

Abstract
Trypanosoma brucei is the only organism known to have evolved a multifunctional RNA polymerase I (pol I) system that is used to express the parasite's ribosomal RNAs, as well as its major cell surface antigens, namely, the variant surface glycoprotein (VSG) and procyclin, which are vital for establishing successful infections in the mammalian host and the tsetse vector, respectively. Thus far, biochemical analyses of the T. brucei RNA pol I transcription machinery have elucidated the subunit structure of the enzyme and identified the class I transcription factor A (CITFA). CITFA binds to RNA pol I promoters, and its CITFA-2 subunit was shown to be absolutely essential for RNA pol I transcription in the parasite. Tandem affinity purification (TAP) of CITFA revealed the subunits CITFA-1 to -6, which are conserved only among kinetoplastid organisms, plus the dynein light chain DYNLL1. Here, by tagging CITFA-6 instead of CITFA-2, a complex was purified that contained all known CITFA subunits, as well as a novel proline-rich protein. Functional studies carried out in vivo and in vitro, as well as a colocalization study, unequivocally demonstrated that this protein is a bona fide CITFA subunit, essential for parasite viability and indispensable for RNA pol I transcription of ribosomal gene units and the active VSG expression site in the mammalian-infective life cycle stage of the parasite. Interestingly, CITFA-7 function appears to be species specific, because expression of an RNA interference (RNAi)-resistant CITFA-7 transgene from Trypanosoma cruzi could not rescue the lethal phenotype of silencing endogenous CITFA-7.
INTRODUCTION
In eukaryotes, RNA polymerase I (pol I) transcribes exclusively the large ribosomal gene units in the nucleolus. The unicellular parasite Trypanosoma brucei is exceptional in this regard because it is the only organism known to have evolved a multifunctional RNA pol I system that is used for rRNA synthesis and for the expression of proteins that are crucial for the parasite's successful interaction with its hosts. T. brucei is a tsetse-borne parasite in sub-Saharan Africa that causes lethal diseases in humans and livestock animals (2). It lives freely in the mammalian bloodstream by virtue of a dense coat of variant surface glycoprotein (VSG) which shields invariant membrane proteins from immune recognition (32) and whose antigenic variation enables the parasite to evade the host's immune system. There are ∼10 million VSG copies on the surface of a bloodstream-form (BF) trypanosome, all of which are expressed from a single gene drawn from a repertoire of up to 2,000 VSG genes (16). To accommodate the dense coat, the active VSG gene, which resides in one of 15 telomeric expression sites (ESs) (11), needs to be transcribed at extremely high rates; it was estimated that VSG RNA synthesis from the active ES exceeds that of a single β-tubulin gene by ∼50-fold (4). This high VSG expression is not only required for antigenic variation but essential to BF viability itself, since VSG silencing led to a rapid block of trypanosome proliferation in culture and clearance of parasites from infected mice (33).
In eukaryotic cells, RNA pol I transcription typically accounts for more than 50% of the total transcriptional activity, although the number of ribosomal gene units is far lower than the number of protein-coding genes. This efficiency of the RNA pol I system appears to be the result of high transcription initiation rates, which have been impressively documented by transmission electron microscopy of so-called “Miller spreads” (reviewed in reference 28). It is therefore likely that only the high efficiency of the RNA pol I system allows the parasite to express enough VSG from a single gene. While in mammals RNA pol I is unable to synthesize functional mRNA (5), the deviating gene expression mechanisms found in trypanosomatids enables T. brucei to use RNA pol I for mRNA synthesis. In trypanosomatids, protein-coding genes are arranged in long tandem arrays which are transcribed polycistronically, with RNA precursors being resolved into individual mRNAs by spliced leader (SL) trans splicing and polyadenylation (reviewed in reference 6). While in other eukaryotes, mRNA capping occurs cotranscriptionally by direct interaction of the capping enzymes with RNA pol II, SL trans splicing, in which the capped, 5′-terminal part of the SL RNA is fused onto the 5′ end of each mRNA, uncouples capping from RNA pol II transcription, thereby enabling T. brucei RNA pol I to express functional mRNA (26, 37).
The multifunctional RNA pol I system of T. brucei is versatile. While in other eukaryotes RNA pol I is confined to the nucleolus, where it transcribes rRNA gene units (RRNAs), the active VSG ES is transcribed outside the nucleolus (3) in the extranucleolar expression site body (ESB), a DNase-resistant compartment that appears to limit productive transcription to a single site (17). In addition to VSG expression, the parasite utilizes RNA pol I in its insect-stage, procyclic form (PF) for transcription of two gene loci (25) which encode two types of the cell surface antigen procyclin. Procylins are important for the parasite to establish successful infections in the tsetse vector (27). The RRNA, VSG ES, and procyclin gene promoters are structurally different, suggesting that they recruit different transcription factors (9). Since the last two promoters are absent in the trypanosomatid organisms Trypanosoma cruzi and Leishmania spp., one would expect to find T. brucei-specific proteins for VSG ES and procyclin gene transcription. However, all proteins involved in T. brucei RNA pol I transcription so far are conserved among all trypanosomatids, suggesting that they fulfill general RNA pol I functions. Alternatively, it is possible that these common transcription factors gained specific functions for protein-coding gene transcription in T. brucei evolution.
Bioinformatic and biochemical analysis of T. brucei RNA pol I revealed 10 of 12 core subunits that are present in every nuclear RNA Pol (13, 19, 35). The notion that the missing ortholog of the essential yeast RPA43 subunit could be replaced in T. brucei by its RNA pol II paralog RPB7 (13) could not be substantiated (22). Instead, an essential RNA pol I subunit, termed T. brucei RPA31 (TbRPA31), which is either a novel subunit or an extremely divergent ortholog of yeast RPA43, was found (18). Recently, the class I transcription factor A (CITFA) was identified; its purification led to the identification of six novel subunits, termed CITFA-1 to -6, plus the dynein light chain DYNLL1 (also known as LC8) (1). Tandem affinity-purified CITFA bound to the 67-bp-long VSG ES promoter in an electrophoretic mobility shift assay, requiring both upstream promoter elements for efficient binding. The gel shift could be competed with RRNA and procyclin gene promoters, indicating that CITFA is a general factor for RNA pol I transcription in T. brucei. Accordingly, silencing CITFA-2 expression was lethal and specifically reduced rRNA and VSG mRNA abundances in BFs. Moreover, in vitro transcription assays unequivocally demonstrated that CITFA-2 was absolutely required for transcription from all three types of RNA pol I promoters (1).
Here we have identified a novel, proline-rich protein that copurified with CITFA-6 from extract. The protein has no known protein domains and is conserved only among trypanosomatids. Our data show that this protein is a bona fide CITFA subunit and essential for RRNA and VSG transcription in BFs. Interestingly, CITFA-7 silencing was rescued by an RNA interference (RNAi)-resistant T. brucei CITFA-7 (TbCITFA-7) transgene but not by the heterologous T. cruzi ortholog (TcCITFA-7). While our data suggest that TcCITFA-7 was generally defective in T. brucei, this experiment confirmed previous observations in mammals that essential functions in RNA pol I transcription diverge very rapidly (10).
