

Abstract
Streptococcus mutans is a commensal member of the healthy plaque biofilm and the primary causative agent of dental caries. The present study is an investigation of SloR, a 25-kDa metalloregulatory protein that modulates genes responsible for S. mutans-induced cariogenesis. Previous studies of SloR homologues in other bacterial pathogens have identified three domains critical to repressor functionality: an N-terminal DNA-binding domain, a central dimerization domain, and a C-terminal FeoA (previously SH3-like) domain. We used site-directed mutagenesis to identify critical amino acid residues within each of these domains of the SloR protein. Select residues were targeted for mutagenesis, and nonconservative amino acid substitutions were introduced by overlap extension PCR. Furthermore, three C-terminally truncated SloR variants were generated using conventional PCR. The repressor functionality and DNA-binding ability of each variant was assessed using CAT reporter gene assays, real-time semiquantitative reverse transcriptase (qRT)-PCR, and electrophoretic mobility shift assays. We identified 12 residues within SloR that cause significant derepression of sloABC promoter activity (P < 0.05) compared to the results for wild-type SloR. Derepression was particularly noteworthy in metal ion-binding site 1 mutants, consistent with the site's importance in gene repression by SloR. In addition, a hyperactive SloR(E169A/Q170A) mutant was identified as having significantly heightened repression of sloABC promoter activity, and experiments with C-terminal deletion mutants support involvement of the FeoA domain in SloR-mediated gene repression. Given these results, we describe the functional domains of the S. mutans SloR protein and propose that the hyperactive mutant could serve as a target for rational drug design aimed at repressing SloR-mediated virulence gene expression.
INTRODUCTION
As a resident member of the healthy plaque biofilm and the principal contributor to dental caries formation in humans, Streptococcus mutans is well adapted to survive in the feast-and-famine conditions of the oral cavity (1). While there are many factors correlated with S. mutans pathogenesis, few play a more crucial role than metal ion homeostasis. Divalent metal ions, such as Fe2+ and Mn2+, serve as important enzyme cofactors and as stabilizing components of proteins in a variety of key metabolic and cariogenic pathways (2, 3). In S. mutans, manganese has been shown to stimulate the conversion of dietary carbohydrates to acid and intracellular polysaccharide storage polymers, both of which are significant contributors to the caries-forming process (4). Moreover, the addition of manganese to drinking water significantly increased caries incidence in rats singly infected with S. mutans (4). Consequently, the sequestering and regulation of metal ions in the highly variable microenvironment of the oral cavity are paramount for the persistence and cariogenicity of S. mutans.
Genome-wide sequence analysis of the S. mutans UA159 wild-type progenitor strain revealed several mechanisms for maintaining desired intracellular metal ion concentrations (5). Ongoing work in our laboratory is focused on SloR, a 25-kDa metalloregulatory protein that has demonstrated roles in both metal ion homeostasis and the regulation of key virulence factors, such as acidogenicity and biofilm formation. SloR has been shown to regulate S. mutans gene expression upon binding to a conserved interrupted palindromic sequence, the SloR recognition element (SRE), in a Mn2+-dependent fashion (6, 7, 8). Bioinformatic analysis has demonstrated a strong correlation between genes that are SloR or Mn2+ responsive and the presence of an SRE consensus sequence upstream from the respective coding sequence (6).
The canonical SRE overlaps the promoter elements that immediately precede the sloABC operon on the S. mutans chromosome, the latter of which coordinate the expression of the downstream sloR gene. Taken together, these genes encode an ATP-binding cassette (ABC) transport system that facilitates S. mutans Fe2+ and Mn2+ acquisition (7). Expression profiling experiments revealed decreased sloABC expression in the presence of elevated levels of Fe2+ or Mn2+ and correlatively increased sloABC expression in the absence of SloR (7, 8). Moreover, SloR binding to the sloABC SRE is supported by the results of in vitro electrophoretic mobility shift assays (EMSAs) (6).
A broader role for SloR in S. mutans-induced cariogenesis is supported by previous research performed in a germfree rat model. We observed a hypercariogenic phenotype in rats that were singly infected with the S. mutans SloR-deficient mutant relative to the phenotype in rats infected with its wild-type progenitor (8). The results of this in vivo study support SloR as a potential target for the design of a therapeutic aimed at alleviating or preventing the onset of dental caries. However, many details about the SloR-SRE binding interaction remain unknown, including the specific amino acid residues that are essential for SloR's overall function and role as a global metalloregulator of S. mutans virulence genes.
In this study, we present a homology model of the S. mutans SloR protein and its proposed interaction with a 22-bp SRE (Fig. 1). We cross-referenced the primary amino acid sequence of SloR with the resolved crystal structures of its DtxR, IdeR, ScaR, and MntR homologues to reveal candidate target amino acid residues that are potentially important for SloR-SRE binding and that reside in each of the three predicted domains of the SloR protein: the N-terminal DNA-binding domain, the central dimerization domain, which houses two metal ion-binding sites, and the C-terminal FeoA domain. Subsequently, a library of SloR mutant strains was generated using overlap extension PCR (OE-PCR) to introduce nonconservative amino acid substitutions at each of the targeted amino acid sites in the SloR coding sequence. We monitored the repressor functionality and DNA-binding ability of the resulting SloR mutant variants in chloramphenicol acetyltransferase (CAT) reporter gene assays, semiquantitative reverse transcriptase (qRT)-PCR experiments, and EMSAs. Our findings elucidate key aspects of the SloR structure-function relationship by characterizing its three functional domains and by identifying the specific amino acid residues that are critical to its role as a metalloregulator.
Fig 1.

Homology model of the S. mutans SloR protein based on the known crystal structure of DtxR from Corynebacterium diphtheriae. Two SloR dimers bind to the 22-bp palindromic SRE sequence on opposite sides of the DNA, forming a double-dimer complex. Shown are the N-terminal DNA-binding domains (DBD) in blue, the dimerization domains (DimD) in red, and the C-terminal FeoA domains (FeoAD) in green. Each SloR monomer contains two metal ion-binding sites to which Mn2+ ions (yellow spheres) can bind. The model predicts that metal ion-binding site 1 is comprised of amino acid residues from the Dim and FeoA domains, whereas residues from DBD and DimD probably contribute to metal ion-binding site 2. Each monomer binds within the major groove of the DNA helix via a winged helix-turn-helix (HTH) motif in the DBD.
MATERIALS AND METHODS
Bacterial strains, plasmids, and primers.
