

Abstract
While a large number of mosquito-transmitted alphaviruses are known to cause serious human diseases, there are no licensed vaccines that protect against alphavirus infections. The alphavirus chikungunya virus (CHIKV) has caused multiple recent outbreaks of chikungunya fever. This virus has the potential to cause a worldwide epidemic and has generated strong interest in development of a prophylactic CHIKV vaccine. We report here on the development of a potent experimental vaccine for CHIKV based on a chimeric vesicular stomatitis virus (VSV) expressing the entire CHIKV envelope polyprotein (E3-E2-6K-E1) in place of the VSV glycoprotein (G). These VSVΔG-CHIKV chimeras incorporated functional CHIKV glycoproteins into the viral envelope in place of VSV G. The chimeric viruses were attenuated for growth in tissue culture but could be propagated to high titers without VSV G complementation. They also generated robust neutralizing antibody and cellular immune responses to CHIKV in mice after a single dose and protected mice against CHIKV infection. VSVΔG-alphavirus chimeras could have general applicability as alphavirus vaccines.
INTRODUCTION
Alphaviruses are found worldwide, and many are associated with serious disease in humans and other vertebrates. They are typically transmitted by mosquitoes to vertebrate hosts and can cause fever, arthritis, and lethal encephalitis (1). There are currently no licensed vaccines that protect against alphavirus infection and disease. Chikungunya virus (CHIKV) is an alphavirus that causes chikungunya fever. The virus was first isolated in 1953 in Tanzania and spread across Africa and Southeast Asia. More recent outbreaks have spread to Europe, and CHIKV infection has been diagnosed in the United States in travelers returning from areas of endemicity (2, 3). CHIKV has generated global public health concern, in part because its spread has been associated with a new mosquito vector, Aedes albopictus, which is widely distributed in Europe and the Americas (2).
CHIKV infection in humans results in high fever, headache, vomiting, skin rash, and crippling arthritis. While most symptoms resolve by about 10 days, the crippling arthritis can persist for months or even years (2). Recent CHIKV epidemics have shown even more severe symptoms, including encephalitis, hemorrhagic disease, and mortality (4).
CHIKV has a single, positive-sense, 11.8-kb RNA genome, which encodes four nonstructural proteins (nsP1 to nsP4) and five structural proteins (C, E3, E2, 6K, and E1) that are cleaved from a precursor to generate the capsid and envelope glycoproteins (5). Neutralizing antibodies (nAbs) play dominant roles in resolving alphavirus infections, and neutralizing epitopes have been mapped within E1 (fusion protein) and E2 (attachment protein) glycoproteins in several alphaviruses (6). However, cellular immunity is also capable of protecting against fatal alphaviral disease (7).
Recent CHIKV epidemics in the La Reunion Island and the Indian subcontinent, Southeast Asia, and Europe have generated substantial interest in development of CHIKV vaccines. A number of successful vaccine approaches based on expression of the E1 and E2 CHIKV glycoproteins have been described recently. These include chimeric alphavirus vectors (8, 9), an adenovirus vector (10), a virus-like particle (VLP) vaccine (11), a DNA vaccine (12), and an internal ribosome entry site (IRES)-based live attenuated CHIKV vaccine (13).
Vesicular stomatitis virus (VSV), a negative-strand RNA virus in the Rhabdoviridae family (Vesiculovirus genus), has been used extensively as an experimental vaccine vector against several viral and bacterial pathogens (14–22). VSV-based vaccine vectors are currently being used in HIV vaccine clinical trials (http://clinicaltrials.gov/ct2/show/NCT01438606).
In this study, we initially wanted to determine if we could construct VSV-based vaccine vectors expressing the entire CHIKV E3-E2-6K-E1 precursor polyprotein. We did this in a full-length VSV construct as well as in a VSV construct lacking the glycoprotein gene (VSVΔG). Interestingly, we found that the VSVΔG vector expressing CHIKV envelope proteins incorporated the CHIKV glycoproteins efficiently into virus particles and propagated without VSV G complementation. This chimeric VSV/alphavirus vector also induced more potent CHIKV immune responses than the full-length vector with VSV G and protected mice from CHIKV challenge after a single dose. Such chimeric viruses could be generally applicable as alphavirus vaccines.
MATERIALS AND METHODS
Cells.
Baby hamster kidney-21 (BHK-21) cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal bovine serum (FBS). Vero cells, derived from African green monkey kidney cells, were maintained in DMEM containing 10% FBS.
Plasmid constructions.
To generate pVSV-CHIKV, we first designed a codon-optimized synthetic E3-E2-6K-E1 gene (CHIKV S27 prototypic African strain) and had it synthesized with flanking XhoI and NheI restriction sites (Genscript, Inc.). This gene was inserted into XhoI-NheI-digested pVSVXN2 vector (23). pVSVΔG-CHIKV was prepared by deleting the VSV G gene from the pVSV-CHIKV by MluI-XhoI digestion, filling in with T4 DNA polymerase (New England BioLabs), and religation. pCAGGS-CHIKV was made by inserting the XhoI-NheI-digested synthetic E3-E2-6K-E1 fragment into corresponding sites of a modified pCAGGS vector (24) containing these sites.
Recombinant virus recovery.
