

Abstract
Hepatitis B virus (HBV) genomes show a high rate of mutations. This can lead to a variety of amino acid changes in the surface and polymerase genes, causing changes in viral protein conformation that can result in diminished antibody binding or decreased secretion of surface antigen (HBsAg). HBV monitoring increasingly relies on HBsAg detection and quantification, and therefore epidemiological data on HBsAg mutations are needed. We therefore analyzed the frequency of HBsAg mutations possibly influencing the quantification of HBsAg (MUPIQHs) in an unselected patient collective. To this end, we determined the HBV surface and polymerase gene sequences of an unselected patient collective of 237 individuals chronically infected with HBV and analyzed the MUPIQHs in these sequences using three different online HBV sequence analysis tools. We found that 17 or 34% of the patients, depending on the online interpretation algorithm used, harbored MUPIQHs and that MUPIQHs were not significantly associated with the duration of disease, treatment, or HBV genotype. Thus, this study shows that a substantial amount of HBV sequences derived from unselected patients chronically infected with HBV carry MUPIQHs, and therefore the reliability of routine quantitative and qualitative HBsAg tests needs to be reevaluated.
INTRODUCTION
Hepatitis B virus (HBV) can cause chronic infection, which can lead to cirrhosis, end-stage liver disease, and hepatocellular carcinoma (1), and it is estimated that more than 240 million persons are chronically infected worldwide (2). HBV genomes show a high rate of mutations because the virus replicates via reverse transcription of an RNA intermediate, and this process lacks proofreading mechanisms (3).
The S open reading frame (ORF), which codes for the hepatitis B surface protein (HBsAg), is overlapped by the polymerase (P) gene. Therefore, mutations in the polymerase gene associated with drug resistance can result in changes in the HBV surface protein (4–7). These, and mutations introduced in the S ORF by other mechanisms, can influence virion secretion (3). They can also reduce binding of HBsAg to anti-HbS antibodies (8), particularly when they occur in the so-called “a” determinant in the major hydrophilic loop (MHL) region of the HBsAg, which is the major antibody neutralization determinant of HBsAg (9). Because the interaction between HBsAg and anti-HbS antibodies is also the basis for routine HBV diagnosis and therapy monitoring by quantitative and qualitative detection of HBsAg (1, 10), changes in HBsAg influencing the interaction with antibodies or secretion of virions might have an impact on the results obtained by these diagnostic assays. This is especially relevant since HBsAg quantitative measurements are now discussed as a predictor to guide treatment decisions (11, 12).
Based on viral genome sequence variability, eight HBV genotypes have been defined by specific mutations and a divergence of >8% in the whole genome, and they have been labeled A to H (13). The clinical impact of the virus genotype in regard to treatment response, to viral mutation patterns associated with drug resistance, and to MHL mutations is not entirely clear (10).
For the rapid online genetic interpretation of HBV sequence data, three internet tools are available free of cost: HIV-grade HBV drug resistance interpretation (DRI), (14), Geno2pheno[HBV] (G2P) (15), and Stanford HBVseq (STAN) (16). All three of these programs have thus far been developed for research use only and to predict patient virus genotype and known drug resistance-associated mutations from P. In addition, G2P and DRI also predict published mutations in S associated with “escape” or with diminished antibody binding.
The aim of the present study was to assess the frequency of mutations in S that were associated either with diminished antibody binding to HBsAg or with reduced secretion of HBsAg, collectively termed MUPIQHs here, and which might thereby influence detection of HBsAg by diagnostic tests in a collective of routinely investigated chronically HBV-infected patients. In addition, we sought to determine the concordance of three online hepatitis B sequence analysis algorithms on the basis of the sequence data collected.
MATERIALS AND METHODS
Patients.
In the present study, one plasma sample from each of 237 individuals who had a chronic infection with HBV as defined by the World Health Organization (WHO) HBV guidelines (2) and who were 18 years or older was included. The samples were collected between September 2007 and September 2009 in the course of routine laboratory HBV tests. Patients were only included if a viral sequence could be gathered from their respective sample. Of the 237 patients, 90 (38%) were female, and 147 (62%) male. The median age was 41 years, with a range of 18 to 75 years. For 183 (77%) patients the duration of the disease was known, showing a median of duration 3 years, with a range of 0 to 42 years. The treatment history was known for 227 (96%) patients, with 165 (70%) patients being treatment naive. The remaining 62 patients received interferon (IFN) (20%), nucleoside/nucleotide analogues (NUKs) (29%), or a combination of both (15%). The treatment duration was available for 62 (26%) patients. The mean IFN treatment duration was 48 ± 4 weeks in all cases used in the calculations. The median duration of therapy with NUKs was 15 months (range, 2 to 130 months). The study was approved by the local ethics committee.