MATERIALS AND METHODS
DNAs.
pCITFA-6-PTP-NEO was described previously (1). pCITFA-7-PTP-NEO and pCITFA-7-HA-BLA were generated by inserting 353 bp of the CITFA-7 coding region (position 56 to position 408) into the ApaI and NotI sites of vectors pC-PTP-NEO (31) and pC-HA-BLA (18), respectively. As for, pTcCITFA-7-HA-bla, 543 bp of the T. brucei CITFA-7 5′ gene flank was fused to the T. cruzi CITFA-7 open reading frame (ORF) by overlap PCR. The product was cloned into the vector pC-HA-BLA through ApaI and NotI sites. The construct was linearized at the MscI site for integration into the T. brucei CITFA-7 locus. The RNAi vector pCITFA-7-T7-stl was generated by inserting 533 bp of the CITFA-7 3′ untranslated region (UTR) (positions 409 to 941 relative to the translation initiation codon) into the vector pT7-stl (1). Allele deletions were achieved by transfecting PCR products in which 100 bp of 5′ and 3′ gene flanks were fused to the hygromycin phosphotransferase coding region.
The following DNA oligonucleotides were used in reverse transcription (RT)-PCR: CITFA-7, 5′-TAGGGCCCAGCAACCTTGCCTGCTATATTACTTGATACC-3′/5′-TAGGTACCTTGCACTCCTGCGTCGGCCGTGAGC-3′; CITFA-2, 5′-ATAACTCGGAGACTGACTCG-3′/5′-AACTGGTGTATTGAACAACG-3′; CITFA-6, 5′-AGAAGCTTACGCGTATGGCTGTACAACCGGGAAAGGTG-3′/5′-AGTCTAGACTCGAGGTTATGTACCTGAATCCAGCTGTC-3′; Tb927.7.2590, 5′-TCGAGCAACTTGAA GAGTTACG-3′/ 5′-TCCTGTGGCTTCTGTTCGTTGG-3′; TFIIB, 5′-CACCTCGACTACCACAGATTT TGTTGGGC-3′/5′-TGGTGATCAACGTCCTCATACGTGG-3′; TcCITFA-7, 5′-CCTCCCTTTAGCTGCGATGCCTAATGTGCGCCGTTGTCGTTGG-3′/5′-TAGCGGCCGCTCCGATGACAACTCATTTCCCTCTTG-3′; VSG221, 5′-AGCTTTTTGGCAACCTCTTTGCCAGG-3′/5′-TGGTAACGCCTGTTTTGCCAGCTATTCC-3′.
Cells.
BFs were cultured in HMI-9 medium as described previously (22). For stable BF transfection, 3 × 107 cells, grown at exponential phase, were washed once in 2 ml Cytomix (120 mM KCl, 0.15 mM CaCl2, 10 mM K2HPO4, 25 mM HEPES-KOH, pH 7.6, 2 mM EDTA, and 5 mM MgCl2), resuspended in 0.4 ml Cytomix, and mixed with 10 μg of linearized plasmid DNA or 5 μg of PCR product. Cells were electroporated at 1.2 kV, 186 Ω, 50 μF on a BTX ECM600 electroporator, immediately resuspended in 50 ml of 37°C prewarmed medium, and dispensed in 1-ml aliquots into two 24-well plates. After incubation overnight, 1 ml of antibiotic-containing medium was added to each well. Cells were selected using G418, phleomycin, hygromycin, or blasticidin at final concentrations of 2.5, 1, 1, and 2 μg/ml, respectively. PF culturing and transfection were carried out as described previously (8, 14).
In vitro transcription.
Preparation of transcriptionally active extracts and cotranscription reactions with the BES promoter template VSG-trm and the RNA pol II-recruiting control template SLins19 was carried out as described previously (14). For immunodepletion of CITFA-2 and CITFA-7 from transcription reactions, extracts were prepared from cells exclusively expressing PTP–CITFA-2 (1) or CITFA-7–PTP and depleted with IgG beads (GE Healthcare). In parallel, extracts were mock treated without beads. Transcription signals were obtained by primer extension of the 32P-end-labeled oligonucleotides Tag-PE and SLtag, which hybridize to unrelated oligonucleotide insertions of VSG-trm and SLins19 RNAs, respectively. Primer extension products were separated on denaturing 50% urea–6% polyacrylamide gels and detected by autoradiography.
RNA analysis.
Total RNA preparation by the hot phenol method and Northern blot and primer extension analyses were carried out as previously described (1, 18). Reverse transcription of total RNA was done using SuperScript II reverse transcriptase (Invitrogen), following the manufacturer's recommendations.
Antibodies.
For the generation of immune sera, full-length CITFA-6 and CITFA-7 were recombinantly expressed in Escherichia coli with an N-terminal glutathione S-transferase (GST) tag and purified by glutathione affinity chromatography. The GST fusion proteins were used for immunizing rats as published previously (29). In addition to these immune sera, immunoblot detections were carried out with polyclonal antibodies directed against CITFA-2 (1), TFIIB (29), and the spliceosomal protein U2A′ (also known as U2-40K [20]). The PTP tag was detected either with a monoclonal anti-protein C epitope antibody (Roche) or with the peroxidase antiperoxidase reagent (Sigma). For hemagglutinin (HA) tag detection, a rat monoclonal anti-HA antibody was used (Roche).
Protein analysis.
Extract preparation and CITFA-6-P and CITFA-7-PTP tandem affinity purifications were conducted according to the standard protocol (31). Purified proteins were separated on SDS–10 to 20% polyacrylamide gradient gels and stained with Coomassie blue (Gelcode Coomassie stain; Thermo Fisher Scientific). For protein identification, protein bands were excised from gels, digested with trypsin, and subjected to liquid chromatography-tandem mass spectrometry. The resulting data were analyzed using the TurboSEQUEST program of the BioWorks 3.1 software package (Thermo Fisher Scientific). Coimmunoprecipitations (co-IPs) were carried out as previously published (18), while BF extract was prepared as recently described (21). Sucrose sedimentation was performed exactly as previously done with procyclic extract (1).
Immunofluorescence microscopy.