Recombinant VSVs (rVSVs) were recovered from pVSV-CHIKV and pVSVΔG-CHIKV as described previously (25, 26). In brief, BHK-21 cells were infected at a multiplicity of infection (MOI) of 10 with vTF-7.3 (27), a vaccinia virus recombinant expressing T7 RNA polymerase. The cells were then transfected with rVSV plasmids (pVSV) together with support plasmids, pBS-N, pBS-P, pBS-L, and pBS-G, encoding VSV proteins. VSV-CHIKV was recovered by transferring the transfected cell supernatants onto fresh BHK-21 cells at 48 h posttransfection and collecting the supernatant containing the virus after another 48 h. Virus stock was prepared from individual plaques grown in BHK-21 cells and stored at −70°C. To recover VSV G-complemented VSVΔG-CHIKV, transfection supernatant was transferred to cells that were transfected with pCAGGS-G (28) 1 day prior, and supernatant containing the virus was collected after 48 h. The virus was further plaque purified on BHK-G cells (26), and VSV G-complemented stock was prepared and stored at −70°C. A part of VSVΔG-CHIKV recovery supernatant was also plaque purified without VSV G complementation on BHK-21 cells and further passaged on BHK-21 cells to generate a non-G-complemented VSVΔG-CHIKV stock.
pCAGGS transfection.
Ten micrograms of the appropriate pCAGGS vector diluted in 0.6 ml of OptiMEM (Invitrogen, CA) was mixed with 30 μl of Lipofectamine reagent (Invitrogen, CA), also diluted in 0.6 ml of OptiMEM, and incubated at room temperature for 30 min. Confluent monolayers of BHK-21 cells in 10-cm dishes were washed once with phosphate-buffered saline (PBS) and once with OptiMEM. OptiMEM (4.8 ml) was then added, followed by addition of the DNA-Lipofectamine mix and incubation for 5 h at 37°C. Subsequently, the transfection mix was removed from the plate and replaced with DMEM containing 10% FBS and incubated overnight at 37°C.
Indirect immunofluorescence microscopy.
BHK-21 cells on coverslips were infected with recombinant wild-type (rwt)-VSV, VSV-CHIKV, or VSVΔG-CHIKV. At 4 h postinfection, cells were washed two times with PBS and fixed with 3% paraformaldehyde at room temperature for 20 min. Cells were then washed with PBS containing 10 mM glycine (PBS-glycine) and incubated with a 1:200 dilution of either VSV G monoclonal antibodies ([MAbs] I1 and I14) (29) or an anti-CHIKV antibody (4-week serum from CHIKV-La Reunion [LR] strain-infected mouse). Following this, cells were washed with PBS-glycine and incubated with 1:500 diluted goat anti-mouse Alexa Fluor 488 IgG (Molecular Probes, Eugene, OR). Cells were then washed twice with PBS-glycine, mounted on slides using ProLong Gold antifade reagent with 4′,6′-diamidino-2-phenylindole (DAPI) (Molecular Probes, Eugene, OR), and imaged with a Nikon Eclipse 80i fluorescence microscope using a 40× objective.
Metabolic labeling and SDS-PAGE of purified viruses and cell lysates.
To label infected cells, BHK-21 cells in 3-cm dishes were infected with recombinant viruses or mock infected at an MOI of 20. At 5 h postinfection, cells were washed with methionine-free DMEM followed by labeling with 100 μCi of [35S]methionine in 1 ml of methionine-free DMEM for 30 min at 37°C. Cells were then washed twice with PBS and lysed with detergent lysis buffer (1% Nonidet P-40, 0.4% deoxycholate, 50 mM Tris-HCl, pH 8.0, 62.5 mM EDTA) on ice for 5 min. Lysates were collected and clarified at 16,000 × g for 2 min at 4°C. To label rVSVs, BHK-21 cells on 6-cm dishes were infected with rwt-VSV, VSV-CHIKV, or VSVΔG-CHIKV at an MOI of 100. After adsorption for 1 h, virus was removed and replaced with DMEM containing 5% FBS. After 2 h, cells were washed two times with prewarmed methionine-free DMEM. Subsequently, labeling medium (90% methionine-free DMEM, 9% serum-free DMEM, 1% FBS) containing 300 μCi of [35S]methionine was added to each dish and incubated overnight at 37°C. Virus supernatants were collected after 20 h, clarified at 16,000 × g for 5 min to remove cell debris, layered onto a 20% sucrose gradient (in 10 mM Tris, pH 7.4), and centrifuged for 1 h at 38,000 rpm at 4°C. Virus pellet was dissolved in Tris-EDTA (TE) buffer.
Labeled viruses and cell lysates were analyzed on a 4 to 12% Bis-Tris NuPAGE gel (Invitrogen, CA), and protein bands were imaged using a Fujifilm BAS 1800 imaging system.
One-step growth curve.
BHK-21 cells were infected with rwt-VSV, VSV-CHIKV, or VSVΔG-CHIKV at an MOI of 10. After virus adsorption for 30 min, cells were washed twice with PBS, and DMEM containing 5% FBS was added. Supernatant samples were collected at 0, 2, 4, 6, 8, and 24 h postadsorption, and titers were determined by plaque assay on BHK-21 cells.
CHIKV pseudotype generation.
VSVΔG-eGFP1 pseudotyped with CHIKV envelope proteins was generated by transfecting BHK cells with pCAGGS-CHIKV, followed by infection with VSV G-complemented VSVΔG-eGFP1 (18). At 1 h postinfection, input virus was removed, cells were washed twice with PBS, and DMEM containing 5% FBS was added. Supernatant containing VSVΔG-eGFP1 pseudotyped with CHIKV envelope proteins was collected at 24 h, and virus titers were determined on BHK-21 cells by counting the number of enhanced green fluorescent protein (eGFP)-expressing cells.
Animal experiments.
