

Abstract
In response to stress, the heart undergoes a pathological remodeling process associated with hypertrophy and the reexpression of a fetal gene program that ultimately causes cardiac dysfunction and heart failure. In this study, we show that A-kinase-anchoring protein (AKAP)–Lbc and the inhibitor of NF-κB kinase subunit β (IKKβ) form a transduction complex in cardiomyocytes that controls the production of proinflammatory cytokines mediating cardiomyocyte hypertrophy. In particular, we can show that activation of IKKβ within the AKAP-Lbc complex promotes NF-κB-dependent production of interleukin-6 (IL-6), which in turn enhances fetal gene expression and cardiomyocyte growth. These findings provide a new mechanistic hypothesis explaining how hypertrophic signals are coordinated and conveyed to interleukin-mediated transcriptional reprogramming events in cardiomyocytes.
INTRODUCTION
Ventricular myocyte hypertrophy is the primary response whereby the heart responds to stress due to hemodynamic overload, myocardial infarction, or neurohumoral activation (1). It is associated with a nonmitotic growth of cardiomyocytes, increased organization of myofibers, and the reexpression of an embryonic gene program. These events alter cardiac contractility, calcium handling, and myocardial energetics and lead to maladaptive changes that, in the long term, reduce cardiac output and cause heart failure (2, 3).
Most stimuli known to promote cardiomyocyte hypertrophy activate G protein-coupled receptors (GPCRs), including α1- and β-adrenergic receptor (ARs), type I angiotensin II receptors (AT1-Rs), and endothelin I receptors (ET1-Rs) (4–8). It is now clear that the multiple signaling pathways activated by these receptors converge at a limited number of nuclear transcription factors that ultimately regulate the expression of genes associated with hypertrophy (9). In this context, targeting the signaling complexes coordinating the activity of such transcriptional regulators emerges as a primary strategy for new therapeutic approaches aimed at preventing myocardial dysfunction.
The transcription factor NF-κB has recently been recognized as a mediator of the growth responses induced by a variety of prohypertrophic agonists (10, 11). Under resting conditions, NF-κB is retained in the cytosol through an interaction with an inhibitor called IκB (inhibitor of NF-κB) (12). Upon stimulation, phosphorylation of IκB by the inhibitor of IκB kinase (IKK) complex, which includes IKKα, IKKβ, and a regulatory protein termed IKKγ (12, 13), targets IκB for polyubiquitination and subsequently for its degradation by the 26S proteasome (14). This permits the translocation of NF-κB to the nucleus, where it can activate the transcription of target genes.
While it is now clear that inhibition of NF-κB signaling in cardiomyocytes strongly reduces the hypertrophic responses activated by neurohumoral and biomechanical stimuli, including adrenergic agonists, angiotensin-II (Ang-II), proinflammatory cytokines, and aortic banding (15–22), it is currently poorly understood how prohypertrophic signals controlling NF-κB transcriptional activity are integrated and coordinated within cardiomyocytes to promote cardiomyocyte growth.
It has become increasingly clear that anchoring and scaffolding proteins play a pivotal role in coordinating intracellular signals in space and time (23, 24). A-kinase-anchoring proteins (AKAPs) are prototypic examples of proteins that compartmentalize signaling complexes at precise subcellular sites (25). In this context, we and others have previously characterized a cardiac anchoring protein, termed AKAP-Lbc, that possesses intrinsic RhoA guanine nucleotide exchange factor (GEF) activity (26). In cardiomyocytes, AKAP-Lbc activation occurs following stimulation of α1-ARs through a signaling pathway that requires the α subunit of the heterotrimeric G protein G12 (26, 27). Silencing AKAP-Lbc expression in rat neonatal ventricular myocytes (NVMs) strongly inhibits both α1-AR-mediated RhoA activation and hypertrophic responses (27, 28). This suggests that this anchoring protein coordinates a transduction pathway activated by the α1-AR that includes Gα12, AKAP-Lbc, and RhoA, which promotes cardiomyocyte hypertrophy. However, it is currently unknown which Rho effector molecules mediate the hypertrophic effect of AKAP-Lbc in cardiomyocytes.
In the present study, we provide evidence that AKAP-Lbc and IKKβ form a signaling complex that efficiently relays signals from α1-ARs on to NF-κB in rat NVMs. Stimulation of AKAP-Lbc Rho-GEF activity promotes activation of a transduction pathway involving RhoA, its effector Rho kinase, and anchored IKKβ. Interestingly, this newly identified AKAP-Lbc signaling network promotes NF-κB-dependent production of interleukin-6 (IL-6), which in turn enhances fetal gene expression and cardiomyocyte growth. These findings indicate that AKAP-Lbc functions to enhance the efficiency of interleukin-mediated hypertrophic signaling.
MATERIALS AND METHODS
Expression constructs.
The Flag-IKKβ-CMV2, Flag-IKKβ-K44M-CMV2, Flag-Iκbα-CMV2, and Flag-Iκbα-S34/36A-CMV2 vectors were obtained from Addgene. The entire IKKβ sequence was PCR amplified from the Flag-IKKβ-CMV2 vector using a 5′ primer containing the sequence encoding the hemagglutinin (HA) tag and subcloned at XbaI/HindIII into the pRK5 vector to generate the HA-IKKβ-pRK5 construct. The IKKβ fragments encoding amino acids 1 to 307 and 308 to 756 were PCR amplified from the HA-IKKβ-pRK5 vector and subcloned at BamHI/SalI into the pET30a vector and at Xba/HindIII into the pRK5 vector to generate protein fragments fused with the S tag and the HA tag, respectively.
The cDNA encoding the first 55 amino acids of Iκbα was PCR amplified from a mouse heart cDNA library and subcloned at SalI/NotI into pGEX4T3 vector to generate a protein fragment fused with glutathione S-transferase (GST).
The Flag-tagged AKAP-Lbc W2328L mutant was generated by standard PCR-directed mutagenesis using the Flag-AKAP-Lbc vector (29) as a template. The Flag-tagged AKAP-Lbc S1565A/W2328L double mutant was generated by excising a Psp1406I/NotI fragment from the Flag-AKAP-Lbc W2328L vector and by subcloning it into the Flag-AKAP-Lbc S1565A construct. For rescue experiments, we generated a silencing-resistant mutant of AKAP-Lbc (AKAP-Lbc*) by introducing silent mutations at codons 2151, 2152, and 2154 in the cDNA encoding Flag-AKAP-Lbc by standard PCR-directed mutagenesis (27). The Flag-tagged silencing-resistant form of the AKAP-Lbc W2328L mutant was generated by excising a HindIII fragment from the Flag-AKAP-Lbc* vector and by subcloning it into the Flag-AKAP-Lbc W2328L construct (27).
The AKAP-Lbc W2328L-mCherry construct was generated by excising a BspEI/HpaI fragment from the Flag-AKAP-Lbc W2328L construct and by subcloning it into the AKAP-Lbc-mCherry-pcDNA3.1 vector (generous gift from G. Carnegie, University of Illinois, Chicago). pAB286.1 and pSD28-GFP lentiviral transfer vectors encoding double-stranded hairpin oligonucleotides directed against the human or the rat form of AKAP-Lbc as well as vectors encoding Flag-AKAP-Lbc, AKAP-Lbc S1565A-GFP, GST-AKAP-Lbc-1923-2336, HA-α1b-AR, and Flag-RhoA N19T were described previously (26, 27, 29).
MS.
Three milligrams of protein derived from mouse ventricular lysates prepared as described previously (30) was incubated with 5 μg of GST or GST-AKAP-Lbc-1388-1922 fragments for 4 h at 4°C. Proteins associated with the beads were eluted and separated by SDS-PAGE as indicated above. Proteins were visualized by silver staining and bands were excised. Proteins were identified by tandem mass spectrometry (MS/MS) by the proteomic facility of the Faculty of Biology and Medicine of the University of Lausanne.
Expression and purification of recombinant proteins in bacteria.
The GST fusion proteins of the Rho binding domain (RBD) of rhotekin and of the AKAP-Lbc fragment encompassing residues 1923 to 2336 were expressed using the bacterial expression vector pGEX-4T1 in the BL21(DE3) strain of Escherichia coli and purified. Exponentially growing bacterial cultures were incubated for 16 h at 16°C with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and subsequently subjected to centrifugation. Pelleted bacteria were lysed in buffer A (20 mM Tris, pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1% [wt/vol] Triton X-100, 1 μg/ml aprotinin, 2 μg/ml leupeptin, 2 μg/ml pepstatin, 0.1 mM phenylmethylsulfonyl fluoride [PMSF]), sonicated, and centrifuged at 38,000 × g for 30 min at 4°C. After incubating the supernatants with glutathione-Sepharose beads (Pharmacia) for 2 h at 4°C, the resin was washed five times with 10 bed volumes of buffer A and stored at 4°C. Beads containing GST-RBD were used immediately for the rhotekin RBD pulldown assay.
The His6-tagged fusion protein of the IKKβ fragment encompassing residues 307 to 756 was expressed using the bacterial expression vector pET30 in BL21(DE3) bacteria and purified. Bacterial extracts containing His6-tagged fusion proteins were prepared in buffer B (20 mM HEPES, pH 7.8, 500 mM NaCl, 10 mM imidazole, 1 mM benzamidine, 2 μg/ml leupeptin, 2 μg/ml pepstatin). After a 1-min sonication, the lysates were centrifuged at 38,000 × g for 30 min at 4°C. The His6-tagged fusion proteins were purified by incubating the supernatant with nickel-nitrilotriacetic acid chelating resin (Amersham Pharmacia Biotech) for 1 h at 4°C. The resin was then washed 5 times with 10 bed volumes of buffer B and stored at 4°C. His6-tagged fusion proteins were eluted from the resin with 20 mM HEPES, pH 7.8, 500 mM NaCl, 300 mM imidazole, 1 mM benzamidine, 2 μg/ml leupeptin, 2 μg/ml pepstatin for 1 h at room temperature, dialyzed, and stored at −20°C. The protein content of the eluates was assessed by Coomassie staining of SDS-PAGE gels.