The bacterial strains and plasmids used in this study are described in Table 1. Oligonucleotide primers were purchased from Sigma-Aldrich (St. Louis, MO) and are presented in Table S1 in the supplemental material.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristicsa | Source or reference |
|---|---|---|
| Strains | ||
| Streptococcus mutans | ||
| UA159 | Wild type, serotype c | ATCC 700610 |
| GMS584 | UA159 derived, sloR deficient, Emr | 24 |
| GMS182 | GMS584 transformed with pJRH182, Emr Kmr | This study |
| GMS186 | GMS584 transformed with pJRH186, Emr Kmr | This study |
| Escherichia coli | ||
| DH5α | F− supE44 ΔlacU169 ϕ80dlacZΔM15 hsdR17 recA1 endA1 gyrA96 thi-1 relA | Bethesda Research Laboratories, Gaithersburg, MD |
| XL10 Gold | Tetr Δ(mcrA)183 Δ(mcrCB-hsdSMR-mrr)173 endA1 supE44 thi-1 recA1 gyrA96 relA1 lac hte [F′ proAB lacIqZΔM15Tn10 (Tetr) Amy Camr] | Stratagene |
| Plasmids | ||
| pES-03 | pET101/D-TOPO (Invitrogen) derivative expressing wild-type SloR, Ampr | This study |
| pJL84 | Promoterless chloramphenicol acetyltransferase (cat) reporter plasmid, Kmr Emr | 39 |
| pJRH182 | pJL84 derivative containing a PsloABC-cat fusion | This study |
| pJRH186 | pJL84 derivative containing a PsloR-cat fusion | This study |
| pDL277 | E. coli-streptococcal shuttle vector, Spr | 20 |
| pJRH277 | pDL277 derived, harbors wild-type sloR gene, Spr | This study |
| pJRH277D7A | pJRH277 derivative expressing SloR(D7A) | This study |
| pJRH277S34A | pJRH277 derivative expressing SloR(S34A) | This study |
| pJRH277P36R | pJRH277 derivative expressing SloR(P36R) | This study |
| pJRH277S39A | pJRH277 derivative expressing SloR(S39A) | This study |
| pJRH277E40A | pJRH277 derivative expressing SloR(E40A) | This study |
| pJRH277S34A/S39A/M41V | pJRH277 derivative expressing SloR(S34A/S39A/M41V) | This study |
| pJRH277S34A/P36R/S39A/E40A | pJRH277 derivative expressing SloR(S34A/P36R/S39A/E40A) | This study |
| pJRH277H76A | pJRH277 derivative expressing SloR(H76A) | This study |
| pJRH277E80A | pJRH277 derivative expressing SloR(E80A) | This study |
| pJRH277E99A | pJRH277 derivative expressing SloR(E99A) | This study |
| pJRH277E102A | pJRH277 derivative expressing SloR(E102A) | This study |
| pJRH277H103A | pJRH277 derivative expressing SloR(H103A) | This study |
| pJRH277E99A/E102A/H103A | pJRH277 derivative expressing SloR(E99A/E102A/H103A) | This study |
| pJRH277C123A | pJRH277 derivative expressing SloR(C123A) | This study |
| pJRH277H125A | pJRH277 derivative expressing SloR(H125A) | This study |
| pJRH277D160A | pJRH277 derivative expressing SloR(D160A) | This study |
| pJRH277E169A | pJRH277 derivative expressing SloR(E169A) | This study |
| pJRH277Q170A | pJRH277 derivative expressing SloR(Q170A) | This study |
| pJRH277E169A/Q170A | pJRH277 derivative expressing SloR(E169A/Q170A) | This study |
| pJRH277L179Q | pJRH277 derivative expressing SloR(L179Q) | This study |
| pJRH277ΔR141-Y217 | pDL277 derivative expressing SloR(ΔR141-Y217) | This study |
| pJRH277ΔQ170-Y217 | pDL277 derivative expressing SloR(ΔQ170-Y217) | This study |
| pJRH277ΔL181-Y217 | pDL277 derivative expressing SloR(ΔL181-Y217) | This study |
Emr, erythromycin resistant; Kmr, kanamycin resistant; Ampr, ampicillin resistant; Spr, spectinomycin resistant.
Bacteriological media and growth conditions.
Escherichia coli was grown at 37°C with aeration in Luria medium, and when necessary, kanamycin (100 μg/ml) and/or spectinomycin (100 μg/ml) were included (Sigma). S. mutans was grown at 37°C and 5% CO2 in Todd-Hewitt medium supplemented with 0.3% yeast extract (THYE), and when necessary, erythromycin (10 μg/ml), kanamycin (700 μg/ml), and/or spectinomycin (1,200 μg/ml) were added. For CAT assays and qRT-PCR experiments, S. mutans UA159-, GMS182-, and GMS182-derived strains were grown to mid-logarithmic phase in prewarmed tryptone-vitamin (TV) base medium (9) supplemented with glucose at a final concentration of 1% (wt/vol) as the carbohydrate source.
Purification of the S. mutans SloR protein.
Wild-type SloR was expressed from plasmid pES-03, a derivative of the pET101/D-TOPO expression vector (Invitrogen). Plasmid pES-03 was generated by PCR amplification of the 654-bp sloR open reading frame with Platinum high-fidelity Taq polymerase (Invitrogen) and genomic DNA isolated from S. mutans UA159 according to established protocols (10). The amplicon was cloned into the pET101/D-TOPO vector according to the manufacturer's recommendations (Invitrogen). The cloned sequence was verified by nucleotide sequencing before transforming it into BL21(DE3) E. coli. Expression of SloR was induced by adding 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) to cell cultures that had been grown to mid-logarithmic phase (optical density at 600 nm [OD600] of 0.5) at 37°C in Luria broth. Postinduction incubation continued for 2.5 h at 37°C. Following centrifugation, the cell pellets were resuspended in a 100 mM Tris-HCl (pH 7.5) buffer containing 50 mM NaCl, disrupted by sonication, and centrifuged to remove cellular debris. The supernatant was applied to a DEAE anion exchange column and eluted with a gradient of 100 to 400 mM NaCl in 20 mM Tris-HCl, pH 7.5. Fractions containing SloR were identified on SDS-PAGE gels, pooled, and dialyzed extensively against a 20 mM Tris-HCl (pH 7.5) buffer containing 150 mM NaCl. The dialyzed protein was then loaded onto a heparin column and eluted by applying a gradient of 200 to 1,800 mM NaCl in 20 mM Tris-HCl, pH 7.5. SloR-containing fractions were identified by SDS-PAGE, pooled, concentrated, and dialyzed against a 25 mM HEPES (pH 7.5) buffer containing 200 mM NaCl and 10% glycerol. The purified protein (>90% homogeneity) was flash-frozen in liquid nitrogen and stored at −20°C.
CD spectroscopy.
Circular dichroism (CD) measurements were performed on a Jasco J-815 CD spectrometer with a 0.1-mm path-length cell. Purified SloR samples prepared as described above were dialyzed in a CD buffer containing 5 mM HEPES, 100 mM NaF, and 10% (vol/vol) glycerol in metal-free water at pH 7 using Slide-a-Lyzer mini-dialysis units (Fisher Scientific). Following dialysis, the protein concentration was recorded at OD277 on a Varian Cary 6000i UV-Vis-NIR (UV–visible–near-infrared) spectrometer, and the sample was subsequently stored at −20°C. Spectra were obtained for protein samples maintained at 4°C for 0, 24, and 48 h and compared with spectra obtained at 25°C to assess SloR stability. Separately, manganese was added to sample aliquots at concentrations of 0.1, 1, 10, and 125 μM and incubated for 1 h at either 4°C or 25°C before CD spectra were collected. The CD measurements (175 to 260 nm) were obtained at a time constant of 4 s, bandwidth of 1.00 nm, data pitch of 0.5 nm, and scanning speed of 10 nm/min. Each spectrum generated represents an average of 4 complete scans. The cell was washed thoroughly with metal-free water three times between samples and cleaned daily for 1 h with 2% Helmenex. Several spectra were truncated at approximately 183 nm due to significant noise at the lower wavelengths. Spectra were analyzed by Dichroweb deconvolution for secondary structure estimation as previously described (11–19).
