

Abstract
Accurate detection of transcriptional regulatory elements is essential for high-quality genome annotation, metabolic reconstruction, and modeling of regulatory networks. We developed a computational approach for reconstruction of regulons operated by transcription factors (TFs) from large protein families and applied this novel approach to three TF families in 10 Desulfovibrionales genomes. Phylogenetic analyses of 125 regulators from the ArsR, Crp/Fnr, and GntR families revealed that 65% of these regulators (termed reference TFs) are well conserved in Desulfovibrionales, while the remaining 35% of regulators (termed singleton TFs) are species specific and show a mosaic distribution. For regulon reconstruction in the group of singleton TFs, the standard orthology-based approach was inefficient, and thus, we developed a novel approach based on the simultaneous study of all homologous TFs from the same family in a group of genomes. As a result, we identified binding for 21 singleton TFs and for all reference TFs in all three analyzed families. Within each TF family we observed structural similarities between DNA-binding motifs of different reference and singleton TFs. The collection of reconstructed regulons is available at the RegPrecise database (http://regprecise.lbl.gov/RegPrecise/Desulfovibrionales.jsp).
INTRODUCTION
A comparative genomics approach is widely used for computational identification of regulatory elements, such as transcription factor (TF) binding sites (TFBSs), and reconstruction of transcriptional regulation in related microbial genomes (1–3). The approach relies on the assumption that if a particular genomic feature is preserved in the course of evolution, it is highly probable that this feature has a specific functional role in a cell. Specifically, in reconstruction of transcriptional regulation, we assume that as long as a TF-encoding gene is conserved in a set of closely related species, the regulation of respective genes via cognate TFBSs also tends to be maintained (4).
The genes controlled by the same TF and their cis-acting regulatory elements comprise a regulon. A set of genes from multiple related genomes regulated by orthologous TFs constitutes a regulog (5). The comparative genomics methods confidently predict conserved regulog members with TFBSs that are present upstream of multiple orthologous genes. Nonconserved members of a regulog lack either gene orthologs or TFBSs in several genomes. Assessment of nonconserved candidate regulog members requires additional evidence, such as the functional concurrence of candidate regulated genes or colocalization of a candidate target gene with a TF-encoding gene. A large group of bacterial TFs is encoded by species-specific genes that lack orthologs in related genomes. These singleton TFs likely arose due to horizontal gene transfer in the evolution of individual microbial species (6, 7).
The standard comparative genomics approach is hardly applicable for reconstruction of regulons controlled by species-specific TFs that lack orthologs in closely related genomes. In this study, we directly addressed this problem and developed a novel computational approach for genomic reconstruction of regulons controlled by singleton TFs, i.e., regulators that are present in only one species in a group of related bacteria. In this work, we use an operational definition of a singleton TF to be a regulator that is present in one or two species, since for a group of two genomes, the comparative genomics approach could not be applied confidently.
The proposed approach is based on three assumptions. First, we assume a constrained diversity of binding sites within a TF family. Since all members of a TF family have one structural type of DNA-binding domain, they are expected to share at least some characteristics of their DNA-binding motifs. This assumption has been used earlier for the discovery of TF-binding motifs with familial binding profiles (8). Second, we assume a structural similarity of TF-binding motifs from the same TF family. Palindromic symmetry of prokaryotic TF-binding motifs has been used in different strategies of the TFBS search (9–11). We compare the palindromic symmetry of known TF-binding motifs to the symmetry of candidate TFBSs using relative entropy as a measure of similarity. Third, we assume colocalization of a singleton TF-encoding gene and its target genes on the chromosome. Many local regulators in Escherichia coli are adjacent to operons that they regulate (12). Since many local regulators were demonstrated to undergo horizontal gene transfer either at the interspecies (6) or at the intraspecies (7) level and horizontal gene transfer seems to be an important mechanism of singleton TF origin, we expect to find TFBSs for singleton regulators in a close genomic neighborhood. This tendency has been used in the sequence-based method for TFBS discovery tested on groups of homologous prokaryotic TFs (13).
In this study, all three assumptions were utilized in the development of a novel computational approach for in silico reconstruction of singleton TF regulons in microbial genomes. We applied this approach to infer singleton regulons in three large TF families in 10 microorganisms from the Desulfovibrionales order, known for their involvement in biocorrosion and potential use in bioremediation (14, 15). Our results demonstrated that a significant fraction of regulons controlled by singleton TFs could be inferred by the proposed approach.
MATERIALS AND METHODS
The overall scheme of the computational approach for reconstruction of both reference and singleton regulons within a single TF family is shown in Fig. 1. Briefly, the work flow consists of three main steps: (i) identification of TF family representatives and phylogenetic analysis to select the orthologous groups of TFs (reference TFs) and the singleton TFs, (ii) comparative genomic reconstruction of reference TF regulons, and (iii) identification of binding sites for singleton TFs.
Fig 1.

Work flow for reconstruction of reference and singleton TF regulons within a single TF family.
Identification and phylogenetic analysis of transcription factors.
We tested our approach on the group of 10 genomes of Desulfovibrionales uploaded from the MicrobesOnline database (16): Desulfovibrio vulgaris Hildenborough, Desulfovibrio vulgaris Miyazaki F, Desulfovibrio alaskensis G20, Desulfovibrio desulfuricans ATCC 27774, Desulfovibrio magneticus RS-1, Desulfovibrio salexigens DSM 2638, Desulfovibrio piger ATCC 29098, Lawsonia intracellularis PHE/MN1-00, Desulfomicrobium baculatum DSM 4028, and Desulfohalobium retbaense DSM 5692. We excluded the Desulfovibrio vulgaris DP4 genome from the study because it is very similar to that of D. vulgaris Hildenborough.