Cell culture and transfections.
HEK293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum and gentamicin (100 μg/ml) and transfected at 50 to 60% confluence in 100-mm dishes using the calcium phosphate method. For the overexpression of constructs containing the full-length AKAP-Lbc, HEK293 cells were transfected at 80% confluence in 100- or 35-mm dishes using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. After transfection, cells were grown for 48 h in DMEM supplemented with 10% fetal calf serum before harvesting. The total amount of transfected DNA was 10 to 24 μg/100-mm dish and 1 to 4 μg/35-mm dish.
Rat NVMs were prepared from 2-day-old Sprague-Dawley rats and cultured on 25-, 35-, or 100-mm dishes coated with 0.2% gelatin solution in maintenance medium (80% DMEM, 20% Medium 199, and 1% penicillin-streptomycin solution [Invitrogen]) containing 10% fetal calf serum and 5% horse serum. For luciferase assays, cardiomyocytes were seeded at 2 × 105 cells/25-mm dish. For indirect immunofluorescence experiments, cardiomyocytes were seeded on coated glass coverslips at 2 × 105 cells/35-mm well, whereas for immunoprecipitation experiments cardiomyocytes were seeded at 7 × 106 cells/100-mm dish. Cardiomyocyte culture purity was >95% as assessed by immunocytochemistry using an anti-α-actinin monoclonal antibody.
Production of lentiviruses.
Vesicular stomatitis virus G protein (VSV-G)-pseudotyped lentiviruses were produced by cotransfecting 293-T cells with 20 μg of the pSD28-GFP or pAB286.1 vector containing AKAP-Lbc short hairpin RNA (shRNA) cassettes, 15 μg of pCMVDR8.91 (31), and 5 μg of pMD2.VSVG (31) using the calcium phosphate method. Culture medium was replaced by serum-free DMEM at 12 h after transfection. Cell supernatants were collected 48 h later, filtered through a 0.45-μm filter unit, concentrated using Centricon-Plus-70 MW 100,000 columns (Millipore), and resuspended in PBS. Virus titers were determined by infecting 293-T cells using serial dilutions of the viral stocks and by scoring the number of either GFP-positive cells (at 72 h after infection) or puromycin-resistant clones (at 6 days after infection). Titers determined using these methods were between 8 × 108 and 1.3 × 109 transducing units (TU)/ml.
Lentiviral infection.
HEK293 cells were infected at 60% confluence using pAB286.1-based lentiviruses encoding wild-type or mutated AKAP-Lbc shRNAs at a multiplicity of infection (MOI) of 10 in the presence of 8 μg/ml Polybrene. Two days after infection, puromycin was added to the culture medium at a final concentration of 2 μg/ml. After 4 days of selection, puromycin-resistant cells were collected and amplified in selective medium containing puromycin at a final concentration of 2 μg/ml.
Rat neonatal ventricular cardiomyocytes were infected 24 h after plating using either pAB286.1- or pSD28-based lentiviruses encoding wild-type or mutated AKAP-Lbc shRNAs at an MOI of 50 in maintenance medium containing 5% horse serum and 8 μg/ml of Polybrene. Twenty-four hours after infection, cardiomyocytes were incubated in maintenance medium for an additional 48 h. For rescue experiments, cardiomyocytes were transfected 24 h after infection with the cDNA encoding the human forms of Flag-AKAP-Lbc or the Flag-AKAP-Lbc W2328L mutant using Lipofectamine 2000. Transfection was performed in maintenance medium containing 5% horse serum in the absence of antibiotics for a period of 6 h. Cells were then incubated in maintenance medium for an additional 48 h.
GST pulldown experiments.
For in vitro GST pulldowns, 2 and 10 nM bacterially purified His6-tagged fragments encompassing residues 307 to 756 of IKKβ were incubated with glutathione-Sepharose beads (Amersham Biosciences) coupled to GST or to GST fusion proteins of the AKAP-Lbc fragment encompassing residues 1388 to 1922 in 0.5 ml of buffer C (20 mM Tris, pH 7.4, 150 mM NaCl, 1% [wt/vol] Triton X-100, 5 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mM PMSF) for 4 h at 4°C. The beads were then washed five times with buffer C containing 500 mM NaCl, resuspended in SDS-PAGE sample buffer (65 mM Tris, 2% SDS, 5% glycerol, 5% β-mercaptoethanol, pH 6.8), and boiled for 3 min at 95°C. Eluted proteins were analyzed by SDS-PAGE and Western blotting.
For GST pulldown experiments performed from lysates, HEK293 cells grown in 100-mm dishes were lysed in 1 ml of buffer C and centrifuged at 100,000 × g for 30 min at 4°C. Glutathione-Sepharose beads (Amersham Biosciences) coupled to the different GST fusion proteins were incubated with 1.5 mg of proteins derived from the cell lysates in a total volume of 1 ml overnight at 4°C. The beads were then washed five times with buffer C and the proteins eluted and analyzed as indicated above.
Immunoprecipitation experiments.
For immunoprecipitation experiments, HEK293 cells grown in 100-mm dishes and expressing various constructs were lysed in 1 ml of buffer C. Cell lysates were incubated for 1 h at 4°C on a rotating wheel. The solubilized material was centrifuged at 100,000 × g for 30 min at 4°C, and the supernatants were incubated for 4 h at 4°C with 20 μl of anti-Flag-M2 affinity resin (Sigma) to immunoprecipitate overexpressed Flag-tagged proteins. Following a brief centrifugation on a benchtop centrifuge, the pelleted beads were washed five times with buffer C and twice with PBS, and proteins were eluted in SDS-PAGE sample buffer by boiling samples for 3 min at 95°C. Eluted proteins were analyzed by SDS-PAGE and Western blotting. For immunoprecipitation of endogenous AKAP-Lbc complexes, rat NVMs were lysed in 1 ml of buffer D (20 mM HEPES, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0,1% sodium deoxycholate, 5 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mM PMSF). Soluble proteins were isolated by centrifugation as indicated above and dialyzed overnight in buffer C at 4°C. Immunoprecipitations were performed by incubating 3 mg of lysate with 4 μg of affinity-purified rabbit polyclonal anti-AKAP-Lbc antibodies (Covance).
Rhotekin Rho binding domain (RBD) pulldown assay.
HEK293 cells grown in 100-mm dishes were transfected with 8 μg of the cDNA encoding the HA-tagged α1b-AR in the absence or presence of 16 μg of the cDNAs encoding Flag-AKAP-Lbc or Flag-AKAP-Lbc W2328L. Twenty-four hours after transfection, cells were incubated in DMEM without serum for an additional 24 h. Cells were then treated for 15 min with 10−4 M phenylephrine and lysed in RBD lysis buffer (50 mM Tris, pH 7.2, 150 mM NaCl, 1% [wt/vol], Triton X-100, 0.1% sodium deoxycholate, 30 mM MgCl2, 1 mM dithiothreitol [DTT], 10% glycerol, 1 mM benzamidine, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mM PMSF). Lysates were subjected to centrifugation at 38,000 × g for 10 min at 4°C and incubated with 60 μg of RDB beads for 1 h at 4°C. Beads were then washed three times with RBD buffer without sodium deoxycholate, resuspended in SDS sample buffer, and analyzed by SDS-PAGE.
Autospot peptide synthesis.
Peptide arrays were synthesized on cellulose paper using an Auto-Spot Robot ASP 222 (AbiMed, Langenfeld, Germany). After synthesis, the amino termini were acetylated with 2% acetic acid anhydride in dimethyl formamide. The peptides were then deprotected by a 1-h treatment with dichloromethane-trifluoroacetic acid (1:1) solution containing 3% tri-isopropylsilane and 2% water. Arrays were subsequently washed 5 times in TBS-Tween and used for the overlay assay.
Solid-phase overlay assay.
Peptide arrays were incubated with TBS-Tween containing 5% (wt/vol) nonfat dry milk and 1% bovine serum albumin (BSA) for 1 h at room temperature and then with 10 nM His6/S-tagged IKKβ-307-756 fragment in TBS-Tween containing 5% nonfat dry milk and 0.1% BSA for 16 h at room temperature. After three washes in TBS-Tween, the blots were incubated for 2 h with horseradish peroxidase (HRP)-conjugated S protein (1:5,000 dilution; Jackson Laboratory) in TBS-Tween containing 5% (wt/vol) nonfat dry milk. Membranes were then washed three times in TBS-Tween and subjected to autoradiography.
Luciferase activity assays.
HEK293 cells or rat NVMs grown in 25-mm dishes and infected using lentiviruses as indicated above were transfected with 50 ng of NF-κB-firefly reporter plasmid and 950 ng of the Renilla-CMV plasmid using Lipofectamine 2000. Luciferase activity was measured in 20 μl of cell lysates using the dual-luciferase assay (Promega).
IKKβ activity assays.