PCR and sequencing.
Archived plasma samples obtained for routine diagnostic purposes were retrospectively analyzed. DNA was purified from samples using the QIAamp MinElute media kit (Qiagen, Hilden, Germany). Quantitative PCR was performed on a COBAS TaqMan HBV test platform (Roche, Basel, Switzerland). HBV strains present in the samples were sequenced according to a previously described protocol (18) using an ABI 3130 XL genetic analyzer (Applied Biosystems, Foster City, CA). Sequence and base polymorphism analysis was done using SeqScape software, v.2.7.
Bioinformatic analysis.
The overlapping surface and polymerase reading frames were analyzed using the internet tools HIV grade HBV drug resistance interpretation (DRI) (14) and Geno2pheno[HBV] (G2P) (15). The sequences were genotyped by analyzing P, while S was analyzed for mutations possibly associated with diminished antibody binding. Changes of note detected in the sequences were recorded. All mutations associated with reduced antibody binding detected using these two systems are designated MARABs. Only HBsAg mutations shown by other authors to impact either qualitative or quantitative detection of HBsAg were included (19–21). In addition, all relevant mutations in the P sequence were detected and interpreted using the DRI, G2P, and Stanford HBVseq (STAN) algorithms (16). Sequences were genotyped by all three algorithms. All sequences were also tested with the STAN quality assurance subprogram and found to be free of frameshifts and stop codons. All MARABs, together with mutations previously shown to reduce HBsAg secretion, and with certain mutations arising in HBsAg as a consequence of drug resistance-associated P mutations, are designated MUPIQHs here.
Statistics.
Statistical analysis was performed using SPSS and GraphPad Prism. A P value of ≤0.05 was considered statistically significant. Samples from patients where single characteristics were unknown were excluded from the respective calculation. Concordance between the two bioinformatics tools used to analyze MARABs was assessed using Cohen's weighed kappa (22). In this model, results range from 0 (no concordance) to 1 (perfect concordance). The relationship between antiviral therapy, type of therapy, and genotype with MARABs was determined by eta coefficients, and significance was tested by chi-square tests.
RESULTS
Analysis of patient samples for HBs and polymerase mutations.
In a first step, we analyzed how frequent HBsAg mutations associated with reduced antibody binding to HBsAg (MARAB) occur in the HBV strain sequences retrieved from the samples included in the study. This was performed by the analysis algorithms DRI and G2P. We only included mutations in our analysis for which recent literature shows an impact on qualitative, quantitative, or both HBsAg assays (19–21). With the use of DRI, MARABs were detected in the HBsAg region in 63 (27%) of the 237 sequences analyzed, and the analysis by G2P identified such mutations in 30 (13%) sequences. An overview of the individual mutations identified and their frequency is presented in Fig. 1.
Fig 1.
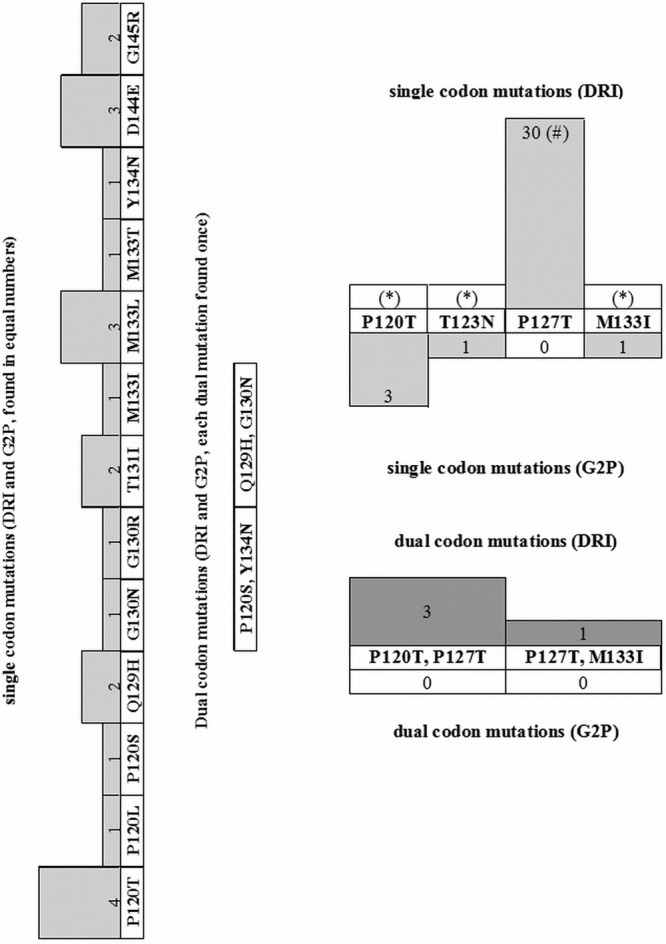
HBV surface gene mutations associated with diminished antibody binding (MARAB) identified in the study sequences, according to the two HBV sequence analysis algorithms. DRI, HIV-grade HBV drug resistance interpretation; G2P, Geno2pheno[HBV]. The numbers indicate the number of sequences. (Left) Mutations for which predictions by the algorithms were concordant. (Right) Mutations for which predictions by the algorithms were discordant. (*), Counted as a single mutation in G2P and a double mutation in DRI.