Cloning of the S. mutans sloR gene and construction of C-terminally truncated SloR protein variants.
A 1,084-bp amplicon containing the wild-type sloR promoter and coding sequence was PCR amplified with primers sloR.EcoRI.F and sloR.SacI.R (see Table S1 in the supplemental material) and cloned into the EcoRI and SacI sites of the shuttle vector pDL277 (20). The resulting recombinant plasmid, pJRH277, was confirmed by restriction enzyme digestion, PCR with primers M13F and M13R, and Sanger sequencing on an ABI3130 genetic analyzer with the same M13F and M13R primer set. In addition, three C-terminally truncated SloR variants, SloR(ΔR141-Y217), SloR(ΔQ170-Y217), and SloR(ΔL181-Y217), were PCR amplified from the UA159 chromosome with the appropriate primer pairs (see Table S1) and cloned into the EcoRI and SacI sites of pDL277. The resulting recombinant plasmids, pJRH277ΔR141-Y217, pJRH277ΔQ170-Y217, and pJRH277ΔL181-Y217 (Table 1), were confirmed by restriction enzyme digestion and by Sanger sequencing with primers M13F and M13R.
Site-directed mutagenesis of the wild-type SloR protein.
Mutant variants of the wild-type SloR protein were generated by overlap extension PCR according to the method of Ho et al. (21). Plasmid pJRH277 (Table 1) was isolated and purified on mini-spin columns (Qiagen) and used as a template for PCR amplification. Plasmids bearing single, double, triple, or quadruple mutations in the sloR coding sequence and the degenerate primers used to introduce these mutations are described in Table 1 and in Table S1 in the supplemental material, respectively. We also used conventional PCR to generate three SloR variants with deletions in the C-terminal FeoA domain (Table 1). All mutant variants were cloned into the EcoRI and SacI sites of the pDL277 expression plasmid (Table 1). The resulting recombinant plasmids were confirmed by restriction enzyme digestion and by Sanger sequencing with primers M13F or M13R (see Table S1). The pDL277- and pJRH277-derived plasmids were then used to transform the S. mutans PsloABC-cat fusion strain GMS182 (Table 1) in the presence of competence-stimulating peptide (CSP) according to established protocols (22). Transformants were selected on THYE agar supplemented with spectinomycin and confirmed by cell lysate PCR (23) with primers M13F and M13R.
To confirm that the mutations did not disrupt the secondary structure of SloR, we analyzed the primary amino acid sequence of all 20 SloR mutant variants using the PSIPRED protein structure prediction server (http://bioinf.cs.ucl.ac.uk/psipred/).
Construction of the S. mutans PsloABC-cat fusion strain, GMS182.
To examine the effect of SloR mutations on sloABC expression, the 238-bp promoter region of the sloABC operon was PCR amplified from the S. mutans UA159 genome with primers PsloA.CAT.F and PsloA.CAT.R (see Table S1 in the supplemental material) and directionally cloned into the BamHI and SacI sites immediately upstream from a promoterless chloramphenicol acetyltransferase (cat) gene on plasmid pJL84 (Table 1; also see Fig. S1 in the supplemental material). Cloning was confirmed by a SacI/SphI double digest and by Sanger sequencing with primers phnA-AS and cat.R (see Table S1). The resulting pJRH182 plasmid (Table 1; also see Fig. S1) was then CSP transformed into S. mutans GMS584, an isogenic SloR-deficient mutant strain (24), where it integrated into the chromosome (see Fig. S1). Integration of the PsloABC-cat reporter fusion via double-crossover homologous recombination was confirmed by PCR with primers phnA-AS and cat.R and by Sanger sequencing with primer phnA-AS (see Table S1).
Preparation of whole-cell lysates for CAT assays and EMSAs.
The GMS182 and GMS182-derived PsloABC-cat fusion strains were grown to mid-exponential phase in 30 ml of prewarmed TV broth supplemented with 1% (wt/vol) glucose. Cells were harvested by centrifugation at 8,000 × g for 10 min in a FiberLite F13S-14x50cy rotor and then washed with 30 ml of 10 mM Tris-Cl, pH 7.8. Cells were resuspended in 750 μl of 10 mM Tris-Cl, pH 7.8, transferred to an ice-cold 2-ml screw-cap tube containing 300 μl of 0.1-mm zirconium beads, and mechanically disrupted at 4°C for 30 s at a speed of 5.0 in a BIO 101 FastPrep machine. The tubes were then centrifuged at 18,000 × g for 10 min at 4°C, and whole-cell lysates were collected as supernatants and stored at −20°C.
CAT assays.
CAT specific activity was assayed spectrophotometrically according to the method of Azemun et al. (25). The assay was performed in triplicate for each of three biological replicates (separate lysates) for each bacterial strain, and CAT specific activity was determined with a Spectronic Genesys 2 UV-Vis spectrophotometer (Fisher Scientific). Specifically, a 930-μl reaction mixture containing 100 mM Tris-Cl, pH 7.8, 0.4 mg/ml 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB), and 0.1 mM acetyl-coenzyme A (Sigma) was mixed with 50 μl of whole-cell lysate in each of four disposable polystyrene cuvettes. An amount of 20 μl of 5 mM chloramphenicol (Cm; dissolved in H2O) was added to three test reaction mixtures, whereas no chloramphenicol was added to the fourth reaction mixture, which served as a background control. The absorbance of each reaction mixture at OD412 (t = 0 min) was measured, after which the reaction mixtures were incubated at 37°C for 2 min. After incubation, absorbance was measured again at OD412 (t = 2 min). To determine the net absorbance change due to CAT specific activity, the absorbance change of the background cuvette was subtracted from the average absorbance change of the three technical replicates. To determine the rate of the Cm-dependent DTNB reaction in nmoles/min, the net absorbance over 2 min was divided by 0.0136. To express the CAT specific activity as a function of total protein concentration, the calculated rate was divided by the total protein concentration, which was determined with a Pierce BCA (bicinchonic acid) assay kit according to the recommendations of the supplier (Fisher Scientific). CAT specific activity was expressed in nanomoles of chloramphenicol acetylated per min per mg of total protein.
EMSAs.