Mice, housed under biosafety level 2 (BSL-2) conditions in microisolator cages, were inoculated with recombinant VSV vectors using 106 PFU per mouse by the intramuscular (i.m.) route in a volume of 50 μl of serum-free DMEM administered into the right hind leg muscle. For the challenge study, 3-week-old female C57BL/6 mice were obtained from Jackson Laboratories, Maine, and acclimatized for 4 days before immunization. Blood was collected at 30 days postimmunization (dpi) from retro-orbital sinus. These animals were challenged with the wild-type CHIKV-LR strain derived from a cDNA clone (3) using 104 PFU per mouse by the subcutaneous (s.c.) route in the left rear footpad. Foot-swelling measurements were taken on the day of challenge and for 10 days following using a caliper to measure the height of the foot at the ball. Animals from each immunization group were divided into two subgroups and were bled on alternate days postchallenge to measure viral titers by plaque assay on Vero cells. Two animals from each group were sacrificed at 4 days postchallenge (38 dpi) for histopathology analysis. UV inactivation of virus was performed using two exposures of virus to 5 × 105 μJ in a UV Stratalinker 1800 (Stratagene).
To look at cellular immune responses, 6- to 8-week-old female C57BL/6 mice were inoculated with virus after a 1-week acclimatization and sacrificed by anesthesia overdose at 7 dpi for isolating splenocytes.
Yale University and/or the University of Texas Medical Branch (UTMB) Institutional Animal Care and Use Committee approved all animal experiments.
Virus neutralization assays.
The VSVΔG-eGFP1/CHIKV pseudotype neutralization assay was similar to that described previously for Nipah virus pseudotypes (30). Briefly, approximately 50 infectious pseudotype particles were mixed with pooled and serially diluted serum samples from each immunization group, added to a monolayer of BHK-21 cells in a 96-well plate, and incubated for 20 to 24 h at 37°C. Infection was determined by visualizing the number of eGFP-expressing cells using an Olympus CK-40 microscope equipped with epifluorescence. Absence of infection in duplicate wells for each sample was scored as 100% neutralization.
A VSV neutralization assay was performed as described previously (21). The VSV neutralization titers were defined as the highest dilution of serum that could completely neutralize infectivity of 100 PFU of VSV on BHK-21 cells.
CHIKV plaque reduction neutralization tests were performed using standard methods (31) and the CHIKV-LR strain.
ELISPOT assay.
A gamma interferon (IFN-γ) enzyme-linked immunospot (ELISPOT) assay kit (BD Biosciences) was used to quantify T-cell activation following immunization. Splenocytes were isolated from immunized mice at 7 dpi by disrupting the spleen between frosted ends of two microscope slides. The red blood cells were removed by using red blood cell lysis buffer (eBioscience Inc., San Diego, CA), and splenocytes were collected in RPMI 1640 medium containing 10% FBS after passage through a strainer. A total of 2 × 105 splenocytes per well were then added to a 96-well plate precoated with mouse IFN-γ antibody and incubated with or without CHIKV E1 (HSMTNAVTI) or E2 (IILYYYELY) peptides at a concentration of 20 μg/ml for 24 h at 37°C. Thereafter, cells were first washed with water and then with wash buffer (supplied in the kit), followed by incubation with biotinylated anti-mouse IFN-γ antibody at a 1:2,000 dilution for 2 h at room temperature. Following this, cells were again washed with wash buffer; streptavidin-horseradish peroxidase (HRP) conjugate (supplied in the kit) was added at a 1:100 dilution and incubated at room temperature for 1 h. The plate was developed according to the manufacturer's protocol using 3-amino-9-ethyl-carbazole (AEC) chromogen diluted in AEC substrate buffer. The reaction was stopped by adding water, and the plate was air dried before spot-forming cells (SFC) were counted.
RESULTS
Construction and characterization of recombinant VSVs expressing chikungunya virus envelope proteins.
To generate a VSV recombinant virus expressing CHIKV surface glycoproteins, a synthetic gene encoding the CHIKV E3-E2-6K-E1 polyprotein was inserted between the G and L genes of VSV (Indiana) in the plasmid vector pVSVXN2 (23), and the resulting plasmid pVSV-CHIKV was used to recover VSV-CHIKV (Fig. 1A) using established procedures (25). A VSV recombinant expressing CHIKV envelope proteins in the absence of VSV G (Fig. 1A) was also recovered by complementation with VSV G (26) and was designated VSVΔG-CHIKV. Initially, this recombinant was grown in cells expressing VSV G because we assumed that CHIKV glycoproteins would not be incorporated efficiently into VSV particles or promote efficient infection. As we show below, the VSVΔG-CHIKV recombinant does propagate in the absence of VSV G complementation.
Fig 1.

Characterization of recombinant VSVs encoding the CHIKV E3-E2-6K-E1 polyprotein. (A) Diagram of rVSV genomes with the insertion of CHIKV envelope polyprotein in full-length VSV and VSVΔG vectors. The gene order is shown in the 3′-to-5′ direction on the negative-sense RNA genome. (B) Expression of CHIKV envelope proteins detected by indirect immunofluorescence microscopy using anti-VSV G MAb or anti-CHIKV mouse serum in infected (as indicated) BHK-21 cells. Photomicrographs show cell nuclei stained with DAPI in all fields. (C) [35S]methionine-labeled crude cell lysates of infected BHK-21 cells analyzed by SDS-PAGE showing VSV proteins and CHIKV envelope proteins. VSV proteins are indicated to the left, and CHIKV envelope proteins and molecular masses of VSV proteins are indicated to the right. (D) [35S]methionine-labeled and purified virions analyzed by SDS-PAGE. Proteins incorporated into the virions are indicated.
Expression of CHIKV proteins in VSV recombinants.
