

Abstract
Cytotoxic necrotizing factor 1 (CNF1) and hemolysin (HlyA1) are toxins produced by uropathogenic Escherichia coli (UPEC). We previously showed that these toxins contribute to the inflammation and tissue damage seen in a mouse model of ascending urinary tract infection. CNF1 constitutively activates small Rho GTPases by deamidation of a conserved glutamine residue, and HlyA1 forms pores in eukaryotic cell membranes. In this study, we used cDNA microarrays of bladder tissue isolated from mice infected intraurethrally with wild-type CP9, CP9cnf1, or CP9ΔhlyA to further evaluate the role that each toxin plays in the host response to UPEC. Regardless of the strain used, we found that UPEC itself elicited a significant change in host gene expression 24 h after inoculation. The largest numbers of upregulated genes were in the cytokine and chemokine signaling and Toll-like receptor signaling pathways. CNF1 exerted a strong positive influence on expression of genes involved in innate immunity and signal transduction and a negative impact on metabolism- and transport-associated genes. HlyA1 evoked an increase in expression of genes that encode innate immunity factors and a decrease in expression of genes involved in cytoskeletal and metabolic processes. Multiplex cytokine and myeloperoxidase assays corroborated our finding that a strong proinflammatory response was elicited by all strains tested. Bladders challenged intraurethrally with purified CNF1 displayed pathology similar to but significantly less intense than the pathology that we observed in CP9-challenged mice. Our data demonstrate substantial roles for CNF1 and HlyA1 in initiation of a strong proinflammatory response to UPEC in the bladder.
INTRODUCTION
Urinary tract infections (UTIs) are the most common bacterial infection in women and affect over 50% of women at some time throughout their lifetime (1). The clinical manifestations of UTIs vary from mild to severe, and common symptoms include dysuria, hematuria, pyuria, urinary frequency and urgency, suprapubic pain, and fever. The annual cost in the United States for diagnosis and treatment of UTIs is over $3 billion (2). Uncomplicated UTIs occur when commensal bacteria from the gastrointestinal (GI) tract transit to the periurethral area or vaginal introitus. The bacteria are then introduced into the urethra and ascend into the bladder to cause cystitis and, in more severe cases, into the kidneys to cause pyelonephritis (3–5). The etiological agents of 85% of uncomplicated UTIs are uropathogenic Escherichia coli (UPEC), a subgroup of extraintestinal pathogenic E. coli (1). UPEC expresses virulence factors that specifically facilitate colonization of and replication within the urinary tract.
Cytotoxic necrotizing factor 1 (CNF1) is an ∼115-kDa toxin that is expressed by ∼40% of UPEC isolates and up to 30% of diarrheal E. coli isolates (6). CNF1 constitutively activates small Rho-family GTPases via deamidation of glutamine 63 of RhoA and glutamine 61 in Rac1 and Cdc42 (7, 8). Constitutive activation of these small GTPases by CNF1 causes changes in vitro that include actin cytoskeletal rearrangement, multinucleation and cell spreading, formation of lamellipodia and filopodia, apoptosis of urothelial cells, decreased polymorphonuclear leukocyte (PMN) phagocytic capacity, and activation of nuclear factor-κB (NF-κB) (9–13). In addition, constitutive activation of Rac1, Cdc42, and RhoA in cultured epithelial cells targets them for ubiquitylation and proteasomal degradation. In cultured HEp-2 cells, CNF1 activates RhoA, which is then ubiquitylated and degraded; once activated RhoA is degraded, Rac1-GTP becomes dominant and activates antiapoptotic and prosurvival networks in a phosphoinositide 3-kinase/Akt/NF-κB-dependent manner (13, 14). In a mouse model of ascending UTI, our laboratory previously showed that a UPEC isolate that expresses CNF1 elicits significantly more interstitial and submucosal edema and neutrophil infiltration in the bladder than an isogenic cnf1-negative strain, despite the fact that both strains colonize the urinary tract equivalently (13–16). Conversely, previous work from Johnson et al. suggested that CNF1 does not contribute to UPEC colonization of or pathological alterations in the bladder (17). However, the inocula used in their study were nearly 100-fold higher than those used in our study, so the effects of CNF1 could have been masked by those of lipopolysaccharide (LPS) and other UPEC virulence factors.
Hemolysin (HlyA) is a 107-kDa extracellular pore-forming cytolysin that belongs to the repeats in toxin (RTX) family of bacterial toxins. HlyA requires posttranslational acylation of lysines at residues 564 and 690 for activation and oligomerizes to form pores in eukaryotic cell membranes in a calcium-dependent manner (18). Pore formation in the membranes of numerous host cell types, including red blood cells and bladder epithelial cells, causes cell lysis. At subcytotoxic concentrations, HlyA causes calcium oscillations in epithelial cells via a mechanism that requires pore formation (19). Wiles et al. reported that a sublethal dose of HlyA induces dephosphorylation of the serine/threonine kinase Akt in human bladder epithelial cells and subsequent modulation of downstream signaling cascades, including a possible reduction in NF-κB transcription (20). Our laboratory previously showed that UPEC strain CP9 causes damage to and exfoliation of 5637 bladder epithelial cells grown in a three-dimensional organoid model in an HlyA1-dependent manner (21). In addition, we found that CP9 causes hemorrhage and urothelial shedding in an HlyA1-dependent manner in the first 24 h of infection in a mouse ascending UTI model (16). Russo et al. determined that HlyA-expressing CP9 causes PMN lysis/necrosis at a high multiplicity of infection in vitro and PMN lysis/necrosis and lung injury in vivo in a rat model of E. coli pneumonia (22). The four-gene operon that encodes the CP9 HlyA with the greatest activity, hly1CABD, is cotranscribed with cnf1; cnf1 is downstream of an untranslated intergenic sequence (23). HlyA and CNF1 are often coexpressed in clinical UPEC isolates, and strains that express both toxins are more commonly isolated from patients with hemorrhagic UTIs (24–29).
Microarray analyses of host RNA isolated from the bladders of UPEC-challenged mice at various times postinoculation have identified numerous cellular pathways that are altered in the presence of UPEC (30–32). Common themes among these studies are that the bladder transcriptional response to UPEC occurs rapidly and that it is dominated by genes that function in proinflammatory, prosurvival, and antiapoptotic pathways. In this study, we extended these findings to more fully evaluate changes in the transcriptional landscape of the murine bladder 24 h after UPEC challenge. Further, we used microarray analysis of bladders challenged with UPEC strains deficient in CNF1 or HlyA1 to specifically address the roles of these toxins in the host response to UTI. We report here that all of the isogenic UPEC strains used in this study elicited a robust proinflammatory response that was independent of the toxins. Nevertheless, we also found that CNF1 and HlyA1 specifically increased expression of a number of genes that encode pattern recognition receptors, many of which interact with LPS. In addition, we provide cytokine and chemokine data that support our microarray findings. Finally, we demonstrate that a portion of the edema and epithelial damage observed in the bladders of UPEC-challenged mice is directly attributable to CNF1. Taken together, our findings show that both CNF1 and HlyA1 contribute significantly to the altered transcriptional response and the resultant pathology associated with ascending UTI.
MATERIALS AND METHODS
Strains and media.
The UPEC strains used in this study are CP9 (HlyA expressing [HlyA+], CNF1 expressing [CNF1+]) (33), CP9cnf1 (HlyA+, CNF1 negative [CNF1−]) (15), and CP9ΔhlyA1::cat (reduced HlyA, CNF1+) (21). The wild-type strain CP9 is a serotype O4:H5:K54 strain that was isolated from the bloodstream of a patient with pyelonephritis (33). The strain also expresses P (class I PapG adhesin), Prs (class III PapG adhesin), and type I pili. Bacteria were routinely cultured in Luria-Bertani (LB) broth or on LB agar (Difco, Sparks, MD) at 37°C. Liquid cultures were grown to the appropriate density at 37°C without aeration for 48 h for animal infections.
Mouse ascending UTI model.
