutant strain did not result in systemic spread or symptoms of disease. Thus, the yopK mutant reached the MLN that constitute the critical barrier for preventing systemic infection by both commensals and pathogens (42, 43) but failed to disseminate further. Analysis of initial colonization of the different intestinal organs using IVIS and immunofluorescence staining showed that Y. pseudotuberculosis lacking YopK colonized PPs and MLN more rapidly than the wild-type strain. Accordingly, upon infection with the yopK mutant strain, there was also an earlier PMN response in the PPs, where PMN recruitment was obvious at day 1 after infection. No foci or associated PMN recruitment were detected for the wild-type strain at this early time point of infection. This discrepancy indicates a different infection pattern for the yopK mutant that might be connected to its deficient spreading capacity, but it can also be a consequence of abnormal effector delivery by this mutant. In vitro experiments have shown that the yopK mutant strain overdelivers Yop effectors into interacting target cells (20, 25, 26, 28), resulting in hypercytotoxicity and enhanced blockage of phagocytosis (20, 28). Hence, it is possible that this phenotype facilitates initial entry into the PPs and also contributes to the very early recruitment of PMNs to this site. Furthermore, at later time points when the wild-type strain already had established colonization, it was detected as large foci in PPs, while the yopK mutant mainly resided in smaller foci. This discrepancy concerning foci size might also be a result of the less-controlled effector delivery of the yopK mutant. However, it did not affect the ability of the yopK mutant to pass from the PPs to the MLN; instead, it appeared to be important for the establishment of colonization leading to subsequent systemic spread.
In accordance with previous reports (22, 29), we show that YopK is required for Y. pseudotuberculosis to spread systemically in the mouse. Our results demonstrate that effective avoidance of PMN defense is critical for this, since a yopK mutant is able to cause full disease with systemic spread and typical symptoms in the absence of these innate immune cells. Similar to that seen for YopK, virulence of a yopM mutant was restored in the absence of PMNs (44). It is therefore likely that the ability of Yersinia to cause disease in the presence of recruited PMNs involves virulence mechanisms at many levels where different Yop proteins contribute with specific virulence traits adding up to a completely PMN-resistant phenotype for this pathogen.
Furthermore, infection experiments using nondepleted and PMN-depleted mice showed that the pathogenesis pattern for the Y. pseudotuberculosis wild-type strain was not more severe in the absence of PMNs. This indicates that this pathogen possesses very efficient defense mechanisms, allowing it to coexist with these specialized innate immune cells that are dedicated to eliminate invading bacteria. Interestingly, the total bioluminescence signals emitted from mice infected with the wild-type strain were significantly lower in the absence of PMNs compared to when PMNs were present. This was not seen in mice infected with the yopK mutant strain. This might suggest that PMN infiltration improves the colonization efficiency of wild-type Y. pseudotuberculosis. This view is, however, not in accordance with previous studies of Yersinia infections in PMN-depleted adult mice (44, 45). However, in those studies bacterial counts in deeper organs were determined, and the infections were either intraperitoneal or intravenous. Duran et al. (8), in evaluating the colonization of intestinal organs, observed equal bacterial loads in the PPs of PMN-depleted and nondepleted mice following intraperitoneal infection. Hence, one possible reason for the discrepancy might be that our data are from orally infected mice. The possible aiding effect of PMNs for Yersinia colonization is interesting, especially since these cells constitute the major target cell for this pathogen. It is possible that the associated release of inflammatory factors, especially hydrolytic proteins from arriving PMNs, affects the environment in a way that benefits colonization and further invasion of the PMN-resistant pathogen.
The yopK mutant appeared to colonize the PMN-depleted mice more efficiently than wild-type Y. pseudotuberculosis. Besides a more rapid colonization of MLN, the yopK mutant also seemed to cause faster systemic spread in PMN-depleted mice compared to the wild-type strain; there were detectable bacterial loads in the livers of PMN-depleted mice at 3 days after infection with the mutant compared to 5 days for wild-type Y. pseudotuberculosis. Hence, it is possible that the phenotype seen in the absence of YopK regarding more rapid initial colonization might also be relevant in systemic spread in mice lacking PMNs.