A search for TFs from the Crp/Fnr, ArsR, and GntR families was conducted using public programmatic access to the MicrobesOnline database (16). Crp/Fnr family members were identified as proteins that belong to the COG0664 family and possess a Crp-type DNA-binding domain (IPR001808). ArsR family members were proteins with an annotated ArsR-type DNA-binding domain (IPR01845). GntR family members had an annotated GntR-type DNA-binding domain (IPR000524). Diverse regulators from the GntR family were divided into three subfamilies according to the previously described classification (17).
We used the ClustalX (version 2.1) program (18) for protein alignment construction and phylogenetic analysis. Phylogenetic trees were constructed by the neighbor-joining method with default parameters, with calculation of bootstraps from 1,000 replications. Orthologous groups of TFs were identified as high-confidence clusters on the phylogenetic tree that have more than 40% pairwise protein identity within the cluster. On the basis of the size, orthologous TF groups were classified as reference groups (three or more orthologous proteins) or singleton groups (containing one or two TFs). The details on the regulon reconstruction for the TFs from these two groups are described below.
Reconstruction of reference regulons.
We used the comparative genomics approach implemented in the RegPredict web server (19). For the three groups of TFs (CooA [20], HcpR [21], and LldR [22]), binding motifs were described earlier; thus, we used a procedure of regulon inference with known positional weight matrices (PWMs). These initial PWMs were used for identification of similar TFBSs in the analyzed group of genomes. Novel lineage-specific PWMs were constructed with TFBSs identified by the initial search in genomes of Desulfovibrionales and used for final regulon description.
For the rest of the reference orthologous TF groups, we applied a procedure of de novo regulon inference (19). Briefly, we selected sets of putative operons in a genomic neighborhood of each TF gene and collected their upstream regions (from nucleotides [nt] −400 to +50 with respect to the translation start). Each set included the TF-encoding operon, two upstream operons, and two downstream operons. We used operon predictions from the MicrobesOnline database (16). A common palindromic motif with the highest information content was identified in each set of upstream sequences by using the Discover Profiles tool of the RegPredict server. For each novel motif, a specific PWM was constructed and further used for a whole-genome site search (19). A gene was considered to be a member of a TF regulon if putative TFBSs were found upstream of the gene and upstream of its orthologs in several other genomes or it was included in the operon bearing such a conserved binding site. Strong nonconserved TFBSs were considered true positives if they were identified upstream of genes that are functionally related to known members of the regulon. All identified TFBSs were aligned and used for refinement of a TF-binding motif and final regulon reconstruction. These TF-binding motifs were used as a reference for singleton regulon reconstruction.
Reconstruction of singleton regulons.
We identified candidate TFBSs in the neighborhood of a TF-encoding gene using the sequence and structural similarities between a candidate TFBS and reference TF motifs for regulators from the same TF family. The structural similarity between a candidate TFBS and known TF motifs was evaluated by use of the symmetry profiles constructed with the sets of TFBSs from the reference regulons reconstructed in the Desulfovibrionales genomes in this study.
Construction of symmetry profiles.
We used the sets of TFBSs of reference regulators to construct symmetry profiles to compare the symmetry of candidate binding sites with the symmetry of each reference TF-binding motif. As a measure of site symmetry, we used the relative entropy. Each site in the set was compared with its reverse-complemented counterpart, and the symmetry score (W) was calculated as follows:
| (1) |
where N is the site length, q(k) is the observed frequency of symmetrical nucleotides at position k, and q(A), q(C), q(G), and q(T) are the observed frequencies of the A, C, G, and T nucleotides in the set of binding sites, respectively. The lowest symmetry score observed in the training set of sites was chosen as a threshold for symmetry scoring of candidate binding sites.
Construction of familial PWMs and symmetry profiles.
In addition to the reference TF-binding motifs, we used PWMs composed of known TF-family motifs that we call familial PWMs. We used an alignment of TFBSs from all reference regulons of a TF family to build a single familial PWM. The Crp/Fnr familial PWM was built on the basis of the alignment of TFBSs from the CooA, DvMF_1708, HcpR, Desal_2066, and MreC regulogs. The ArsR familial PWM was constructed using TFBSs of the ArsR, ArsR2, SahR, and SmtB TFs. The first and the last nucleotide of SmtB binding sites were removed to ensure that all sites in the set had identical lengths (20 bp). Symmetry profiles for familial PWMs were constructed as described above. For the GntR TF family, a familial PWM could not be constructed because of the great dissimilarity between reference motifs.
Identification of binding sites for singleton TFs.
The procedure for reconstruction of singleton TF regulons consisted of the following three steps. First, candidate singleton TFBSs with the known reference and familial PWMs were identified using the RegPredict web server with the following search parameters: a site score threshold of 3.0 and a search range of −250 to 1 nt with respect to the start codon but excluding the coding region of upstream genes. We collected a set of candidate TFBSs found in upstream regions of the operon encoding the singleton TF, two upstream operons, and two downstream operons. At the second step, these candidate TFBSs were evaluated using the symmetry profile for the corresponding reference TF regulon motif, and sites with a score below the threshold were rejected. For the Crp/Fnr- and ArsR-family regulators, these two steps were performed for each reference regulon motif and for the familial motif. For singleton regulators from the GntR family, members of each subfamily were analyzed only with the PWM and the symmetry profile of regulators from the same subfamily. Specifically, for analysis of the FadR subfamily of singleton TFs, the DVU2644 and LldR motifs were used. For analysis of MocR subfamily members, the DVU0030 and DVU2953 motifs were used.