Animal experiments were approved by the Uniformed Services University of the Health Sciences (USUHS) Institutional Animal Care and Use Committee and were carried out in accordance with the Guide for the Care and Use of Laboratory Animals, 8th ed. (34). Static cultures of CP9 and the CP9 isogenic strains were grown for 48 h at 37°C. Bacterial cultures were harvested by centrifugation (10,000 × g for 5 min at 4°C) and resuspended in sterile endotoxin-free phosphate-buffered saline (PBS; PromoCell, Heidelberg, Germany) at a concentration of 109 CFU/ml. Mice were inoculated intraurethrally as previously described with a few modifications (15, 16, 35). Briefly, 4-week-old female C3H/HeOuJ mice (Jackson Laboratory, Bar Harbor, ME) were quarantined for a minimum of 1 week prior to inoculation and were allowed food and water ad libitum. Mice were anesthetized with isoflurane USP (Clipper Distribution Company LLC, St. Joseph, MO) delivered by a vaporizer (Vet Equip Inc., Pleasanton, CA) set at 2% isoflurane and 0.8 liter O2 per minute. Mice were catheterized with sterile 0.66-mm (internal diameter) polyethylene tubing (Becton, Dickinson, Sparks, MD). A urine sample was collected prior to inoculation to test for preexisting bacteriuria. Mice with bacterial counts greater than 102 CFU/ml, as determined by dilution plating, were eliminated from the study. The inoculum was delivered via a 3-ml syringe mounted on an infusion pump (Harvard Apparatus, Holliston, MA). The syringe was attached to a 15-cm piece of pressure tubing (Argon Medical Devices, Plano, TX) connected to a 1/2-in., 30-gauge needle. The needle was inserted into the catheter, and 27 μl of inoculum (2.5 × 107 CFU/ml UPEC or 1 to 2 μg CNF1) was delivered at flow rate of 1 μl per second (80% force). Control mice received 27 μl of sterile endotoxin-free PBS. After 24 h, mice were anesthetized with isoflurane and catheterized to collect a urine sample. Mice were euthanized by cervical dislocation while under anesthesia, and the bladder and kidneys were aseptically removed. Urine samples were processed for bacterial enumeration, determination of the intensity of pyuria, and measurement of myeloperoxidase. Bladders were processed for microarray analysis, histology, and measurement of proinflammatory cytokines.
RNA extraction for microarray analysis.
Groups of 7 mice were challenged intraurethrally with CP9, CP9cnf1, CP9ΔhlyA1::cat, or PBS as described above, and a group of 10 naïve mice was used as the untreated reference. Mice were euthanized at 24 h postinoculation, and bladders were harvested and flash frozen in liquid nitrogen prior to storage at −80°C. Bladders from naïve animals were similarly harvested. Total RNA was isolated from the bladders with TRIzol reagent as previously described (36). After the RNA was precipitated, the samples were purified and concentrated on RNeasy columns (Qiagen Inc., Valencia, CA). On-column DNA digestion was conducted with an RNase-free DNase kit (Qiagen Inc.), according to the manufacturer's protocol. RNA quality and quantity were assessed with an Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA). Total RNA quality was assigned an RNA integrity number (RIN), and any sample with a RIN below the minimum threshold of 6.0 was eliminated from the study. RNA was stored at −80°C until use.
Microarray analysis.
Standard two-color microarray hybridizations were conducted at the Johns Hopkins University Deep Sequencing and Microarray Core. Fluorescently labeled cRNA was generated from 100 ng of total RNA from each sample using an Agilent low-input QuickAmp protocol (Agilent Technologies); the protocol simultaneously amplifies and incorporates cyanine 3 (Cy3) for experimental cRNA or cyanine 5 (Cy5) for reference cRNA. The resultant labeled probes were hybridized to Agilent whole-mouse-genome microarrays (4 × 44K; Agilent Technologies). For hybridization, individual samples from mice challenged with CP9, CP9ΔhlyA1, CP9cnf1, or PBS were hybridized against a reference sample; the reference sample consisted of pooled RNA isolated from the bladders of 10 mice that received no treatment. Posthybridization, microarrays were washed and scanned using an Agilent G2565CA microarray scanner system (Agilent Technologies). Spot signals were extracted with Agilent feature extraction software, and data were loaded into the Stanford Microarray Database (SMD) (37). For the analysis, the log2 R/G normalized (mean) inverted ratio data, where R is the red (Cy5) signal intensity and G is the green (Cy3) signal intensity, were retrieved from SMD by unique Biosequence identifiers (BioIDs); use of the inverted ratio accounted for the dye swap utilized in the labeling and hybridization. Spots with obvious abnormalities or numerous missing data points were excluded from the analysis. The Significance Analysis for Microarray (SAM) program (version 2.20) was used to identify significant genes that showed a minimum 2-fold change in expression. Two-class unpaired analyses were used to directly compare data from PBS-treated animals with data from either CP9-, CP9ΔhlyA1-, or CP9cnf1-infected animals. Next, data from CP9-infected animals were directly compared to data from CP9ΔhlyA1- or CP9cnf1-infected animals, and data from CP9cnf1-infected animals were compared to data from CP9ΔhlyA1-infected animals. The false discovery rates (FDRs) of the pairwise comparisons were 0.0 except for the CP9-versus-CP9cnf1, CP9-versus-CP9ΔhlyA1, and CP9cnf1-versus-CP9ΔhlyA1 comparisons, which had FDRs of 2.8, 8.0, and 19.28, respectively (Table 1). A complete list of those BioIDs whose expression was different among the various comparison groups is available in Table S1 in the supplemental material. Ontological classifications of genes were categorized using the Database for Annotation, Visualization, and Integrated Discovery (DAVID; version 6.7), available from NIH at http://david.abcc.ncifcrf.gov/home.jsp (38). The distribution of data across arrays and among arrays was analyzed with complete linkage clustering within the hierarchical clustering algorithm of the Cluster (version 2.11.0.0) program. The resultant heat maps were visualized with Treeview software (version 1.60). Data were further analyzed with the Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Database (39), Gene Ontology (GO), and Protein KnowledgeBase (UniProtKB) (40–42).
Table 1.
Number of unique Biosequence genes up- or downregulated in each comparison
| Comparison | No. of unique Biosequence IDsa |
FDR | ||
|---|---|---|---|---|
| Total | Upregulated | Downregulated | ||
| PBS vs: | ||||
| CP9 | 1,447 | 933 | 514 | 0 |
| CP9cnf1 | 738 | 638 | 100 | 0 |
| CP9ΔhlyA1 | 993 | 746 | 247 | 0 |
| CP9 vs: | ||||
| CP9cnf1 | 577 | 239 | 338 | 2.8 |
| CP9ΔhlyA1 | 352 | 230 | 122 | 8.0 |
| CP9cnf1 vs CP9ΔhlyA1 | 126 | 118 | 8 | 19.28 |
ID, identifier.
Proinflammatory cytokine analysis.
The proinflammatory cytokines interleukin-6 (IL-6), macrophage inflammatory protein 2 (MIP-2; CXCL-2), keratinocyte-derived cytokine (KC; CXCL-1), gamma interferon (IFN-γ), and tumor necrosis factor alpha (TNF-α) were measured by multiplex immunoassay with a mouse cytokine/chemokine panel in a magnetic bead format (Milliplex MAG; Millipore, Billerica, MA). Groups of 8 (UPEC) or 4 (PBS) mouse bladders were collected at 24 h postinoculation, snap-frozen in liquid nitrogen, and stored at −80°C until needed. Bladders were homogenized in 300 μl of lysis buffer (PBS [pH 7.4], 0.2% Tween 20, and complete mini-EDTA-free protease inhibitor cocktail tablets [Roche, Indianapolis, IN]) and pelleted by centrifugation at 10,000 × g for 10 min at 4°C. A volume of 25 μl of the supernatant was used for the assay. Signals were measured with a Luminex 100 system (Luminex Corp., Austin, TX), and data were acquired with the MasterPlex CT program (version 1.0; MiraiBio, South San Francisco, CA) and analyzed with the MasterPlex QT program (version 2.0; MiraiBio). Data were analyzed with the Kruskal-Wallis test and Dunn's multiple-comparison posttest (GraphPad Prism software, version 5.03).
Analysis of pyuria.
Mouse urine was collected by intraurethral catheterization prior to and 24 h after inoculation and stored on ice. Pyuria was assessed qualitatively with colorimetric urine test strips (Chemstrip 5 OB; Roche). For analysis of leukocytes, 4 μl of urine was spotted onto test pads. After 60 s, the extent of color development on the test pads was compared to color-coded standards provided by the manufacturer.