EMSAs were carried out according to a previously described protocol (6, 24). A 364-bp DNA amplicon containing the sloABC promoter was PCR amplified with primers sloA.F and sloA.R (see Table S1 in the supplemental material) and purified using a QIAquick PCR purification kit (Qiagen) according to the recommendations of the supplier. The purified PCR products were end labeled with 10 μCi [γ-32P]ATP using 10 U T4 polynucleotide kinase (New England BioLabs). The end-labeling reaction mixture was incubated at 37°C for 30 min and then at 70°C for 20 min before the final volume was brought to 100 μl with sterile distilled H2O. The samples were passed through a TE Select-D G-25 spin column (Roche Applied Science) to remove unincorporated radiolabel. Binding reactions were performed in a 16-μl total volume containing 1 μl of end-labeled sloABC promoter DNA (the equivalent of 38 picograms), whole-cell lysates containing 6.5 μg of total protein prepared from the GMS182 fusion strain and its derivatives as described above, and 3.2 μl of 5× binding buffer (42 mM NaH2PO4, 58 mM Na2HPO4, 250 mM NaCl, 25 mM MgCl2, 50 μg/ml bovine serum albumin, 1 mg sonicated salmon sperm DNA, 50% [vol/vol] glycerol, and 37.5 μM MnCl2). EDTA was added to select reaction mixtures at a final concentration of 15 mM to determine whether SloR binding was metal ion dependent. After incubation at room temperature for 20 min, the samples were resolved on 6% nondenaturing polyacrylamide gels (3 ml 20× bis-Tris borate [pH 7.4], 74 μl 100 mM MnCl2, 1.5 ml 100% glycerol, 43 ml Millipore H2O, 12 ml 30% acrylamide [37.5:1 acrylamide-bis], 300 μl 15% ammonium persulfate [APS], and 90 μl TEMED [N,N,N′,N′-tetramethylethylenediamine]) for 70 min at 285 V. Gels were exposed to Kodak BioMax film for up to 48 h at −80°C in the presence of an intensifying screen. The band shifts were quantified using Kodak 1D Image Analysis software, version 3.6, and expressed as a percentage of the band shift associated with wild-type SloR binding.
RNA isolation and cDNA synthesis.
Total RNA was isolated from S. mutans by following a modified protocol from Chen et al. (26). Briefly, 14-ml cultures of mid-logarithmic-phase S. mutans UA159 and its mutant SloR(P36R), SloR(E80A), SloR(H103A), and SloR(E169A/Q170A) derivatives were centrifuged at 11,000 × g for 5 min prior to resuspending the cell pellet in RNAprotect bacterial reagent (Qiagen) according to the instructions provided by the manufacturer. Cultures were centrifuged again as described above and resuspended in 250 μl of 50 mM Tris–10 mM EDTA buffer. The cell suspensions were then transferred to separate lysing matrix B tubes (MP Biomedicals) containing 10 μl of 10% SDS and 300 μl of acid phenol, placed in a Bio101 FastPrep mechanical disrupter, and subjected to three rounds of disruption with intermittent chilling on ice. The cell slurry was centrifuged at 17,000 × g and 4°C in an AccuSpin micro-17 centrifuge (Fisher Scientific), after which 200 μl of the RNA-containing supernatant was purified and DNase I treated on an RNeasy column (Qiagen) according to the manufacturer's instructions. Total RNA was examined for integrity on a 1% agarose gel and adjusted to a final concentration of 100 ng/μl using a NanoDrop lite spectrophotometer (Thermo Fisher Scientific, Waltham, MA). The total intact RNAs were then reverse transcribed using a Revert-Aid first-strand cDNA synthesis kit according to the manufacturer's instructions (MBI Fermentas). Reaction mixtures without RNA or without reverse transcriptase served as controls.
Real-time qRT-PCR.
Real-time semiquantitative reverse transcriptase PCR (qRT-PCR) was performed with ∼5 ng/μl single-stranded cDNA. The cDNA was reverse transcribed from total intact RNA isolated from S. mutans UA159, GMS182, and its isogenic SloR(P36R), SloR(E80A), SloR(H103A), and SloR(E169A/Q170A) variants. Amplification was performed in a CFX96 real-time system (Bio-Rad) with EvaGreen as the intercalating dye (Bio-Rad) and 500 nM sloA, hk11, or 16S rRNA primers (see Table S1 in the supplemental material). Controls without RT were run in parallel with the same primers for each cDNA template to confirm the absence of contaminating genomic DNA. The thermal cycling program was set as follows: 95°C for 3 min followed by 40 cycles of 95°C for 10 s, 60°C for 10 s, and 72°C for 15 s. Primer efficiency was calculated using a standard curve with various concentrations of UA159 cDNA. Gene expression was normalized against the expression of endogenous control genes (either hk11 or 16SrRNA), whose expression did not change under the experimental test conditions.
RESULTS AND DISCUSSION
The SloR protein is stable at various temperatures and manganese concentrations.
Circular dichroism spectroscopy of the S. mutans SloR protein and its analysis using Dichroweb deconvolution demonstrate that the protein is stable at both 4°C and 25°C (data not shown). Furthermore, various concentrations of manganese from 0.1 to 125 μM did not significantly alter the wild-type SloR spectrum shown in Figure 2, suggesting that the proposed conformational shift in the SloR protein upon metal ion binding does not involve changes in secondary structure. This observation is consistent with previous observations with DtxR, a SloR homologue for which no detectable changes in secondary structure were observed upon metal ion binding (27). The double minima around 220 and 210 nm (Fig. 2) are characteristic of the α-helical content of the SloR protein, and the relatively small amplitude of both the maximum and minimum are characteristic of β-sheets (16).
Fig 2.

Circular dichroism spectroscopy of the wild-type SloR protein. The double minima occurring at 220 nm and 210 nm are characteristic of alpha-helical regions, while the relatively low amplitude of the maxima and minima is characteristic of beta-sheet regions. The spectrum is reported in delta epsilons, or per-residue molar absorption units. SloR ≈ 1 mg/ml buffer (5 mM HEPES [pH 7], 100 mM NaF, 10% [vol/vol] glycerol); temperature, 25°C; path length, 0.1 mm.
The S. mutans SloR protein represses sloABC promoter activity.
To monitor sloABC promoter activity that is subject to SloR control, we cloned the sloABC operon promoter immediately upstream of a promoterless cat gene that is resident on plasmid pJL84 (Table 1). The results of CAT assays revealed PsloABC-cat activity that was 64-fold greater in the absence of wild-type SloR than in its presence (Fig. 3; also see Table S2 in the supplemental material), consistent with previously observed sloABC repression by SloR in qRT-PCR experiments (6, 24). Interestingly, sloR expression that is driven by a second promoter located in the intergenic region between sloC and sloR is not subject to SloR autoregulation (see Table S2). The results of EMSAs support SloR-dependent shifting of the SRE-containing sloABC amplicon that is Mn2+ dependent, given that the EDTA metal ion chelator abrogated the band shift (Fig. 4 and 5).
Fig 3.

Effects of site-specific mutations in the predicted SloR DNA-binding domain (DBD), dimerization domain (DimD), and FeoA domain (FeoAD) on sloABC promoter activity. Shown are the results of CAT assays in the presence of wild-type SloR and its mutant variants expressed in trans. The means ± standard deviations (error bars) from each of three independent experiments, each performed in triplicate, are represented. All of the SloR variants with the exception of SloR(D7A), SloR(D160A), SloR(E169A), SloR(Q170A), and SloR(E169A/Q170A) were significantly compromised in their ability to repress sloABC promoter activity, as indicated by CAT activity that was significantly derepressed compared to that of wild-type (w/t) SloR (*, P < 0.05; Student's t test). Interestingly, the SloR(E169A/Q170A) variant was a significantly better repressor of sloABC promoter activity than wild-type SloR (*, P < 0.05; Student's t test) (see inset).
Fig 4.