BFs grown to exponential phase were centrifuged at 800 × g for 5 min, washed in ice-cold phosphate-buffered saline (PBS), and fixed by adding formaldehyde to a final concentration of 2% for 10 min at room temperature. Fixation was terminated by adding glycine to a final concentration of 125 mM for 5 min. Cells were washed three times with PBS and then resuspended in PBS at a density of 3 × 107 cells/ml. Cells (5 × 106) were spotted onto silanized coverslips (Thermo Fisher Scientific) and allowed to settle for 30 min, after which the coverslips were washed twice in PBS for 5 min each. To reduce spherical aberrations of cells, coverslips were placed in a 24-well plate and centrifuged for 2 min at 800 × g (12). After a 10-min rehydration in PBS, fixed cells were permeabilized in 0.1% NP-40-containing PBS for exactly 5 min, washed twice in PBS, blocked in PBG (PBS containing 0.5% bovine serum albumin [BSA] and 0.2% cold water fish gelatin; Sigma), and incubated consecutively with primary and secondary antibodies diluted in PBG. Coverslips were extensively washed with PBS after each antibody probing and finally mounted on the slide with VectaShield (Vector lab). PTP-tagged proteins were detected with a polyclonal anti-protein A immune serum (Sigma) followed by an Alexa 594-conjugated anti-rabbit secondary antibody (Invitrogen), while HA-tagged CITFA-7 was detected with the above-specified anti-HA antibody and an Alexa 488-conjugated anti-rat secondary antibody (Invitrogen). Immunofluorescence assays of procyclics were done as described previously (15). Images were acquired on a Zeiss AxioVert 200 microscope, and colocalizations were quantified using the Zeiss Axiovision 4.6.3.0 colocalization software program.
RESULTS
Identification of novel CITFA subunit.
CITFA was originally characterized by tandem affinity purification of tagged CITFA-2. To further confirm the nature of the complex, we tandem affinity purified the most conserved subunit, CITFA-6, which had been C-terminally fused with a composite PTP tag consisting of a tandem protein A (ProtA) domain, a TEV protease cleavage site, and the protein C epitope (ProtC [31]). CITFA-1 to -6 and DYNLL1 were recovered in the final eluate in addition to two fainter bands around 35 kDa and a third band at ∼17.5 kDa (Fig. 1A). Liquid chromatography-tandem mass spectrometry revealed that the two larger bands were specific degradation products of CITFA-2, while the 17.5-kDa band was identified as a new protein that was termed CITFA-7 (accession number Tb927.7.2600).
Fig 1.

Identification of CITFA-7. (A) Tandem affinity purification of CITFA-6–PTP. Purified proteins were separated by SDS-PAGE and stained with Coomassie blue. For comparison, 0.002% of crude extract (Inp) and 5% of the TEV protease eluate were coanalyzed. On the right, mass spectrometric protein identifications of CITFA-1 to -7, DYNLL1, and CITFA-2 degradation products (2-deg) are indicated. (B) Reciprocal co-IP of CITFA-7–HA and PTP–CITFA-2 from PF and BF extracts. The immunoblot analysis includes equal amounts of starting material (Inp) and supernatant (S) and a 5-fold enrichment of the precipitate (P). Detection of the RNA pol II transcription factor TFIIB served as a negative control. (C) Multiple sequence alignment of trypanosomatid CITFA-7 orthologs from T. brucei (accession numbers are listed in Table S1 in the supplemental material), T. congolense (Tco), T. vivax (Tv), T. cruzi (Tc), Leishmania major (Lm), L. infantum (Li), and L. braziliensis (Lb). Positions with more than 50% identity or similarity are shaded in black or gray, respectively. Overall identity/similarity values specified at the end of each sequence were determined by pairwise comparison with the T. brucei sequence using the EMBOSS software program (http://www.ebi.ac.uk/emboss/align/) at default settings. Dashes indicate sequence gaps. Numbers in parentheses specify number of residues without significant sequence similarity. Asterisks mark 13 invariant proline positions.
While the apparent size of CITFA-7 was in the range of its predicted mass of 15.1 kDa, its identification was based only on a single, partially tryptic peptide match (data not shown). To confirm that the correct protein was identified, we generated PF and BF cell lines in which CITFA-2 and -7 were PTP and HA tagged, respectively. Reciprocal coimmunoprecipitation (co-IP) assays with extracts prepared from these cells unequivocally demonstrated the interaction of both proteins (Fig. 1B). Except for the very C terminus, CITFA-7's amino acid sequence is highly conserved among members of the Trypanosomatidae but has no sequence similarity to proteins outside this phylogenetic family. Furthermore, CITFA-7 does not contain sequence motifs of known function but does contain 13 proline residues which are absolutely conserved among trypanosomatid orthologs (Fig. 1C).
CITFA-7 is a bona fide subunit of CITFA.
Since CITFA-7 was not detected in CITFA-2 tandem affinity purification (1) and copurified with CITFA-6 in substoichiometric amounts (Fig. 1A), it was possible that it represented a distinct factor that interacted with the CITFA complex. To explore this possibility, we tagged and tandem affinity purified CITFA-7. We first generated a PF cell line, termed TbC7ee, which exclusively expressed CITFA-7 with a C-terminal PTP tag. This was achieved by integrating the plasmid pCITFA-7-PTP-NEO into one CITFA-7 allele and by knocking out the remaining CITFA-7 wild-type allele with the hygromycin phosphotransferase (HYGR) marker (Fig. 2A). As a molecular tool, we raised a specific anti-CITFA-7 immune serum in rats and confirmed that TbC7ee cells did not express wild-type, untagged CITFA-7 (Fig. 2B). Since CITFA-7 is encoded by an essential gene (see below), we concluded that the tag did not interfere with CITFA-7 function. Both anti-ProtC and anti-CITFA-7 antibodies detected wild-type or tagged CITFA-7 in up to four distinct, narrowly spaced bands. This pattern is primarily the result of multiple CITFA-7 phosphorylations, since treatment with alkaline phosphatase resolved the larger bands into the lower band (Fig. 2E). However, a faint band just above the main band was not resolved, suggesting that CITFA-7 also carries a modification other than phosphates.
Fig 2.

CITFA-7 is a bona fide subunit of CITFA. (A) Schematic drawing of a CITFA-7 wild-type (WT) allele and the allele modifications in cell line TbC7ee, which exclusively expresses CITFA-7–PTP and no untagged CITFA-7. The CITFA-7 coding region, HYGR and NEOR genes, and the PTP tag sequence are depicted by open boxes, striped boxes, and a black box, respectively. Gene flanks for RNA processing signals are indicated by smaller gray boxes. (B) Immunoblot analysis of whole-cell lysates of wild-type procyclic cells and of TbC7ee cells using a newly generated polyclonal anti-CITFA-7 immune serum (α CITFA-7) and a commercially available (Roche), monoclonal anti-ProtC antibody (α ProtC). (C) Immunoblot monitoring of CITFA-7–PTP purification (note that the TEV protease digest reduced CITFA-7–PTP to CITFA-7–P). Aliquots of crude extract (Inp), the flowthrough of the IgG affinity chromatography (FT-IgG), the TEV protease elution (TEV), the flowthrough of the anti-ProtC affinity chromatography (FT-ProtC), and the final eluate (Elu) were separated on a 10% SDS-polyacrylamide gel, blotted, and probed with anti-ProtC antibody. The blot was reprobed with immune sera directed against CITFA-2 and CITFA-6. Arrows point to the FT-IgG signals indicating codepletion of these subunits. (D) Protein analysis of CITFA-7 TAP, analogous to CITFA-6 TAP in Fig. 1A. (E) Anti-ProtC immunoblot of the final eluate of CITFA-7–PTP purification. The purified complex was either left untreated or incubated with alkaline phosphatase (AP) (from calf intestine) or with AP and a phosphatase inhibitor cocktail (ph-inh).