To determine if CHIKV proteins were expressed in cells infected with the VSV/CHIKV recombinants, we initially used indirect immunofluorescence microscopy. BHK-21 cells were infected with VSV-CHIKV, VSVΔG-CHIKV, or rwt-VSV (control). Fixed, nonpermeabilized cells were incubated with MAbs to VSV G or with serum from CHIKV-infected mice, followed by secondary antibody (anti-mouse Alexa Fluor 488 conjugate) treatment. As shown in Fig. 1B, cells infected with both recombinants showed surface expression of CHIKV proteins, while rwt-VSV-infected cells showed no CHIKV protein expression. The CHIKV antiserum recognizes both E1 and E2. VSV G protein was expressed on the cell surface of rwt-VSV- and VSV-CHIKV-infected cells, while VSVΔG-CHIKV-infected cells did not express VSV G.
To visualize the expression levels of both E1 and E2 proteins, we performed a 30-min metabolic labeling ([35S]methionine) of BHK-21 cells infected with VSV-CHIKV, VSVΔG-CHIKV, or rwt-VSV. Lysates from infected and mock-infected cells were separated by SDS-PAGE and analyzed on a Fujifilm imaging system (Fig. 1C). Because VSV infection shuts off host protein synthesis, the cell lysate from rwt-VSV-infected cells shows mainly the viral proteins L, G, nucleocapsid (N), phosphoprotein (P) and matrix protein (M) (Fig. 1C, lane 2). VSV-CHIKV-infected cells expressed an additional protein with the mobility expected for CHIKV E1 protein (∼52 kDa) (Fig. 1C, lane 3). Because this was only a 30-min labeling, the CHIKV E2 would still be in its precursor form of E3-E2 (p62). The p62 protein is evident in the VSVΔG-CHIKV-infected cells along with E1 (Fig. 1C, lane 4). However, in the VSV-CHIKV-infected cells, the p62 protein runs just ahead of and overlapping VSV G and cannot be discerned clearly.
CHIKV glycoproteins are incorporated into VSV recombinants.
To determine if the CHIKV glycoproteins were incorporated into VSV particles, BHK-21 cells were infected with rwt-VSV or either VSV/CHIKV recombinant and labeled for 16 h with [35S]methionine. Virus particles purified from the medium were then analyzed by SDS-PAGE (Fig. 1D). rwt-VSV and VSV-CHIKV particles contained the five indicated VSV proteins, while VSVΔG-CHIKV particles lacked VSV G. Both VSV-CHIKV and VSVΔG-CHIKV particles contained a single extra protein band with the mobility of a 52-kDa protein, the mobility expected for both E1 and mature E2. The precursor protein p62 present in cells was absent from the VSVΔG-CHIKV virions as expected because it is cleaved prior to transport to the cell surface.
VSVΔG-CHIKV recombinants grow in the absence of VSV G complementation.
Although VSV is promiscuous in terms of accepting foreign proteins into its envelope (32, 33), VSVΔG recombinants with foreign viral envelopes often grow to titers 100-fold lower than the titer of VSV itself in the absence of complementation with VSV G (34). However, we found that the VSVΔG-CHIKV recombinant typically grew to titers only 2- to 3-fold lower than the titer of rwt-VSV and did not require VSV G complementation. To examine the growth of VSVΔG-CHIKV and VSV-CHIKV in more detail, we performed a one-step growth curve as shown in Fig. 2A. VSV and VSV-CHIKV grew at similar rates and reached equivalent titers, while the VSVΔG-CHIKV appeared to lag initially but eventually reached a titer similar to that of VSV by 24 h. VSVΔG-CHIKV also generated smaller plaques in BHK-21 cells than VSV-CHIKV (Fig. 2B). The plaques generated by VSV-CHIKV are indistinguishable from VSV plaques (data not shown).
Fig 2.
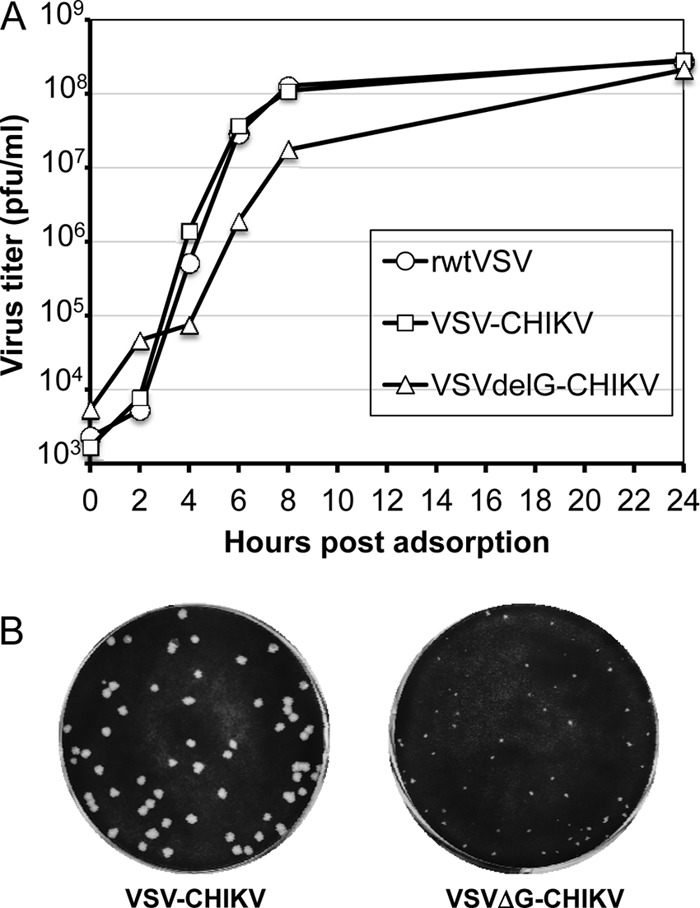
Growth of recombinant VSVs and plaque morphology. (A) BHK-21 cells were infected (MOI of 10) for 30 min with rwt-VSV, VSV-CHIKV, or VSVΔG-CHIKV, and unadsorbed virus was removed by washing cells twice with PBS. Complete growth medium was added, and supernatants were collected at indicated times postadsorption. Virus titers were determined by plaque assay on BHK-21 cells. (B) Plaque morphology of VSV-CHIKV and VSVΔG-CHIKV at 2 days postinfection of BHK-21 cells.