At the third step, the sets of candidate sites were merged and a candidate site with the highest symmetry score was considered a true binding site. If there were several different candidate sites with similar symmetry scores or no candidate sites were found, two additional criteria were considered. The first additional criterion was cotranscription of a TF-encoding gene with other genes in one operon. A candidate site upstream of the multigene operon that included a singleton regulator was considered the most probable candidate.
The second additional criterion was based on the observation that similar regulators may regulate similar biological processes from the same functional category. For instance, a vast majority of Fur TF family members regulate metal homeostasis (23), while many regulators from the LacI/GalR family control the catabolism of various carbohydrates (24). If the first criterion could not discriminate between several candidates, gene ontology annotations of upstream and downstream neighbors of a singleton TF gene were used to select the proximal genes of similar function and to repeat the PWM-based site search with a decreased threshold of 2.5.
After identification of a singleton regulator binding site(s), a symmetrized PWM was constructed for each regulator from those sites' sequences and their reverse complements. Additional binding sites were identified by whole-genome search with the symmetrized PWM using the RegPredict server as described earlier (19).
Motif logos were constructed using the Weblogo tool (25). Predicted regulons were deposited in the RegPrecise database (26; http://regprecise.lbl.gov/RegPrecise/Desulfovibrionales.jsp).
RESULTS
Phylogenetic analysis and classification of TF families in Desulfovibrionales.
To test a novel TF regulon inference approach, we chose a group of 10 deltaproteobacteria from the Desulfovibrionales order and three large TF families, Crp/Fnr, ArsR, and GntR. In total, 125 proteins were found in these genomes within the Crp/Fnr, ArsR, and GntR families (Table 1). The numbers of TFs within the three analyzed families varied significantly between individual genomes, (see Table S1 in the supplemental material). For instance, L. intracellularis and D. salexigens possess 1 and 20 proteins within the three TF families, respectively. For GntR-family TFs, 26 proteins were from the FadR subfamily, 15 proteins were from the MocR subfamily, 6 proteins were from the HutC subfamily, and 6 proteins did not belong to any known subfamilies.
Table 1.
Classification of TFs in the Desulfovibrionales
| TF family | No. of TFs (no. of orthologous groups) |
||
|---|---|---|---|
| Reference | Singleton | Total | |
| Crp/Fnr | 24 (5) | 10 | 34 |
| ArsR | 30 (4) | 8 | 38 |
| GntR | 28 (5) | 25 | 53 |
To identify orthologous groups of transcriptional regulators, we constructed phylogenetic trees for each studied TF family (Fig. 2 to 4). Among 125 proteins, 81 TFs (65%) were assigned to orthologous groups which included three or more TFs. The remaining 44 regulators were found to be either species specific (34 regulators) or possess a single ortholog within the Desulfovibrionales (totally 10 regulators). Typically, every organism had only one representative per orthologous group. The only exception was the ArsR orthologous group of regulators associated with arsenic resistance cassettes. In this group, a second copy of a regulatory gene emerged in three genomes, possibly as a result of recent duplication or horizontal transfer of the cassettes. For reconstruction of regulons controlled by these TFs, we started from the comparative genomic analysis of regulators from the orthologous groups (reference TFs) and then proceeded with regulon inference for the remaining group of species-specific regulators (singleton TFs).
Fig 2.
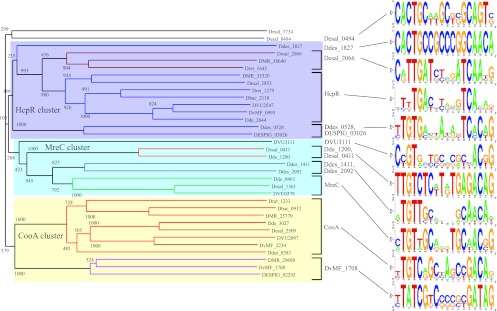
Neighbor-joining tree of the Crp/Fnr-family TFs. The numbers represent bootstrap values obtained from 1,000 replicates. Orthologous groups of TFs are marked by colors. Groups of TFs that have common TF-binding motifs are marked by brackets.
Fig 3.
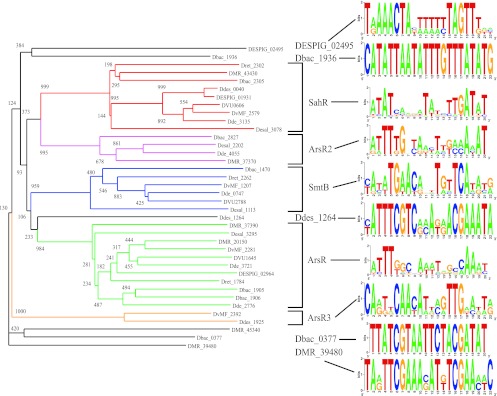
Neighbor-joining tree of the ArsR-family TFs. The numbers represent bootstrap values obtained from 1,000 replicates. Orthologous groups of TFs are marked by colors. Groups of TFs that have common TF-binding motifs are marked by brackets.
Fig 4.
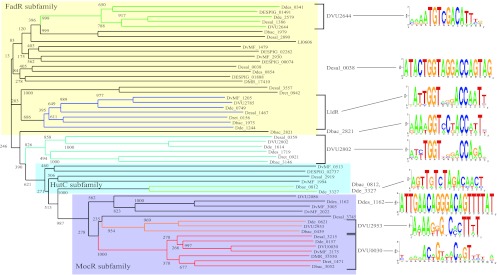
Neighbor-joining tree of the GntR-family TFs. The numbers represent bootstrap values obtained from 1,000 replicates. Orthologous groups of TFs are marked by colors. Groups of TFs that have common TF-binding motifs are marked by brackets.