Monitoring the PTP tandem affinity purification by immunoblotting showed that CITFA-7–PTP, which was reduced to CITFA-7–P in the TEV protease elution, was efficiently purified in both chromatography steps (Fig. 2C, upper panel). When the blot shown in Fig. 2C was reprobed with a polyclonal anti-CITFA-2 antibody (1), it was apparent that CITFA-2 had been efficiently codepleted from the extract (Fig. 2C, middle panel, see arrow). To analyze whether this finding extended to other CITFA subunits, we raised a specific polyclonal anti-CITFA-6 immune serum in rats (see Fig. S1 in the supplemental material). Reprobing the blot with this antiserum showed codepletion of CITFA-6 as well (Fig. 2C, lower panel). As follows, the CITFA complex is quantitatively associated with tagged CITFA-7. Furthermore, the protein profile of the CITFA-7 purification revealed that all other CITFA subunits were copurified in apparently stoichiometric amounts, with no other major protein bands detectable (Fig. 2D). We concluded that CITFA-7 is a bona fide subunit of the CITFA complex. It appears that tagging of CITFA-2 and to a lesser extent tagging of CITFA-6 destabilized CITFA-7 binding to the CITFA complex, leading to partial losses of CITFA-7 in the corresponding purifications.
CITFA-7 is essential for trypanosome viability and RNA pol I-mediated gene expression.
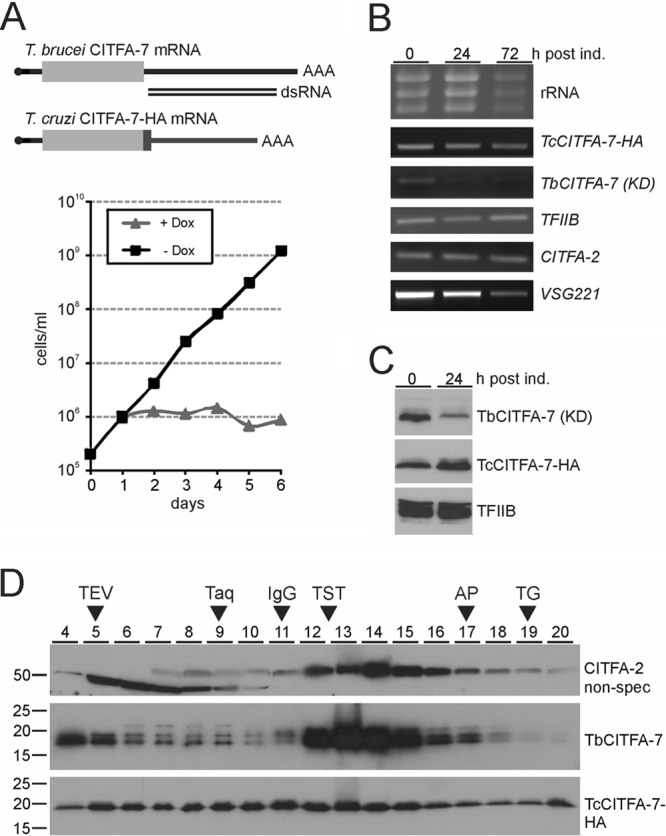
To address whether CITFA-7 is an essential component of CITFA, we silenced CITFA-7 expression in BFs by conditional, doxycycline-induced expression of CITFA-7 double-stranded RNA (dsRNA) (36), targeting the 3′ UTR of CITFA-7 mRNA through the RNAi pathway. We obtained a single clonal cell line the proliferation of which ceased after 1 day of doxycycline induction, whereas the noninduced cells proliferated logarithmically throughout the 3-day period of the experiment (Fig. 3A). The lethal phenotype appeared to be due to CITFA-7 silencing because the CITFA-7 mRNA level was strongly reduced 24 h after doxycycline induction whereas mRNA abundances of the gene downstream and adjacent to CITFA-7 and of CITFA-2, CITFA-6, and the TFIIB control were not affected (Fig. 3B). To unequivocally demonstrate that the lethal effect was specific to CITFA-7 silencing as opposed to a nonspecific off-target siRNA effect, we manipulated one CITFA-7 allele in the “RNAi” cell line to create an RNAi-resistant CITFA-7 mRNA which fully restored cell growth in the presence of doxycycline (Fig. 3A). The gene modification was achieved by targeted integration of a plasmid into the CITFA-7 locus that fused an HA tag sequence and the 3′ flank of the gene encoding the largest RNA pol I subunit RPA1 to one CITFA-7 allele. As shown in Fig. 3C, the abundance of HA-tagged CITFA-7 did not decrease upon doxycycline induction. We therefore concluded that CITFA-7 is essential for the viability of BFs. The essentiality of CITFA-7 appears to extend to PFs as well, because a corresponding knockdown in this life cycle stage halted culture growth after 3 days (see Fig. S2 in the supplemental material).
Fig 3.

CITFA-7 silencing is lethal, leading to a specific loss of RNA pol I-synthesized transcripts. (A) Growth curves of a clonal BF cell line in the absence (-Dox, blue) and presence (+Dox, red) of doxycyline, which, in this line, induces the synthesis of CITFA-7 dsRNA that targets the 3′ UTR of the corresponding mRNA. The lethal phenotype in the presence of doxycycline was rescued (Rescue +Dox, green) by fusing an HA tag sequence and the RPA1 3′ UTR to an endogenous CITFA-7 allele. In the schematic of wild-type and modified CITFA-7 mRNAs (not to scale), the coding region is depicted as a gray box and wild-type UTRs as black lines, the capped spliced leader is drawn in cyan, and the poly(A) tail indicated by AAA. In the modified mRNA, HA coding sequence and modified 3′ UTR are drawn in green. (B) Semiquantitative RT-PCR analysis of CITFA-7, CITFA-2, and CITFA-6 mRNA levels in total RNA prepared from cells in which CITFA-7 was silenced for the specified periods. In addition, the mRNA abundance of the gene Tb927.7.2590, which is the downstream, tandemly linked gene of CITFA-7, and TFIIB, as an independent control, were determined. post ind., postinduction. (C) Anti-HA immunoblot analysis of whole-cell lysates prepared from rescued cells that were induced by doxycycline for the specified periods and expressed CITFA-7–HA from an RNAi-resistant mRNA. U2A′ detection on the same blot served as a loading control. (D) Analysis of total RNA prepared from CITFA-7-silenced cells. rRNA was detected by ethidium bromide staining, VSG mRNA and mRNA of the spliceosomal protein SmD1 by Northern blotting, and SL RNA and U2 snRNA by primer extension analysis.