VSV/CHIKV recombinants induce CHIKV nAbs in C57BL/6 mice.
To determine if the VSV recombinants expressing CHIKV glycoproteins were immunogenic, we initially performed a pilot study to test the immunogenicity of VSV-CHIKV, VSVΔG-CHIKV (G-complemented), or VSVΔG-CHIKV (noncomplemented) in C57BL/6 mice. A single i.m. immunization (106 PFU) with these VSV/CHIKV recombinants induced antibodies that neutralized a VSVΔG-eGFP1/CHIKV pseudotype (data not shown). Moreover, the noncomplemented VSVΔG-CHIKV elicited a CHIKV nAb titer that was at least as good as that of the G-complemented virus or VSV-CHIKV.
The immune response requires viral RNA synthesis.
To determine if the nAb response to VSVΔG-CHIKV was dependent on viral replication, we examined immune responses to UV-inactivated virus particles. As shown in Fig. 3, UV inactivation completely eliminated the antibody response to G-complemented or noncomplemented VSVΔG-CHIKV. This result indicates that proteins on incoming virions are not sufficient to generate the immune response and that viral transcription and presumably replication are required to generate sufficient mRNA and protein to induce the immune response. Because the noncomplemented VSVΔG-CHIKV would have the major advantage of not inducing anti-G antibodies, we performed a more detailed analysis of this virus as well as VSV-CHIKV to assess induction of immune responses and protective efficacy in a CHIKV challenge model.
Fig 3.

UV inactivation prevents induction of CHIKV nAbs. Mice were immunized with noncomplemented (NC) or G-complemented (GC) VSVΔG-CHIKV without or with UV inactivation (NC-UV and GC-UV, respectively), and pooled sera from day 30 postimmunization were analyzed for induction of VSVΔG-eGFP/CHIKV pseudotype nAbs. The representative curve shows percent neutralization at the indicated serum dilutions. All assays were performed in duplicate.
Immunization and challenge study.
Fig. 4A shows the timeline of the immunization/challenge study using 3.5-week-old C57BL/6 mice. Two groups of eight mice each were immunized i.m. with VSV-CHIKV or VSVΔG-CHIKV, and a control group (n = 8) received rwt-VSV. Blood was collected from the animals at day 30 postimmunization and used to determine the CHIKV nAb response from each group. As shown in Fig. 4B, while both VSV/CHIKV recombinants elicited 100% neutralization of VSV/CHIKV pseudotypes (homologous S27 strain) at a low dilution, VSVΔG-CHIKV induced a superior response at dilutions greater than 1:160. However, the differences between the two vaccine groups did not reach statistical significance. Control rwt-VSV-immunized mice had no CHIKV nAb. To assess the nAb responses to the heterologous CHIKV-LR strain (challenge virus), plaque reduction neutralization tests (PRNT) were also performed using these serum samples. As shown in Table 1, all rVSV/CHIKV-immunized animals had 80% PRNT (PRNT80) titers of ≥80, with the VSVΔG-CHIKV group showing a higher nAb titer on average (ranging from 160 to >640) than the VSV-CHIKV group (titer ranging from 80 to 320). Control mice that received rwt-VSV showed titers below the detection limit (<20). Antivector nAb titers directed against VSV G were also measured using pooled serum samples from each immunization group (data not shown). Mice immunized with rwt-VSV and VSV-CHIKV had VSV nAb titers of 1:2,560, while mice immunized with VSVΔG-CHIKV did not generate detectable VSV nAbs (<1:20).
Fig 4.

VSV-CHIKV and VSVΔG-CHIKV induce VSVΔG-eGFP/CHIKV pseudotype nAbs in mice. (A) Timeline for vaccination challenge study for testing efficacy of rVSV/CHIKV vectors. Mice were immunized (day 0) i.m. with 106 PFU of indicated virus and challenged at about 5 weeks postimmunization (day 34) with 104 PFU of CHIKV-LR strain by the s.c. route in the hind leg footpad. rwt-VSV immunization was used as a control. (B) Percent neutralization of VSV/CHIKV pseudotype using pooled serum samples from day 30 postimmunization with rwt-VSV, VSV-CHIKV, or VSVΔG-CHIKV. All assays were performed in duplicate.
Table 1.
Titers of CHIKV-specific neutralizing antibodies at day 30 postimmunization with different VSV recombinants
| Mouse no. | PRNT80a |
||
|---|---|---|---|
| rwt-VSV | VSV-CHIKV | VSVΔG-CHIKV | |
| 1 | <20 | 320 | 640 |
| 2 | <20 | 320 | >640 |
| 3 | <20 | 160 | >640 |
| 4 | <20 | 320 | 320 |
| 5 | <20 | 320 | 160 |
| 6 | <20 | 80 | 320 |
| 7 | <20 | 80 | 160 |
| 8 | <20 | 320 | 320 |
Groups of 3.5-week-old C57BL/6 mice were immunized i.m. with 106 PFU of indicated viruses. Neutralizing antibody titers were determined in day 30 postimmunization serum samples using PRNT80.
rVSV/CHIKV-immunized mice are protected against CHIKV infection.