Binding of wild-type (w/t) SloR and its metal ion-binding site 2 mutant variants to the sloABC promoter in EMSA. EMSAs were performed with a [γ-32P]ATP end-labeled 364-bp sloABC promoter-containing amplicon (the equivalent of 38 picograms) and whole-cell lysates containing 6.5 μg of total protein prepared from the GMS182 fusion strain and its derivatives. The shift in the lane without SloR (SloR−) is likely the result of nonspecific interactions between the target DNA and proteins present in the whole-cell lysate. This level of background was accounted for in subsequent quantitative analyses. When appropriate, EDTA was added at a final concentration of 15 mM. Reaction mixtures were run on 6% nondenaturing polyacrylamide gels and exposed for up to 48 h at −80°C in the presence of an intensifying screen. All of the metal ion-binding site 2 variants with the exception of SloR(E99A) were 30% to 90% compromised in their ability to bind DNA compared to wild-type SloR's binding, consistent with the predicted role for site 2 in activating DNA binding.
Fig 5.

Binding of wild-type SloR and its C-terminally truncated mutant variants to the sloABC promoter in EMSA. EMSAs were performed with a [γ-32P]ATP end-labeled 364-bp sloABC promoter-containing amplicon (the equivalent of 38 picograms) and whole-cell lysates containing 6.5 μg of total protein prepared from the GMS182 fusion strain and its derivatives. When appropriate, EDTA was added at a final concentration of 15 mM. Reaction mixtures were run on 6% nondenaturing polyacrylamide gels and exposed for up to 48 h at −80°C in the presence of an intensifying screen. All three SloR deletion variants demonstrate reduced DNA binding (<40%) compared to wild-type SloR's binding. However, all retain some baseline affinity for DNA binding, suggesting that the FeoA domain, while important for DNA binding, does not appear to be a requirement for DNA binding.
Mutagenesis of the SloR N terminus supports its involvement in DNA binding.
To determine the impact of nonconservative amino acid substitutions in the predicted DNA-binding domain (DBD) of SloR on sloABC promoter activity, we performed CAT assays, real-time qRT-PCR experiments, and/or EMSAs with the SloR(S34A), SloR(P36R), SloR(S39A), SloR(E40A), SloR(S34A/S39A/M41V), and SloR(S34A/P36R/S39A/E40A) mutant variants alongside the SloR wild-type protein. All six variants demonstrated significant derepression (P < 0.01) of CAT activity compared to its repression by wild-type SloR (Fig. 3), although the derepression did not reach the level observed in the complete absence of SloR. Given that the targeted amino acids are predicted to interact with conserved SRE nucleotides according to our homology model, we hypothesize that the derepression observed in CAT reporter gene studies reflects a weakened ability of the SloR protein variants to recognize and bind specific bases in the SRE sequence. Consistent with this hypothesis are the results of qRT-PCR experiments that confirmed derepression of select DBD mutant variants of up to 5-fold and EMSA experiments that demonstrated compromised band shifts (between 20 and 75% of the results for the wild type) for five of the six variants, with the exception of SloR(S39A) (see Fig. S2 in the supplemental material). Despite the limitations of having used whole-cell lysates in the EMSAs, the results we report here are robust, because the SloR-deficient cell lysates generally did not give rise to a band shift and the band shifts were fully abolished upon the addition of EDTA, consistent with the metal ion dependency of SloR-specific DNA binding. To minimize the binding of nonspecific proteins to the labeled target DNA in these assays, we also included sonicated salmon sperm DNA as a nonspecific competitor. Taking these results together, we propose that Ser34, Pro36, Ser39, and Glu40 are all involved in SloR recognition of and binding to the SRE palindrome that is located upstream and proximal to the sloABC promoter, as nonconservative mutagenesis of these residues partially inactivated the DNA-binding or repressor function of SloR.
Reports in the literature describe both DtxR and IdeR as making only three base-specific contacts with the DNA backbone (28, 29). Therefore, it is likely that single nonconservative mutations in residues that bind specific SRE nucleotides would significantly compromise the DNA recognition ability of SloR. Our CAT assay results support this hypothesis, as we observed significant derepression of sloABC promoter activity for all six SloR variants with mutations in the DBD (Fig. 3). Interestingly, disruption of base-specific contacts within the SloR protein did not affect SloR's ability to bind DNA for all variants, as the band shift associated with SloR(S39A) binding to DNA shared nearly equal intensity (91.5%) with that of wild-type SloR binding in EMSA experiments (see Fig. S2 in the supplemental material). X-ray crystallography studies are under way with SloR wild-type and mutant variants in the presence of duplex DNA to validate the involvement of Ser34, Pro36, Ser39, and Glu40 in SloR SRE recognition and binding. In addition, ongoing experiments with mutant variants of the canonical S. mutans SRE will help reveal specific nucleotides within the predicted SRE that interact with the DNA-binding domain of SloR.
Mutagenesis of metal ion-binding site 1 in the SloR dimerization domain supports relatively low-affinity manganese binding.
Metal ion-binding site 1 has been previously described as a high-affinity metal ion-binding site that plays a role in protein stabilization and dimerization of DtxR-like metalloregulators (30, 31). Both DtxR and IdeR possess a pentavalent metal ion-binding site 1 (called the ancillary site), whereas ScaR harbors a nonanalogous, structurally distinct tetravalent metal ion-binding site 1 (called the secondary site) (see Table S3 in the supplemental material) (32). Interestingly, alignment of SloR's primary amino acid sequence with those of its homologues reveals that it shares analogous residues at both the DtxR/IdeR “ancillary” site 1 and the ScaR “secondary” site 1 (see Table S3). That is, SloR harbors the same four secondary-site residues as ScaR while also possessing residues analogous to those of the DtxR and IdeR ancillary sites. Based on the sequence alignment we present in Figure 6 and the homology model shown in Figure 1, we predict that metal ion-binding site 1 in the S. mutans SloR protein is structurally similar to the secondary site of ScaR in Streptococcus gordonii, sharing three residues in common in the dimerization domain (Glu80, Cys123, and His125) and one residue in the FeoA domain (Asp160).
Fig 6.
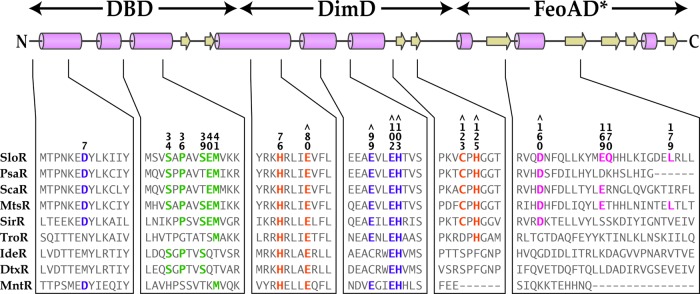
Sequence alignment of the S. mutans SloR protein with members of the DtxR family of metalloregulators. The predicted secondary structure of SloR is shown above the primary amino acid sequence, with the proposed positions of the DNA-binding domain (DBD), dimerization domain (DimD), and FeoA domain (FeoAD*) designated. Cylinders represent alpha helices, and arrows beta strands. Residues selected for mutagenesis are numbered and colored according to domain prediction: DBD residues (green), DimD residues in metal ion-binding site 1 (orange), DimD residues in metal ion-binding site 2 (purple), and residues in the FeoA/SH3-like domain (pink). Residues that we believe contribute functionally to metal ion-binding sites 1 and 2 in the SloR protein are designated by the symbol ⋀.