Due to the differences in the results obtained by both interpretation tools, we have further compared the two interpretation assays for overall concordance in the assignment and frequency of MARABs using Cohen's weighed kappa. DRI and G2P had a Cohen's weighed kappa of 0.56 (standard error of 0.058, 95% confidence interval of 0.444 to 0.672), signifying better-than-average concordance between the two assays. DRI consistently reported more S mutations than G2P, single mutations as well as combinations. The difference was found to be due to position s127, which is rated as a MARAB in DRI and not in G2P. Mutation s127 appears at a relatively high frequency, and this is reflected in the concordance calculation. In Fig. 1, these differences are shown.
Next, the frequency of polymerase drug resistance mutations was assessed in the study samples. The sequences of the polymerase gene derived from the patient samples were analyzed using the DRI, G2P, and STAN interpretation systems. The data revealed that in 22 of the 237 sequences (9%) polymerase mutations which are associated with drug resistance were present. The concordance of mutations identified as relevant for resistance between all interpretation tools was 100%. However, the interpretation of these mutations by the three algorithms differs. Where STAN just lists detected drug resistance-associated mutations, G2P and DRI rate them on a scale of three resistance “levels” (susceptible, possibly resistant, and resistant), and some mutations are assigned a different rating by the respective system. When rating the mutations, G2P and DRI concurred in 9 of 22 cases. A detailed overview of the concordant and discordant results obtained by the two assays is presented in Table 1.
Table 1.
Sequence analysis results for resistance-associated mutations in the polymerase sequence (P) of samples and their rating as determined by DRI and G2P
| Mutation in P associated with drug resistancea | Interpretationb |
No. of strainsc (%) | |
|---|---|---|---|
| DRI | G2P | ||
| T184S | I, entecavir | I, entecavir | 1 (4.5) |
| M204I | R, lamivudine; R, telbivudine | R, lamivudine; R, telbivudine | 7 (32) |
| N236T | R, adefovir | R, adefovir | 1 (4.5) |
| A181V, N236T | R, adefovir | R, adefovir; I, lamivudine | 1 (4.5) |
| A181T, M204I | R, lamivudine; R, telbivudine | R, lamivudine; R, telbivudine; I, tenofovir | 1 (4.5) |
| L180M, M204I | R, lamivudine; R, telbivudine; I, entecavir | R, lamivudine; R, telbivudine | 1 (4.5) |
| L180M, M204V | R, lamivudine; R, telbivudine; I, entecavir | R, lamivudine; R, telbivudine | 6 (27) |
| L180M, T184A, M204V | R, lamivudine; R, telbivudine; R, entecavir | R, lamivudine; R, entecavir; I, telbivudine | 1 (4.5) |
| L180M, T184S, M204V | R, lamivudine; R, telbivudine; R, entecavir | R, lamivudine; R, entecavir; I, telbivudine | 1 (4.5) |
| V173L, L180M, M204V | R, lamivudine; R, telbivudine; I, entecavir | R, lamivudine; I, telbivudine | 2 (9) |
All positions refer to codons in P.
I, intermediate (DRI) or possible (G2P) resistance; R, resistance. Discordant interpretations in the algorithms are indicated by gray shading. DRI, HIV-grade HBV drug resistance interpretation; G2P, Geno2pheno[HBV] result.
That is, the number of strains with the respective mutation.