The PMN defense is critical for hindering systemic infections of bacteria (45), and our data demonstrate that Yersinia requires YopK to circumvent this host defense. YopK might contribute to the PMN resistance by restricting and orchestrating delivery of Yop translocators and effectors to obtain a fine-tuned effect on these target cells. In vitro analyses of Y. pseudotuberculosis-PMN interactions indicate that YopK does not directly inhibit phagocytic uptake or block ROS production. Instead, YopK probably plays a role in the Y. pseudotuberculosis evasion of PMN defense by reducing PMN cell death. We show that Y. pseudotuberculosis lacking YopK has a direct destructive effect on PMNs, observed as increased levels of PMN cell death after prolonged infection. In contrast, the wild-type strain disarms these cells by blocking phagocytosis and oxidative burst and yet by concurrently preventing the onset of PMN death. Bacterium-mediated induction of NET formation by PMNs is known to induce cell death by a pathway that is dependent on ROS production, an event that is important for the clearance of infection and inflammation (30, 46–50). PMN ROS production was clearly seen with the avirulent, plasmid-cured strain, which was associated with PMN NET formation, whereas the wild-type strain efficiently blocked phagocytosis and ROS production, PMN death, and eventually NET formation. The yopK mutant, on the contrary, caused pronounced cell death, despite an effective blockage of internalization and ROS production. In compliance with an absent oxidative burst, NET formation was not induced by the mutant, suggesting that the PMN death caused by YopK-deficient bacteria is a different type of cell death. We further demonstrated that this cell death is not a form of apoptosis. Since we can exclude apoptosis and NETosis, we propose that it is a necrosis-like PMN cell death. However, the exact mechanism for the induction of this PMN death remains to be elucidated. YopK has previously been reported to act by preventing T3SS-mediated inflammasome activation and thereby host recognition (21). However, the level of PMN death was not affected upon inhibition of caspase-1, and therefore an involvement of abnormal activation of the inflammasome is less likely to be responsible for the observed PMN death. One possible explanation involves the translocator YopB, which, in the absence of YopK, is present in elevated levels in target cell membranes (20). The YopB protein can insert into membranes and reportedly has lytic and stress-related effects on target cells; one reported effect is proinflammatory signaling, involving the activation of NF-κB (51, 52). It is thus possible that elevated levels of YopB in the PMN membranes after infection with the yopK mutant are responsible for the destructive effect on PMNs. The possible contribution of YopB to PMN cell death by YopK-deficient bacteria will be addressed in further studies.
The effective avoidance of causing PMN death by fully virulent Y. pseudotuberculosis is very likely to be crucial for this pathogen's resistance to PMN attack and the resolution of the infection. Generally, killing macrophages can be beneficial for pathogens, whereas bacterium-induced PMN death has an opposite effect, since this contributes to the resolution of bacterial infections (46). This seems plausible for infections with Yersinia, where the virulence effector YopJ mediates the induction of apoptosis in macrophages but not in PMNs (53). The control of PMN death has also been reported for other pathogens, such as Mycobacterium tuberculosis, where such a mechanism allows optimal growth, survival, and transmission of the bacteria (54).
Taken together, the presented data suggest that YopK is essential for Yersinia to evade the PMN defense and thereby spread systemically and cause full disease. Our findings suggest a mechanism where YopK functions to prevent unintended Yop delivery that could cause PMN disruption and thereby enhance the inflammatory response to favor the host response.
ACKNOWLEDGMENTS
This study was supported by grants from the Swedish Research Council VR-M (K2008-58X-11222-14-3 [M.F.]) and grants from Laboratory for Molecular Infection Medicine Sweden and the Medial Faculty of Umeå University. D.E. was funded by a postdoctoral fellowship of the Kempe Foundation, and S.E.T. acknowledges financial support from the J. C. Kempe Memorial Fund.
We thank N. Taheri for technical assistance with uptake experiments and K. Ruuth for advice concerning the annexin V/PI flow cytometry.