Further analysis of total RNA prepared from CITFA-7-silenced BFs showed that the RNA pol I transcripts rRNA and VSG mRNA became less abundant upon doxycycline induction (Fig. 3D). In the same samples containing equal amounts of RNA, compensatory increases of relative abundance were observed for the RNA pol II-synthesized SmD1 mRNA, encoding an RNA splicing protein, and SL RNA, as well as the RNA pol III-synthesized U2 snRNA. Since corresponding results were obtained upon loss of CITFA-2 (1) and the RNA pol I subunit RPA31 (18), this profile of RNA abundances strongly indicated that CITFA-7 is essential for RNA pol I transcription.
CITFA-7 colocalizes with CITFA-2 and RNA pol I subunit RPB6z in the nucleolus.
Thus far, the localization of CITFA subunits has not been investigated. To facilitate such an analysis, we generated two BF cell lines in both of which CITFA-7 was C-terminally HA tagged and either RPB6z or CITFA-2 was N-terminally PTP tagged. These tags were all functional, since exclusive expression of the tagged versions of these essential proteins did not impair proliferation of cultured trypanosomes (1, 18; this study). In 4′,6-diamidino-2-phenylindole (DAPI) stains of the nucleus, the nucleolus can be recognized as a spherical structure of low DNA density (Fig. 4). As expected, the RNA pol I subunit RPB6z localized consistently to the nucleolus, in most cases with an accumulation in the nucleolar periphery (Fig. 4A), as has been observed before (23). The tagged CITFA-7 subunit exhibited a similar distribution, and colocalization of CITFA-7 and RPB6z was calculated to be approximately 50% for each protein (see Fig. S3 in the supplemental material). This partial overlap is consistent with CITFA being a promoter-binding transcription initiation factor and not associated with transcribing RNA pol I. As expected, colocalization of CITFA-7 and CITFA-2 (Fig. 4B) showed a greater overlap. The colocalization signal between these two proteins comprised 80% of the CITFA-2 signal and 63% of the CITFA-7 signal (see Fig. S4), supporting our conclusion that CITFA-7 is a bona fide subunit of the CITFA complex. The greater, noncolocalized CITFA-7 signal may indicate that not all CITFA complexes carry the CITFA-2 subunit (see below).
Fig 4.
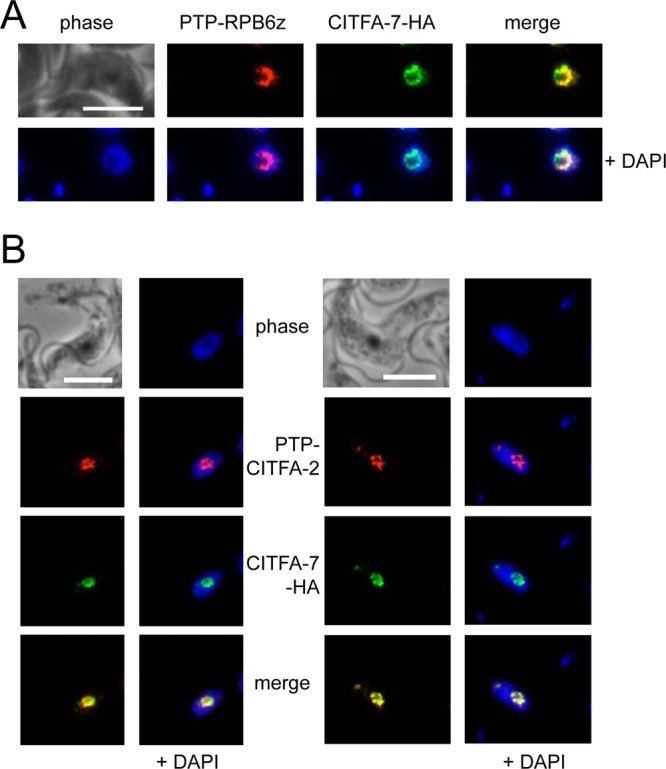
CITFA-7 colocalizations. (A) Colocalization of PTP-RPB6z (red) with CITFA-7–HA (green). (B) Colocalization of PTP–CITFA-2 (red) and CITFA-7–HA (green). White bars represent 10 μm.
In our hands, detection of the ESB was difficult, and we observed extranucleolar spots in less than 10% of cells (n = 174). Interestingly, these spots were observed with both RPB6z and CITFA subunit localizations. In these rare cases, CITFA-7 colocalized with either RPB6z or CITFA-2 outside the nucleolus, raising the possibility that the transcription initiation factor CITFA is concentrated in both the nucleolus and ESB (Fig. 4B, second panel set; also data not shown).
CITFA-7 depletion from extract abolishes RNA Pol I transcription in vitro.
To verify the essential transcription function of CITFA-7, we depleted CITFA-7 from extracts prepared from TbC7ee cells. Cotranscription of the VSG ES promoter template VSG-trm with the RNA pol II-recruiting template SLins19, harboring the SL RNA gene promoter, showed that depletion of CITFA-7 from extract virtually abolished VSG-trm transcription without affecting SLins19 transcription (Fig. 5, compare lanes 5 and 6). This result mirrored depletion of CITFA-2 from extract prepared from cells that exclusively expressed tagged CITFA-2 (lanes 1 and 2). Previously, we reported that RNA pol I transcription in CITFA-2-depleted extract was only partially restored through the addition of CITFA complex purified via tagged CITFA-2 (1); here, this result was reproduced in both CITFA-2- and CITFA-7-depleted extracts (lanes 3 and 7). In contrast, adding back CITFA which was purified by tagged CITFA-7 resulted in full restoration of VSG-trm transcription (lanes 4 and 8). Detection of the CITFA-2 and CITFA-7 proteins in depleted and restored extracts showed that the amounts of proteins added back to extract were comparable to the respective protein levels in mock-depleted extracts (Fig. 5, middle and bottom panel). Moreover, these immunoblots revealed that CITFA-2 depletion did not codeplete CITFA-7 (lane 2) whereas CITFA-7 depletion led to efficient codepletion of CITFA-2 (lane 6). Since the major difference between CITFA-2 and CITFA-7 purifications was the presence of CITFA-7 in the complex, these results confirmed that CITFA-7 is an essential component of CITFA.
Fig 5.