To determine the potential impact of nonconservative amino acid substitutions in the predicted metal ion-binding site 1 of SloR on sloABC expression and SRE binding, we performed CAT assays, qRT-PCR experiments, and/or EMSAs with the SloR(H76A), SloR(E80A), SloR(C123A), and SloR(H125A) mutant variants. SloR(H76A) and SloR(E80A) demonstrated the highest levels of derepression in CAT assays among all SloR variants tested (Fig. 3). Furthermore, the mutants demonstrated up to 4.5-fold derepression in qRT-PCR experiments, substantially attenuating the repressor functionality of SloR (data not shown). Although the results of EMSA experiments demonstrated compromised DNA binding for SloR(H76A) and SloR(E80A) (57% and 68% of the results for the wild type, respectively), DNA binding was not completely abrogated (see Fig. S3 in the supplemental material). Interestingly, these findings are unlike those deriving from mutational analysis of metal-binding site 1 in the DtxR and IdeR proteins, which required two alanine substitutions to abolish the repressor function of these metalloregulators (31, 33). In addition, consistent metal ion occupation at site 1 in ScaR crystallography studies support this site as having a high affinity for Mn2+ (32). In contrast, our results suggest that SloR metal ion-binding site 1 has a relatively low affinity for metal ion-binding, requiring only one alanine substitution to abolish repressor function. Given the high levels of derepression that we observed for SloR(E80A) in CAT and qRT-PCR assays, we predict that this residue likely plays a critical role in Mn2+ binding at site 1 of SloR.
Although His76 in ScaR does not appear to be appropriately positioned to bind metal (32), it is positioned to interact with residues in both metal-binding sites 1 and 2, suggesting that it may play a role in mediating interactions between the two metal ion-binding sites of the ScaR protein (32). While mutational analysis of the ScaR protein has not been reported, our results reveal the importance of His76 in SloR repressor functionality, supporting its proposed role in mediating metal ion binding at both sites.
SloR(C123A) and SloR(H125A) also demonstrated significant derepression of sloABC promoter activity (P < 0.01) in CAT assays (Fig. 3) but not to the same degree as SloR(H76A) and SloR(E80A). The results of EMSA experiments support a 15% reduction in SRE binding by SloR(C123A) and SloR(H125A) compared to that of wild-type SloR (see Fig. S3 in the supplemental material). In contrast, an alanine substitution at SloR residue Asp160 (which binds metal at site 1 in ScaR) had no effect on SloR SRE binding or on sloABC promoter activity (Fig. 3; also see Fig. S4 in the supplemental material), suggesting that SloR might rely on different FeoA domain residues for coordinating metal ion binding at site 1 than ScaR. In addition, alanine substitutions at SloR residues Glu169 and Gln170 had no significant impact on the repressor functionality of SloR (Fig. 3), supporting the hypothesis that SloR does not possess an ancillary metal ion-binding site analogous to that of DtxR and IdeR (see Table S3 in the supplemental material). Based on these results, we propose that metal ion-binding site 1 in the SloR protein is similar to site 1 in ScaR, consisting of residues Glu80, Cys123, His125, and a potential fourth residue from the FeoA domain. In addition, we propose that His76 may play a crucial role in mediating metal ion binding at both metal ion-binding sites in SloR.
Mutagenesis of metal ion-binding site 2 in the SloR dimerization domain supports its role in DNA binding.
Metal ion-binding site 2, the primary site, has been previously described as a low-affinity metal ion-binding site that plays a role in DNA binding in other DtxR-like proteins (the second step of repressor activation) (30, 31). The amino acid residues at site 2 are conserved across members of the DtxR family (see Table S3 in the supplemental material), and previous mutational analysis of DtxR has shown that introducing nonconservative amino acid substitutions at the primary site gives rise to a nonfunctional repressor protein (33).
We observed significant derepression (P < 0.05) of sloABC promoter activity in CAT assays for each SloR variant bearing a mutation in metal ion-binding site 2 compared to the repression by wild-type SloR, with the exception of the SloR(D7A) variant (Fig. 3). Moreover, the results of qRT-PCR experiments confirm derepression of variants with mutations in metal ion-binding site 2 by at least 5-fold compared to the results for wild-type SloR. In EMSA experiments, we observed compromised DNA binding (as low as 30% of the results for the wild type) for each SloR variant, with the exception of SloR(E99A) (Fig. 4), consistent with the predicted role for site 2 in activating DNA binding. Given these results, we hypothesize that SloR's metal ion-binding site 2 consists of residues Glu99, Glu102, and His103, along with another possible residue from the α1 helix in the DNA-binding domain. Interestingly, the addition of EDTA to the EMSA binding reaction mixtures relieved DNA binding for all five SloR site 2 variants (Fig. 4), suggesting retention of Mn2+ binding at site 1. Based on our results and previous observations with other SloR homologues, we propose that Mn2+ binding at site 2 in the S. mutans SloR protein activates the conformational changes that are necessary for optimal SloR DNA binding. However, the fact that SloR variants bearing nonconservative amino acid substitutions at site 2 still bind DNA in a metal ion-dependent manner suggests that baseline levels of DNA binding can occur if only site 1 is metal ion occupied.
Mutagenesis of the C-terminal FeoA domain supports its involvement in metal ion binding at site 1.
The C-terminal SH3-like domain (referred to as the FeoA domain in ScaR and SloR) is a small, highly flexible domain present in select members of the DtxR family of metalloregulators that plays a role in coordinating metal ion binding at site 1 (32, 34, 35, 36). To reveal which residues in the FeoA domain of SloR contribute to its ability to bind metal at metal ion-binding site 1, we generated nonconservative alanine substitutions at residues Asp160, Glu169, and Gln170. In addition, we made a nonconservative substitution at the hydrophobic residue Leu179 (L179Q), which is analogous to the Phe179 residue in DtxR shown previously to interact with Phe128, a highly conserved residue in the dimerization domain (28).
We did not observe significant derepression (P < 0.05) of CAT activity for any of the tested SloR variants with mutations in the FeoA domain, with the exception of SloR(L179Q) (Fig. 3). Consistent with this finding are the results of EMSA experiments (see Fig. S4 in the supplemental material) that supported DNA binding nearly equal to or greater than that of wild-type SloR for all of the FeoA mutant variants except SloR(L179Q), which demonstrated reduced SRE binding (45% of the wild-type binding). These results, in conjunction with the SloR homology model presented in Figure 1, suggest that Leu179 in the S. mutans SloR protein may play an important role in stabilizing the FeoA domain by interacting with the dimerization domain. The lack of derepression observed for the SloR(E169A), SloR(Q170A), and SloR(E169A/Q170A) variants (Fig. 3) suggests that SloR probably does not contain an ancillary metal ion-binding site like that which is present in DtxR and IdeR (see Table S3 in the supplemental material). However, because mutations at Asp160 (conserved in ScaR) likewise did not result in derepression (Fig. 3), we hypothesize that SloR may be using different FeoA domain residues than those used by ScaR to coordinate Mn2+ binding at its secondary site (site 1). X-ray crystallography of the SloR protein, along with further mutational analysis of potentially critical FeoA domain residues, is necessary to reveal which residues, if any, bind Mn2+ at metal ion-binding site 1.