Transfected HEK293 cells grown in 100-mm dishes or rat NVMs grown in 60-mm dishes were lysed in buffer E (20 mM Tris, pH 7.4, 150 mM NaCl, 1% [wt/vol] Triton X-100, 10 mM NaF, 10 mM Na-pyrophosphate, 1 mM Na-orthovanadate, 1 mM glycerophosphate, 5 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mM PMSF). Cell lysates were incubated for 10 min at 4°C on a rotating wheel. The solubilized material was centrifuged at 100,000 × g for 30 min at 4°C. Supernatants were incubated with 4 μl of rabbit polyclonal anti-IKKβ antibodies (Cell Signaling Technology) and 20 μl of protein A-Sepharose beads (Amersham) for 2 h at 4°C to immunoprecipitate endogenous IKKβ. Following a centrifugation on a benchtop centrifuge, the pelleted beads were washed three times with buffer E and twice with a buffer containing 50 mM Tris (pH 7.4) and 5 mM MgCl2. Immunoprecipitates containing IKKβ were incubated with 0.5 μg of purified GST-IκBα-1-55. Reactions were carried out in 50 mM Tris, pH 7.4, 5 mM MgCl2, and 1 mM ATP-Na2 for 30 min at 30°C and ended by the addition of SDS-PAGE sample buffer and loaded on SDS-PAGE gels.
SDS-PAGE and Western blotting.
Samples denatured in SDS-PAGE sample buffer were separated on acrylamide gels and electroblotted onto nitrocellulose membranes. The blots were incubated with primary antibodies and horseradish-conjugated secondary antibodies (Amersham). The following affinity-purified primary antibodies were used for immunoblotting: affinity-purified rabbit polyclonal anti-AKAP-Lbc (0.1 mg/ml; 1:1,000 dilution; Covance), mouse monoclonal anti-Flag (4.9 mg/ml; 1:2,000 dilution; Sigma), rabbit polyclonal anti-GFP (400 μg/ml; 1:1,000 dilution; Roche), mouse monoclonal anti-HA (1:5,000 dilution; Sigma), rabbit polyclonal anti-HA (1:1,000 dilution; Sigma), goat polyclonal anti-GST (1:4,000 dilution; Roche), rabbit polyclonal anti-IKKβ (1:1000 dilution; Cell Signaling Technologies), rabbit polyclonal anti-phospho-IκBα (serine 32 and serine 34) (1:1,000 dilution; Cell Signaling Technologies), rabbit polyclonal anti-IκBα (1:1,000 dilution; Cell Signaling Technologies), mouse monoclonal anti-RhoA (1:250; Santa Cruz Biotechnologies), mouse monoclonal antiactin (1:1,000 dilution; Sigma), mouse monoclonal anti-histidine tag (100 μg/ml; 1:1,000 dilution; Qiagen), and S protein-HRP (1:5,000 dilution; Novagen).
Fluorescence microscopy.
Infected cardiomyocytes were grown for 24 h in the presence of 5% horse serum and for an additional 48 h in the absence of serum. Cells were then incubated for 24 h with or without 10−4 M phenylephrine and washed twice with PBS. They were then fixed for 10 min in PBS–3.7% formaldehyde, permeabilized for 5 min with 0.2% (wt/vol) Triton X-100 in PBS, and blocked for 1 h in PBS–1% BSA. The expression of α-actinin and Flag-AKAP-Lbc constructs was assessed by incubating cardiomyocytes for 1 h with a 1:500 dilution of mouse monoclonal antibodies against α-actinin (Sigma) and rabbit monoclonal antibodies against the Flag epitope (Sigma), respectively, followed by a 1-h incubation with rhodamine-conjugated anti-mouse and aminomethylcoumarin acetate (AMCA)-conjugated anti-rabbit secondary antibodies (Jackson ImmunoResearch). The cells were mounted using Prolong (Molecular Probes). Intrinsic GFP fluorescence and immunofluorescent staining were visualized using a Zeiss Axiophot fluorescence microscope.
Single-cell real-time PCR.
Rat NVMs were infected with control lentiviruses encoding GFP or lentiviruses encoding GFP and shRNAs directed against AKAP-Lbc at an MOI of 50. Cells were subsequently transfected with control vectors or plasmids encoding AKAP-Lbc-mCherry or AKAP-Lbc-W2328L-mCherry. GFP-positive (i.e., lentivirus-infected) and GFP/mCherry-double-positive (i.e., lentivirus-infected and AKAP-Lbc-transfected) rat NVMs were sorted by fluorescent-activated cell sorting (FACS). Groups of 10 cells were eluted in 9 μl of lysis buffer (1.2% Triton X-100, 10 mM DTT, 1× DNase buffer [Promega]) at 4°C. Lysed cells were incubated with 5 U of DNase I for 30 min at 37°C. After DNase I inactivation using a DNA-free kit (Ambion), lysates were submitted to reverse transcription (RT) using random hexamers and superscript II reverse transcriptase for 1 h at 37°C. The cDNAs were then preamplified using a TaqMan PreAmp Master Mix kit (Applied Biosystems) and TaqMan gene expression assays for the atrial natriuretic factor, β-myosin heavy chain, interleukin-6, and β2-microglobulin (Applied Biosystems) for 14 cycles using a 7500 Fast instrument from Applied Biosystems. Finally, preamplified cDNAs were diluted 1:20 and amplified using the TaqMan gene expression assays described above for 60 cycles using a 7500 Fast instrument (Applied Biosystems).
Statistical analysis.
Statistical significance was analyzed using an analysis of variance (ANOVA) test followed by Tukey posttests with Bonferroni corrections.
RESULTS
AKAP-Lbc anchors IKKβ in cardiomyocytes.
To identify novel AKAP-Lbc-interacting proteins potentially involved in cardiomyocyte hypertrophy, we initially performed a proteomic screen for AKAP-Lbc binding partners expressed in cardiac tissues. In this context, a GST fragment encompassing the GEF domain of AKAP-Lbc (residues 1922 to 2336) was used as bait in pulldown experiments using mouse heart lysates. Associated protein complexes were separated by SDS-PAGE and identified by tandem mass spectrometry (MS/MS). Using this approach, we could identify peptides from the NF-κB-activating kinase IKKβ (Fig. 1A). Immunoblots using IKKβ-specific antibodies confirmed that IKKβ associates with the GST-AKAP-Lbc-1922-2336 fragment but not with GST (Fig. 1B, top). We next could determine that the kinase activity of IKKβ is not required for this interaction, as shown by the fact that deletion of the entire kinase domain from IKKβ does not affect the ability of the kinase to coimmunoprecipitate with the Flag-AKAP-Lbc-1922-2336 fragment from HEK293 cells (Fig. 2A, top). In line with these findings, the IKKβ-specific inhibitor BMS-345541 does not influence the ability of Flag-AKAP-Lbc-1922-2336 to associate with HA-tagged IKKβ (Fig. 2B, top).
Fig 1.

AKAP-Lbc anchors IKKβ. (A) Pulldown experiments were performed by incubating GST or the GST-AKAP fragment encompassing residues 1923 to 2336 with mouse heart extracts. Proteins were resolved by SDS-PAGE and silver staining and identified by MS/MS spectrometry. (B) Pulldown experiments were performed as indicated in panel A. IKKβ binding was detected by immunoblotting (IB) (top). GST fusion proteins were visualized by Ponceau S staining (bottom). (C) GST and the GST-AKAP-Lbc-1923-2336 fragment were used as bait in pulldown experiments with increasing concentrations of the purified S-tagged 307-756 fragment of IKKβ. Binding of the S-tagged IKKβ fragment was detected by immunoblotting (top). GST fusion proteins were visualized using Ponceau S staining (bottom). (D) Lysates from HEK293 cells transfected with HA-IKKβ in combination with control vector or the plasmid encoding Flag-AKAP-Lbc were immunoprecipitated (IP) using anti-Flag antibodies. Proteins in the lysates and immunoprecipitates were identified by immunoblotting using antibodies against the HA tag or the Flag tag as indicated. (E) Lysates from HEK293 cells transfected with AKAP-Lbc-GFP in combination with control vector or the plasmid encoding Flag-IKKβ were immunoprecipitated using anti-Flag antibodies. Proteins in the lysates and immunoprecipitates were identified by immunoblotting using antibodies against GFP or the Flag tag as indicated. (F) Rat NVMs were serum starved for 24 h and treated with 10−4 M PE for the indicated periods of time. Extracts were subsequently subjected to immunoprecipitation with either nonimmune IgGs or affinity-purified anti-AKAP-Lbc antibodies. Western blots of the immunoprecipitates and the cell extracts were revealed using either anti-IKKβ (top) or affinity-purified anti-AKAP-Lbc polyclonal antibodies (bottom). (G) Rat NVM extracts were subjected to immunoprecipitation as described for panel F. Western blots of the immunoprecipitates and the cell extracts were revealed using either anti-RhoA monoclonal antibodies (top) or affinity-purified anti-AKAP-Lbc polyclonal antibodies (bottom).
Fig 2.

Interaction between AKAP-Lbc and IKKβ does not require IKKβ kinase activity. (A) HEK293 cells were transfected with HA-IKKβ-1-307, HA-IKKβ-308-756, and HA-IKKβ in combination with the empty pFlag vector or the plasmid carrying the Flag-tagged fragment of AKAP-Lbc encompassing residues 1388 to 1922. Cell lysates were subjected to immunoprecipitation with anti-Flag antibodies. Western blots of the immunoprecipitates and of the cell extracts were revealed using anti-HA polyclonal antibodies to detect HA-IKKβ (top and middle) or anti-Flag monoclonal antibodies to detect the Flag-1388-1922 fragment of AKAP-Lbc (bottom). (B) HEK293 cells were transfected with HA-tagged IKKβ in combination with either empty vector or the plasmid carrying the Flag-tagged fragment of AKAP-Lbc encompassing residues 1923 to 2336. Forty-eight hours after transfection, cells were incubated for 8 h in the absence or presence of 10 μM BMS-345541 and subsequently lysed. Cell lysates were subjected to immunoprecipitation with anti-Flag antibodies. Western blots of the immunoprecipitates and of the cell extracts were revealed using anti-HA polyclonal antibodies to detect HA-IKKβ (top and middle) or anti-Flag monoclonal antibodies to detect the Flag-1923-2336 fragment (bottom).