Footnotes
Published ahead of print 22 October 2012
REFERENCES
- 1. Perry RD, Fetherston JD. 1997. Yersinia pestis–etiologic agent of plague. Clin. Microbiol. Rev. 10:35–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carniel E, Mollaret HH. 1990. Yersiniosis. Comp. Immunol. Microbiol. Infect. Dis. 13:51–58 [DOI] [PubMed] [Google Scholar]
- 3. Hanski C, Kutschka U, Schmoranzer HP, Naumann M, Stallmach A, Hahn H, Menge H, Riecken EO. 1989. Immunohistochemical and electron microscopic study of interaction of Yersinia enterocolitica serotype O8 with intestinal mucosa during experimental enteritis. Infect. Immun. 57:673–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marra A, Isberg RR. 1997. Invasin-dependent and invasin-independent pathways for translocation of Yersinia pseudotuberculosis across the Peyer's patch intestinal epithelium. Infect. Immun. 65:3412–3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Koornhof HJ, Smego RA, Jr, Nicol M. 1999. Yersiniosis. II. The pathogenesis of Yersinia infections. Eur. J. Clin. Microbiol. Infect. Dis. 18:87–112 [DOI] [PubMed] [Google Scholar]
- 6. Galan JE, Wolf-Watz H. 2006. Protein delivery into eukaryotic cells by type III secretion machines. Nature 444:567–573 [DOI] [PubMed] [Google Scholar]
- 7. Viboud GI, Bliska JB. 2005. Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu. Rev. Microbiol. 59:69–89 [DOI] [PubMed] [Google Scholar]
- 8. Durand EA, Maldonado-Arocho FJ, Castillo C, Walsh RL, Mecsas J. 2010. The presence of professional phagocytes dictates the number of host cells targeted for Yop translocation during infection. Cell Microbiol. 12:1064–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang P, Summer WR, Bagby GJ, Nelson S. 2000. Innate immunity and pulmonary host defense. Immunol. Rev. 173:39–51 [DOI] [PubMed] [Google Scholar]
- 10. Mayer-Scholl A, Averhoff P, Zychlinsky A. 2004. How do neutrophils and pathogens interact? Curr. Opin. Microbiol. 7:62–66 [DOI] [PubMed] [Google Scholar]
- 11. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. 2004. Neutrophil extracellular traps kill bacteria. Science 303:1532–1535 [DOI] [PubMed] [Google Scholar]
- 12. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, Brinkmann V, Jungblut PR, Zychlinsky A. 2009. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 5:e1000639 doi:10.1371/journal.ppat.1000639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brinkmann V, Laube B, Abu Abed U, Goosmann C, Zychlinsky A. 2010. Neutrophil extracellular traps: how to generate and visualize them. J. Vis Exp. 36:e1724 doi.10.3791/1724/jove.com/video/1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grosdent N, Maridonneau-Parini I, Sory MP, Cornelis GR. 2002. Role of Yops and adhesins in resistance of Yersinia enterocolitica to phagocytosis. Infect. Immun. 70:4165–4176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ruckdeschel K, Roggenkamp A, Schubert S, Heesemann J. 1996. Differential contribution of Yersinia enterocolitica virulence factors to evasion of microbicidal action of neutrophils. Infect. Immun. 64:724–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Songsungthong W, Higgins MC, Rolan HG, Murphy JL, Mecsas J. 2010. ROS-inhibitory activity of YopE is required for full virulence of Yersinia in mice. Cell Microbiol. 12:988–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Spinner JL, Cundiff JA, Kobayashi SD. 2008. Yersinia pestis type III secretion system-dependent inhibition of human polymorphonuclear leukocyte function. Infect. Immun. 76:3754–3760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Visser LG, Annema A, van Furth R. 1995. Role of Yops in inhibition of phagocytosis and killing of opsonized Yersinia enterocolitica by human granulocytes. Infect. Immun. 63:2570–2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Visser LG, Seijmonsbergen E, Nibbering PH, van den Broek PJ, van Furth R. 1999. Yops of Yersinia enterocolitica inhibit receptor-dependent superoxide anion production by human granulocytes. Infect. Immun. 