Measurement of urine MPO.
The myeloperoxidase (MPO) released by neutrophils was detected by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's protocol (Hycult Biotech, Plymouth Meeting, PA). Urine was collected prior to and at 24 h after inoculation. Prechallenge urine samples were used undiluted, and postchallenge urine samples were diluted in sample buffer. Data were analyzed with the Kruskal-Wallis test and Dunn's multiple-comparison posttest (GraphPad Prism software, version 5.03).
Purification and characterization of CNF1.
The protein 6× His-CNF1 was expressed in E. coli M15(pREP4)(pCNF24) and purified as previously described (12, 43). To assess the purity of the CNF1 preparation and the presence of LPS, CNF1 (2 μg) and LPS (E. coli serotype O55:B5, 5 μg) were electrophoresed in 4 to 20% Tris-glycine gradient gels (Novex; Life Technologies) and stained with oriole fluorescent gel stain (Bio-Rad, Hercules, CA) or a modified silver stain (Bio-Rad) according to the manufacturer's directions. Western blot analysis of the CNF1 preparation was done with polyclonal goat anti-CNF (12), rabbit anti-goat IgG (H+L)-horseradish peroxidase (Bio-Rad), and an Amersham ECL Plus Western blotting detection system (GE Healthcare Biosciences, Piscataway, NJ). Bacterial endotoxin in the CNF1 preparation was detected with the Limulus amebocyte lysate (LAL) assay (Hycult Biotech) according to the manufacturer's directions.
Histopathology.
The bladder and kidneys from infected and control mice were aseptically removed and fixed in 10% phosphate-buffered formalin (Fisher Scientific, Pittsburgh, PA) for a minimum of 12 h. The fixed tissue was paraffin embedded and sectioned to 5 μm thick (Histoserv, Germantown, MD). Sections were stained with hematoxylin-eosin (H&E) for morphology and Giemsa for localization of bacteria. Four stained sections per organ per animal were prescreened. A veterinary pathologist screened single organ sections in a blinded manner for four histologic parameters: necrosis/epithelial damage, edema, hemorrhage, and neutrophil infiltration. Each parameter was scored as follows: 0, no lesions; 1, mild; 2, minimal; 3, moderate; 4, marked; and 5, severe. The scores for each group of mice were averaged. Data were analyzed by one-way analysis of variance and Tukey's multiple-comparison posttest (GraphPad Prism software, version 5.03).
Microarray data accession number.
All microarray data generated by this study are publically available on the Gene Expression Omnibus website under accession number GSE37959 (www.ncbi.nih.nlm.gov/geo/query/acc.cgi?acc=GSE37959).
RESULTS AND DISCUSSION
Global changes in gene expression occur in the bladder in response to UPEC.
Our previous studies demonstrated that intraurethral inoculation of the mouse bladder with UPEC strain CP9 caused edema, hemorrhage, neutrophil infiltration, and epithelial damage by 24 h postinoculation (15, 16). By 3 and 5 days postinoculation, we observed edema and modest neutrophil inflammation. A CP9ΔhlyA1 mutant induced less hemorrhage and urothelial damage than the parent strain after 24 h but similar edema and neutrophil infiltration throughout the 5 days (16). In contrast, a CP9cnf1 mutant elicited less edema and neutrophil infiltration over 5 days (15). Thus, we concluded that HlyA1 induces hemorrhage and urothelial shedding in the first 24 h of infection, while CNF1 causes edema and neutrophil infiltration throughout the course of the infection. The goal of this study was to determine the specific contributions of CNF1 and HlyA1 to the overall host response to UPEC in the bladder. We chose to evaluate changes in the bladder transcriptome 24 h after inoculation because our previous data showed that both toxins contributed to the pathology that we observed at 24 h. We challenged seven mice per group intraurethrally with CP9, CP9cnf1, CP9ΔhlyA1, or PBS and harvested the bladders 24 h later. Urine colonization, which we used as a surrogate for bladder colonization, was equivalent among the three groups of mice (data not shown). We used pooled bladder RNA from 10 untreated mice as the reference sample for normalization. Bladder RNA was isolated, amplified, labeled, and hybridized to whole-mouse-genome microarrays. Spot data were extracted and loaded into the Stanford Microarray Database (44), and we used SAM pairwise analysis to identify BioIDs that were changed 2-fold or more between experimental groups. The SAM output data, which showed significant 2-fold or greater changes between mice challenged with PBS and CP9, PBS and CP9cnf1, PBS and CP9ΔhlyA1, CP9 and CP9cnf1, CP9 and CP9ΔhlyA1, and CP9cnf1 and CP9ΔhlyA1, are provided in Table S1 in the supplemental material.
We clustered all of the BioIDs whose expression changed 2-fold or more under at least one condition (2,200 unique BioIDs) with a complete linkage algorithm. As shown in Fig. 1A and B, the complete linkage clustering resulted in a dendrogram with two major nodes: one that contained the mice inoculated with PBS and one that contained the mice inoculated with any one of the UPEC strains. We observed slight changes in gene expression in the bladders of PBS-inoculated mice compared to the bladders of mice that had not been catheterized (Fig. 1B, PBS control). These data are consistent with our observations of very mild inflammation that is likely induced by the catheterization process itself. In sharp contrast, the gene expression profiles in the bladders of UPEC-challenged mice indicated that infection caused significant numbers of genes to be up- or downregulated compared to the gene regulation in the PBS-inoculated mice. A total of 1,447 BioIDs were differentially expressed when we challenged mice with wild-type CP9, and 738 BioIDs and 993 BioIDs were differentially expressed in mice challenged with CP9cnf1 and CP9ΔhlyA1, respectively (Table 1).
Fig 1.
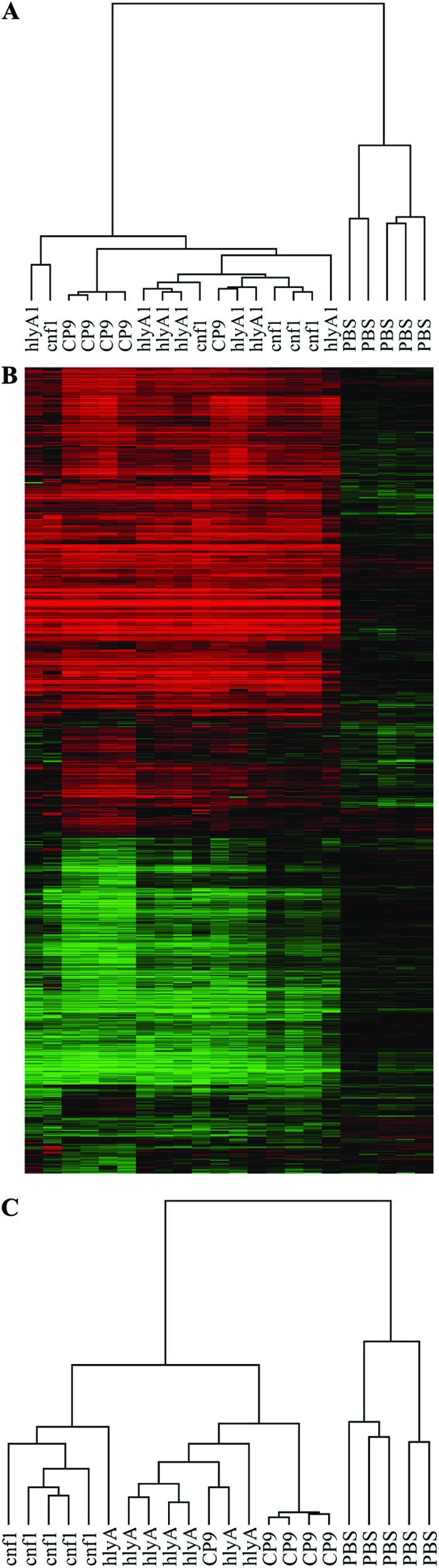
UPEC elicits significant host transcriptional changes in the bladder. Complete linkage cluster (A) and heat map (B) of all of the 2,200 unique BioIDs that were significantly different in any of the five comparisons in Table S1 in the supplemental material. Red, an increase in gene expression relative to the control; green, a decrease in gene expression relative to the control. (C) Complete linkage cluster of 783 BioIDs from CP9-versus-CP9cnf1 and/or CP9-versus-CP9ΔhlyA1 comparisons. For complete linkage clusters, gene expression relative to the reference control (normal bladder RNA) is shown for RNA isolated from the bladder of each mouse challenged with CP9 (CP9), CP9cnf1 (cnf1), CP9ΔhlyA1 (hlyA), or PBS (PBS).