In vitro pulldown assays revealed that AKAP-Lbc and IKKβ associate through a direct interaction, as shown by the fact that GST–AKAP-Lbc-1922-2336 binds a purified S-tagged C-terminal fragment of IKKβ encompassing residues 307 to 756 (Fig. 1C, upper, lanes 2 and 3).
To determine whether full-length AKAP-Lbc is able to associate with IKKβ, coimmunoprecipitation experiments were performed from HEK293 cells expressing recombinant Flag-tagged AKAP-Lbc and HA-tagged IKKβ. Our results indicate that HA-IKKβ was copurified with Flag-AKAP-Lbc (Fig. 1D, top). In line with these findings, reciprocal coimmunoprecipitation experiments revealed that AKAP-Lbc-GFP copurified with Flag-tagged IKKβ (Fig. 1E, upper panel). Finally, this protein-protein interaction was validated in rat NVMs when endogenous IKKβ was detected in AKAP-Lbc-immunoprecipitated complexes (Fig. 1F, upper panel, lane 3). The ability of AKAP-Lbc to coimmunoprecipitate with IKKβ is detected in serum-starved cardiomyocytes and is not affected by stimulation of cells with 10−4 M phenylephrine (PE), suggesting that AKAP-Lbc and IKKβ interact in a constitutive manner (Fig. 1F, upper panel, lanes 6 and 9). Collectively these findings indicate that AKAP-Lbc represents a novel cardiac IKKβ-anchoring protein.
Mapping studies next were used to define the interactive surface on AKAP-Lbc. We initially assessed the ability of HA-IKKβ to coimmunoprecipitate with a family of six consecutive Flag-tagged AKAP-Lbc fragments encompassing the entire AKAP-Lbc sequence (Fig. 3A). Our results indicate that interaction occurs only with a region included between residues 1923 and 2336 containing the Dbl homology (DH) and pleckstrin homology (PH) domains, suggesting that the GEF module represents the only IKKβ binding region on the anchoring protein (Fig. 3B, top). To precisely identify the IKKβ interaction site(s) on AKAP-Lbc, a purified S-tagged fragment of IKKβ encompassing residues 307 to 756 was used as a probe to screen a solid-phase peptide array of overlapping 21-residue peptides (each displaced by three amino acids) spanning the AKAP-Lbc region between residues 1923 and 2348 (Fig. 3C). Binding of the S-tagged IKKβ fragment was detected using HRP-conjugated S protein. Specific binding was detected at two sites between residues 2190 and 2107 and residues 2322 and 2336 of AKAP-Lbc (Fig. 3C). The first site is located within the last α-helix of the DH domain and contributes only modestly to the total IKKβ binding, whereas the second site corresponds to an α-helical region located at the end of the PH domain and represents the main IKKβ binding site (Fig. 3C). In line with these findings, mutation of tryptophan 2328, located in the middle of the second binding site, to leucine strongly impaired the ability of Flag-tagged AKAP-Lbc to associate with endogenous IKKβ in HEK293 cell lysates, as assessed by coimmunoprecipitation (Fig. 3D, top). Tryptophan was mutated to a leucine in order to suppress the aromatic properties of the side chain while preserving the hydrophobic character. In conclusion, our mapping analysis indicates that the region included between residues 2322 and 2336 of AKAP-Lbc represents the main anchoring site for IKKβ.
Fig 3.

Mapping of the IKKβ binding site(s) on AKAP-Lbc. (A) Schematic representation of the protein domain organization of AKAP-Lbc. The PKA binding domain (PKA), the PKN- and PKD-interacting region, and the Dbl (DH) and pleckstrin (PH) homology domains are shown. (B) HEK293 cells were transfected with HA-tagged IKKβ in combination with Flag-tagged fragments of AKAP-Lbc encompassing residues 1 to 503, 504 to 1000, 1001 to 1387, 1388 to 1922, 1923 to 2336, and 2337 to 2817. Cell lysates were subjected to immunoprecipitation with anti-Flag antibodies. Western blots of the immunoprecipitates and of the cell extracts were revealed using anti-HA polyclonal antibodies to detect HA-IKKβ (top and middle) or anti-Flag monoclonal antibodies to detect the Flag-tagged AKAP-Lbc fragments (bottom). (C) Peptide array analysis of the IKKβ binding region on AKAP-Lbc. The array was incubated with 10 nM S-tagged IKKβ-307-756 fragment. Solid-phase binding was assessed using HRP-conjugated S protein. The IKKβ binding peptides are numbered. (D) HEK293 cell lysates transfected with control vector or plasmids encoding Flag-AKAP-Lbc or Flag-AKAP-Lbc W2328L were immunoprecipitated using anti-Flag antibodies. Proteins in the extracts and immunoprecipitates were identified by immunoblotting using antibodies against IKKβ or the Flag tag, as indicated. Data are representative of three independent experiments.
The AKAP-Lbc/IKKβ complex mediates α1-AR-induced NF-κB activation in cardiomyocytes.
We have previously shown that AKAP-Lbc integrates signals from α1-ARs (27, 28, 32). Based on these findings, we raised the hypothesis that AKAP-Lbc, through the recruitment of IKKβ, could mediate α1-AR-induced activation of NF-κB. To address this question, we used RNA interference to determine whether reduced expression of AKAP-Lbc perturbed the ability of α1b-ARs to promote NF-κB transcriptional activity in HEK293 cells. Short hairpin RNAs (shRNAs) specific to AKAP-Lbc or control shRNAs were delivered into HEK293 cells using lentiviral vectors (27). Gene silencing was confirmed upon immunoblot analysis of cell lysates using AKAP-Lbc-specific antibodies (Fig. 4B, upper). Infected cells were transfected with empty vector or the plasmid encoding the HA-tagged α1b-AR in combination with NF-κB–luciferase and Renilla luciferase reporter constructs. After a 24-h serum starvation, cells were incubated for 8 h in the absence or presence of 10−4 M epinephrine (EPI), and NF-κB transcriptional activity was assessed with the luciferase reporter assay. Our results indicate that AKAP-Lbc silencing impairs by 57% the ability of α1b-ARs to induce NF-κB activation following epinephrine stimulation (Fig. 4A). The inhibitory effect due to silencing was totally reversed when HEK293 cells were rescued with a silencing-resistant form of AKAP-Lbc (Fig. 4A). Importantly, this was not observed when cells were rescued with the AKAP-Lbc W2328L mutant that cannot anchor IKKβ (Fig. 4A).
Fig 4.

AKAP-Lbc-IKKβ complex mediates α1-AR-induced NF-κB activation in cardiomyocytes. (A) HEK293 cells were infected using control lentiviruses or lentiviruses encoding AKAP-Lbc shRNAs and subsequently transfected with NF-κB–luciferase and Renilla luciferase reporter constructs in combination with either control vector (no receptor), the vector encoding the HA-tagged α1b-AR alone, or the vector encoding the HA-tagged α1b-AR together with the plasmids encoding the silencing-resistant mutants of AKAP-Lbc (AKAP-Lbc*) or AKAP-Lbc W2328L (AKAP-Lbc* W2328L). After a 24-h serum starvation, cells were treated with epinephrine (EPI) for 8 h or left untreated. Firefly luciferase activity was normalized to Renilla luciferase activity. Results are the means ± standard errors (SE) from 4 to 10 independent experiments. *, P < 0.05. (B) Expression of AKAP-Lbc, HA-α1b-AR, and actin in the lysates was assessed by Western blotting using specific antibodies as indicated. (C) Rat NVMs were infected using control lentiviruses or lentiviruses encoding wild-type or mutated AKAP-Lbc shRNAs and subsequently transfected with NF-κB–luciferase and Renilla luciferase reporter constructs. Seventy-two hours after infection, cells were incubated for 8 h in the absence or presence of 10−4 M PE. Firefly luciferase activity was normalized to Renilla luciferase activity. Results are the means ± SE from five independent experiments. *, P < 0.05. (D) Expression of AKAP-Lbc and actin in the lysates was assessed by Western blotting using specific antibodies as indicated. (E) Rat NVMs were serum starved for 24 h and subsequently treated with 10−4 M PE for the indicated periods of time. Expression of IκBα and actin in the lysates was assessed by Western blotting using specific antibodies as indicated. (F) Rat NVMs were infected as indicated in panel C. Seventy-two hours after infection, cells were incubated for 15 min in the absence or presence of 10−4 M PE. Expression of IκBα, AKAP-Lbc, and actin in the lysates was assessed by Western blotting using specific antibodies as indicated. (G) Quantitative analysis of the expression of IκBα in the cell lysates was obtained by densitometry. Untr., untreated. The amount of IκBα was normalized to the actin content of cell extracts. Results are the means ± SE from four independent experiments. *, P < 0.05.
In a similar set of experiments, we could show that the ability of α1b-ARs to induce IKKβ activation is severely compromised in AKAP-Lbc-silenced cells, as assessed using a kinase assay that measured the ability of immunoprecipitated endogenous IKKβ to induce the phosphorylation of a purified GST-fused protein fragment encompassing the first 55 amino acids of IκBα. Rescuing HEK293 cells with AKAP-Lbc, but not with its W2328L mutant, fully restored the ability of α1b-AR-induced IKKβ activation (Fig. 5A, top, and B). Collectively, these findings suggest that the inhibition of the IKKβ-NF-κB pathway induced by AKAP-Lbc silencing is strictly dependent on reduced AKAP-Lbc expression and not due to an off-target effect, and that the ability of α1b-AR to induce IKKβ activity and NF-κB activation requires the integrity of the AKAP-Lbc/IKKβ complex.