We further investigated whether and to what extent drug resistance mutations in P occur in the patient samples which potentially influence antibody binding and/or HBsAg secretion in the corresponding S sequence. Since neither of the interpretation algorithms includes these mutations, they were assessed according to previously published data (23). The analysis of the 22 sequences with resistance mutations in P revealed that 19 of these sequences contained mutations which potentially influence antibody binding and/or HBsAg secretion in the corresponding S sequence, namely, pA181T, pM204I, and pM204V. Adding these sequences to those with MARABs as detected by the algorithms brings the total of sequences with mutations possibly influencing quantitative detection of HBsAg (MUPIQHs) to 72 (30% of all sequences analyzed) when using DRI and to 44 (19% of all sequences analyzed) when using G2P. An overview of the mutations in P detected in our sequences is shown in Table 1.
Genotyping and assignment of sequences to patients' country of origin.
We then analyzed the HBV sequences for their genotype. Alignment and phylogenetic analysis using the respective P nucleotide sequence was performed, and the samples were analyzed by three interpretation systems. The concordance of predicted genotypes between the three interpretation systems used was 100%. Overall, 37 samples were assigned to genotype A, 17 to genotype B, 23 to genotype C, 155 to genotype D, and 5 to genotype E. The patients' country of origin could be determined by chart review in 186 cases. The association between country group of origin and genotype is presented in Fig. 2.
Fig 2.

HBV genotypes of the 237 individuals included in the study, grouped according to the patients' region of origin. Patient countries of origin were assigned to one of four groups: Western (W.) Europe, Eastern (E.) Europe, Africa, and Asia.
Correlation between MARAB, patient data, and treatment.
Finally, we analyzed whether the MARABs in the HBV sequences of the routinely investigated patients show a correlation with specific aspects, including duration of disease, treatment, and HBV genotype.
First, we tested whether the duration from diagnosis of HBV infection until the time point of observation correlated with the number of MARABs in the sequence of the patient's HBV strains. The data for the time of diagnosis of HBV infection was available for 183 patients. We could not find a correlation between those two factors (P = 0.370 and r = 0.067 for DRI and P = 0.830 and r = 0.016 for G2P [Spearman rank correlation, two-tailed P value]). We then investigated whether there is a correlation between the patients' antiviral treatment and the presence of MARABs in the sequences of the patients' HBV strains. For our analyses, we summarized individuals who had received IFN (including IFN-α2a, IFN-α2b, pegylated IFN-α2a, or pegylated IFN-α2b) into one group (designated IFN), patients who had received lamivudine, telbivudine, or adefovir into a second group (nucleoside/nucleotide analogues [NUKs]), and patients who had received both in any combination of these into a third group (IFN and NUKs). Treatment data were available for 227 patients. Of these, 165 (73%) were treatment naive when tested, and 62 patients (27%) had experienced anti-HBV treatment before the samples were collected. No significant difference in MARABs detected by the different algorithms was observed when the HBV strains of the 62 individuals who had received any kind of treatment were compared to those of the 165 treatment-naive individuals (eta = 0.060 and P = 0.189 for DRI and eta = 0.410 and P = 0.273 for G2P [chi-square test]). We further compared the presence of MARABs in HBV sequences obtained from the individual treatment groups. No difference in the frequency of MARABs was associated with the type of treatment, IFN, NUKs or both, respectively (eta = 0.186 and P = 0.565 for DRI and eta = 0.089 and P = 0.976 for G2P [chi-square test]). We then tested whether the duration of treatment correlated with the amount of MARABs detected in sequences. Data for the duration of treatment were available for all 62 treated patients, and no correlation of MARABs with duration of treatment was found (P = 0.819 and r = 0.030 for DRI and P = 0.190 and r = −0.169 for G2P [Spearman rank correlation, two-tailed P value]). We finally investigated whether the HBV genotype is associated with the occurrence of MARABs. The frequency of relevant MARAB mutations detected by any of the algorithms did not differ significantly between the different genotypes only applicable for G2P, because DRI includes the serotype variant P127T (eta = 0.085 and P = 0.733 for G2P [chi-square test]).
DISCUSSION
The present study revealed that in a substantial proportion of HBV strains detected in patients with chronic HBV infections, mutations associated with reduced antibody binding to HBsAg can be identified.