To determine if the VSV/CHIKV recombinants can protect against CHIKV-induced viremia and disease, the immunized C57BL/6 mice were subjected to s.c. challenge in their rear left footpad with 104 PFU of CHIKV-LR strain at 34 days postimmunization and followed for signs of disease over 10 subsequent days. Four mice from each immunization group were bled on alternate days postchallenge to assess viremia. Control animals that received rwt-VSV showed viremia during the first 2 days after challenge while none of the rVSV/CHIKV-vaccinated mice had detectible viremia (Table 2). CHIKV-induced disease was also monitored for 10 days postchallenge by following signs of local inflammation as measured by swelling of the inoculated foot. As shown in Fig. 5A, rwt-VSV-immunized control animals displayed substantial footpad swelling that was sustained for 10 days after challenge. In contrast, mice in both of the rVSV/CHIKV-vaccinated groups separately showed significantly less (P < 0.001, by analysis of variance [ANOVA]) foot swelling, which returned to prechallenge levels by day 8 or 9 postchallenge (Fig. 5A). While the control animals showed a transient weight loss or failed to gain weight after challenge, VSV/CHIKV-immunized mice did not show any weight loss and gained weight (Fig. 5B). However, the differences in weight loss did not reach statistical significance because of the high degree of animal-to-animal variability.
Table 2.
Viremia in vaccinated and control mice after challenge with CHIKV
| Immunization group | Virus titer (log10 PFU/ml ± SD)a |
|||
|---|---|---|---|---|
| Day 1 | Day 2 | Day 3 | Day 4 | |
| rwt-VSV | 2.55 ± 0.56 (2/4) | 2.85 ± 0.47 (4/4) | <0.9 (0/4) | 2.0 ± 0.0 (1/4) |
| VSV-CHIKV | <0.9 (0/4) | <0.9 (0/4) | <0.9 (0/4) | <0.9 (0/4) |
| VSVΔG-CHIKV | <0.9 (0/4) | <0.9 (0/4) | <0.9 (0/4) | <0.9 (0/4) |
Mice were challenged with 104 PFU of CHIKV-LR administered s.c. in the rear left footpad. Four mice from each immunization group were bled on alternate days postchallenge, i.e., either on days 1 and 3 postchallenge or on days 2 and 4 postchallenge. Values in parentheses indicate the number of positive mice/total number of mice in group.
Fig 5.

Footpad inflammation and body weight loss profiles in immunized mice following CHIKV challenge. (A) Foot swelling, measured as the vertical height of the hind feet at the ball, was monitored in rwt-VSV-, VSV-CHIKV-, or VSVΔG-CHIKV-immunized mice, for 10 days after s.c. challenge with 104 PFU of CHIKV-LR isolate. Error bars represent standard deviations. (B) Body weight of mice in control rwt-VSV-, VSV-CHIKV-, and VSVΔG-CHIKV-immunized groups were measured on the day of challenge and monitored daily for 9 days postchallenge. The graph represents average percent prechallenge body weight in each group over time. Error bars represent standard deviation.
Two animals from each group were sacrificed at day 4 postchallenge for histopathologic evaluation (data not shown). The brain, lung, heart, kidney, bowel, spleen, and muscle demonstrated no significant histopathologic differences among the groups. It is possible that we might have noted more significant histopathologic change at later times after infection. This has been noted by others in the muscle at day 7 following challenge (35). We did, however, note microgranulomas in the liver of all groups, and the vaccine groups showed a 75% reduction in the number of microgranulomas compared to the rwt-VSV control group. These microgranulomas most likely represent a nonspecific reaction to hepatic injury.
VSV/CHIKV recombinants induce cellular immune responses in C57BL/6 mice.
Because VSV-based vaccine vectors are known to be potent inducers of both humoral and cellular immune responses (36, 37), we also looked at the CHIKV-specific cellular immune response to the VSV/CHIKV vectors. A previous study, using a multidose DNA vaccine expressing consensus CHIKV E1 or E2 sequence, reported induction of CHIKV-specific CD8+ cytotoxic T-lymphocyte (CTL) responses using peptide pools and predicted two dominant CD8+ epitopes, one in E1 protein and one in E2 protein (38). We used synthetic peptides corresponding to these predicted epitopes (HSMTNAVTI on E1 and IILYYYELY on E2) in an IFN-γ ELISPOT assay. Our studies (Fig. 6) showed that the VSV/CHIKV vectors induced CHIKV cellular immune responses by day 7 after a single immunization. Although the response to the E1 epitope was higher than the response to the E2 epitope in both vaccinated groups, VSVΔG-CHIKV induced a significantly stronger (P < 0.01, ANOVA) response to the E2 epitope than VSV-CHIKV.
Fig 6.

Induction of cellular immune response in rVSV/CHIKV-immunized mice. Splenocytes, harvested from uninfected naïve mice (n = 2) or mice immunized with 106 PFU of VSV-CHIKV (n = 5) or VSVΔG-CHIKV (n = 5) at day 7 after i.m. immunization, were stimulated with peptides corresponding to CHIKV E1 (HSMTNAVTI) or E2 (IILYYYELY) CD8+ epitopes for 24 h followed by detection of IFN-γ spot-forming cells (SFC) using an ELISPOT assay. The numbers of SFC from unstimulated control samples have been subtracted. Error bars represent standard deviation. The asterisk indicates a significant difference (P < 0.01, ANOVA) between VSV-CHIKV and VSVΔG-CHIKV groups in response to E2 peptide.