Fig 5.

Formation of the AKAP-Lbc/IKKβ complex is required for AKAP-Lbc-mediated IKKβ activation. (A) HEK293 cells were infected using control lentiviruses or lentiviruses carrying AKAP-Lbc shRNAs and subsequently transfected with either the vector encoding the HA-tagged α1b-AR alone or the vector encoding the HA-tagged α1b-AR together with the plasmids encoding the silencing resistant mutants of AKAP-Lbc (AKAP-Lbc*) or AKAP-Lbc W2328L (AKAP-Lbc* W2328L). After a 24-h serum starvation, cells were treated with phenylephrine (PE) for 15 min or left untreated. IKKβ immunocomplexes were isolated from cell extracts and incubated with GST-IκBα-1-55 and ATP. Phospho-GST-IκBα-1-55 was detected by immunoblotting using anti-phosphoserine 32/34-specific antibodies (top). (B) Quantitative analysis of phosphorylated GST-IκBα-1-55 was obtained by densitometry. The amount of phospho-GST-IκBα-1-55 was normalized to the total amount of GST-IκBα-1-55 and IKKβ. Results are expressed as means ± SE from 3 experiments. *, P < 0.05. The amounts of GST-IκBα-1-55, IKKβ, and Flag-AKAP-Lbc constructs and HA-α1b-AR were detected by Western blotting using specific antibodies as indicated. (C) HEK293 cells were transfected with the empty pFlag vector or the plasmids encoding Flag-AKAP-Lbc-S1565A or Flag-AKAP-Lbc-S1565A/W2328L. After a 24-h serum starvation, IKKβ immunocomplexes were isolated from cell extracts and used to phosphorylate purified GST-IκBα-1-55 as indicated for panel A. (D) Quantitative analysis of phosphorylated GST-IκBα-1-55 was obtained as indicated for panel B. Results are expressed as means ± SE from 4 experiments. *, P < 0.05.
In line with these results, we showed that mutation of tryptophan 2328 strongly reduces the ability of a mutant form of AKAP-Lbc displaying constitutive Rho-GEF activity (AKAP-Lbc S1565A) (29, 33) to induce IKKβ activation when overexpressed in HEK293 cells (Fig. 5C, top, and D).
Importantly, AKAP-Lbc also mediates NF-κB activation in cardiomyocytes, as shown by the fact that silencing of AKAP-Lbc expression in primary cultures of rat NVMs using two independent shRNAs reduced phenylephrine (PE)-induced NF-κB transcriptional activity by 60 to 65% (Fig. 4C). No inhibition of NF-κB activation was observed in cells infected with control lentiviruses encoding the mutated AKAP-Lbc shRNA (Fig. 4C).
NF-κB is maintained in an inactive conformation in the cytoplasm through an interaction with its inhibitor, IκB. Upon activation, IKKβ directly phosphorylates IκB on serines 32 and 36. This targets IκB for polyubiquitination and degradation by the 26S proteasome, which permits the translocation of NF-κB to the nucleus (14). In this context, our results indicate that treatment of rat NVMs with 10−4 M PE induces a transient degradation of endogenous IκBα as assessed by immunoblotting (Fig. 4E). The expression of IκBα is reduced by 50% at 15 min after stimulation and is totally recovered at 45 min after stimulation due to the newly synthesized IκBα (Fig. 4E, top). Interestingly, AKAP-Lbc silencing reduced by 61 to 64% the ability of PE to induce IκBα degradation (Fig. 4F, top, lanes 4 and 6, and G), confirming the notion that the AKAP-Lbc/IKKβ complex mediates α1b-AR-induced NF-κB activation in cardiomyocytes through the regulation of IκBα degradation. In control experiments, no inhibition of IκBα degradation was observed in cells infected with control lentiviruses encoding the mutated AKAP-Lbc shRNA (Fig. 4F, top, lane 8, and G).
AKAP-Lbc promotes the activation of IKKβ through an RhoA-Rho kinase pathway.
Previous findings indicate that Rho kinase, a direct effector of RhoA, can activate IKKβ (34). Knowing that AKAP-Lbc is a guanine nucleotide exchange factor that can directly activate RhoA, we investigated the possibility that AKAP-Lbc stimulates NF-κB signaling through an RhoA-Rho kinase-IKKβ-dependent pathway. In agreement with our hypothesis, inhibition of RhoA, Rho kinase, IKKβ, or NF-κB using a dominant-negative mutant of RhoA (RhoA T19N), the Rho kinase-specific inhibitor Y-27362, a dominant-negative mutant of IKKβ, or the IκBα (32Ala/36Ala) mutant (14) (which functions as a supersuppressor of NF-κB activation), respectively, significantly reduced the transcriptional activation of NF-κB induced by the overexpression of the AKAP-Lbc S1565A mutant (Fig. 6A).
Fig 6.

AKAP-Lbc promotes the activation of IKKβ through a RhoA-Rho kinase-dependent pathway. (A) HEK293 cells were transfected with NF-κB–luciferase and Renilla luciferase reporter constructs in combination with either control vector, the plasmid encoding Flag-AKAP-Lbc S1565A, or the plasmid encoding Flag-AKAP-Lbc S1565A together with the cDNAs encoding the Flag-tagged dominant-negative (DN) mutants of RhoA, IKKβ, or IκBα. After a 24-h serum starvation, cells were incubated in the absence or presence of 10 μM Y27632, RO318220, SB203580, and PD98059 for 8 h. Firefly luciferase activity was normalized to Renilla luciferase activity. Results are the means ± SE from 4 to 10 independent experiments. *, P < 0.05. (B) Expression of Flag-AKAP-Lbc and the Flag-tagged dominant-negative (DN) mutants of RhoA, IKKβ, and IκBα in the lysates was assessed by Western blotting using specific antibodies as indicated. (C) HEK293 cells were transfected with the vectors encoding GFP or AKAP-Lbc-S1565A-GFP in combination with the cDNAs encoding the empty Flag vector or the plasmid encoding the Flag-RhoA DN mutant. After a 24-h serum starvation, cells were incubated for 2 h with or without 10 μM Y27632. IKKβ immunocomplexes were isolated from cell extracts and incubated with GST-IκBα-1-55 and ATP. Phospho-GST-IκBα-1-55 was detected by immunoblotting using anti-phosphoserine 32/34-specific antibodies (top). The amounts of GST-IκBα-1-55, IKKβ, Flag-RhoA DN, AKAP-Lbc-S1565A-GFP, and GFP were detected by Western blotting using specific antibodies as indicated. (D) Quantitative analysis of phosphorylated GST-IκBα-1-55 was obtained by densitometry. The amount of phospho-GST-IκBα-1-55 was normalized to the total amount of GST-IκBα-1-55 and IKKβ. Results are expressed as means ± SE from 3 different experiments. *, P < 0.05. (E) Rat NVMs were infected using control lentiviruses or lentiviruses carrying wild-type or mutated AKAP-Lbc shRNAs. Ninety-six hours after infection, cells were incubated for 15 min in the absence or presence of 10−4 M PE. Endogenous IKKβ was immunoprecipitated, and its kinase activity was determined as indicated for panel C. Expression of AKAP-Lbc was assessed using specific antibodies. (F) Quantitative analysis of phosphorylated GST-IκBα-1-55 was obtained as indicated for panel D. *, P < 0.05.
Previous findings have shown that AKAP-Lbc interacts with several kinases, including PKCη, p38α, and MEK1, that also could modulate NF-κB signaling (28, 32, 35). Importantly, control experiments revealed that inhibition of these kinases using the specific inhibitors RO318220, SB203580, and PD098059, respectively, did not affect in a significant manner AKAP-Lbc-mediated NF-κB activation (Fig. 6A). These findings suggest that RhoA, Rho kinase, and IKKβ mediate NF-κB activation downstream of AKAP-Lbc.
To provide a formal demonstration that the RhoA/Rho kinase pathway links AKAP-Lbc to the activation of IKKβ, we determined whether overexpression of the GFP-tagged AKAP-Lbc S1565A active mutant in HEK293 could enhance the activity of endogenous IKKβ and whether this effect could be reduced by the inhibition of RhoA and Rho kinase. IKKβ activity was determined using a kinase assay that measured the ability of immunoprecipitated endogenous IKKβ to induce the phosphorylation of a purified GST-fused protein fragment encompassing the first 55 amino acids of IκBα. Interestingly, inhibition of both RhoA and Rho kinase impaired by 58 and 69%, respectively, the ability of AKAP-Lbc to induce IKKβ activation (Fig. 6C, row 1, lanes 5 and 6, and D). In line with these findings, the ability of PE to stimulate IKKβ activity in rat NVMs was reduced by 65 and 70% by the silencing of AKAP-Lbc expression and by the inhibition of Rho kinase using Y27632, respectively (Fig. 6E, top, lanes 4 and 8, and F). In control experiments, no inhibition of IKKβ activity was observed in rat NVMs infected with control lentiviruses carrying the mutated AKAP-Lbc shRNA (Fig. 6E, upper, lane 6, and F).
Collectively, these findings suggest that the AKAP-Lbc/RhoA/Rho kinase pathway mediates α1-AR-induced IKKβ activation in cardiomyocytes.
The AKAP-Lbc/IKKβ complex mediates α1-AR-induced cardiomyocyte hypertrophy.