Thus far, the epidemiology and frequency of MARABs in Europe and the United States is not clear. Previous analyses about the frequency of S ORF mutants leading to reduced antibody binding mostly only focused on the detection of the so-called “escape” mutations such as sG145R, sT131I, or sP120T. These mutations are known for their interference with antibody binding through disruption of the “a” determinant structure (24, 25) and are more frequent in risk groups in the United States and Western Europe, such as chronic carriers of HBV and immunosuppressed individuals (26, 27). In the present study, 1.7% of the sequences of the chronically HBV-infected patients showed such escape mutations. This low frequency of classical escape mutations occurring in the present HBV sequences is in accordance with the low frequency of these mutations described for the overall chronically HBV-infected European and U.S. population (27). For other MARAB mutations, the epidemiological situation in Europe is unclear. Our data now reveal that these other MARAB mutations were found much more frequently, in 13 or 27% of our study population, respectively, depending on the interpretation system used. Thus, the frequency of these mutations is clearly higher than that of the classical escape mutations. However, while it has been shown that HBV strains carrying the classical escape mutants can escape from the host antibody response elicited by HBV infection or vaccination (23) and thus have clinical implications (24), this has not yet been shown for the other MARABs and needs to be further elucidated.
Aside from the clinical implications, mutations in the S region of an HBV strain may have an effect on the results obtained by HBsAg detection tests used for routine diagnosis. The mutations in S can roughly be divided into three categories. The first category comprises the MARABs described previously (13–16, 28, 29), which are detected by the G2P and DRI algorithms and which cause a reduction in anti-HbS antibody binding, thereby possibly impairing the HBsAg detection in the patient's samples (3, 30, 31). Second, defined mutations may arise in S as a consequence of drug resistance associated mutations in P because the two genes share an overlapping reading frame (4, 5) and may also influence HBs antibody binding. Finally, mutations in S have been described that do not affect the interaction between HBsAg and antibodies but do impair HBsAg secretion and may thereby lead to reduced HBsAg levels in the patient's blood. These S mutations also occur as a consequence of gene overlap with P and are predominantly selected during antiviral therapy (6, 7, 32). When we included published mutations from these three categories, collectively termed MUPIQHs (3, 5, 6, 20, 27, 31, 33) in our analysis, we found a relatively high total frequency of such mutations. The presence of MUPIQHs in patient HBV strains may influence the diagnostic detection and quantification of HBsAg tests. Qualitative measurement of HBsAg is used for routine diagnosis of HBV infection, and a loss of HBsAg is associated with a favorable outcome (34). Quantitative measurement of this antigen is supposed to play an increasingly important role in prediction and monitoring of treatment response, (11) and is under investigation as a marker for NUK treatment endpoints (23). MUPIQHs may lead to a decrease in binding of test antibodies to HBsAg in the patients' samples, and it has already been described that some mutations may thereby render HBsAg undetectable by certain commercial qualitative HBsAg detection assays (8, 24, 30, 31, 33). We show here that many of the patient HBV strains carry such MUPIQHs, and this may provide a substantial diagnostic problem for the detection of HBsAg in patient samples.
For the quantitative measurement of HBsAg, two platforms are mostly used for the quantification of HBsAg: the Abbott Architect (Abbott Diagnostics, Abbott Park, IL) and the Elecsys HBsAg II (Roche Diagnostics GmbH, Mannheim, Germany) (35, 36). With respect to MUPIQHs, these platforms were tested for their ability to detect an array of HBsAg mutants (23, 28, 29, 37), and both were able to detect most of the mutant HBsAg (28). The study design, however, tested for detection only and not for accurate quantification. Recently, it was proven that MUPIQHs can affect HBsAg quantitation by using wild-type virus HBsAg compared to HBsAg with MUPIQ point mutants quantified using the Architect and Elecsys assays, (21). Many of the mutations examined in the present study were also detected in the sequences from our patient samples, suggesting that 17 (7%) samples would lead to HBsAg quantification problems, as described previously (21). Another recent study shows that in occult HBV infection, where S mutations are common, only 7 of 20 mutant HBsAg variants were detected in both quantitative assays. In addition, the assays also did not quantify the mutant HBV strains correctly, which showed mutations similar to those we identified in the present study (8). Another study using standard concentrations of HBsAg mutants for analysis shows that the relative light units used to measure qualitative HBsAg varies widely in various mutant HBsAg samples, although the concentration of HBsAg was the same for all samples (37). On the basis of these data and also taking into account the high frequency of mutations we observed in the present study, the introduction of HBsAg quantitative measurement as a predictor of treatment response or a treatment endpoint should be carefully evaluated with regard to all MUPIQHs. In addition, the sequence analysis of the patient HBV strains for MUPIQHs may aid in the interpretation of quantitative HBsAg data.