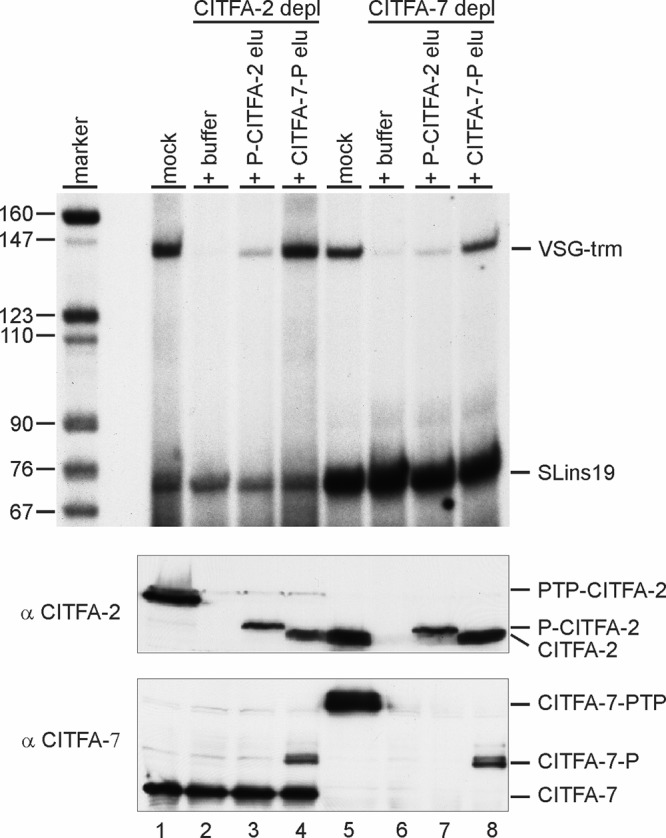
CITFA-7 is essential for RNA pol I transcription. Cotranscriptions of VSG ES promoter template VSG-trm and RNA pol II control template SLins19 in extracts that were mock depleted (mock), immunodepleted (depl) of CITFA-2 or CITFA-7, and reconstituted with buffer or final eluates (elu) of PTP–CITFA-2 or CITFA-7–PTP TAPs (top panel). Anti-CITFA-2 (middle panel) and anti-CITFA-7 (bottom panel) immunoblots of depleted and reconstituted extracts detecting endogenous, PTP-tagged, and P-tagged (after TEV protease digest) proteins are shown.
The increased functionality of the CITFA-7-purified complex also became apparent in an electrophoretic mobility shift assay. The PTP–CITFA-2-purified complex, harboring very little CITFA-7, bound a VSG ES promoter much less efficiently than the CITFA-7–PTP-purified complex (see Fig. S5 in the supplemental material). This result may indicate that CITFA-7 has a function in binding of CITFA to the VSG ES promoter (see Discussion).
The transcriptional function of T. brucei CITFA-7 is species specific.
Despite the fact that the multifunctional RNA pol I system is not present in other trypanosomatids, such as T. cruzi, where RNA pol I exclusively transcribes RRNA copies as in other eukaryotes, all RNA pol I and CITFA subunits found in T. brucei are conserved among all trypanosomatids. This lack of a T. brucei-specific factor raises the possibility that one or more of the T. brucei factors gained a function during evolution that is specific for VSG ES and procyclin gene expression. To test this possibility for CITFA-7, we introduced a C-terminally HA-tagged T. cruzi CITFA-7 transgene into the T. brucei CITFA-7 RNAi cell line that conditionally expressed dsRNA of the CITFA-7 3′ UTR (Fig. 6A). In contrast to the equivalently tagged, RNAi-resistant T. brucei CITFA-7–HA (Fig. 3A), T. cruzi CITFA-7–HA could not rescue trypanosome proliferation (Fig. 6A), although the expression of the heterologous transgene was not affected at the mRNA (Fig. 6B) and protein (Fig. 6C) levels. The RNA analysis of the BF cell line also showed that the abundances of both RNA pol I transcripts, rRNA and VSG mRNA, were strongly reduced after TbCITFA-7 silencing (Fig. 6B), indicating that TcCITFA-7–HA was generally unable to function in the T. brucei background. Although it is surprising that a conserved transcription factor from a related organism of the same genus is dysfunctional, this species specificity of function is a hallmark of the RNA pol I system typically not found in the RNA pol II and III systems. It was previously shown that even between the more closely related human and murine systems, the factor that binds the ribosomal core promoter, termed selectivity factor 1 (SL1) in humans and TIF-IB in the mouse, are each dysfunctional in the other's system (10).
Fig 6.
Expression of a T. cruzi CITFA-7-HA transgene did not rescue trypanosome proliferation from a T. brucei CITFA-7 knockdown. (A) Culture growth of BFs of noninduced (-Dox) and TbCITFA-7-silenced (+Dox) cells that express TcCITFA-7-HA. (B) Ethidium bromide staining of rRNA and semiquantitative RT-PCR analysis of listed mRNAs at 0, 24, and 72 h of doxycycline induction. Note that TbCITFA-7 mRNA was targeted by the knockdown (KD). (C) Immunoblot analysis of whole-cell lysates prepared from noninduced cells or cells that were induced for 24 h. TbCITFA-7 and TFIIB were detected with polyclonal antibodies against the endogenous proteins, and TcCITFA-7–HA was detected with a monoclonal anti-HA antibody. (D) Sedimentation of extract by ultracentrifugation in a 10 to 40% linear sucrose gradient. Fractions 4 to 20, taken from top to bottom, were analyzed by immunoblotting. For comparison, sedimentations of TEV protease (29 kDa), Taq DNA polymerase (95 kDa), IgG (150 kDa, 6.6S), the TRF4-SNAPc-TFIIA transcription factor complex (TST) (∼230 kDa) (30), apoferritin (AP) (444 kDa, 17S), and thyroglobin (TG) (660 kDa, 19S) were analyzed in parallel gradients (arrowheads).
In a final step, we wanted to find out the specific defect of TcCITFA-7 in T. brucei. Localization of the HA-tagged protein by indirect immunofluorescence (IF) microscopy found it to be concentrated in the nucleolus, as expected for a CITFA subunit (see Fig. S6 in the supplemental material). Another possibility was that TcCITFA-7 did not assemble into a full CITFA complex. To test this, we sedimented transcription extract from noninduced BF cells in a linear sucrose gradient. TbCITFA-7 was detected at the top of the gradient as an unbound protein and, as part of the CITFA complex, concentrated around its peak in fraction 13 (Fig. 6D, middle panel). The sedimentation profile of TbCITFA-2 in the same gradient closely resembled a previous sedimentation analysis (1), with unbound protein peaking in fraction 8 and CITFA-associated protein peaking in fraction 14 (Fig. 6D, top panel). Interestingly, the latter finding revealed a shift of sedimentation by one fraction down the gradient from the CITFA-7 peak, which suggested that there are two distinct CITFA complexes in the extract: an inactive complex without CITFA-2 and an active one with CITFA-2. This notion is consistent with our finding that a substantial CITFA-7 signal did not colocalize with CITFA-2 in our IF analysis (Fig. 4B; see also Fig. S4). In contrast to the T. brucei CITFA subunits, TcCITFA-7–HA was found spread throughout the gradient without exhibiting the strong sedimentation peak around fractions 13 and 14. This sedimentation pattern showed that TcCITFA-7–HA was not efficiently incorporated into a full CITFA complex. Formation of partial CITFA complexes and recruitment of heat shock proteins to these incomplete protein assemblies may account for the signal spread. An alternative explanation for this finding is self-aggregation of TcCITFA-7–HA in extract. However, this is less likely, because self-aggregation is typically observed upon protein overexpression, which we avoided by integrating the TcCITFA-7-HA transgene into the endogenous CITFA-7 locus. Furthermore, the formation of partial CITFA complexes may explain the correct nucleolar localization of TcCITFA-7–HA. In sum, the sedimentation analysis suggested that inefficient formation of a fully assembled CITFA complex may be the reason why TcCITFA-7–HA could not rescue silencing of endogenous CITFA-7. In addition, TcCITFA-7 appeared to carry fewer posttranslational modifications than TbCITFA-7 (Fig. 2B and C, 3C, and 6C and D), which may have contributed to the failure of this heterologous protein to function in the T. brucei system.