We used the KEGG Pathway Database (39) to assign the identified genes to functional pathways. We found that the pathways that contained the largest numbers of genes that were more highly expressed in UPEC-challenged bladders are involved in signaling and recruitment of innate immune effectors. In particular, the largest numbers of upregulated genes were in the cytokine-cytokine receptor, chemokine signaling, Jak-STAT signaling, and the Toll-like receptor (TLR) signaling pathways. Genes that encode the proinflammatory cytokines IL-6, IL-17, and IFN-γ, as well as receptors for the cytokines IL-2, IL-10, and TNF-α, were increased in expression when UPEC was present. The upregulated genes in the chemokine signaling pathway included 14 chemokine (CC, CXC) ligands and four chemokine (CC, CXC) receptors. Transcription of the TLR1- and TLR6-encoding genes as well as the gene that encodes the downstream adaptor protein MyD88 was also higher in the UPEC-challenged bladders. TLR1 and TLR6 can dimerize to bind bacterial surface lipopeptides. This dimerization can lead to upregulation of downstream effectors, such as cytokines and chemokines, through activation of NF-κB. The cytokines and chemokines recruit neutrophils and other immune mediators to the site of infection. Our finding that CP9 infection of the murine bladder causes substantial changes in the mouse transcriptome by 24 h postchallenge supports previous observations that the clinical UPEC isolates CFT073, NU14, and UTI89 induced significant innate immune responses by 1.5 to 24 h after bladder challenge (30–32). In particular, Duell et al. showed that genes that encode proinflammatory cytokines and chemokines were upregulated by 2 and 24 h after challenge with CFT073, and the changes in expression of these genes at 24 h were similar in magnitude to the changes that we observed (30). Our data and the data from other recent studies clearly support the pathology that we previously observed in the bladders of mice challenged with UPEC (15, 16).
HlyA1 and CNF1 cause specific transcriptional changes in the bladder.
Since our major goal was to determine the contributions of HlyA1 and/or CNF1 to the host response, we next focused our analysis on the transcriptional changes in the bladder that were modulated by these toxins. To accomplish this, we identified statistically significant 2-fold or greater changes in gene expression when CP9-infected bladders were directly compared to CP9ΔhlyA1- or CP9cnf1-challenged bladders. This analysis revealed a total of 783 unique BioIDs that were expressed differently between bladders challenged with the wild-type strain and those challenged with either mutant strain. Complete linkage clustering of these data once again revealed that PBS-challenged bladders clustered on a separate branch of the dendrogram (Fig. 1C). Among the bladders from infected animals, we found that UPEC-infected bladders were further divided into two smaller nodes that efficiently segregated CP9cnf1-infected animals away from the other groups; 6/7 CP9ΔhlyA1-challenged bladders and 5/5 CP9-challenged bladders distinctly clustered away from CP9cnf1-infected bladders (Fig. 1C). Thus, the transcriptional responses to CP9 and CP9ΔhlyA1 were more similar to one another, while the response to CP9cnf1 was more divergent. This observation suggests that CNF1 plays a more dramatic role than HlyA1 in host signaling responses in the first 24 h of infection.
To address the role of each toxin, we focused our analysis more narrowly on those genes whose transcription was altered 3-fold or more in the CP9-versus-CP9ΔhlyA1 or CP9-versus-CP9cnf1 comparisons. We chose to use a minimum 3-fold change in transcription as a means to identify the most biologically relevant changes. The identified genes were assigned to GO biological process categories (40–42) (Fig. 2; see Table S2 in the supplemental material). The relative impact of HlyA1 on gene expression was less pronounced than that of CNF1 in terms of the number of differentially expressed genes; HlyA1 affected expression of 86 genes, and CNF1 altered expression of 184 genes. These findings correlate well with the cluster data described above (Fig. 1C). Genes involved in cytoskeletal (n = 6), metabolic (n = 9), and transport (n = 6) processes were negatively impacted by HlyA1 (Fig. 2, CP9 versus CP9ΔhlyA1 UP [upregulated]), while genes that play a role in innate immunity (n = 10) and, to a lesser extent, extracellular matrix (n = 4) were positively influenced by HlyA1 (Fig. 2, CP9 versus CP9ΔhlyA1 DOWN). Our finding that HlyA1 exerted a lesser effect on gene expression than CNF1 was anticipated due to the mechanism of action of HlyA1, which is to form pores in host cell membranes and cause cell lysis. We theorize that the observed transcriptional changes attributed to HlyA1 occurred in cells exposed to a subcytotoxic dose of HlyA1 and/or in cells that were proximal to lysed cells. In the former case, alterations in gene expression could be caused by signals that are transduced as a result of the calcium oscillations that occur in response to low and/or fluctuating levels of HlyA that insert into the membrane. In the latter case, the proximal cells could respond to changes in the surrounding milieu that occurred because the affected cells lysed and released their cellular contents, which, in turn, caused a concomitant influx of inflammatory mediators.
Fig 2.

CNF1 alters the expression of more genes than HlyA1. Genes whose expression changed 3-fold or more in the CP9-versus-CP9cnf1 and/or CP9-versus-CP9ΔhlyA1 comparisons were separated into GO biological process categories. The total numbers of genes altered in each comparison are as follows: 91 genes upregulated in CP9 versus CP9cnf1, 93 genes downregulated in CP9 versus CP9cnf1, 49 genes upregulated in CP9 versus CP9ΔhlyA1, and 37 genes downregulated in CP9 versus CP9ΔhlyA1.
CNF1 positively influenced the expression of genes involved in innate immunity (n = 24), signal transduction (n = 10), and metabolism (n = 8) (Fig. 2, CP9 versus CP9cnf1 DOWN) and negatively impacted genes in other metabolic (n = 13) and transport (n = 12) processes (Fig. 2, CP9 versus CP9Δcnf1 UP). As mentioned above, our previous studies demonstrated a significant role for CNF1 in the induction of a proinflammatory response in the bladder (15). CNF1 deamidates RhoA, Rac1, and Cdc42, which leads to constitutive activation of these small Rho-family GTPases. The activated GTPases can then regulate downstream effectors that play a role in proinflammatory cytokine expression, actin rearrangement, apoptosis, and cell spread.
To determine whether genes involved in innate immunity are represented at a higher frequency in our data sets than in the whole microarray data set, we assigned 100 randomized BioIDs from the whole data set to biological process categories. The microarrays that we used had a total of 42,404 probes, of which 10,511 did not have corresponding official gene symbols and 14,932 were duplicate probes for the same gene. We randomized the remaining 16,961 unique gene symbols and assigned 100 of the genes to GO categories according to the same criteria that were used to generate Table 2 (see Table S2 in the supplemental material). We found that the majority of the genes were involved in signaling (n = 16), protein processing (n = 11), regulation (n = 10), transport (n = 9), transcription (n = 9), or metabolism (n = 12) or were not assigned (n = 24). Only one gene was involved in innate immunity, which we believe indicates that approximately 1% of the unique genes represented on the microarray encode products that play a role in innate immunity. Taken together with the data presented in Fig. 2 and Table S2 in the supplemental material, the predominance of genes involved in innate immunity appears to be correlated with UPEC infection of the bladder and, in particular, with infection with a CNF1-producing UPEC strain.
Table 2.