Reconstruction of reference TF regulons.
The comparative genomics approach implemented in the RegPredict web server (19) was utilized for reconstruction of the reference TF regulons for 14 orthologous groups of regulators from the Crp/Fnr, ArsR, and GntR families (Table 2). For three reference TF regulogs, the regulon reconstruction procedure was started from the previously known DNA-binding motifs for CooA (20), HcpR (21), and LldR (22), whereas the remaining 11 regulogs were reconstructed by the de novo regulon reconstruction procedure, as described in Materials and Methods. The majority of reconstructed reference TF regulons contain only one or two target operons (Table 2). Typically, these local TF regulons have a TF-encoding gene, which is either autoregulated or located near a TF-regulated operon. Two remarkable exceptions are the HcpR and SahR regulons, which include up to five target operons per genome (see Table S1 in the supplemental material). The phylogenetic distribution of the reference TFs in Desulfovibrionales is nonuniform. The SahR regulon is the most widely distributed among the Desulfovibrionales genomes, being present in nine genomes, whereas four reference TFs are present in only three genomes (Table 2). The reconstructed reference TF regulons were further used for the analysis of singleton TF regulons.
Table 2.
Size and distribution of reference TF regulons in Desulfovibrionales genomes
| Family and TF regulon | No. of candidate TF-regulated operons/genome (total no. of genes in TF-regulated operons) |
Functional role | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| D. vulgaris Hildenborough | D. vulgaris Miyazaki F | D. alaskensis G20 | D. desulfuricans ATCC 27774 | D. piger ATCC 29098 | D. salexigens DSM 2638 | D. magneticus RS-1 | D. baculatum DSM 4028 | D. retbaense DSM 5692 | ||
| Crp/Fnr | ||||||||||
| CooA | 1 (2) | 1 (2) | 1 (2) | 1 (2) | 1 (2) | 1 (2) | 1 (2) | 1 (2) | Carbon monoxide dehydrogenase | |
| HcpR | 3 (5) | 2 (3) | 4 (6) | 4 (6) | 4 (8) | 4 (5) | 3 (3) | Nitrosative stress response | ||
| MreC | 1 (8) | 1 (7) | 1 (6) | Uranium reduction | ||||||
| DvMF_1708 | 1 (5) | 1 (1) | 1 (4) | |||||||
| Desal_2066 | 2 (2) | 2 (3) | 2 (2) | |||||||
| ArsR | ||||||||||
| ArsR | 2 (3) | 1 (1) | 3 (5) | 2 (2) | 3 (4) | 3 (7) | 2 (4) | 2 (3) | Arsenic resistance | |
| ArsR2 | 1 (4) | 1 (4) | 1 (1) | 1 (1) | Arsenic resistance | |||||
| SahR | 4 (6) | 3 (5) | 5 (8) | 3 (5) | 3 (5) | 3 (5) | 1 (2) | 1 (3) | 2 (4) | Methionine metabolism |
| SmtB | 1 (1) | 1 (1) | 1 (1) | 1 (2) | 1 (2) | 1 (2) | Heavy metal resistance | |||
| GntR | ||||||||||
| LldR | 1 (3) | 1 (2) | 1 (2) | 1 (3) | 3 (4) | 1 (3) | Lactate utilization | |||
| DVU0030 | 2 (3) | 2 (3) | 2 (3) | 2 (4) | 1 (1) | 2 (3) | 2 (3) | Amino acid transport | ||
| DVU2644 | 1 (4) | 1 (3) | 1 (2) | 2 (4) | 2 (3) | Amino acid metabolism | ||||
| DVU2802 | 1 (7) | 1 (4) | 1 (5) | 1 (8) | 1 (5) | 1 (8) | ||||
| DVU2953 | 2 (2) | 2 (5) | 2 (2) | |||||||
Table 3.
Reconstruction of singleton TF regulons
| Family and regulon | No. of sites |
|||
|---|---|---|---|---|
| Sites identified with PWM and symmetry profilea | Good sites | Additional sites in genome | Total | |
| Crp/Fnr | ||||
| DVU3111 | 4 | 2 | 1 | 3 |
| Dde_1200 | 5 | 1 | 0 | 1 |
| Ddes_0528 | 1 | 1 | 1 | 2 |
| Ddes_1411 | 5 | 3 | 3 | 6 |
| Ddes_1827 | 1b | 1b | 0 | 1 |
| Ddes_2092 | 1 | 1 | 0 | 1 |
| Desal_0411 | 1 | 1 | 0 | 1 |
| Desal_0494 | 1 | 1 | 1 | 2 |
| DESPIG_03026 | 2 | 2 | 0 | 2 |
| ArsR | ||||
| DvMF_2392 | 0 | 0 | 2b | 2 |
| Ddes_1264 | 4 | 2 | 0 | 2 |
| Ddes_1925 | 0 | 0 | 2b | 2 |
| DESPIG_02495 | 2 | 1 | 2 | 3 |
| DMR_39480 | 2 | 1 | 1 | 2 |
| Dbac_0377 | 2 | 1 | 0 | 1 |
| Dbac_1936 | 6 | 1 | 0 | 1 |
| GntR | ||||
| Dbac_0812 | 1b | 1 | 1 | 2 |
| Dbac_2821 | 2 | 2 | 1 | 3 |
| Dde_3327 | 1b | 1 | 0 | 1 |
| Ddes_1162 | 2 | 1 | 0 | 1 |
| Desal_0038 | 1 | 1 | 0 | 1 |
Identified at the 1st step.
Sites identified using additional criteria.
Reconstruction of singleton TF regulons.