In the present study we also demonstrated that the online interpretation algorithms (DRI and G2P) used show a good concordance in the detection of MARABs. The assignment of MARABs differs only for three amino acid variations between the two analysis tools, of which our analysis only takes one into account. The mutation sP127T is assigned as relevant for diminished antibody binding to HBsAg by DRI but not by G2P. One study (38) describes the mutation P127T, and there is evidence that it can cause HBsAg to not be detected by some assays (19). This mutation, a HBsAg serotype determinant (39), occurred at a relatively high frequency as one of a cluster of mutations usually associated with altered antigenicity. When the algorithms for genotyping and detecting drug resistance mutations in P (STAN, G2P, and DRI) were used, however, they concurred in 100% of the cases.
Different HBV genotypes were detected in HBV-infected patients presenting in Viennese hospitals, and these are strongly associated with the epidemiological situation in the countries of origin of the patients (40–42). The dominance of genotype D sequences in our collective is mostly due to the fact that many of the HBV-infected patients seen in Vienna, Austria, originated from Eastern Europe, where this genotype is predominant (20, 42).
One limitation of the present study is the fact that patients were only included if a sequence could be amplified from their virus strains. This excludes patients with HBsAg in their blood but no detectable or amplifiable DNA. Therefore, the patient collective is not completely random but, due to the great variation in patient characteristics, we think it is still representative.
In conclusion, the present data show that a relatively high frequency of MUPIQHs exists in HBV strains circulating in European countries. The present findings may have significant implications for the interpretation of routine qualitative and quantitative HBsAg detection assays and underline that careful evaluation of diagnostic tests regarding mutants influencing anti-HBsAg antibody binding or secretion is needed.
ACKNOWLEDGMENTS
Sample sequencing was financially supported by research grants from Gilead Sciences GmbH, Vienna, Austria, and from Novartis Austria GmbH, Vienna, Austria.
We thank Barbara Dalmatiner for excellent technical assistance.
Footnotes
Published ahead of print 31 October 2012
REFERENCES
- 1.Liang TJ. 2009. Hepatitis B: the virus and disease. Hepatology 49:S13–S21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization 2012. Hepatitis B fact sheet. World Health Organization, Geneva, Switzerland: http://www.who.int/mediacentre/factsheets/fs204/en/ [Google Scholar]
- 3.Chotiyaputta W, Lok AS. 2009. Hepatitis B virus variants. Nat. Rev. Gastroenterol. Hepatol. 6:453–462 [DOI] [PubMed] [Google Scholar]
- 4.Sloan RD, Ijaz S, Moore PL, Harrison TJ, Teo CG, Tedder RS. 2008. Antiviral resistance mutations potentiate hepatitis B virus immune evasion through disruption of its surface antigen a determinant. Antivir. Ther. 13:439–447 [PubMed] [Google Scholar]
- 5.Torresi J, Earnest-Silveira L, Deliyannis G, Edgtton K, Zhuang H, Locarnini SA, Fyfe J, Sozzi T, Jackson DC. 2002. Reduced antigenicity of the hepatitis B virus HBsAg protein arising as a consequence of sequence changes in the overlapping polymerase gene that are selected by lamivudine therapy. Virology 293:305–313 [DOI] [PubMed] [Google Scholar]
- 6.Warner N, Locarnini S. 2008. The antiviral drug selected hepatitis B virus rtA181T/sW172* mutant has a dominant-negative secretion defect and alters the typical profile of viral rebound. Hepatology 48:88–98 [DOI] [PubMed] [Google Scholar]