Subset of genes whose expression changed 3-fold or morea
| GO categoryb | Gene symbol | Protein | Fold change in gene expression |
|||||
|---|---|---|---|---|---|---|---|---|
| Upregulated |
Downregulated |
|||||||
| PBS vs CP9 | CP9 vs CP9cnf | CP9 vs CP9ΔhlyA | PBS vs CP9 | CP9 vs CP9cnf | CP9 vs CP9ΔhlyA | |||
| Extracellular matrix | Itgam | Integrin αM (CD11b, CR-3) | 8.83 | 0.33 | 0.42 | |||
| Lama3 | Laminin subunit α-3 | 0.32 | ||||||
| Mmp3 | MMP-3 (stromelysin-1) | 13.88 | 0.22 | 0.27 | ||||
| Thbs4 | Thrombospondin | 17.23 | 0.25 | |||||
| Tnc | Tenascin | 0.32 | 0.30 | |||||
| Upk3a | Uroplakin-3a | 3.36 | ||||||
| Innate immunity | C1qtnf9 | C1q TNF-related protein 9 | 3.25 | 0.24 | ||||
| C3ar1 | C3a receptor | 0.26 | ||||||
| Ccl20 | CCL-20 | 4.31 | 4.35 | |||||
| Ccl24 | CCL-24 | 0.22 | ||||||
| Ccr2 | CCR-2 | 7.5 | 0.23 | |||||
| Cd180 | CD180 | 0.26 | ||||||
| Cd5l | CD5 antigen-like | 20.32 | 0.19 | 0.24 | ||||
| Ctss | Cathepsin S | 0.33 | ||||||
| Cxcl13 | CXCL13 | 19.54 | 0.09 | 0.09 | ||||
| F5 | Coagulation factor V | 7.26 | 0.39 | 0.29 | ||||
| Hspb7 | Heat shock protein beta 7 | 0.26 | 0.30 | |||||
| Icos | Inducible T-cell costimulator | 42.9 | 0.22 | 0.20 | ||||
| Il17c | IL-17c | 5.13 | ||||||
| Lair1 | Leukocyte-associated Ig-like receptor (CD305) | 12.29 | 0.22 | |||||
| Ly86 | Lymphocyte antigen 86 | 0.32 | ||||||
| Marco | Macrophage receptor MARCO | 50.94 | 0.09 | 0.18 | ||||
| Saa1 | Serum amyloid | 23.68 | 0.33 | |||||
| Sod3 | Superoxide dismutase [Cu-Zn] | 0.25 | ||||||
| Spon2 | Spondin-2 (mindin) | 0.32 | ||||||
| Timp1 | Tissue inhibitor of metalloprotease | 11.27 | 0.39 | 0.33 | ||||
| Tlr1 | Toll-like receptor 1 | 10.59 | 0.21 | |||||
| Tnfrsf11b | TNF receptor superfamily | 8.22 | 0.34 | |||||
| Signal transduction | Cwh43 | PGAP2-interacting protein | 34.34 | 0.08 | ||||
| Gm12185 | T-cell specific GTPase | 0.32 | ||||||
| Gm5431 | Novel interferon-inducible GTPase family member | 0.17 | ||||||
| Olfr916 | Olfactory receptor MOR168-1 | 0.27 | ||||||
Expression of genes was altered 3-fold or more in the CP9-versus-CP9cnf1 and/or CP9-versus-CP9ΔhlyA1 comparisons. The fold changes for genes represented by more than one spot on the array were averaged.
GO categories were manually assigned on the basis of information obtained from the UniProt database.
HlyA1 and CNF1 cause increased expression of genes involved in innate immune responses.
Alone or in combination, HlyA1 and CNF1 caused a 3-fold or greater change in expression of 33 genes whose products play a role in innate immunity (see Table S2 in the supplemental material). Of these, only two genes and three genes, respectively, were negatively impacted by the presence of HlyA1 and CNF1 (CP9 versus CP9cnf UP or CP9 versus CP9ΔhlyA1 UP). In contrast, we found that expression of 8 genes was downregulated in the absence of CNF1 and HlyA1 (CP9 versus CP9cnf DOWN and CP9 versus CP9ΔhlyA1 DOWN), while transcription of an additional 18 and 3 genes was negatively affected when CNF1 or HlyA1, respectively, was absent (CP9 versus CP9cnf DOWN or CP9 versus CP9ΔhlyA1 DOWN). Thus, CNF1 exerted a strong positive effect on transcription of genes involved in the innate immune response. The genes that encode spondin-1 (mindin) and CD180, both of which play a role in recognition of LPS, were negatively impacted when HlyA1 was deleted, which implies a positive effect of HlyA1 on expression of these genes. HlyA1 also increased transcription of CD180 (RP-105), and CNF1 exerted a positive effect on expression of Ly86 (MD-1). MD-1 and RP-105 associate with one another on the surface of B cells to stimulate antibody production and on the surface of antigen-presenting cells to downregulate the TLR4–MD-2-mediated response to LPS (49). Expression of the macrophage scavenger receptor MARCO was increased 51-fold when CP9 was introduced into the bladder and reduced 5-fold when HlyA1 was absent and 10-fold when CNF1 was absent. Thus, UPEC enhanced MARCO expression in a CNF1- and HlyA1-dependent manner. MARCO is overproduced by certain macrophage populations in a TLR-dependent manner when the macrophages are exposed to bacterial stimuli (50). Further, MARCO is a scavenger receptor that mediates nonopsonic phagocytosis and can bind LPS (50). Taken together, these data show that CNF1 and HlyA1 specifically caused increased expression of genes whose products function in recognition and binding of LPS and signaling of downstream effectors. Herlax et al. demonstrated that HlyA1 that is associated with LPS is more stable and less likely to self-aggregate (51). Binding of LPS-HlyA1 to pattern recognition receptors on the cell surface could bring HlyA1 into closer contact with the cell membrane on which it acts. Further, CNF1 is found in vitro in outer membrane vesicles (OMVs) that contain LPS (45, 52). If CNF1 is released from the bacterial cell in OMVs in vivo, association of the LPS-OMV-CNF1 complex with pattern recognition receptors on host cells could facilitate the interaction between CNF1 and its cell surface receptor laminin receptor protein (45, 47, 52, 53).
UPEC elicits a robust proinflammatory response in the bladder.
Our microarray data demonstrated that introduction of CP9, CP9cnf1, or CP9ΔhlyA1 into the bladder caused a dramatic increase in transcription of genes that encode the proinflammatory cytokines IL-6, MIP-2 (CXCL-2), KC (CXCL-1), and TNF-α (Table 3). We observed a 2.5-fold increase in expression of the gene that encodes KC in the bladders of mice challenged with CP9cnf1 compared to those of mice inoculated with CP9, as well as a slightly less than 2-fold increase in expression of the genes that encode IL-6 and MIP-2 (Table 3). To determine whether these increases in transcription caused concomitant increases in the amount of protein, we measured IL-6, MIP-2, KC, IFN-γ, and TNF-α in bladder homogenates from mice challenged with each strain with the Milliplex magnetic bead system (Millipore). As shown in Fig. 3, we observed similar levels of each cytokine in all bladders challenged with UPEC, regardless of the strain. We also found that bladders from the PBS-treated group contained significantly lower levels of the proinflammatory cytokines than those from the group challenged with CP9 or CP9cnf1. Bladders from CP9- and CP9cnf1-challenged mice had similar levels of each cytokine, while bladders from CP9ΔhlyA1-infected mice had lower, though not statistically significantly different, levels of each cytokine than those challenged with CP9 and CP9cnf1. These data show that introduction of UPEC, regardless of the strain, into the bladder caused an increase in expression of genes that encode proinflammatory cytokines, as well as a significant increase in the levels of these cytokines. The microarray data suggest that CNF1 may exert a slight suppressive effect on transcription of the genes that encode IL-6, MIP-2, and KC by 24 h postinoculation (Table 3). However, given that the levels of these cytokines were approximately equal by 24 h after inoculation with any of the UPEC strains, we theorize that the small increases in gene expression when CNF1 was absent did not immediately cause increases in cytokine protein levels.
Table 3.
Increases in transcription of proinflammatory cytokine genes
| Gene symbol | Cytokine | Fold change for PBS challenge vs challenge with: |
||
|---|---|---|---|---|
| CP9 | CP9cnf | CP9ΔhlyA1 | ||
| Il6 | IL-6 | 288 | 680 | 246 |
| Cxcl2 | MIP-2 (CXCL-2) | 81 | 179 | 115 |
| Cxcl1 | KC (CXCL-1) | 145a | 367a | 273 |
| Ifng | IFN-γ | 64b | 63b | 61b |
| Tnf | TNF-α | 27 | 34 | 35 |
The fold change in gene expression between CP9- and CP9 cnf1-challenged bladders was 2.5. All other fold changes in gene expression between CP9 and either mutant failed to reach our requirement for statistical significance.