A modified comparative genomics approach was developed for reconstruction of the singleton TF regulons (see Materials and Methods). By utilizing this approach, we were able to reconstruct 21 out of 44 singleton TF regulons from the Crp/Fnr, ArsR, and GntR families in the Desulfovibrionales (Table 3). At the first step, we used the reference and familial PWMs to identify candidate TFBSs in the neighborhood of singleton TF genes. As result, the best TFBS candidates were tentatively assigned to 16 singleton regulators by using the TFBS symmetry scores, whereas for the remaining 5 singleton regulators, two or more candidate TFBSs were chosen at this step because of their high similarity to each other. At the next step, additional candidate TFBSs were identified for eight singleton TFs by whole-genome scanning using the candidate site(s) determined at the first step.
For reconstruction of five singleton TF regulons, we used additional criteria. For the Crp/Fnr-family regulator Ddes_1827, no candidate binding sites were found with the reference TFBS profiles. Since the gene encoding this regulator is probably cotranscribed with two other genes, this putative operon was expected to be autoregulated. Newly identified singleton TFBS motifs of the Crp/Fnr family were used to search for a candidate binding site upstream of the Ddes_1827 gene. As result, the Desal_0494 motif was applied to identify a putative binding site of Ddes_1827. For reconstruction of two ArsR-family singleton TFs, DvMF_2392 and Ddes_1925, we used an additional functional consideration based on the observation that many ArsR-like TFs control arsenic resistance. Indeed, the arsenic resistance genes were located directly downstream from these singleton TF genes and a common 22-bp palindromic motif was identified in their upstream gene regions (see Fig. S6 in the supplemental material).
A distinctive feature of the GntR family is the presence of several TF subfamilies with very different binding motifs. Since our collection of reference TF regulons included only representatives from the FadR and MocR subfamilies, we were unable to use their motifs for the inference of HutC-like singleton TF regulons. Thus, the functional criterion was used for regulon reconstruction for two HutC-subfamily TFs, Dde_3327 and Dbac_0812, that are encoded by genes colocalized with the phosphonate metabolism operons (see Fig. S8 in the supplemental material). The search for a common binding motif within the upstream region of these two operons identified a 16-bp inverted repeat, and an additional copy of this repeat was found upstream of an operon encoding the phosphonate transporter in D. baculatum.
In summary, singleton TF regulons were identified for 9 out of 10 singleton TFs from the Crp/Fnr family (see Table S2 in the supplemental material), 7 out of 8 singleton TFs from the ArsR family (see Table S3 in the supplemental material), and 5 out of 26 singleton TFs from the GntR family (see Table S4 in the supplemental material). Brief descriptions of the reconstructed singleton regulons for each TF family are given in the following sections. More details on the reconstruction of both reference and singleton TF regulons are available in the supplemental material.
Crp/Fnr-family singleton TF regulons.
Ten representatives of the Crp/Fnr family were classified as singleton TFs (Table 1). Analysis of the genomic context of two singleton TFs, Dde_1200 and Desal_0411, suggests that they are located within operons containing paralogs of genes from the reconstructed MreC reference regulon (see Fig. S1 in the supplemental material). The 18-bp palindromic DNA motif identified upstream of the mreB homologs was assigned as a candidate motif for these singleton TFs. The motifs identified for both singleton TFs and MreC have the same 18-bp structure and partially similar consensus sequences (Fig. 2). The only difference between these reconstructed regulons is that in both singleton regulons, the TF-encoding genes are not subject to autoregulation, whereas in the MreC regulon, the mreC gene is a part of the MreC-regulated operon.
The DVU3111 singleton TF is encoded in a unique genomic locus that also includes a paralog of the mreA transporter, a ferredoxin, and a hypothetical oxidoreductase (see Fig. S1 in the supplemental material). A common 18-bp DNA motif identified upstream of these genes demonstrated significant similarity with the MreC binding motif and was assigned as a putative binding motif for DVU3111.
The phylogenetic and genome context analyses of two singleton TFs, Ddes_2092 and Ddes_1411, suggest that these regulators are close paralogs that are adjacent to hypothetical arylsulfotransferases (see Fig. S1 in the supplemental material). However, these paralogous loci contain different transporter genes: Ddes_2092 is colocalized with a transporter from the sodium/sulfate family, whereas Ddes_1411 is clustered with a major facilitator superfamily (MFS)-type transporter. The identified 18-bp DNA motif for the Ddes_2092 and Ddes_1411 singleton TFs has the consensus sequence WTGTTGCNNNNGCAACAW, which is very similar to the MreC motif.
Genomic context analysis of two singleton TFs, DESPIG_03026 and Ddes_1827, suggests that they are located in putative operons with the hydroxylamine reductase hcp genes (see Fig. S2 in the supplemental material). Analysis of upstream regions of these operons identified 18-bp palindromic TFBSs that have some similarity to the HcpR binding motif. A similar candidate TFBS was identified upstream of Ddes_0528, which encodes another singleton TF. A whole-genome search with the Ddes_0528 PWM identified an additional target gene of the respective singleton TF, Ddes_1164, encoding a hypothetical cupin domain-containing protein, which is orthologous to a previously identified member of the reference HcpR regulon, Dret_2382. We conclude that the reference HcpR regulon and the above-mentioned three HcpR-like singleton regulons in Desulfovibrionales have similar functional roles in the response to nitrosative stress.
A candidate TFBS for the Desal_0494 singleton TF was identified in the upstream region of the downstream Desal_0493 gene encoding a GlnB-like protein.
ArsR-family singleton TF regulons.