67:1245–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thorslund SE, Edgren T, Pettersson J, Nordfelth R, Sellin ME, Ivanova E, Francis MS, Isaksson EL, Wolf-Watz H, Fallman M. 2011. The RACK1 signaling scaffold protein selectively interacts with Yersinia pseudotuberculosis virulence function. PLoS One 6:e16784 doi:10.1371/journal.pone.0016784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brodsky IE, Palm NW, Sadanand S, Ryndak MB, Sutterwala FS, Flavell RA, Bliska JB, Medzhitov R. 2010. A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe 7:376–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Holmstrom A, Rosqvist R, Wolf-Watz H, Forsberg A. 1995. Virulence plasmid-encoded YopK is essential for Yersinia pseudotuberculosis to cause systemic infection in mice. Infect. Immun. 63:2269–2276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mulder B, Michiels T, Simonet M, Sory MP, Cornelis G. 1989. Identification of additional virulence determinants on the pYV plasmid of Yersinia enterocolitica W227. Infect. Immun. 57:2534–2541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Straley SC, Bowmer WS. 1986. Virulence genes regulated at the transcriptional level by Ca2+ in Yersinia pestis include structural genes for outer membrane proteins. Infect. Immun. 51:445–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aili M, Isaksson EL, Carlsson SE, Wolf-Watz H, Rosqvist R, Francis MS. 2008. Regulation of Yersinia Yop-effector delivery by translocated YopE. Int. J. Med. Microbiol. 298:183–192 [DOI] [PubMed] [Google Scholar]
- 26. Dewoody R, Merritt PM, Houppert AS, Marketon MM. 2011. YopK regulates the Yersinia pestis type III secretion system from within host cells. Mol. Microbiol. 79:1445–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garcia JT, Ferracci F, Jackson MW, Joseph SS, Pattis I, Plano LR, Fischer W, Plano GV. 2006. Measurement of effector protein injection by type III and type IV secretion systems by using a 13-residue phosphorylatable glycogen synthase kinase tag. Infect. Immun. 74:5645–5657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Holmstrom A, Petterson J, Rosqvist R, Hakansson S, Tafazoli F, Fallman M, Magnusson KE, Wolf-Watz H, Forsberg A. 1997. YopK of Yersinia pseudotuberculosis controls translocation of Yop effectors across the eukaryotic cell membrane. Mol. Microbiol. 24:73–91 [DOI] [PubMed] [Google Scholar]
- 29. Trulzsch K, Sporleder T, Igwe EI, Russmann H, Heesemann J. 2004. Contribution of the major secreted Yops of Yersinia enterocolitica O:8 to pathogenicity in the mouse infection model. Infect. Immun. 72:5227–5234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. 2007. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 176:231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bolin I, Wolf-Watz H. 1984. Molecular cloning of the temperature-inducible outer membrane protein 1 of Yersinia pseudotuberculosis. Infect. Immun. 43:72–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bolin I, Norlander L, Wolf-Watz H. 1982. Temperature-inducible outer membrane protein of Yersinia pseudotuberculosis and Yersinia enterocolitica is associated with the virulence plasmid. Infect. Immun. 37:506–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tepper RI, Coffman RL, Leder P. 1992. An eosinophil-dependent mechanism for the antitumor effect of interleukin-4. Science 257:548–551 [DOI] [PubMed] [Google Scholar]
- 34. Deleuil F, Mogemark L, Francis MS, Wolf-Watz H, Fallman M. 2003. Interaction between the Yersinia protein tyrosine phosphatase YopH and eukaryotic Cas/Fyb is an important virulence mechanism. Cell Microbiol. 5:53–64 [DOI] [PubMed] [Google Scholar]
- 35. Heesemann J, Laufs R. 1985. Double immunofluorescence microscopic technique for accurate differentiation of extracellularly and intracellularly located bacteria in cell culture. J. Clin. Microbiol. 22:168–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ermert D, Urban CF, Laube B, Goosmann C, Zychlinsky A, Brinkmann V. 2009. Mouse neutrophil extracellular traps in microbial infections. J. Innate Immun. 1:181–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. 1995. A novel assay for apoptosis: flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein-labeled annexin V. J. Immunol. Methods 184:39–51 [DOI] [PubMed] [Google Scholar]