Fold changes in IFN-γ gene expression are averages of values from two different spots on the microarray. A comparison of each spot across the three treatment groups revealed that the differences in gene expression were not statistically significantly different.
Fig 3.

Proinflammatory cytokines (A to E) and myeloperoxidase (F) are present in the bladders or urine by 24 h after challenge with UPEC. The proinflammatory cytokines IL-6 (A), MIP-2 (CXCL-2) (B), KC (CXCL-1) (C), IFN-γ (D), and TNF-α (E) were measured by use of the Milliplex MAG mouse cytokine/chemokine panel and Luminex 100 MultiAnalyte platform in a magnetic bead format. The limit of detection was 2.2 pg/ml for each cytokine. (F) The level of myeloperoxidase in mouse urine was detected by ELISA. Prechallenge urine samples within an experiment were pooled. Horizontal black bars represent the median. Data were analyzed by the Kruskal-Wallis test with Dunn's multiple-comparison posttest. *, P < 0.05; **, P < 0.005.
Because a major role of proinflammatory cytokines is to recruit neutrophils to the site of infection, we qualitatively determined the level of pyuria in urine collected from mice challenged with each of the UPEC strains with a urine dipstick test. We found that 50% and 40%, respectively, of mice challenged with CP9 exhibited severe and moderate pyuria. The intensity of pyuria was similar for mice challenged with CP9cnf1. In contrast, 20% and 75%, respectively, of mice challenged with CP9ΔhlyA1 exhibited severe and moderate pyuria. Moderate pyuria was detected in only 10% of PBS-inoculated mice. To more quantitatively assess the influx of neutrophils, we measured the level of MPO in the urine collected from mice challenged with the three strains. MPO is released from azurophilic granules of neutrophils once they are activated. We found that MPO was present at statistically significantly higher levels in the urine of UPEC-challenged mice than in the urine of PBS-inoculated mice (Fig. 3F). Though not significantly different from the MPO level in urine from CP9- or CP9cnf1-challenged mice, the level of MPO in the urine of CP9ΔhlyA1-inoculated mice was lower (Fig. 3F). Taken together, these data suggest that HlyA1 plays a role in shedding of neutrophils into the urine of infected mice. Histological analyses of bladders from mice infected with the three strains indicated that strain CP9ΔhlyA1 elicited less urothelial destruction than the HlyA1+ strains (16). Further, Dhakal and Mulvey recently demonstrated that sublytic doses of HlyA1 can activate host serine proteases that cleave the scaffold protein paxillin. Degradation of paxillin, in turn, led to exfoliation of bladder epithelial cells (48). When the urothelial layer is destroyed and excreted in the urine, any cells that have infiltrated the urothelial layer (i.e., neutrophils) would be excreted as well.
We did not observe suppression of cytokine expression and/or neutrophil recruitment by our UPEC strains, regardless of HlyA1 production, by 6 h (data not shown) or 24 h after inoculation, as has previously been reported by others (46, 54–58). These groups measured the cytokine IL-6 and/or IL-8 after infection of different strains of bladder epithelial cell lines with clinical isolates (NU14, F11, UTI89) or with laboratory strains (HB101, MG1655) and showed that the laboratory strains induced significantly more cytokine release than the clinical isolates (54–57). In addition, Loughman and Hunstad observed a reduction in neutrophil recruitment to the mouse bladder after challenge with UPEC strain UTI89 or CFT073 compared to that after challenge with the laboratory strain MG1655 (58). We believe that the reasons for these differences between our study and other studies are 2-fold. First, we assessed the host responses to challenge only with smooth UPEC strains (CP9 and isogenic derivatives); we did not compare the host responses to challenge with smooth clinical isolates versus nonisogenic rough laboratory strains. Lipid A, a TLR4 ligand that is well-known to elicit a very strong innate immune response, is more accessible on rough strains than it is on smooth strains. Billips et al. and Hunstad et al. demonstrated that UPEC strains that contain mutations in genes involved in LPS biosynthesis, particularly O- and core-polysaccharide antigen synthesis, induced more IL-6 and IL-8 from urothelial cells in vitro than the isogenic smooth parent strains (46, 57). Thus, though they and others found that the type of LPS expressed by the challenge strain (rough versus smooth) was not the only factor that contributed to immune suppression, rough LPS expressed by the more immune-stimulatory laboratory strains clearly played a role. Second, we conducted our study 24 h after infection rather than 1 to 8 h after challenge. The immune suppression observed by others was limited to the first 4 to 8 h postchallenge in vitro and in vivo (46, 54, 56). In addition, the role of HlyA in immune suppression remains unclear. Hilbert et al. showed that IL-6 and IL-8 release by urothelial 5637 cells was suppressed in an HlyA-dependent manner in vitro but that HlyA did not impact the release of IL-1β or IL-10 in vivo after 24 h (56). We also did not observe a significant role for HlyA1 in the establishment of the inflammatory response in our mouse UTI model at 24 h (46, 54, 56).
CNF1 causes edema and recruitment of neutrophils to the bladder.
Because our microarray data showed a substantial role for CNF1 in the non-cytokine-mediated induction of a proinflammatory response, we inoculated mouse bladders with purified recombinant CNF1 and analyzed bladder sections for pathology. Prior to inoculation of mouse bladders, we quantified the amount of LPS in our CNF1 preparation because LPS itself elicits a robust inflammatory response that could potentially confound interpretation of our results. We found that the CNF1 preparation contained 9.9 ng/ml LPS, a concentration that is 10,000-fold lower than the concentration of LPS (100 μg/ml) that is typically introduced into the bladder to stimulate an inflammatory response (59, 60). We inoculated 12 mice per group intraurethrally with 1 or 2 μg CNF1 and sacrificed them 24 h later. We sectioned and stained bladder tissue with H&E, and a veterinary pathologist scored the samples in a blinded manner. Mice challenged with CP9 served as a positive control for severe pathology. We observed significant edema, neutrophil infiltration, and urothelial damage, as well as moderate hemorrhage, throughout the bladders of mice challenged with CP9 (Fig. 4A, B, and G). These findings are similar to our previously published observations (15, 16). A dose of 2 μg CNF1 elicited moderate edema and urothelial damage only in localized regions of the bladder (Fig. 4C, D, and G). Neutrophil infiltration and hemorrhage were negligible in the bladders of mice inoculated with CNF1. The 1-μg dose of CNF1 did not cause any significant pathology in the bladders (data not shown). These data demonstrate that CNF1 alone elicits a mild to moderate localized inflammatory response that is independent of LPS and other bacterial factors and corroborate our observations of the decreased inflammation that occurs when CNF1 is absent. One explanation for our finding that inoculation with purified CNF1 caused only localized inflammation is that, unlike intact bacteria, CNF1 does not bind tightly to the urothelial layer and replicate. The relatively small bolus of toxin is likely to remain in a local area for a short period of time before being voided with urine.
Fig 4.

Bladders from CP9- and CNF1-inoculated mice exhibit significant pathology. Female C3H/HeOuJ mice were challenged with CP9 (A and B), 2 μg CNF1 (C and D), or PBS (E and F). Each panel is a representative sample from its respective treatment group. Magnifications, ×40 (A, C, and E) and ×400 (B, D, and F). The rectangles in panels A, C, and E correspond to the selection shown at ×400. Symbols: ●, edema; ▼, epithelial damage. (G) Histological scores of mouse bladders challenged with CP9, CNF1, or PBS. The four histological parameters scored were epithelial damage, edema, hemorrhage, and neutrophil infiltration, and each was assigned a score of 0 to 5. The scoring scale is as follows: 0, no lesion; 1, minimal; 2, mild; 3, moderate; 4, marked; 5, severe.
Conclusion.
In this report, we showed that introduction of UPEC into the bladders of mice caused significant alterations in host gene expression by 24 h after inoculation and that a majority of the changes occurred independently of CNF1 and/or HlyA1. However, expression of a subset of genes was dramatically affected by CNF1 and, to a lesser extent, by HlyA1. As evidenced by significant increases in proinflammatory cytokine and MPO levels, all strains elicited a significant proinflammatory response that was enhanced in the bladders of mice challenged with HlyA1-positive strains. These findings support and extend our previous studies that demonstrated specific roles for CNF1 and HlyA1 in the pathology that occurs as a result of UPEC inoculation into the bladder.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grant AI038281 from the National Institute for Allergy and Infectious Diseases (to A.D.O.) and the United States Army Veterinary Corps (to M.A.S.).