Eight ArsR-family proteins from Desulfovibrionales genomes were identified as singleton regulators by the phylogenetic analysis (Table 1). Two orthologous ArsR-like regulatory genes were found near operons that encode the arsenite metallochaperone ArsD, the arsenite-transporting ATPase ArsA, and the S-adenosylmethionine-dependent arsenic methyltransferase. A common binding motif was identified upstream of these arsenic resistance operons and both regulatory genes (see Fig. S6 in the supplemental material). There is no membrane protein associated with the ArsA ATPase in the DvMF_2392 regulon, but the ArsR regulon in the same genome includes the DvMF_2282 gene, encoding a membrane protein similar to ArsP. The ArsP membrane protein was found to be associated with the ArsA-ArsD transporting system in different bacterial species (27). Another singleton regulator, Ddes_1264, is cotranscribed with a gene encoding a P-type ATPase, similar to transporters from the SmtB regulon (see Fig. S5 in the supplemental material). Two similar strong 22-bp palindromes were identified upstream of this operon.
Candidate TFBSs were found upstream of three other ArsR-family singleton TFs, DMR_39480, Dbac_0377, and Dbac_1936. Three nearly identical 22-bp palindromes were found upstream of the operon encoding the singleton TF DESPIG_02495 and a fusion of acyl coenzyme A synthase and MFS transporter proteins that may be involved in secondary metabolism (see Fig. S5 in the supplemental material). Dbac_0377 is predicted to regulate a thioredoxin and an acetyl-CoA synthase-like enzyme. Other singleton regulons contain either enzymes of unknown specificity (hydrolase and flavin mononucleotide reductase) or hypothetical proteins (see Fig. S5 in the supplemental material).
In summary, three of the ArsR-family singleton TFs are predicted to regulate the resistance to arsenate or other heavy metal ions, but the role of five other singleton TFs in this family is unclear. DESPIG_02495 likely regulates a protein containing the acyl-CoA transferase domain.
GntR-family singleton TF regulons.
All members of the GntR family possess similar N-terminal winged helix-turn-helix DNA-binding domains, while C-terminal ligand-binding domains are highly variable. Several subfamilies have been inferred within the family, on the basis of different types of C-terminal domains (28). We assigned 14, 6, and 5 GntR-family singleton TFs to the FadR, HutC, and MocR subfamilies, respectively (Fig. 4).
Candidate binding motifs of two FadR-subfamily reference TFs, LldR and DVU2644, have different lengths and consensus sequences. To identify singleton TF-binding sites in the FadR subfamily, we used the two reference TF-binding motifs and searched for similar sites using relaxed thresholds. Candidate sites similar to the LldR-binding motif were found upstream of the two FadR-subfamily regulatory genes, Desal_0038 and Dbac_2821 (see Fig. S7 in the supplemental material). We propose that Desal_0038 probably regulates the glycerate kinase-encoding operon; however, we could not exclude the possibility of regulation of a divergently transcribed gene encoding a chemotaxis protein. Dbac_2821 likely regulates an operon encoding choline dehydrogenase, betaine aldehyde dehydrogenase, and a glycine-betaine transporter.
A candidate TFBS for the MocR-subfamily singleton TF Ddes_1162 was identified in the common upstream region of this TF gene and the Ddes_1163 gene encoding a pyridoxamine 5′-phosphate oxidase-like protein.
Two genes encoding similar HutC-subfamily singleton TFs, Dde_3327 and Dbac_0812, are adjacent to the phosphonate metabolism operons (see Fig. S8 in the supplemental material). Regulons for these two TFs were reconstructed as described earlier. The TFBSs of these two regulators are similar to the known binding motif of the phosphonate metabolism regulator PhnF from Mycobacterium smegmatis (29). Since no reference regulon was found in Desulfovibrionales, a PhnF binding motif from M. smegmatis was used for singleton reconstruction. Binding sites weakly similar to PhnF were predicted upstream of both operons, and an additional TFBS was found upstream of a phosphonate transport operon in D. baculatum (see Fig. S8 in the supplemental material).
DISCUSSION
We present a novel approach aimed at solving a challenging problem of reconstruction of singleton regulons, i.e., regulons controlled by species-specific TFs present in only one or two genomes. In contrast to the highly conserved regulatory systems, it is impossible to apply a conventional comparative genomics approach to infer singleton TF regulons. The proposed approach is based on the simultaneous analysis of a whole TF family in a group of closely related genomes. We applied this approach to three TF families, Crp/Fnr, ArsR, and GntR, in 10 bacterial genomes from the Desulfovibrionales lineage. As a result, we were able to reconstruct almost half (21 out of 44) of all singleton TF regulons from these families. The results of the singleton TF regulon reconstruction approach differed between the three analyzed TF families, demonstrating advantages and limitations of this method.
The initial step of the approach requires a TF family to have several orthologous groups to infer reference TF regulons by a conventional comparative genomics technique. The TF families that we selected for this study have representatives in several analyzed genomes, providing enough material for comparative reconstruction of the respective reference TF regulons. Indeed, five reference TF regulons were characterized in each of the Crp/Fnr and GntR families, and four reference TF regulons were inferred in the ArsR family. In all three families, the TF-binding motifs and gene functional roles of the reconstructed reference regulons served as a basis for the subsequent inference of singleton TF regulons.
The success of the singleton TF reconstruction procedure greatly depends on the variability of TF-binding DNA motifs within a TF family. The highest percentage of successfully reconstructed singleton regulons has been achieved for the Crp/Fnr family. In this family, a TF-binding motif of all reference regulons has the same structure, an 18-nt palindrome, and a notable similarity at the sequence level (Fig. 2). It allowed us to build a common profile and use it to search for candidate binding sites of the Crp/Fnr-family singleton TFs. A similar TF-binding motif specific for the whole Crp/Fnr family has been found in alphaproteobacteria (30). The Crp/Fnr family in the Desulfovibrionales provides an example of TF-binding DNA motifs extremely conserved across the whole family, the property that played a critical role in the singleton reconstruction procedure.