Several lines of evidence indicate that the transcription factor NF-κB is a crucial mediator of cardiomyocyte hypertrophy (11, 16, 20). While it is now clear that NF-κB can be activated by a variety of GPCRs, including α1-ARs, AT1-Rs, and ET-1-Rs, the signaling complexes that integrate hypertrophic signals from these receptors to coordinate the activation of NF-κB are poorly characterized.
Based on these results and on our present findings indicating that the AKAP-Lbc/IKKβ complex mediates α1-AR-induced NF-κB activation in cardiomyocytes, we investigated the possibility that this complex controls the transduction of hypertrophic signals downstream of α1-ARs. To address this question, we performed experiments that combined silencing of the endogenous anchoring protein in rat NVMs and rescue with human AKAP-Lbc forms that are refractory to the shRNA (27). Rat NVMs infected using control lentiviruses expressing GFP or with lentiviruses encoding GFP and AKAP-Lbc shRNAs were subsequently transfected with the cDNA encoding the Flag-tagged form of AKAP-Lbc or AKAP-Lbc W2328L, which is unable to anchor IKKβ. Cells were stimulated for 24 h with 10−4 M PE, and induction of cardiomyocytes was hypertrophically assessed by measuring cell size as well as sarcomere assembly and reorganization. Silencing of AKAP-Lbc expression reduced by 61% the PE-induced increase in cardiomyocyte surface area compared to that of NVMs infected with control lentiviruses (Fig. 7 and 8). This effect was associated with an inhibition of PE-induced sarcomere reorganization as visualized by staining cardiomyocytes using antibodies against α-actinin and the Z-disk-associated protein ZASP, respectively (Fig. 7). Interestingly, rescue with wild-type AKAP-Lbc completely restored α1-AR-mediated hypertrophy and sarcomere reorganization, whereas rescue with the AKAP-Lbc W2328L mutant did not (Fig. 7 and 8). These findings are consistent with the idea that the anchoring of IKKβ to AKAP-Lbc is required for transducing hypertrophic signals downstream of α1-ARs.
Fig 7.

AKAP-Lbc-IKKβ complex mediates α1-AR-induced cardiomyocyte hypertrophy and sarcomere reorganization. Rat NVMs were infected with lentiviruses carrying GFP (control) or both GFP and AKAP-Lbc shRNAs and subsequently transfected with the empty Flag vector or the cDNAs encoding human AKAP-Lbc and AKAP-Lbc W2328L. Forty-eight hours after transfection, cells were incubated for 24 h with 10−4 M PE. Cells were then fixed, permeabilized, and incubated with mouse anti-α-actinin (A) or anti-ZASP (B) antibodies followed by rhodamine-conjugated donkey anti-mouse secondary antibodies to visualize the sarcomeres. The expression of Flag-AKAP-Lbc was detected using rabbit anti-Flag polyclonal antibodies and AMCA-conjugated donkey anti-rabbit secondary antibodies, whereas GFP expression was visualized directly by fluorescent excitation at 490 nm.
Fig 8.

Quantitation of the hypertrophic effect mediated by the AKAP-Lbc-IKKβ complex. Mean cell surface area (±SE) of cardiomyocytes infected and transfected as indicated in Fig. 7 and treated for 24 h with or without 10−4 M PE. The cell surface area was determined on a total of 150 to 200 GFP-positive cardiomyocytes derived from 6 independent experiments. *, P < 0.05.
It has been shown previously that the PH domain of AKAP-Lbc also contributes to the regulation of the GEF activity of the anchoring protein (36) and to the recruitment of PKCη (28). We therefore determined the impact of the W2328L mutation on the ability of AKAP-Lbc to activate RhoA and bind PKCη. In this respect, control experiments revealed that the W2328L mutation does not alter the ability of α1-AR to promote AKAP-Lbc Rho-GEF activity, as shown using the rhotekin pulldown assay (Fig. 9A, top), or the capacity of AKAP-Lbc to recruit PKCη, as assessed by coimmunoprecipitation (Fig. 9B, top). This indicates that the α-helix encompassing residues 2320 to 2338 is selectively involved in binding IKKβ, and that the impact of the W2328L mutation on the hypertrophic responses induced by AKAP-Lbc is the consequence of an impaired IKKβ anchoring and not of additional nonspecific effects of the mutation.
Fig 9.
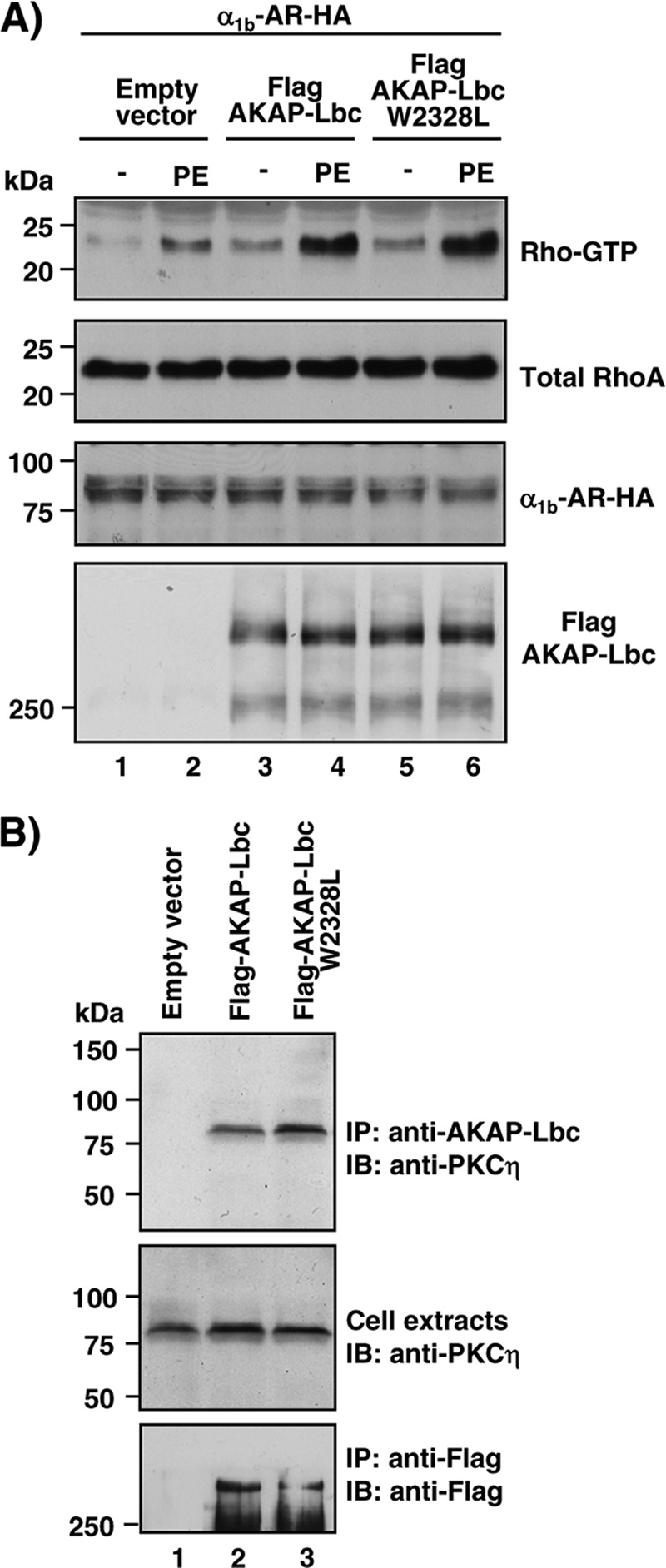
AKAP-Lbc and the AKAP-Lbc W2328L mutant display similar RhoA-activating and PKCη binding properties. (A) HEK293 cells were transfected with the HA-tagged α1b-AR in combination with either empty vector or the plasmids encoding the Flag-tagged forms of AKAP-Lbc or AKAP-Lbc W2328L. After a 24-h serum starvation, cells were treated with 10−4 M PE for 15 min or left untreated. Cell lysates were incubated with GST-RBD beads. The bound RhoA was detected with a monoclonal anti-RhoA antibody (top). The relative amounts of total RhoA, HA-α1b-AR, and Flag-AKAP-Lbc proteins in the cell lysates were assessed by Western blotting as indicated. Results are representative of three independent experiments. (B) HEK293 cells were transfected with the empty Flag vector or the plasmids encoding the Flag-tagged forms of AKAP-Lbc or AKAP-Lbc W2328L. Cell lysates were subjected to immunoprecipitation with anti-Flag antibodies. Western blots of the immunoprecipitates and of the cell extracts were revealed using anti-PKCη polyclonal antibodies to detect endogenous PKCη (top and middle) or anti-Flag monoclonal antibodies to detect the Flag-tagged AKAP-Lbc proteins (bottom). Results are representative of three independent experiments.
Recent evidence suggests that α1-ARs can promote IL-6 expression in cardiomyocytes through NF-κB-mediated transcription (37), and that IL-6 can induce hypertrophy when infused in adult rats (38). This raises the possibility that IL-6 contributes to the growth responses induced by α1-ARs. To test this hypothesis, we determined the impact of inhibiting IL-6 using neutralizing antibodies on the ability of α1-ARs to promote fetal gene transcription. As shown in Fig. 10A, incubation of rat NVMs with IL-6 neutralizing antibodies reduced PE-induced transcription of atrial natriuretic factor (ANF) and β-myosin heavy chain (β-MHC) by 54 and 60%, respectively, suggesting that IL-6 represents a key mediator of the hypertrophic responses induced by α1-ARs.
Fig 10.