We thank Stephen Darnell for expert technical assistance.
Milliplex studies were done at the USUHS Biomedical Instrumentation Core.
The opinions or assertions contained herein are the private ones of the authors and are not to be construed as official or reflecting the views of the U.S. Department of Defense, the Uniformed Services University of the Health Sciences, or the National Institutes of Health.
Footnotes
Published ahead of print 22 October 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00605-12.
REFERENCES
- 1. Griebling TL, Litwin MS, Saigal CS. 2007. Urinary tract infection in women, p 589–619 In Urologic Diseases in America: 2007. U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Washington, DC [Google Scholar]
- 2. Litwin MS, Saigal CS, Yano EM, Avila C, Geschwind SA, Hanley JM, Joyce GF, Madison R, Pace J, Polich SM, Wang M. 2005. Urologic Diseases in America Project: analytical methods and principal findings. J. Urol. 173:933–937 [DOI] [PubMed] [Google Scholar]
- 3. Foxman B. 2010. The epidemiology of urinary tract infection. Nat. Rev. Urol. 7:653–660 [DOI] [PubMed] [Google Scholar]
- 4. Foxman B, Brown P. 2003. Epidemiology of urinary tract infections: transmission and risk factors, incidence, and costs. Infect. Dis. Clin. North Am. 17:227–241 [DOI] [PubMed] [Google Scholar]
- 5. Kaper JB, Nataro JP, Mobley HLT. 2004. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2:123–140 [DOI] [PubMed] [Google Scholar]
- 6. Hofman P, Le Negrate G, Mograbi B, Hofman V, Brest P, Alliana-Schmid A, Flatau G, Boquet P, Rossi B. 2000. Escherichia coli cytotoxic necrotizing factor-1 (CNF-1) increases the adherence to epithelia and the oxidative burst of human polymorphonuclear leukocytes but decreases bacteria phagocytosis. J. Leukoc. Biol. 68:522–528 [PubMed] [Google Scholar]
- 7. Lerm M, Selzer J, Hoffmeyer A, Rapp UR, Aktories K, Schmidt G. 1999. Deamidation of Cdc42 and Rac by Escherichia coli cytotoxic necrotizing factor 1: activation of c-Jun N-terminal kinase in HeLa cells. Infect. Immun. 67:496–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schmidt G, Sehr P, Wilm M, Selzer J, Mann M, Aktories K. 1997. Gln63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature 387:725–729 [DOI] [PubMed] [Google Scholar]
- 9. Caprioli A, Falbo V, Roda LG, Ruggeri FM, Zona C. 1983. Partial purification and characterization of an Escherichia coli toxic factor that induces morphological cell alterations. Infect. Immun. 39:1300–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davis JM, Rasmussen SB, O'Brien AD. 2005. Cytotoxic necrotizing factor type 1 production by uropathogenic Escherichia coli modulates polymorphonuclear leukocyte function. Infect. Immun. 73:5301–5310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fiorentini C, Arancia G, Caprioli A, Falbo V, Ruggeri FM, Donelli G. 1988. Cytoskeletal changes induced in HEp-2 cells by the cytotoxic necrotizing factor of Escherichia coli. Toxicon 26:1047–1056 [DOI] [PubMed] [Google Scholar]
- 12. Mills M, Meysick KC, O'Brien AD. 2000. Cytotoxic necrotizing factor type 1 of uropathogenic Escherichia coli kills cultured human uroepithelial 5637 cells by an apoptotic mechanism. Infect. Immun. 68:5869–5880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Miraglia AG, Travaglione S, Meschini S, Falzano L, Matarrese P, Quaranta MG, Viora M, Fiorentini C, Fabbri A. 2007. Cytotoxic necrotizing factor 1 prevents apoptosis via the Akt/IkB kinase pathway: role of nuclear factor-kB and Bcl-2. Mol. Biol. Cell 18:2735–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hoffmann C, Aktories K, Schmidt G. 2007. Change in substrate specificity of cytotoxic necrotizing factor unmasks proteasome-independent down-regulation of constitutively active RhoA. J. Biol. Chem. 282:10826–10832 [DOI] [PubMed] [Google Scholar]
- 15. Rippere-Lampe KE, O'Brien AD, Conran R, Lockman HA. 2001. Mutation of the gene encoding cytotoxic necrotizing factor type 1 (cnf1) attenuates the virulence of uropathogenic Escherichia coli. Infect. Immun. 69:3954–3964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smith YC, Rasmussen SB, Grande KK, Conran RM, O'Brien AD. 2008. Hemolysin of uropathogenic Escherichia coli evokes extensive shedding of the uroepithelium and hemorrhage in bladder tissue within the first 24 hours after intraurethral inoculation of mice. Infect. Immun. 76:2978–2990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johnson DE, Drachenberg C, Lockatell CV, Island MD, Warren JW, Donnenberg MS. 2000. The role of cytotoxic necrotizing factor-1 in colonization and tissue injury in a murine model of urinary tract infection. FEMS Immunol. Med. Microbiol. 28:37–41 [DOI] [PubMed] [Google Scholar]
- 18. Stanley P, Packman LC, Koronakis V, Hughes C. 1994. Fatty acylation of two internal lysine residues required for the toxic activity of Escherichia coli hemolysin. Science 266:1992–1996 [DOI] [PubMed] [Google Scholar]
- 19. Koschinski A, Repp H, Unver B, Dreyer F, Brockmeier D, Valeva A, Bhakdi S, Walev I. 2006. Why Escherichia coli alpha-hemolysin induces calcium oscillations in mammalian cells—the pore is on its own. FASEB J. 20:973–975 [DOI] [PubMed] [Google Scholar]
- 20. Wiles TJ, Dhakal BK, Eto DS, Mulvey MA. 2008. Inactivation of host Akt/protein kinase B signaling by bacterial pore-forming toxins. Mol. Biol. Cell 19:1427–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smith YC, Grande KK, Rasmussen SB, O'Brien AD. 2006. Novel three-dimensional organoid model for evaluation of the interaction of uropathogenic Escherichia coli with terminally differentiated human urothelial cells. Infect. Immun. 74:750–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Russo TA, Davidson BA, Genagon SA, Warholic NM, Macdonald U, Pawlicki PD, Beanan JM, Olson R, Holm BA, Knight PR., III 2005. E. coli virulence factor hemolysin induces neutrophil apoptosis and necrosis/lysis in vitro and necrosis/lysis and lung injury in a rat pneumonia model. Am. J. Physiol. Lung Cell. Mol. Physiol. 289:L207–L216 [DOI] [PubMed] [Google Scholar]
- 23. Landraud L, Gibert M, Popoff MR, Boquet P, Gauthier M. 2003. Expression of cnf1 by Escherichia coli J96 involves a large upstream DNA region including the hlyCABD operon, and is regulated by the RfaH protein. Mol. Microbiol. 47:1653–1667 [DOI] [PubMed] [Google Scholar]
- 24. Blanco J, Blanco M, Alonso MP, Blanco JE, Gonzalez EA, Garabal JI. 1992. Characteristics of haemolytic Escherichia coli with particular reference to production of cytotoxic necrotizing factor type 1 (CNF1). Res. Microbiol. 143:869–878 [DOI] [PubMed] [Google Scholar]
- 25. Cavalieri SJ, Bohach GA, Snyder IS. 1984. Escherichia coli alpha-hemolysin: characteristics and probable role in pathogenicity. Microbiol. Rev. 48:326–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Falbo V, Famiglietti M, Caprioli A. 1992. Gene block encoding production of cytotoxic necrotizing factor 1 and hemolysin in Escherichia coli isolates from extraintestinal infections. Infect. Immun. 60:2182–2187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Landraud L, Gauthier M, Fosse T, Boquet P. 2000. Frequency of Escherichia coli strains producing the cytotoxic necrotizing factor (CNF1) in nosocomial urinary tract infections. Lett. Appl. Microbiol. 30:213–216 [DOI] [PubMed] [Google Scholar]
- 28. Real JM, Munro P, Buisson-Touati C, Lemichez E, Boquet P, Landraud L. 2007. Specificity of immunomodulator secretion in urinary samples in response to infection by alpha-hemolysin and CNF1-bearing uropathogenic Escherichia coli. Cytokine 37:22–25 [DOI] [PubMed] [Google Scholar]