A lower level of motif conservation has been observed for TFs from the ArsR family, where the binding motifs have the same structural characteristics but differ from each other at the sequence level (Fig. 3). As a result, only one of eight singleton TFs was reconstructed by sequence similarity to reference TF regulon motifs. At the same time, conservation of the motif structure (20- or 22-nt inverted repeat) was successfully used to identify ArsR-family binding motifs for seven other singleton regulators.
Analysis of the GntR family revealed a dramatic difference between motifs from the same TF subfamilies. Neither the structure nor the nucleotide sequence was conserved between reference TF regulons, with the exception of LldR and DVU2802. As a result, only two singleton TF regulons were reconstructed by utilizing the similarity with the LldR binding motif. For other regulators from the FadR subfamily, the lack of TF-binding motif conservation made the reconstruction of singleton TF regulons impossible. A significant diversity of sequence and structure of TF-binding motifs across various GntR subfamilies has been reported for other taxonomic groups (17, 28).
The functional similarity of target genes for related TFs typically plays a supplementary role in making higher-confidence regulon predictions. Nevertheless, in a number of cases, conservation of a general functional role of regulated genes becomes a primary source of evidence for reconstruction of singleton TF regulons. For example, additional functional support was obtained for three HcpR-like singleton regulons that were originally reconstructed using the reference HcpR motif. At the same time, analysis of entire TF families provides higher confidence even for the reference TF regulons.
A different situation was observed in the ArsR family, where conservation of the general regulon function, arsenic resistance, had a critical significance. Two singleton regulatory genes from the ArsR family were found upstream of the operons encoding different arsenic resistance proteins that have no orthologs in the ArsR reference regulons. We propose that these operons are coregulated with upstream regulatory genes and were able to infer a new TF-binding motif by comparison of upstream regions (see Fig. S7 in the supplemental material).
Another interesting example of using a functional criterion is MreC-like singletons. Only two of them (Dde_1200 and Desal_0411) have genes similar to those of MreC reference regulon members. These regulons also contain genes encoding divalent-anion/Na symporter (DASS)-family transporters. Another singleton, Ddes_2092, regulates a transporter of the DASS family but lacks any homologous or functionally related genes shared with the reference MreC regulog. Thus, in this case, the only functional support comes from the first pair of singletons via the DASS-family transporter gene.
In summary, we were able to reconstruct regulons for 33 out of 34 TFs from the Crp/Fnr family, 37 out of 38 TFs from the ArsR family, and 32 out of 53 TFs from the GntR family. Species-specific TFs comprise nearly 35% of all TFs from the Crp/Fnr, ArsR, and GntR families in the Desulfovibrionales. The TF-family-based comparative genomic approach proposed in this study demonstrates that a significant portion of species-specific singleton TF regulons can be reconstructed by simultaneous analysis of the entire TF family in closely related genomes. Two possible applications of the proposed novel approach include (i) computational reconstruction of species-specific regulons and (ii) inference of TF-binding motifs for ubiquitous regulons for subsequent analysis by standard comparative genomics. The similarity of TFBS motifs and the functional roles of target-regulated genes across a whole TF family can be utilized as an additional strong criterion in the regulon reconstruction procedure. Overall, this work lays the foundation for the design of novel TF-family-centric algorithms for the automated and accurate reconstruction of transcriptional regulons.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the Office of Science, Office of Biological and Environmental Research, of the U.S. Department of Energy under contracts DE-AC02-05CH11231 with Lawrence Berkeley National Laboratory (ENIGMA SFA), and DE-SC0004999 with Sanford-Burnham Medical Research Institute and Lawrence Berkeley National Laboratory. D.A.R. was also supported by the Russian Foundation for Basic Research (10-04-01768).
Footnotes
Published ahead of print 19 October 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01977-12.