α1-ARs induce hypertrophic gene transcription through the AKAP-Lbc-IKKβ complex and IL-6. (A) Real-time PCR analysis of ANF and β-MHC expression was performed on total RNA preparations obtained from NVMs that were serum starved for 48 h and subsequently incubated for 24 h in the absence or presence of 10−4 M PE and IL-6 neutralizing antibodies (IL-6 Ab). Results are expressed as means ± SE from eight independent experiments. *, P < 0.05. (B) Single-cell real-time PCR analysis of ANF, β-MHC, and IL-6 expression was performed on total RNA preparations obtained from FACS-sorted NVMs that were infected as indicated in the legend to Fig. 5A and subsequently transfected with rescue constructs encoding mCherry-tagged AKAP-Lbc or AKAP-Lbc W2328L. Results are expressed as means ± SE from six independent experiments. *, P < 0.05.
Based on these results, we next determined whether the AKAP-Lbc/IKKβ complex could control the transcription of IL-6 and the subsequent induction of hypertrophic genes in response to α1-AR activation. To address this point, rat NVMs were infected with lentiviruses encoding AKAP-Lbc shRNAs and GFP and were subsequently transfected with the plasmids encoding mCherry fusion protein of AKAP-Lbc or AKAP-Lbc W2328L. After 24 h of stimulation with 10−4 M PE, GFP-positive and GFP/mCherry-double-positive cardiomyocytes were sorted by FACS, and hypertrophic gene expression was determined by real-time PCR. Our results indicate that AKAP-Lbc silencing strongly inhibits PE-mediated induction of IL-6, ANF, and β-MHC (Fig. 10B). Interestingly, while rescue with wild-type AKAP-Lbc fully restored α1-AR-mediated transcription of IL-6 and fetal genes, rescue with the AKAP-Lbc W2328L mutant did not (Fig. 10B). Collectively, these findings strongly suggest that the AKAP-Lbc/IKKβ complex plays a crucial role in mediating α1-AR-induced transcription of the prohypertrophic cytokine IL-6 and activation of the hypertrophic gene program.
DISCUSSION
During the last few years, several studies using rat NVMs as a model system have shown that NF-κB is required for the hypertrophic effects induced by numerous GPCR agonists, including catecholamines, Ang-II, and ET-1 (15, 17, 19). Moreover, in vivo studies confirmed that inhibition of NF-κB signaling significantly reduces cardiac hypertrophy induced by chronic infusion of GPCR agonist, pressure overload, and myocardial infarction (16, 20–22, 39). While NF-κB is now recognized as a key transcription factor mediating cardiac hypertrophy and remodeling, it is currently poorly understood how hypertrophic signals emanating from membrane receptors are integrated and transmitted to NF-κB.
In the present study, we demonstrate that the RhoA-selective exchange factor AKAP-Lbc forms a complex with the kinase IKKβ, which transduces hypertrophic signals from α1-ARs down to NF-κB (Fig. 11). We previously showed that AKAP-Lbc Rho-GEF activity is enhanced in response to α1-AR stimulation through a pathway that requires Gα12 (27). Our current data now support a model where activated AKAP-Lbc promotes the formation of RhoA-GTP and the activation of Rho kinase, which, in turn, enhances the activity of anchored IKKβ (Fig. 11). This leads to the phosphorylation and degradation of IκBα and to the activation of NF-κB. Finally, activated NF-κB induces the transcription of the IL-6 gene and the subsequent stimulation of IL-6-mediated pathways controlling fetal gene transcription and cardiomyocyte hypertrophy (Fig. 11). Overall, these findings provide a new mechanistic hypothesis explaining how anchoring proteins convey hypertrophic signals to interleukin-mediated transcriptional reprogramming events in cardiomyocytes.
Fig 11.
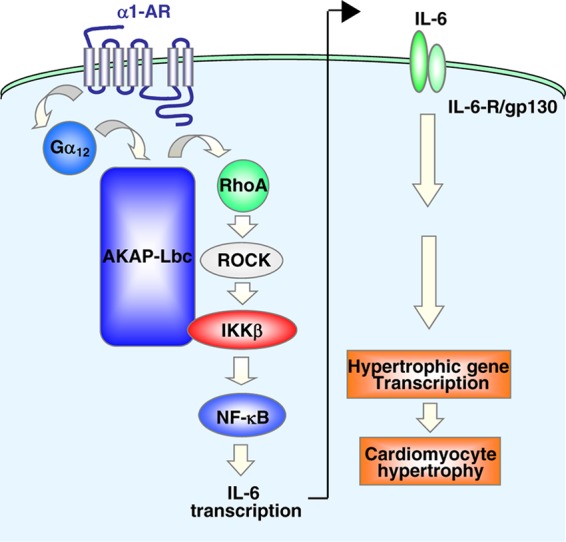
Model showing the role of the AKAP-Lbc–IKKβ complex in the regulation of cardiomyocyte hypertrophy through the NF-κB–IL-6 signaling network. See the text for details.
Intriguingly, no NF-κB-responsive elements have been identified in the promoter regions of genes associated with cardiomyocyte hypertrophy. Therefore, it was postulated that NF-κB mediates its growth-promoting effects through indirect mechanisms. In this context, it has been recently shown that, in rat NVMs, NF-κB can activate the transcription of the IL-6 gene in response to α1-AR stimulation (37). Our results now indicate that the AKAP-Lbc/IKKβ complex functions as a key mediator of α1-AR-mediated NF-κB activation and IL-6 transcription (Fig. 4 and 10B) and that inhibition of IL-6 significantly impairs the hypertrophic responses induced by α1-ARs (Fig. 10A). Therefore, it appears that NF-κB can regulate cardiomyocyte hypertrophy at least in part through the regulation of IL-6 production. These findings provide a molecular explanation to previous observations showing that IL-6 can mediate cardiac hypertrophy when infused in adult rats (38).
Recent findings indicate that NF-κB can exert part of its hypertrophic effects through a physical interaction with the transcription factor NFAT (39). This suggests that NF-κB can control cardiomyocyte growth by directly influencing the activity and nuclear shuttling of prohypertrophic transcription factors (39) and, as shown here, by enhancing the transcription of the prohypertrophic cytokine IL-6 (Fig. 10A).
Our current findings indicate that AKAP-Lbc mediates the activation of anchored IKKβ through RhoA and Rho kinase (Fig. 6). This raises the question of whether these signaling proteins can also form a complex with AKAP-Lbc. While we can show that AKAP-Lbc and RhoA can coimmunoprecipitate from rat NVMs (Fig. 1G, upper), we were not able to detect Rho kinase in AKAP-Lbc immunoprecipitates (data not shown). This suggests that Rho kinase is not physically associated with the complex. In this situation, the transduction of signals from activated RhoA and AKAP-Lbc-anchored IKKβ might be mediated by a local pool of Rho kinase that could be maintained in the vicinity of the AKAP-Lbc complex. Alternatively, the interaction between endogenous AKAP-Lbc and Rho kinase might be too labile or transient to be detected by coimmunoprecipitation.
In addition to the RhoA-Rho kinase-IKKβ pathway described here, other signaling cascades coordinated by AKAP-Lbc have been shown to play a role in the hypertrophic responses induced by α1-ARs. In fact, it has been recently shown that AKAP-Lbc can also assemble a signaling complex involved in protein kinase D (PKD) activation (40). Activated PKD is released from the AKAP-Lbc complex and translocates to the nucleus, where it phosphorylates histone deacetylase 5 (HDAC5). This promotes HDAC5 nuclear export, which favors chromatin-remodeling events that were proposed to enhance transcriptional activation of hypertrophic genes (28).
Interestingly, interfering with the ability of AKAP-Lbc to interact with PKD impairs the ability of α1-ARs to promote cardiomyocyte hypertrophy (28). This suggests that, taken individually, the IKKβ- and PKD-mediated transduction cascades organized by the AKAP-Lbc complex are both necessary but are not sufficient to mediate the growth response to PE. One possibility is that activation of the hypertrophic response requires both chromatin derepression through PKD-mediated HDAC5 nuclear export and activation of hypertrophic gene transcription through NF-κB activation.
Overall, these findings highlight the importance of AKAP-based signaling complexes in the regulation of NF-κB (41) and interleukin-mediated hypertrophic signaling. Based on these results, one could speculate that targeting the binding interface between AKAP-Lbc and IKKβ represents an approach to attenuate cardiomyocyte hypertrophy.
In conclusion, the implications of our findings are 2-fold. First, they identify the AKAP-Lbc/IKKβ activation complex as a crucial mediator of NF-κB activation and IL-6 transcription in cardiomyocytes. Second, they identify AKAP-Lbc-mediated IL-6 transcription as a key event controlling cardiomyocyte hypertrophy.
ACKNOWLEDGMENTS
We acknowledge Monique Nenniger-Tosato for excellent technical assistance in preparing primary cultures of rat NVMs and G. Carnegie (University of Illinois, Chicago) for providing the vector encoding mCherry-AKAP-Lbc.
This work was supported by grants 3100A0-138289 (to D.D.) of the Fonds National Suisse de la Recherche Scientifique and by a grant of the Muschamp Foundation (to D.D.).