- 38. Echeverry A, Schesser K, Adkins B. 2007. Murine neonates are highly resistant to Yersinia enterocolitica following orogastric exposure. Infect. Immun. 75:2234–2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Handley SA, Dube PH, Revell PA, Miller VL. 2004. Characterization of oral Yersinia enterocolitica infection in three different strains of inbred mice. Infect. Immun. 72:1645–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Logsdon LK, Mecsas J. 2006. The proinflammatory response induced by wild-type Yersinia pseudotuberculosis infection inhibits survival of yop mutants in the gastrointestinal tract and Peyer's patches. Infect. Immun. 74:1516–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Urban CF, Lourido S, Zychlinsky A. 2006. How do microbes evade neutrophil killing? Cell Microbiol. 8:1687–1696 [DOI] [PubMed] [Google Scholar]
- 42. Macpherson AJ, Smith K. 2006. Mesenteric lymph nodes at the center of immune anatomy. J. Exp. Med. 203:497–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Voedisch S, Koenecke C, David S, Herbrand H, Forster R, Rhen M, Pabst O. 2009. Mesenteric lymph nodes confine dendritic cell-mediated dissemination of Salmonella enterica serovar Typhimurium and limit systemic disease in mice. Infect. Immun. 77:3170–3180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ye Z, Kerschen EJ, Cohen DA, Kaplan AM, van Rooijen N, Straley SC. 2009. Gr1+ cells control growth of YopM-negative yersinia pestis during systemic plague. Infect. Immun. 77:3791–3806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Conlan JW. 1997. Critical roles of neutrophils in host defense against experimental systemic infections of mice by Listeria monocytogenes, Salmonella typhimurium, and Yersinia enterocolitica. Infect. Immun. 65:630–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. DeLeo FR. 2004. Modulation of phagocyte apoptosis by bacterial pathogens. Apoptosis 9:399–413 [DOI] [PubMed] [Google Scholar]
- 47. Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, Waldmann H. 2011. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 7:75–77 [DOI] [PubMed] [Google Scholar]
- 48. Sanmun D, Witasp E, Jitkaew S, Tyurina YY, Kagan VE, Ahlin A, Palmblad J, Fadeel B. 2009. Involvement of a functional NADPH oxidase in neutrophils and macrophages during programmed cell clearance: implications for chronic granulomatous disease. Am. J. Physiol. Cell Physiol. 297:C621–C631 [DOI] [PubMed] [Google Scholar]
- 49. Watson RW, Redmond HP, Wang JH, Condron C, Bouchier-Hayes D. 1996. Neutrophils undergo apoptosis following ingestion of Escherichia coli. J. Immunol. 156:3986–3992 [PubMed] [Google Scholar]
- 50. Zhang B, Hirahashi J, Cullere X, Mayadas TN. 2003. Elucidation of molecular events leading to neutrophil apoptosis following phagocytosis: cross-talk between caspase 8, reactive oxygen species, and MAPK/ERK activation. J. Biol. Chem. 278:28443–28454 [DOI] [PubMed] [Google Scholar]
- 51. Viboud GI, Bliska JB. 2001. A bacterial type III secretion system inhibits actin polymerization to prevent pore formation in host cell membranes. EMBO J. 20:5373–5382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Viboud GI, So SS, Ryndak MB, Bliska JB. 2003. Proinflammatory signaling stimulated by the type III translocation factor YopB is counteracted by multiple effectors in epithelial cells infected with Yersinia pseudotuberculosis. Mol. Microbiol. 47:1305–1315 [DOI] [PubMed] [Google Scholar]
- 53. Spinner JL, Seo KS, O'Loughlin JL, Cundiff JA, Minnich SA, Bohach GA, Kobayashi SD. 2010. Neutrophils are resistant to Yersinia YopJ/P-induced apoptosis and are protected from ROS-mediated cell death by the type III secretion system. PLoS One 5:e9279 doi:10.1371/journal.pone.0009279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Blomgran R, Desvignes L, Briken V, Ernst JD. 2012. Mycobacterium tuberculosis inhibits neutrophil apoptosis, leading to delayed activation of naive CD4 T cells. Cell Host Microbe 11:81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]