Interestingly, the double variant SloR(E169A/Q170A) gave rise to a hyperactive SloR repressor, demonstrating significantly heightened repression of PsloABC-CAT activity in CAT assays (P < 0.05) (Fig. 3, inset) and up to 8-fold repression of sloABC transcription compared to that of the wild type in qRT-PCR experiments (data not shown). Consistent with these findings was the ability of SloR(E169A/Q170A) to bind DNA with affinity equal to that of wild-type SloR in EMSA experiments (see Fig. S4 in the supplemental material). The identification of such a hyperactive SloR variant could provide insight into the role of SloR in S. mutans virulence gene expression. Previous studies in a germfree rat model demonstrated hypercariogenicity of an S. mutans SloR-deficient strain (8), consistent with SloR functioning as a repressor of key genes associated with cariogenesis. In accordance with these observations, we predict a hypocariogenic phenotype for S. mutans strains expressing the hyperactive SloR(E169A/Q170A) mutant. A similar hyperactive DtxR variant with a nonconservative mutation at its C terminus, called DtxR(E175K), was described previously (37). In fact, mice infected with Mycobacterium tuberculosis strains expressing the DtxR(E175K) mutant variant showed reduced signs of tuberculosis infection (38). We predict that the hyperactive SloR(E169A/Q170A) variant may similarly reduce the cariogenic potential of S. mutans by widespread silencing of key virulence genes in the S. mutans genome. These in vivo studies are under way.
C-terminal deletions in the FeoA domain support its involvement in SloR-mediated gene repression.
To determine the potential role of the C terminus in SloR repressor function, we performed CAT assays and EMSAs with the C-terminally truncated mutants SloR(ΔR141-Y217), SloR(ΔQ170-Y217), and SloR(ΔL181-Y217). All three of these deletion variants demonstrated significant derepression (P < 0.05) of sloABC promoter activity in CAT assays (Fig. 3), as well as reduced DNA binding in EMSA experiments (<40% compared to wild-type SloR binding) (Fig. 5). Interestingly, SloR(ΔR141-Y217), which lacks the entire C-terminal FeoA domain, demonstrated higher levels of derepression than the other truncated variants [SloR(ΔQ170-Y217) and SloR(ΔL181-Y217)]. This supports an important role in SloR repressor functionality for residues located between R141 and Q170. Persistent dimerization of the truncated SloR proteins was implicated in EMSA experiments, as all three variants retained some baseline affinity for DNA binding (Fig. 5). From this, we concluded that the FeoA domain, while important for the DNA-binding ability and transcriptional repression of SloR, does not appear to be a requirement for SloR dimerization or DNA binding.
Previous studies with a two-domain (2D) IdeR protein (also lacking a C-terminal domain) revealed similar results (36), with 2D-IdeR demonstrating the ability to dimerize and bind DNA. Wisedchaisri et al. (36) suggest that the C-terminal domain of IdeR plays a role in stabilizing the monomeric form of the repressor and its closely related homologue DtxR. The results of the present study with both full-length SloR and 2D-SloR support a role for the FeoA domain in stabilizing the monomeric form of SloR in the absence of Mn2+. We predict that by deleting the FeoA domain, SloR's affinity for Mn2+ is weakened at metal-binding site 1. However, because the truncated variants did not repress sloABC promoter activity to the same degree as wild-type SloR (Fig. 3), we propose that the FeoA domain is critical for the full repressor function of SloR. Hence, when sufficient Mn2+ is available to bind SloR, the FeoA domain coordinates metal ion binding at site 1, thereby activating the repressor by facilitating dimerization and DNA binding.
Summary.
The present study provides a strong foundation for characterizing the three functional domains of the S. mutans SloR metalloregulatory protein. The results of expression profiling experiments and EMSAs reveal specific amino acid residues within each SloR domain that are essential for SloR repression of sloABC transcription and/or SloR DNA binding. Furthermore, analysis of C-terminally truncated SloR variants underscores the importance of the FeoA domain in SloR repressor functionality and is consistent with the importance of the C-terminal domain in other SloR homologues (32, 34, 35, 36). The identification of a hyperactive mutant is an exciting finding given its potential value for rational drug design.
Further site-directed mutagenesis experiments with the SloR protein will help elucidate other amino acid residues that may be involved in Mn2+ binding and SloR activation. In addition, crystallography studies to resolve the structure of the native wild-type and mutant apo-/holo-SloR proteins are under way to help reveal important residues involved in the DNA binding, dimerization, and metal ion binding of this global metalloregulator of S. mutans virulence attributes.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by funds from the National Institutes of Health (NIH), grant R01 DE014711 to G.A.S., and from the Middlebury College Department of Biology.
We thank Lin Zeng at the University of Florida College of Dentistry for his advice in optimizing the CAT assay, Arthur Glasfeld at Reed College and Christopher Matteri at Middlebury College for their helpful discussions, Gary Nelson for figure preparation, and Frank Spatafora for technical assistance.
Footnotes
Published ahead of print 26 October 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01648-12.
REFERENCES
- 1. Carlsson J. 1983. Regulation of sugar metabolism in relation to feast-and-famine existence of plaque, p 205–211 In Guggenheim B. (ed), Cariology today. Karger, Basel, Switzerland [Google Scholar]
- 2. Rees DC. 2002. Great metalloclusters in enzymology. Annu. Rev. Biochem. 71: 221–246 [DOI] [PubMed] [Google Scholar]
- 3. Reyes-Caballero H, Campanello GC, Giedroc DP. 2011. Metalloregulatory proteins: metal selectivity and allosteric switching. Biophys. Chem. 156: 103–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beighton D. 1982. The influence of manganese on carbohydrate metabolism and caries induction by Streptococcus mutans strain Ingbritt. Caries Res. 16: 189–192 [DOI] [PubMed] [Google Scholar]
- 5. Ajdić D, McShan WM, McLaughlin RE, Savić G, Chang J, Carson MB, Primeaux C, Tian R, Kenton S, Jia H, Lin S, Qian Y, Li S, Zhu H, Najar J, Lai J, White J, Roe BA, Ferretti JJ. 2002. Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proc. Natl. Acad. Sci. U. S. A. 99: 14434–14439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. O'Rourke KP, Shaw JD, Pesesky MW, Cook BT, Roberts S, Pond JP, Spatafora G. 2010. Genome-wide characterization of the SloR metalloregulome in Streptococcus mutans. J. Bacteriol. 192: 1433–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Paik S, Brown A, Munro CL, Cornelissen CN, Kitten T. 2003. The sloABCR operon of Streptococcus mutans encodes a Mn and Fe transport system required for endocarditis virulence and its Mn-dependent repressor. J. Bacteriol. 185: 5967–5975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spatafora G, Moore M, Landgren S, Stonehouse E, Michalek S. 2001. Expression of Streptococcus mutans fimA is iron-responsive and regulated by a DtxR homologue. Microbiology 147: 1559–1610 [DOI] [PubMed] [Google Scholar]