Footnotes
Published ahead of print 22 October 2012
REFERENCES
- 1. Hill JA, Olson EN. 2008. Cardiac plasticity. N. Engl. J. Med. 358:1370–1380 [DOI] [PubMed] [Google Scholar]
- 2. Frey N, Katus HA, Olson EN, Hill JA. 2004. Hypertrophy of the heart: a new therapeutic target? Circulation 109:1580–1589 [DOI] [PubMed] [Google Scholar]
- 3. Towbin JA, Bowles NE. 2002. The failing heart. Nature 415:227–233 [DOI] [PubMed] [Google Scholar]
- 4. Milano CA, Dolber PC, Rockman HA, Bond RA, Venable ME, Allen LF, Lefkowitz RJ. 1994. Myocardial expression of a constitutively active alpha 1B-adrenergic receptor in transgenic mice induces cardiac hypertrophy. Proc. Natl. Acad. Sci. U. S. A. 91:10109–10113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. O'Connell TD, Ishizaka S, Nakamura A, Swigart PM, Rodrigo MC, Simpson GL, Cotecchia S, Rokosh DG, Grossman W, Foster E, Simpson PC. 2003. The alpha(1A/C)- and alpha(1B)-adrenergic receptors are required for physiological cardiac hypertrophy in the double-knockout mouse. J. Clin. Investig. 111:1783–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pare GC, Bauman AL, McHenry M, Michel JJ, Dodge-Kafka KL, Kapiloff MS. 2005. The mAKAP complex participates in the induction of cardiac myocyte hypertrophy by adrenergic receptor signaling. J. Cell Sci. 118:5637–5646 [DOI] [PubMed] [Google Scholar]
- 7. Rockman HA, Wachhorst SP, Mao L, Ross J., Jr 1994. ANG II receptor blockade prevents ventricular hypertrophy and ANF gene expression with pressure overload in mice. Am. J. Physiol. 266:H2468–H2475 [DOI] [PubMed] [Google Scholar]
- 8. Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I. 2004. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat. Cell Biol. 6:499–506 [DOI] [PubMed] [Google Scholar]
- 9. Heineke J, Molkentin JD. 2006. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 7:589–600 [DOI] [PubMed] [Google Scholar]
- 10. Hall G, Hasday JD, Rogers TB. 2006. Regulating the regulator: NF-kappaB signaling in heart. J. Mol. Cell. Cardiol. 41:580–591 [DOI] [PubMed] [Google Scholar]
- 11. Purcell NH, Molkentin JD. 2003. Is nuclear factor kappaB an attractive therapeutic target for treating cardiac hypertrophy? Circulation 108:638–640 [DOI] [PubMed] [Google Scholar]
- 12. Hacker H, Karin M. 2006. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006:re13 doi:10.1126/stke.3572006re13 [DOI] [PubMed] [Google Scholar]
- 13. Scheidereit C. 2006. IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene 25:6685–6705 [DOI] [PubMed] [Google Scholar]
- 14. Perkins ND. 2007. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat. Rev. Mol. Cell Biol. 8:49–62 [DOI] [PubMed] [Google Scholar]
- 15. Cook SA, Novikov MS, Ahn Y, Matsui T, Rosenzweig A. 2003. A20 is dynamically regulated in the heart and inhibits the hypertrophic response. Circulation 108:664–667 [DOI] [PubMed] [Google Scholar]
- 16. Freund C, Schmidt-Ullrich R, Baurand A, Dunger S, Schneider W, Loser P, El-Jamali A, Dietz R, Scheidereit C, Bergmann MW. 2005. Requirement of nuclear factor-kappaB in angiotensin II- and isoproterenol-induced cardiac hypertrophy in vivo. Circulation 111:2319–2325 [DOI] [PubMed] [Google Scholar]
- 17. Hirotani S, Otsu K, Nishida K, Higuchi Y, Morita T, Nakayama H, Yamaguchi O, Mano T, Matsumura Y, Ueno H, Tada M, Hori M. 2002. Involvement of nuclear factor-kappaB and apoptosis signal-regulating kinase 1 in G-protein-coupled receptor agonist-induced cardiomyocyte hypertrophy. Circulation 105:509–515 [DOI] [PubMed] [Google Scholar]
- 18. Kawamura N, Kubota T, Kawano S, Monden Y, Feldman AM, Tsutsui H, Takeshita A, Sunagawa K. 2005. Blockade of NF-kappaB improves cardiac function and survival without affecting inflammation in TNF-alpha-induced cardiomyopathy. Cardiovasc. Res. 66:520–529 [DOI] [PubMed] [Google Scholar]
- 19. Purcell NH, Tang G, Yu C, Mercurio F, DiDonato JA, Lin A. 2001. Activation of NF-kappa B is required for hypertrophic growth of primary rat neonatal ventricular cardiomyocytes. Proc. Natl. Acad. Sci. U. S. A. 98:6668–6673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sorriento D, Santulli G, Fusco A, Anastasio A, Trimarco B, Iaccarino G. 2010. Intracardiac injection of AdGRK5-NT reduces left ventricular hypertrophy by inhibiting NF-kappaB-dependent hypertrophic gene expression. Hypertension 56:696–704 [DOI] [PubMed] [Google Scholar]
- 21. Usui S, Maejima Y, Pain J, Hong C, Cho J, Park JY, Zablocki D, Tian B, Glass DJ, Sadoshima J. 2011. Endogenous muscle atrophy F-box mediates pressure overload-induced cardiac hypertrophy through regulation of nuclear factor-kappaB. Circ. Res. 109:161–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zelarayan L, Renger A, Noack C, Zafiriou MP, Gehrke C, van der Nagel R, Dietz R, de Windt L, Bergmann MW. 2009. NF-kappaB activation is required for adaptive cardiac hypertrophy. Cardiovasc. Res. 84:416–424 [DOI] [PubMed] [Google Scholar]
- 23. Pawson CT, Scott JD. 2010. Signal integration through blending, bolstering and bifurcating of intracellular information. Nat. Struct. Mol. Biol. 17:653–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Scott JD, Pawson T. 2009. Cell signaling in space and time: where proteins come together and when they're apart. Science 326:1220–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Beene DL, Scott JD. 2007. A-kinase anchoring proteins take shape. Curr. Opin. Cell Biol. 19:192–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Diviani D, Soderling J, Scott JD. 2001. AKAP-Lbc anchors protein kinase A and nucleates Galpha 12-selective Rho-mediated stress fiber formation. J. Biol. Chem. 276:44247–44257 [DOI] [PubMed] [Google Scholar]
- 27. Appert-Collin A, Cotecchia S, Nenniger-Tosato M, Pedrazzini T, Diviani D. 2007. The A-kinase anchoring protein (AKAP)-Lbc-signaling complex mediates alpha1 adrenergic receptor-induced cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. U. S. A. 104:10140–10145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Carnegie GK, Soughayer J, Smith FD, Pedroja BS, Zhang F, Diviani D, Bristow MR, Kunkel MT, Newton AC, Langeberg LK, Scott JD. 2008. AKAP-Lbc mobilizes a cardiac hypertrophy signaling pathway. Mol. Cell 32:169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Diviani D, Abuin L, Cotecchia S, Pansier L. 2004. Anchoring of both PKA and 14-3-3 inhibits the Rho-GEF activity of the AKAP-Lbc signaling complex. EMBO J. 23:2811–2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Petitprez S, Zmoos AF, Ogrodnik J, Balse E, Raad N, El-Haou S, Albesa M, Bittihn P, Luther S, Lehnart SE, Hatem SN, Coulombe A, Abriel H. 2011. SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ. Res. 108:294–304 [DOI] [PubMed] [Google Scholar]
- 31. Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. 1996. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272:263–267 [DOI] [PubMed] [Google Scholar]
- 32. Cariolato L, Cavin S, Diviani D. 2011. A-kinase anchoring protein (AKAP)-Lbc anchors a PKN-based signaling complex involved in alpha1-adrenergic receptor-induced p38 activation. J. Biol. Chem. 286:7925–7937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jin J, Smith FD, Stark C, Wells CD, Fawcett JP, Kulkarni S, Metalnikov P, O'Donnell P, Taylor P, Taylor L, Zougman A, Woodgett JR, Langeberg LK, Scott JD, Pawson T. 2004. Proteomic, functional, and domain-based analysis of in vivo 14-3-3 binding proteins involved in cytoskeletal regulation and cellular organization. Curr. Biol. 14:1436–1450 [DOI] [PubMed] [Google Scholar]
- 34. Anwar KN, Fazal F, Malik AB, Rahman A. 2004. RhoA/Rho-associated kinase pathway selectively regulates thrombin-induced intercellular adhesion molecule-1 expression in endothelial cells via activation of I kappa B kinase beta and phosphorylation of RelA/p65. J. Immunol. 173:6965–6972 [DOI] [PubMed] [Google Scholar]
- 35. Smith FD, Langeberg LK, Cellurale C, Pawson T, Morrison DK, Davis RJ, Scott JD. 2010. AKAP-Lbc enhances cyclic AMP control of the ERK1/2 cascade. Nat. Cell Biol. 12:1242–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zheng Y. 2001. Dbl family guanine nucleotide exchange factors. Trends Biochem. Sci. 26:724–732 [DOI] [PubMed] [Google Scholar]
- 37. Perez DM, Papay RS, Shi T. 2009. α1-Adrenergic receptor stimulates interleukin-6 expression and secretion through both mRNA stability and transcriptional regulation: involvement of p38 mitogen-activated protein kinase and nuclear factor-kappaB. Mol. Pharmacol. 76:144–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Melendez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. 2010. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension 56:225–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu Q, Chen Y, Auger-Messier M, Molkentin JD. 2012. Interaction between NFκB and NFAT coordinates cardiac hypertrophy and pathological remodeling. Circ. Res. 110:1077–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Carnegie GK, Smith FD, McConnachie G, Langeberg LK, Scott JD. 2004. AKAP-Lbc nucleates a protein kinase D activation scaffold. Mol. Cell 15:889–899 [DOI] [PubMed] [Google Scholar]
- 41. Shibolet O, Giallourakis C, Rosenberg I, Mueller T, Xavier RJ, Podolsky DK. 2007. AKAP13, a RhoA GTPase-specific guanine exchange factor, is a novel regulator of TLR2 signaling. J. Biol. Chem. 282:35308–35317 [DOI] [PubMed] [Google Scholar]