- 7.Yeh CT, Chien RN, Chu CM, Liaw YF. 2000. Clearance of the original hepatitis B virus YMDD-motif mutants with emergence of distinct lamivudine-resistant mutants during prolonged lamivudine therapy. Hepatology 31:1318–1326 [DOI] [PubMed] [Google Scholar]
- 8.El Chaar M, Candotti D, Crowther RA, Allain JP. 2010. Impact of hepatitis B virus surface protein mutations on the diagnosis of occult hepatitis B virus infection. Hepatology 52:1600–1610 [DOI] [PubMed] [Google Scholar]
- 9.Zanetti AR, Van Damme P, Shouval D. 2008. The global impact of vaccination against hepatitis B: a historical overview. Vaccine 26:6266–6273 [DOI] [PubMed] [Google Scholar]
- 10.Wong GL, Chan HL. 2009. Predictors of treatment response in chronic hepatitis B. Drugs 69:2167–2177 [DOI] [PubMed] [Google Scholar]
- 11.Perrillo R. 2011. Measurement of HBsAg concentration during antiviral therapy of hepatitis B: who, when, and why. Am. J. Gastroenterol. 106:1774–1776 [DOI] [PubMed] [Google Scholar]
- 12.Sonneveld MJ, Rijckborst V, Boucher CA, Hansen BE, Janssen HL. 2010. Prediction of sustained response to peginterferon alfa-2b for hepatitis B e antigen-positive chronic hepatitis B using on-treatment hepatitis B surface antigen decline. Hepatology 52:1251–1257 [DOI] [PubMed] [Google Scholar]
- 13.Norder H, Courouce AM, Coursaget P, Echevarria JM, Lee SD, Mushahwar IK, Robertson BH, Locarnini S, Magnius LO. 2004. Genetic diversity of hepatitis B virus strains derived worldwide: genotypes, subgenotypes, and HBsAg subtypes. Intervirology 47:289–309 [DOI] [PubMed] [Google Scholar]
- 14.Walter H. 2007. HIV-grade HBV drug resistance interpretation: HBVseq sequence analysis. Max von Pettenkofer-Institute, Munich, Germany: http://www.hiv-grade.de/hbv_grade/deployed/grade.pl?program=hbvalg [Google Scholar]
- 15.Beggel B, Büch J, Däumer M, Hoffmann D, Kaiser R, Lengauer T, Schuldenzucker U, Schülter E, Verheyen J. 2009. Geno2pheno [HBV]. Max Planck Institute for Informatics, Saarbrücken, Germany: http://hbv.bioinf.mpi-inf.mpg.de/index.php [Google Scholar]
- 16.Anonymous 2009. Stanford HBVseq sequence analysis. Stanford University, Stanford, CA: http://hivdb.stanford.edu/HBV/HBVseq/development/hbvseq.pl?action=showSequenceForm [Google Scholar]
- 17.Reference deleted.
- 18.Puchhammer-Stockl E, Mandl CW, Kletzmayr J, Holzmann H, Hofmann A, Aberle SW, Heinz FX, Watschinger B, Hofmann H. 2000. Monitoring the virus load can predict the emergence of drug-resistant hepatitis B virus strains in renal transplantation patients during lamivudine therapy. J. Infect. Dis. 181:2063–2066 [DOI] [PubMed] [Google Scholar]
- 19.Alavian SM, Carman WF, Jazayeri SM. 2012. HBsAg variants: diagnostic-escape and diagnostic dilemma. J. Clin. Virol. [Epub ahead of print.] doi:10.1016/j.jcv.2012.04.027. [DOI] [PubMed]
- 20.Echevarria JM, Avellon A. 2006. Hepatitis B virus genetic diversity. J. Med. Virol. 78(Suppl 1):S36–S42 [DOI] [PubMed] [Google Scholar]
- 21.Verheyen J, Neumann-Fraune M, Berg T, Kaiser R, Obermeier M. 2012. The detection of HBsAg mutants expressed in vitro using two different quantitative HBsAg assays. J. Clin. Virol. 54:279–281 [DOI] [PubMed] [Google Scholar]
- 22.Cohen J. 1968. Weighted kappa: nominal scale agreement with provision for scaled disagreement or partial credit. Psychol. Bull. 70:213–220 [DOI] [PubMed] [Google Scholar]
- 23.Lee JM, Ahn SH. 2011. Quantification of HBsAg: basic virology for clinical practice. World J. Gastroenterol. 17:283–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carman WF, Zanetti AR, Karayiannis P, Waters J, Manzillo G, Tanzi E, Zuckerman AJ, Thomas HC. 1990. Vaccine-induced escape mutant of hepatitis B virus. Lancet 336:325–329 [DOI] [PubMed] [Google Scholar]