- 9. Burne RA, Wen ZT, Chen YM, Penders JE. 1999. Regulation of expression of the fructan hydrolase gene of Streptococcus mutans GS-5 by induction and carbon catabolite repression. J. Bacteriol. 181: 2863–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen YM, Clancy KA, Burne RA. 1996. Streptococcus salivarius urease: genetic and biochemical characterization and expression in a dental plaque streptococcus. Infect. Immun. 64: 585–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Compton LA, Johnson WC. 1986. Analysis of protein circular dichroism spectra for secondary structure using a simple matrix multiplication. Anal. Biochem. 155: 155–167 [DOI] [PubMed] [Google Scholar]
- 12. Manavalan P, Johnson WC. 1987. Variable selection method improves the prediction of protein secondary structure from circular dichroism spectra. Anal. Biochem. 167: 76–85 [DOI] [PubMed] [Google Scholar]
- 13. Provencher SW, Glockner J. 1981. Estimation of globular protein secondary structure from circular dichroism. Biochemistry 20: 33–37 [DOI] [PubMed] [Google Scholar]
- 14. Sreerama N, Woody RW. 2000. Estimation of protein secondary structure from CD spectra: comparison of CONTIN, SELCON and CDSSTR methods with an expanded reference set. Anal. Biochem. 287: 252–260 [DOI] [PubMed] [Google Scholar]
- 15. Sreerama N, Woody RW. 1993. A self-consistent method for the analysis of protein secondary structure from circular dichroism. Anal. Biochem. 209: 32–44 [DOI] [PubMed] [Google Scholar]
- 16. Sreerama N, Venyaminov SY, Woody RW. 1999. Estimation of the number of helical and strand segments in proteins using CD spectroscopy. Protein. Sci. 8: 370–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Stokkum IHM, Spoelder HJW, Bloemendal M, Van Grondelle R, Groen FCA. 1990. Estimation of protein secondary structure and error analysis from CD spectra. Anal. Biochem. 191: 110–118 [DOI] [PubMed] [Google Scholar]
- 18. Whitmore L, Wallace BA. 2004. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32: W668–W673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Whitmore L, Wallace BA. 2008. Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89: 392–400 [DOI] [PubMed] [Google Scholar]
- 20. LeBlanc DJ, Lee LN, Abu-Al-Jaibat A. 1992. Molecular, genetic, and functional analysis of the basic replicon of pVA380-1, a plasmid of oral streptococcal origin. Plasmid 28: 130–145 [DOI] [PubMed] [Google Scholar]
- 21. Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77: 51–59 [DOI] [PubMed] [Google Scholar]
- 22. Li YH, Lau PCY, Lee JH, Ellen RP, Cvitkovitch DG. 2001. Natural genetic transformation of Streptococcus mutans growing in biofilms. J. Bacteriol. 183: 897–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hynes WL, Ferretti JJ, Gilmore MS, Segarra RA. 1992. PCR amplification of streptococcal DNA using crude cell lysates. FEMS Microbiol. Lett. 94: 139–142 [DOI] [PubMed] [Google Scholar]
- 24. Rolerson E, Swick A, Newlon L, Palmer C, Pan Y, Keeshan B, Spatafora G. 2006. The SloR/Dlg metalloregulator modulates Streptococcus mutans virulence gene expression. J. Bacteriol. 188: 5033–5044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Azemun P, Stull T, Roberts M, Smith AL. 1981. Rapid detection of chloramphenicol resistance in Haemophilus influenzae. Antimicrob. Agents Chemother. 20: 168–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen YM, Weaver CA, Mendelsohn DR, Burne RA. 1998. Transcriptional regulation of the Streptococcus salivarius 57.I urease operon. J. Bacteriol. 180: 5769–5775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Twigg PD, Parthasarathy G, Guerrero L, Logan TM, Caspar DLD. 2001. Disordered to ordered folding in the regulation of diphtheria toxin repressor activity. Proc. Natl. Acad. Sci. U. S. A. 98: 11259–11264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pohl E, Holmes RK, Hol WG. 1999. Crystal structure complex of a cobalt-activated diphtheria toxin repressor-DNA complex reveals a metal-binding SH3-like domain. J. Mol. Biol. 292: 653–667 [DOI] [PubMed] [Google Scholar]
- 29. Wisedchaisri G, Holmes RK, Hol WG. 2004. Crystal structure of an IdeR-DNA complex reveals a conformational change in activated IdeR for base-specific interactions. J. Mol. Biol. 342: 1155–1169 [DOI] [PubMed] [Google Scholar]
- 30. D'Aquino JA, Tetenbaum-Novatt J, White A, Berkovitch F, Ringe D. 2005. Mechanism of metal ion activation of the diphtheria toxin repressor DtxR. Proc. Natl. Acad. Sci. U. S. A. 102: 18408–18413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chou CJ, Wisedchaisri G, Monfeli RR, Oram DM, Holmes RK, Hol WG, Beeson C. 2004. Functional studies of the Mycobacterium tuberculosis iron-dependent regulator. J. Biol. Chem. 279: 53554–53561 [DOI] [PubMed] [Google Scholar]
- 32. Stoll KE, Draper WE, Kliegman JI, Golynskiy MV, Brew-Appiah RA, Phillips RK, Brown HK, Breyer WA, Jakubovics NS, Jenkinson HF, Brennan RG, Cohen SM, Glasfeld A. 2009. Characterization and structure of the manganese-responsive transcriptional regulator ScaR. Biochem. 48: 10308–10320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ding X, Zeng H, Schiering N, Ringe D, Murphy JR. 1996. Identification of the primary metal ion-activation sites of the diphtheria tox repressor by X-ray crystallography and site-directed mutational analysis. Nat. Struct. Biol. 3: 382–387 [DOI] [PubMed] [Google Scholar]
- 34. Love JF, VanderSpek JC, Murphy JR. 2003. The src homology 3-like domain of the diphtheria toxin repressor (DtxR) modulates repressor activation through interaction with the ancillary metal ion-binding site. J. Bacteriol. 185: 2251–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Love JF, Murphy JR. 2006. Corynebacterium diphtheriae: iron-mediated activation of DtxR and regulation of diphtheria toxin expression, p 726–737 In Fischetti VA, Novick RP, Ferretti JJ, Portnoy DA, Rood JI. (ed), Gram-positive pathogens, vol 1 ASM Press, Washington, DC [Google Scholar]
- 36. Wisedchaisri G, Chou CJ, Wu M, Roach C, Rice AE, Holmes RK, Beeson C, Hol WG. 2007. Crystal structures, metal activation, and DNA-binding properties of two-domain IdeR from Mycobacterium tuberculosis. Biochemistry 46: 436–447 [DOI] [PubMed] [Google Scholar]
- 37. D'Aquino JA, Denninger AR, Moulin AG, D'Aquino KE, Ringe D. 2009. Decreased sensitivity to changes in the concentration of metal ions as the basis for the hyperactivity of DtxR(E175K). J. Mol. Biol. 390: 112–123 [DOI] [PubMed] [Google Scholar]
- 38. Manabe YC, Saviola BJ, Sun L, Murphy JR, Bashai WR. 1999. Attenuation of virulence in Mycobacterium tuberculosis expressing a constitutively active iron repressor. Proc. Natl. Acad. Sci. U. S. A. 96: 12844–12848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zeng L, Wen ZT, Burne RA. 2006. A novel signal transduction system and feedback loop regulate fructan hydrolase gene expression in Streptococcus mutans. Mol. Microbiol. 62: 187–200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.