REFERENCES
- 1. Ravcheev DA, Best AA, Tintle N, Dejongh M, Osterman AL, Novichkov PS, Rodionov DA. 2011. Inference of the transcriptional regulatory network in Staphylococcus aureus by integration of experimental and genomics-based evidence. J. Bacteriol. 193: 3228–3240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rodionov DA. 2007. Comparative genomic reconstruction of transcriptional regulatory networks in bacteria. Chem. Rev. 107: 3467–3497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rodionov DA, Novichkov PS, Stavrovskaya ED, Rodionova IA, Li X, Kazanov MD, Ravcheev DA, Gerasimova AV, Kazakov AE, Kovaleva GY, Permina EA, Laikova ON, Overbeek R, Romine MF, Fredrickson JK, Arkin AP, Dubchak I, Osterman AL, Gelfand MS. 2011. Comparative genomic reconstruction of transcriptional networks controlling central metabolism in the Shewanella genus. BMC Genomics 12(Suppl. 1): S3 doi:10.1186/1471-2164-12-S1-S3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mironov AA, Koonin EV, Roytberg MA, Gelfand MS. 1999. Computer analysis of transcription regulatory patterns in completely sequenced bacterial genomes. Nucleic Acids Res. 27: 2981–2989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alkema WB, Lenhard B, Wasserman WW. 2004. Regulog analysis: detection of conserved regulatory networks across bacteria: application to Staphylococcus aureus. Genome Res. 14: 1362–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Price MN, Dehal PS, Arkin AP. 2008. Horizontal gene transfer and the evolution of transcriptional regulation in Escherichia coli. Genome Biol. 9: R4 doi:10.1186/gb-2008-9-1-r4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Treangen TJ, Rocha EP. 2011. Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genet. 7: e1001284 doi:10.1371/journal.pgen.1001284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sandelin A, Wasserman WW. 2004. Constrained binding site diversity within families of transcription factors enhances pattern discovery bioinformatics. J. Mol. Biol. 338: 207–215 [DOI] [PubMed] [Google Scholar]
- 9. Hertz GZ, Hartzell GW, III, Stormo GD. 1990. Identification of consensus patterns in unaligned DNA sequences known to be functionally related. Comput. Appl. Biosci. 6: 81–92 [DOI] [PubMed] [Google Scholar]
- 10. Kechris KJ, van Zwet E, Bickel PJ, Eisen MB. 2004. Detecting DNA regulatory motifs by incorporating positional trends in information content. Genome Biol. 5: R50 doi:10.1186/gb-2004-5-7-r50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu X, Brutlag DL, Liu JS. 2001. BioProspector: discovering conserved DNA motifs in upstream regulatory regions of co-expressed genes. Pac. Symp. Biocomput. 2001: 127–138 [PubMed] [Google Scholar]
- 12. Hershberg R, Yeger-Lotem E, Margalit H. 2005. Chromosomal organization is shaped by the transcription regulatory network. Trends Genet. 21: 138–142 [DOI] [PubMed] [Google Scholar]
- 13. Sahota G, Stormo GD. 2010. Novel sequence-based method for identifying transcription factor binding sites in prokaryotic genomes. Bioinformatics 26: 2672–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gavrilescu M, Pavel LV, Cretescu I. 2009. Characterization and remediation of soils contaminated with uranium. J. Hazard. Mater. 163: 475–510 [DOI] [PubMed] [Google Scholar]
- 15. Hansen TA. 1994. Metabolism of sulfate-reducing prokaryotes. Antonie Van Leeuwenhoek 66: 165–185 [DOI] [PubMed] [Google Scholar]
- 16. Dehal PS, Joachimiak MP, Price MN, Bates JT, Baumohl JK, Chivian D, Friedland GD, Huang KH, Keller K, Novichkov PS, Dubchak IL, Alm EJ, Arkin AP. 2010. MicrobesOnline: an integrated portal for comparative and functional genomics. Nucleic Acids Res. 38(Suppl 1): D396–D400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vindal V, Suma K, Ranjan A. 2007. GntR family of regulators in Mycobacterium smegmatis: a sequence and structure based characterization. BMC Genomics 8: 289 doi:10.1186/1471-2164-8-289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948 [DOI] [PubMed] [Google Scholar]
- 19. Novichkov PS, Rodionov DA, Stavrovskaya ED, Novichkova ES, Kazakov AE, Gelfand MS, Arkin AP, Mironov AA, Dubchak I. 2010. RegPredict: an integrated system for regulon inference in prokaryotes by comparative genomics approach. Nucleic Acids Res. 38(Suppl 2): W299–W307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rodionov DA, Dubchak I, Arkin A, Alm E, Gelfand MS. 2004. Reconstruction of regulatory and metabolic pathways in metal-reducing delta-proteobacteria. Genome Biol. 5: R90 doi:10.1186/gb-2004-5-11-r90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rodionov DA, Dubchak IL, Arkin AP, Alm EJ, Gelfand MS. 2005. Dissimilatory metabolism of nitrogen oxides in bacteria: comparative reconstruction of transcriptional networks. PLoS Comput. Biol. 1:e55 doi:10.1371/journal.pcbi.0010055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pinchuk GE, Rodionov DA, Yang C, Li X, Osterman AL, Dervyn E, Geydebrekht OV, Reed SB, Romine MF, Collart FR, Scott JH, Fredrickson JK, Beliaev AS. 2009. Genomic reconstruction of Shewanella oneidensis MR-1 metabolism reveals a previously uncharacterized machinery for lactate utilization. Proc. Natl. Acad. Sci. U. S. A. 106: 2874–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee JW, Helmann JD. 2007. Functional specialization within the Fur family of metalloregulators. Biometals 20: 485–499 [DOI] [PubMed] [Google Scholar]
- 24. Nguyen CC, Saier MH., Jr 1995. Phylogenetic, structural and functional analyses of the LacI-GalR family of bacterial transcription factors. FEBS Lett. 377: 98–102 [DOI] [PubMed] [Google Scholar]
- 25. Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res. 14: 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Novichkov PS, Laikova ON, Novichkova ES, Gelfand MS, Arkin AP, Dubchak I, Rodionov DA. 2010. RegPrecise: a database of curated genomic inferences of transcriptional regulatory interactions in prokaryotes. Nucleic Acids Res. 38(Suppl 1): D111–D118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Castillo R, Saier MH., Jr 2010. Functional promiscuity of homologues of the bacterial ArsA ATPases. Int. J. Microbiol. 2010: 187373 doi:10.1155/2010/187373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rigali S, Derouaux A, Giannotta F, Dusart J. 2002. Subdivision of the helix-turn-helix GntR family of bacterial regulators in the FadR, HutC, MocR, and YtrA subfamilies. J. Biol. Chem. 277: 12507–12515 [DOI] [PubMed] [Google Scholar]
- 29. Gebhard S, Cook GM. 2008. Differential regulation of high-affinity phosphate transport systems of Mycobacterium smegmatis: identification of PhnF, a repressor of the phnDCE operon. J. Bacteriol. 190: 1335–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dufour YS, Kiley PJ, Donohue TJ. 2010. Reconstruction of the core and extended regulons of global transcription factors. PLoS Genet. 6: e1001027 doi:10.1371/journal.pgen.1001027 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.