DISCUSSION
Here we have characterized a seventh nondynein subunit of CITFA and termed it, accordingly, CITFA-7. The CITFA complex is essential for transcription from the ribosomal, VSG ES, and procyclin gene promoters and, as shown for the VSG ES promoter, binds to DNA in a sequence-dependent fashion (1). CITFA-7 was not discovered previously because tagging and tandem affinity purification of CITFA-2 copurified only minute amounts of CITFA-7 (1). Nevertheless, our data clearly demonstrated that CITFA-7 is a bona fide CITFA subunit and essential for the proper function of the protein complex. We showed that when CITFA-7 is tagged and depleted from extract, the two other trackable CITFA subunits, 2 and 6, were codepleted from extracts, indicating that these two subunits were quantitatively bound to CITFA-7. Consistent with CITFA-7 being a CITFA subunit, its knockdown specifically affected the abundance of the RNA pol I transcripts rRNA and VSG mRNA. Moreover, our in vitro assays unequivocally demonstrated the importance of CITFA-7 for CITFA function: CITFA-2-purified complex, which comprised all CITFA subunits but CITFA-7, is much less efficient than the CITFA-7-purified complex in restoring RNA pol I transcription in CITFA-depleted extracts and binding to the VSG ES promoter in gel shifts.
What is the specific function of CITFA-7? We do not know. CITFA is inherently difficult to analyze because of its many subunits, its lack of sequence similarity to mammalian and yeast RNA pol I transcription factors, and its lack of known sequence motifs. Mechanistically, RNA pol I transcription initiation is conserved between yeast and mammals (28). It involves the formation of the RNA pol I transcription preinitiation complex (PIC), in which a core promoter-binding factor recruits RNA pol I to the transcription initiation site through interaction with the enzyme-bound factor RRN3. In yeast, this factor was termed core factor, and in humans it was termed SL1. In addition, another distinct factor binds to an upstream promoter element and activates ribosomal DNA (rDNA) transcription by stabilizing PIC formation. The VSG ES promoter extends only 67 bp upstream of the transcription initiation site and consists of two short sequence elements (24, 34). Since both elements are required for efficient binding of purified CITFA (1) and since they are so close to the initiation site, CITFA appears to be the functional equivalent of the yeast core factor and human SL1. According to the known functions of these factors, CITFA-7 may be directly involved in DNA binding or interact with an RRN3-like factor. However, we do not think this is the case. Previously, we have been able to inhibit the transcriptional function of the RNA pol II transcription factor TFIIB by adding a polyclonal anti-TFIIB antibody to extract prior to an in vitro transcription reaction (30). Since RNA Pol II PIC formation requires TFIIB to bind to the promoter, the TATA-binding protein, and the polymerase, it appears that the antibody was able to interfere with these interactions. The function of CITFA was similarly inhibited with an antibody against CITFA-2 (1) but not with the equivalently generated CITFA-7 antibody, which was raised against the recombinant full-length protein (see Materials and Methods; also data not shown). Although we cannot exclude the possibility that the polyclonal antibody did not recognize an interaction domain of CITFA-7, a likely interpretation of this result is that CITFA-7's essential interactions occur within the complex and are inaccessible to antibody blockage. If this is true, CITFA-7's main function would be in correct assembly of CITFA, a notion that is supported by our finding that CITFA-7, in contrast to CITFA-2, is part of all CITFA complexes in extract.
Finally, we confirmed for trypanosomes that the RNA pol I system is prone to rapid divergence between closely related species. Expression of the heterologous CITFA-7 gene from T. cruzi could not rescue the knockdown of the endogenous CITFA-7 gene. Our data strongly indicated that TcCITFA-7 was unable to assemble into a functional CITFA complex and showed that, in comparison to TbCITFA-7, it carried fewer posttranslational modifications. Interestingly, the defect between the murine and human RNA pol I systems was attributed to the rapid divergence of the rDNA promoter sequence and the DNA-protein interaction with the core factors (10), while our results suggest that TcCITFA-7 was unable to undergo correct complex assembly and obtain potentially important posttranslational modifications. These more dramatic defects in trypanosomes are likely the consequence of changes in amino acid sequences which in trypanosomatid transcription factors have diverged much more rapidly than, for example, in trypanosomatid RNA processing factors (6, 7). Nevertheless, our approach of reconstituting CITFA function in vivo with heterologous subunits or subunit domains has the potential to uncover VSG or procyclin gene-specific functions of this essential RNA pol I transcription factor complex.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Barbara Burleigh (Harvard University) for providing T. cruzi genomic DNA and to Sung Hee Park for help in generating the anti-CITFA-6 immune serum.
This project was funded by NIH grant R01 AI059377 (to A.G.).
Footnotes
Published ahead of print 26 October 2012
Supplemental material for this article may be found at http://ec.asm.org/.