- 25.Seddigh-Tonekaboni S, Waters JA, Jeffers S, Gehrke R, Ofenloch B, Horsch A, Hess G, Thomas HC, Karayiannis P. 2000. Effect of variation in the common “a” determinant on the antigenicity of hepatitis B surface antigen. J. Med. Virol. 60:113–121 [DOI] [PubMed] [Google Scholar]
- 26.Szomor KN, Dencs A, Garai E, Rusvai E, Berencsi G, Takacs M. 2008. Mutation spectra of the surface-protein-coding region of the HBV genome in HBV-vaccinated and non-vaccinated individuals in Hungary. Arch. Virol. 153:1885–1892 [DOI] [PubMed] [Google Scholar]
- 27.Weber B. 2005. Genetic variability of the S gene of hepatitis B virus: clinical and diagnostic impact. J. Clin. Virol. 32:102–112 [DOI] [PubMed] [Google Scholar]
- 28.Muhlbacher A, Weber B, Burgisser P, Eiras A, Cabrera J, Louisirirotchanakul S, Tiller FW, Kim HS, Jv Helden Bossi V, Echevarria JM. 2008. Multicenter study of a new fully automated HBsAg screening assay with enhanced sensitivity for the detection of HBV mutants. Med. Microbiol. Immunol. 197:55–64 [DOI] [PubMed] [Google Scholar]
- 29.Popp C, Krams D, Beckert C, Buenning C, Queiros L, Piro L, Luciani M, Roebbecke M, Kapprell HP. 2011. HBsAg blood screening and diagnosis: performance evaluation of the Architect HBsAg qualitative and Architect HBsAg qualitative confirmatory assays. Diagn. Microbiol. Infect. Dis. 70:479–485 [DOI] [PubMed] [Google Scholar]
- 30.Hsu CW, Yeh CT. 2011. Emergence of hepatitis B virus S gene mutants in patients experiencing hepatitis B surface antigen seroconversion after peginterferon therapy. Hepatology 54:101–108 [DOI] [PubMed] [Google Scholar]
- 31.Zuckerman JN, Zuckerman AJ. 2003. Mutations of the surface protein of hepatitis B virus. Antivir. Res. 60:75–78 [DOI] [PubMed] [Google Scholar]
- 32.Zoulim F, Locarnini S. 2009. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology 137:1593–1608 [DOI] [PubMed] [Google Scholar]
- 33.Hollinger FB. 2007. Hepatitis B virus genetic diversity and its impact on diagnostic assays. J. Viral Hepat 14(Suppl 1):11–15 [DOI] [PubMed] [Google Scholar]
- 34.McMahon BJ. 2009. The natural history of chronic hepatitis B virus infection. Hepatology 49:S45–S55 [DOI] [PubMed] [Google Scholar]
- 35.Sonneveld MJ, Rijckborst V, Boucher CA, Zwang L, Beersma MF, Hansen BE, Janssen HL. 2011. A comparison of two assays for quantification of hepatitis B surface antigen in patients with chronic hepatitis B. J. Clin. Virol. 51:175–178 [DOI] [PubMed] [Google Scholar]
- 36.Wursthorn K, Jaroszewicz J, Zacher BJ, Darnedde M, Raupach R, Mederacke I, Cornberg M, Manns MP, Wedemeyer H. 2011. Correlation between the Elecsys HBsAg II assay and the Architect assay for the quantification of hepatitis B surface antigen (HBsAg) in the serum. J. Clin. Virol. 50:292–296 [DOI] [PubMed] [Google Scholar]
- 37.Health Protection Agency 2008. Evaluation of Abbott Architect HbS Ag assay. Health Protection Agency, London, United Kingdom: http://www.hpa.org.uk/webc/HPAwebFile/HPAweb_C/1317133423782 [Google Scholar]
- 38.Tong WB, He JL, Sun L. 2009. Analysis on mutation of S gene and P gene of hepatitis B virus in two counties of Sichuan Province. Zhongguo Yi Miao He Mian Yi 15:45–48 (In Chinese.) [PubMed] [Google Scholar]
- 39.Kramvis A, Kew M, Francois G. 2005. Hepatitis B virus genotypes. Vaccine 23:2409–2423 [DOI] [PubMed] [Google Scholar]
- 40.Lin CL, Kao JH. 2011. The clinical implications of hepatitis B virus genotype: recent advances. J. Gastroenterol. Hepatol 26(Suppl 1):123–130 [DOI] [PubMed] [Google Scholar]
- 41.Mahtab MA, Rahman S, Khan M, Karim F. 2008. Hepatitis B virus genotypes: an overview. Hepatobiliary Pancreat. Dis. Int. 7:457–464 [PubMed] [Google Scholar]
- 42.Schaefer S. 2007. Hepatitis B virus taxonomy and hepatitis B virus genotypes. World J. Gastroenterol. 13:14–21 [DOI] [PMC free article] [PubMed] [Google Scholar]