DISCUSSION
We report here the development of experimental CHIKV vaccines based on VSV vectors expressing the envelope proteins of CHIKV. These were either a full-length VSV-CHIKV vector or a VSVΔG-CHIKV vector that lacks the VSV G gene. Both of these vectors generated robust nAb responses to CHIKV in mice after a single dose and protected mice from CHIKV challenge. Most interestingly, we found that the VSVΔG-CHIKV recombinant incorporated functional CHIKV envelope proteins into the viral envelope and propagated in the absence of VSV G complementation. Although the VSVΔG-CHIKV recombinant grew more slowly than the full-length vector and made only small plaques, it generated a stronger nAb response to CHIKV than the full-length VSV-CHIKV vector. The stronger antibody responses probably resulted from a combination of greater expression of CHIKV glycoproteins in the absence of VSV G, greater display of the CHIKV proteins on the surface of the viral particles, and/or lack of antigenic competition from VSV G.
Chimeric alphaviruses expressing structural protein genes from CHIKV and nonstructural protein genes of either Venezuelan or eastern equine encephalitis virus (VEEV or EEEV, respectively) have been successfully used as experimental vaccines for CHIKV (9). These viruses also generated potent CHIKV nAbs in mice and protection from challenge after a single dose. It is interesting that the titers generated following a single-dose VSVΔG-CHIKV vector were comparable to those reported for the VEEV/EEEV-CHIKV chimeras (9).
Passive antibody transfer studies have shown that antibody alone can confer immunity to CHIKV challenge (11, 13, 39). The role of cellular immunity in clearance of CHIKV infection is not well studied. However, recent studies have shown that strong CHIKV-specific T-cell activation occurs early in infection and likely plays a role in early control of viral replication prior to generation of nAbs (35, 40, 41). We showed here that a single dose of VSV/CHIKV recombinants induced potent CD8 T-cell responses to dominant epitopes in CHIKV E1 and E2 proteins. Recall of memory T cells generated by vaccination with VSV/CHIKV vectors could therefore complement the robust B-cell responses and aid in viral clearance and protection from CHIKV-induced disease.
A major advantage of the VSVΔG vector system is that it does not induce any antivector nAb since it lacks the VSV G protein. Hence, it could be used in multiple vaccine applications. Based on our experience here with CHIKV, it is likely that VSVΔG vectors could be a general chimeric vaccine platform for other medically important alphaviruses such as VEEV, EEEV, and western equine encephalitis virus (WEEV), which are widespread in the Americas. Although the VSVΔG-CHIKV chimeric virus grew more slowly than wild-type VSV, it eventually reached titers nearly equivalent to those of VSV and thus could easily be scaled up for large-scale vaccine production. We have also found that the VSVΔG-CHIKV recombinant grows in Vero cells to titers equivalent to those obtained in BHK-21 cells. This is important because Vero cells are an approved cell line for vaccine production.
VSV-based vaccine vectors given by the i.m. route typically cause no measurable pathogenesis in mice. They are trafficked to draining lymph nodes and rapidly eliminated without causing systemic infection (42, 43). We also observed that the VSV-CHIKV and VSVΔG-CHIKV viruses caused no pathogenesis when given intramuscularly. Interestingly, our preliminary studies have shown that the VSVΔG-CHIKV vectors are nonpathogenic but immunogenic when given by the intranasal route, while the VSV-CHIKV vector or the VSV vector alone induces substantial pathogenesis (weight loss). A more extensive analysis of pathogenesis and vector spread using multiple routes of inoculation will, of course, be required before VSVΔG-CHIKV vectors could be moved into clinical trials.
Our studies have demonstrated that a VSV-vectored vaccine provides protection against a CHIKV strain (La Reunion) that is about 2% divergent in sequence (44) compared to the vaccine strain (S27). Based on these studies and others showing vaccine cross-protection among more distantly related arthrogenic alphaviruses (35, 45), the VSV-vectored vaccine is likely to provide protection against all three CHIKV clades. Vaccine cross-protection within the encephalitic alphaviruses has also been reported (46, 47). Given the potent immune responses generated by the VSVΔG-CHIKV recombinant, it will be important in the future to examine possible cross-protection against other arthrogenic viruses as well as the general applicability of the platform for other alphaviruses.
ACKNOWLEDGMENTS
This work was supported by grant numbers U54 AI057158 and U54 AI057156 from NIAID/NIH, and its contents are solely the responsibility of the authors and do not necessarily represent the official views of the RCE Programs Office, NIAID, or NIH.
We thank John Schell and Kapil Bahl for advice on the T-cell studies and Alex Ryder for assistance with statistical analysis.