REFERENCES
- 1. Brandenburg J, et al. 2007. Multifunctional class I transcription in Trypanosoma brucei depends on a novel protein complex. EMBO J. 26:4856–4866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brun R, Blum J. 2012. Human African trypanosomiasis. Infect. Dis. Clin. North Am. 26:261–273 [DOI] [PubMed] [Google Scholar]
- 3. Chaves I, et al. 1998. Subnuclear localization of the active variant surface glycoprotein gene expression site in Trypanosoma brucei. Proc. Natl. Acad. Sci. U. S. A. 95:12328–12333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ehlers B, Czichos J, Overath P. 1987. RNA turnover in Trypanosoma brucei. Mol. Cell. Biol. 7:1242–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grummt I, Skinner JA. 1985. Efficient transcription of a protein-coding gene from the RNA polymerase I promoter in transfected cells. Proc. Natl. Acad. Sci. U. S. A. 82:722–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Günzl A. 2010. The pre-mRNA splicing machinery of trypanosomes: complex or simplified? Eukaryot. Cell 9:1159–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Günzl A. 2012. RNA polymerases and transcription factors of trypanosomes, p 1–27 In Bindereif A. (ed), RNA metabolism in trypanosomes, vol 28 Springer Press, Berlin, Germany [Google Scholar]
- 8. Günzl A, Bindereif A, Ullu E, Tschudi C. 2000. Determinants for cap trimethylation of the U2 small nuclear RNA are not conserved between Trypanosoma brucei and higher eukaryotic organisms. Nucleic Acids Res. 28:3702–3709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Günzl A, Vanhamme L, Myler PJ. 2007. Transcription in trypanosomes: a different means to the end, p 177–208 In Barry JD, Mottram JC, McCulloch R, Acosta-Serrano A. (ed), Trypanosomes—after the genome. Horizon Press, Pittsburgh, PA [Google Scholar]
- 10. Heix J, Zomerdijk JC, Ravanpay A, Tjian R, Grummt I. 1997. Cloning of murine RNA polymerase I-specific TAF factors: conserved interactions between the subunits of the species-specific transcription initiation factor TIF-IB/SL1. Proc. Natl. Acad. Sci. U. S. A. 94:1733–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hertz-Fowler C, et al. 2008. Telomeric expression sites are highly conserved in Trypanosoma brucei. PLoS One 3:e3527 doi:10.1371/journal.pone.0003527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kawahara T, et al. 2008. Two essential MYST-family proteins display distinct roles in histone H4K10 acetylation and telomeric silencing in trypanosomes. Mol. Microbiol. 69:1054–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kelly S, Wickstead B, Gull K. 2005. An in silico analysis of trypanosomatid RNA polymerases: insights into their unusual transcription. Biochem. Soc. Trans. 33:1435–1437 [DOI] [PubMed] [Google Scholar]
- 14. Laufer G, Schaaf G, Bollgönn S, Günzl A. 1999. In vitro analysis of alpha-amanitin-resistant transcription from the rRNA, procyclic acidic repetitive protein, and variant surface glycoprotein gene promoters in Trypanosoma brucei. Mol. Cell. Biol. 19:5466–5473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Luz Ambrosio D, et al. 2009. Spliceosomal proteomics in Trypanosoma brucei reveal new RNA splicing factors. Eukaryot. Cell 8:990–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marcello L, Barry JD. 2007. Analysis of the VSG gene silent archive in Trypanosoma brucei reveals that mosaic gene expression is prominent in antigenic variation and is favored by archive substructure. Genome Res. 17:1344–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Navarro M, Gull K. 2001. A pol I transcriptional body associated with VSG mono-allelic expression in Trypanosoma brucei. Nature 414:759–763 [DOI] [PubMed] [Google Scholar]
- 18. Nguyen TN, Schimanski B, Günzl A. 2007. Active RNA polymerase I of Trypanosoma brucei harbors a novel subunit essential for transcription. Mol. Cell. Biol. 27:6254–6263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nguyen TN, Schimanski B, Zahn A, Klumpp B, Günzl A. 2006. Purification of an eight subunit RNA polymerase I complex in Trypanosoma brucei. Mol. Biochem. Parasitol. 149:27–37 [DOI] [PubMed] [Google Scholar]
- 20. Palfi Z, Bindereif A. 1992. Immunological characterization and intracellular localization of trans-spliceosomal small nuclear ribonucleoproteins in Trypanosoma brucei. J. Biol. Chem. 267:20159–20163 [PubMed] [Google Scholar]
- 21. Park SH, Nguyen TN, Günzl A. 2012. Development of an efficient in vitro transcription system for bloodstream form Trypanosoma brucei reveals life cycle-independent functionality of class I transcription factor A. Mol. Biochem. Parasitol. 181:29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Park SH, Nguyen TN, Kirkham JK, Lee JH, Günzl A. 2011. Transcription by the multifunctional RNA polymerase I in Trypanosoma brucei functions independently of RPB7. Mol. Biochem. Parasitol. 180:35–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Penate X, et al. 2009. RNA pol II subunit RPB7 is required for RNA pol I-mediated transcription in Trypanosoma brucei. EMBO Rep. 10:252–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pham VP, Qi CC, Gottesdiener KM. 1996. A detailed mutational analysis of the VSG gene expression site promoter. Mol. Biochem. Parasitol. 75:241–254 [DOI] [PubMed] [Google Scholar]
- 25. Roditi I, Clayton C. 1999. An unambiguous nomenclature for the major surface glycoproteins of the procyclic form of Trypanosoma brucei. Mol. Biochem. Parasitol. 103:99–100 [DOI] [PubMed] [Google Scholar]
- 26. Rudenko G, Chung HM, Pham VP, Van der Ploeg LH. 1991. RNA polymerase I can mediate expression of CAT and neo protein-coding genes in Trypanosoma brucei. EMBO J. 10:3387–3397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ruepp S, et al. 1997. Survival of Trypanosoma brucei in the tsetse fly is enhanced by the expression of specific forms of procyclin. J. Cell Biol. 137:1369–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Russell J, Zomerdijk JC. 2005. RNA-polymerase-I-directed rDNA transcription, life and works. Trends Biochem. Sci. 30:87–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schimanski B, Brandenburg J, Nguyen TN, Caimano MJ, Günzl A. 2006. A TFIIB-like protein is indispensable for spliced leader RNA gene transcription in Trypanosoma brucei. Nucleic Acids Res. 34:1676–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schimanski B, Nguyen TN, Günzl A. 2005. Characterization of a multisubunit transcription factor complex essential for spliced-leader RNA gene transcription in Trypanosoma brucei. Mol. Cell. Biol. 25:7303–7313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schimanski B, Nguyen TN, Günzl A. 2005. Highly efficient tandem affinity purification of trypanosome protein complexes based on a novel epitope combination. Eukaryot. Cell 4:1942–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schwede A, Jones N, Engstler M, Carrington M. 2011. The VSG C-terminal domain is inaccessible to antibodies on live trypanosomes. Mol. Biochem. Parasitol. 175:201–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sheader K, et al. 2005. Variant surface glycoprotein RNA interference triggers a precytokinesis cell cycle arrest in African trypanosomes. Proc. Natl. Acad. Sci. U. S. A. 102:8716–8721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vanhamme L, Pays A, Tebabi P, Alexandre S, Pays E. 1995. Specific binding of proteins to the noncoding strand of a crucial element of the variant surface glycoprotein, procyclin, and ribosomal promoters of Trypanosoma brucei. Mol. Cell. Biol. 15:5598–5606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Walgraffe D, et al. 2005. Characterization of subunits of the RNA polymerase I complex in Trypanosoma brucei. Mol. Biochem. Parasitol. 139:249–260 [DOI] [PubMed] [Google Scholar]
- 36. Wirtz E, Leal S, Ochatt C, Cross GAM. 1999. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 99:89–101 [DOI] [PubMed] [Google Scholar]
- 37. Zomerdijk JC, Kieft R, Borst P. 1991. Efficient production of functional mRNA mediated by RNA polymerase I in Trypanosoma brucei. Nature 353:772–775 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.