- 29. Yamamoto S, Tsukamoto T, Terai A, Kurazono H, Takeda Y, Yoshida O. 1995. Distribution of virulence factors in Escherichia coli isolated from urine of cystitis patients. Microbiol. Immunol. 39:401–404 [DOI] [PubMed] [Google Scholar]
- 30. Duell BL, Carey AJ, Tan CK, Cui X, Webb RI, Totsika M, Schembri MA, Derrington P, Irving-Rodgers H, Brooks AJ, Cripps AW, Crowley M, Ulett GC. 2012. Innate transcriptional networks activated in bladder in response to uropathogenic Escherichia coli drive diverse biological pathways and rapid synthesis of IL-10 for defense against bacterial urinary tract infection. J. Immunol. 188:781–792 [DOI] [PubMed] [Google Scholar]
- 31. Mysorekar IU, Mulvey MA, Hultgren SJ, Gordon JI. 2002. Molecular regulation of urothelial renewal and host defenses during infection with uropathogenic Escherichia coli. J. Biol. Chem. 277:7412–7419 [DOI] [PubMed] [Google Scholar]
- 32. Reigstad CS, Hultgren SJ, Gordon JI. 2007. Functional genomic studies of uropathogenic Escherichia coli and host urothelial cells when intracellular bacterial communities are assembled. J. Biol. Chem. 282:21259–21267 [DOI] [PubMed] [Google Scholar]
- 33. Russo TA, Guenther JE, Wenderoth S, Frank MM. 1993. Generation of isogenic K54 capsule-deficient Escherichia coli strains through TnphoA-mediated gene disruption. Mol. Microbiol. 9:357–364 [DOI] [PubMed] [Google Scholar]
- 34. Committee for the Update of the Guide for the Care and Use of Laboratory Animals 2011. Guide for the care and use of laboratory animals, 8th ed The National Academies Press, Washington, DC [Google Scholar]
- 35. Hagberg L, Engberg I, Freter R, Lam J, Olling S, Svanborg-Ed,n C. 1983. Ascending, unobstructed urinary tract infection in mice caused by pyelonephritogenic Escherichia coli of human origin. Infect. Immun. 40:273–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Merrell DS, Butler SM, Qadri F, Dolganov NA, Alam A, Cohen MB, Calderwood SB, Schoolnik GK, Camilli A. 2002. Host-induced epidemic spread of the cholera bacterium. Nature 417:642–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sherlock G, Hernandez-Boussard T, Kasarskis A, Binkley G, Matese JC, Dwight SS, Kaloper M, Weng S, Jin H, Ball CA, Eisen MB, Spellman PT, Brown PO, Botstein D, Cherry JM. 2001. The Stanford Microarray Database. Nucleic Acids Res. 29:152–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huang da W, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4:44–57 [DOI] [PubMed] [Google Scholar]
- 39. Kanehisa M, Goto S. 2000. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28:27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. 2000. Gene Ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25:25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jain E, Bairoch A, Duvaud S, Phan I, Redaschi N, Suzek BE, Martin MJ, McGarvey P, Gasteiger E. 2009. Infrastructure for the life sciences: design and implementation of the UniProt website. BMC Bioinformatics 10:136 doi:10.1186/1471-2105-10-136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. UniProt Consortium 2012. Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucl. Acid Res. 40:D71–D75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grande KK, Meysick KC, Rasmussen SB, O'Brien AD. 2009. Cytotoxic necrotizing factor type 1-neutralizing monoclonal antibody NG8 recognizes three amino acids in a C-terminal region of the toxin and reduces toxin binding to HEp-2 cells. Infect. Immun. 77:170–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hubble J, Demeter J, Jin H, Mao M, Nitzberg M, Reddy TB, Wymore F, Zachariah ZK, Sherlock G, Ball CA. 2009. Implementation of GenePattern within the Stanford Microarray Database. Nucleic Acids Res. 37:D898–D901 doi:10.1093/nar/gkn786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Davis JM, Carvalho HM, Rasmussen SB, O'Brien AD. 2006. Cytotoxic necrotizing factor type 1 (CNF1) delivered by outer membrane vesicles of uropathogenic Escherichia coli attenuates polymorphonuclear leukocyte antimicrobial activity and chemotaxis. Infect. Immun. 78:4401–4408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Billips BK, Schaeffer AJ, Klumpp DJ. 2008. Molecular basis of uropathogenic Escherichia coli evasion of the innate immune response in the bladder. Infect. Immun. 76:3891–3900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chung JW, Hong SJ, Kim KJ, Goti D, Stins MF, Shin S, Dawson VL, Dawson TM, Kim KS. 2003. 37-kDa laminin receptor precursor modulates cytotoxic necrotizing factor 1-mediated RhoA activation and bacterial uptake. J. Biol. Chem. 278:16857–16862 [DOI] [PubMed] [Google Scholar]
- 48. Dhakal BK, Mulvey MA. 2012. The UPEC pore-forming toxin α-hemolysin triggers proteolysis of host proteins to disrupt cell adhesion, inflammatory, and survival pathways. Cell Host Microbe 11:58–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yoon SI, Hong M, Wilson IA. 2011. An unusual dimeric structure and assembly for TLR4 regulator RP105-MD-1. Nat. Struct. Mol. Biol. 18:1028–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Areschoug T, Gordon S. 2009. Scavenger receptors: role in innate immunity and microbial pathogenesis. Cell. Microbiol. 11:1160–1169 [DOI] [PubMed] [Google Scholar]
- 51. Herlax V, de Alaniz MJ, Bakas L. 2005. Role of lipopolysaccharide on the structure and function of alpha-hemolysin from Escherichia coli. Chem. Phys. Lipids 135:107–115 [DOI] [PubMed] [Google Scholar]
- 52. Kouokam JC, Wai SN, Fallman M, Dobrindt U, Hacker J, Uhlin BE. 2006. Active cytotoxic necrotizing factor 1 associated with outer membrane vesicles from uropathogenic Escherichia coli. Infect. Immun. 74:2022–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim KJ, Chung JW, Kim KS. 2005. 67-kDa laminin receptor promotes internalization of cytotoxic necrotizing factor 1-expressing Escherichia coli K1 into human brain microvascular endothelial cells. J. Biol. Chem. 280:1360–1368 [DOI] [PubMed] [Google Scholar]
- 54. Billips BK, Forrestal SG, Rycyk MT, Johnson JR, Klumpp DJ, Schaeffer AJ. 2007. Modulation of host innate immune response in the bladder by uropathogenic Escherichia coli. Infect. Immun. 75:5353–5360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hilbert DW, Pascal KE, Libby EK, Mordechai E, Adelson ME, Trama JP. 2008. Uropathogenic Escherichia coli dominantly suppress the innate immune response of bladder epithelial cells by a lipopolysaccharide- and Toll-like receptor 4-independent pathway. Microb. Infect. 10:114–121 [DOI] [PubMed] [Google Scholar]
- 56. Hilbert DW, Paulish-Miller TE, Tan CK, Carey AJ, Ulett GC, Mordechai E, Adelson ME, Gygax SE, Trama JP. 2012. Clinical Escherichia coli isolates utilize alpha-hemolysin to inhibit in vitro epithelial cytokine production. Microb. Infect. 14:628–638 [DOI] [PubMed] [Google Scholar]
- 57. Hunstad DA, Justice SS, Hung CS, Lauer SR, Hultgren SJ. 2005. Suppression of bladder epithelial cytokine responses by uropathogenic Escherichia coli. Infect. Immun. 73:3999–4006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Loughman JA, Hunstad DA. 2011. Attenuation of human neutrophil migration and function by uropathogenic bacteria. Microb. Infect. 13:555–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Saban R, D'Andrea MR, Andrade-Gordon P, Derian CK, Dozmorov I, Ihnat MA, Hurst RE, Simpson C, Saban MR. 2007. Regulatory network of inflammation downstream of proteinase-activated receptors. BMC Physiol. 7:3 doi:10.1186/1472-6793-7-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Song J, Bishop BL, Li G, Grady R, Stapleton A, Abraham SN. 2009. TLR4-mediated expulsion of bacteria from infected bladder epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 106:14966–14971 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.