Footnotes
Published ahead of print 17 October 2012
REFERENCES
- 1. Griffin D. 2006. Alphaviruses, p 1023–1067 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Powers AM, Logue CH. 2007. Changing patterns of chikungunya virus: re-emergence of a zoonotic arbovirus. J. Gen. Virol. 88:2363–2377 [DOI] [PubMed] [Google Scholar]
- 3. Tsetsarkin KA, Chen R, Sherman MB, Weaver SC. 2011. Chikungunya virus: evolution and genetic determinants of emergence. Curr. Opin. Virol. 1:310–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schwartz O, Albert ML. 2010. Biology and pathogenesis of Chikungunya virus. Nat. Rev. Microbiol. 8:491–500 [DOI] [PubMed] [Google Scholar]
- 5. Simizu B, Yamamoto K, Hashimoto K, Ogata T. 1984. Structural proteins of Chikungunya virus. J. Virol. 51:254–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Strauss JH, Strauss EG. 1994. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 58:491–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Paessler S, Yun NE, Judy BM, Dziuba N, Zacks MA, Grund AH, Frolov I, Campbell GA, Weaver SC, Estes DM. 2007. Alpha-beta T cells provide protection against lethal encephalitis in the murine model of VEEV infection. Virology 367:307–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang E, Kim DY, Weaver SC, Frolov I. 2011. Chimeric Chikungunya viruses are nonpathogenic in highly sensitive mouse models but efficiently induce a protective immune response. J. Virol. 85:9249–9252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang E, Volkova E, Adams AP, Forrester N, Xiao SY, Frolov I, Weaver SC. 2008. Chimeric alphavirus vaccine candidates for Chikungunya. Vaccine 26:5030–5039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang D, Suhrbier A, Penn-Nicholson A, Woraratanadharm J, Gardner J, Luo M, Le TT, Anraku I, Sakalian M, Einfeld D, Dong JY. 2011. A complex adenovirus vaccine against Chikungunya virus provides complete protection against viraemia and arthritis. Vaccine 29:2803–2809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Akahata W, Yang ZY, Andersen H, Sun S, Holdaway HA, Kong WP, Lewis MG, Higgs S, Rossmann MG, Rao S, Nabel GJ. 2010. A virus-like particle vaccine for epidemic Chikungunya virus protects nonhuman primates against infection. Nat. Med. 16:334–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mallilankaraman K, Shedlock DJ, Bao H, Kawalekar OU, Fagone P, Ramanathan AA, Ferraro B, Stabenow J, Vijayachari P, Sundaram SG, Muruganandam N, Sarangan G, Srikanth P, Khan AS, Lewis MG, Kim JJ, Sardesai NY, Muthumani K, Weiner DB. 2011. A DNA vaccine against Chikungunya virus is protective in mice and induces neutralizing antibodies in mice and nonhuman primates. PLoS Negl. Trop. Dis. 5:e928 doi:10.1371/journal.pntd.0000928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Plante K, Wang E, Partidos CD, Weger J, Gorchakov R, Tsetsarkin K, Borland EM, Powers AM, Seymour R, Stinchcomb DT, Osorio JE, Frolov I, Weaver SC. 2011. Novel chikungunya vaccine candidate with an IRES-based attenuation and host range alteration mechanism. PLoS Pathog. 7:e1002142 doi:10.1371/journal.ppat.1002142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chattopadhyay A, Park S, Delmas G, Suresh R, Senina S, Perlin DS, Rose JK. 2008. Single-dose, virus-vectored vaccine protection against Yersinia pestis challenge: CD4+ cells are required at the time of challenge for optimal protection. Vaccine 26:6329–6337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jones SM, Feldmann H, Stroher U, Geisbert JB, Fernando L, Grolla A, Klenk HD, Sullivan NJ, Volchkov VE, Fritz EA, Daddario KM, Hensley LE, Jahrling PB, Geisbert TW. 2005. Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat. Med. 11:786–790 [DOI] [PubMed] [Google Scholar]
- 16. Kahn JS, Roberts A, Weibel C, Buonocore L, Rose JK. 2001. Replication-competent or attenuated, nonpropagating vesicular stomatitis viruses expressing respiratory syncytial virus (RSV) antigens protect mice against RSV challenge. J. Virol. 75:11079–11087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kapadia SU, Rose JK, Lamirande E, Vogel L, Subbarao K, Roberts A. 2005. Long-term protection from SARS coronavirus infection conferred by a single immunization with an attenuated VSV-based vaccine. Virology 340:174–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kapadia SU, Simon ID, Rose JK. 2008. SARS vaccine based on a replication-defective recombinant vesicular stomatitis virus is more potent than one based on a replication-competent vector. Virology 376:165–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roberts A, Buonocore L, Price R, Forman J, Rose JK. 1999. Attenuated vesicular stomatitis viruses as vaccine vectors. J. Virol. 73:3723–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roberts A, Reuter JD, Wilson JH, Baldwin S, Rose JK. 2004. Complete protection from papillomavirus challenge after a single vaccination with a vesicular stomatitis virus vector expressing high levels of L1 protein. J. Virol. 78:3196–3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rose NF, Marx PA, Luckay A, Nixon DF, Moretto WJ, Donahoe SM, Montefiori D, Roberts A, Buonocore L, Rose JK. 2001. An effective AIDS vaccine based on live attenuated vesicular stomatitis virus recombinants. Cell 106:539–549 [DOI] [PubMed] [Google Scholar]
- 22. Schwartz JA, Buonocore L, Roberts A, Suguitan A, Jr, Kobasa D, Kobinger G, Feldmann H, Subbarao K, Rose JK. 2007. Vesicular stomatitis virus vectors expressing avian influenza H5 HA induce cross-neutralizing antibodies and long-term protection. Virology 366:166–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schnell MJ, Buonocore L, Whitt MA, Rose JK. 1996. The minimal conserved transcription stop-start signal promotes stable expression of a foreign gene in vesicular stomatitis virus. J. Virol. 70:2318–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199 [DOI] [PubMed] [Google Scholar]
- 25. Lawson ND, Stillman EA, Whitt MA, Rose JK. 1995. Recombinant vesicular stomatitis viruses from DNA. Proc. Natl. Acad. Sci. U. S. A. 92:4477–4481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schnell MJ, Johnson JE, Buonocore L, Rose JK. 1997. Construction of a novel virus that targets HIV-1-infected cells and controls HIV-1 infection. Cell 90:849–857 [DOI] [PubMed] [Google Scholar]
- 27. Fuerst TR, Niles EG, Studier FW, Moss B. 1986. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 83:8122–8126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Okuma K, Matsuura Y, Tatsuo H, Inagaki Y, Nakamura M, Yamamoto N, Yanagi Y. 2001. Analysis of the molecules involved in human T-cell leukaemia virus type 1 entry by a vesicular stomatitis virus pseudotype bearing its envelope glycoproteins. J. Gen. Virol. 82:821–830 [DOI] [PubMed] [Google Scholar]
- 29. Lefrancois L, Lyles DS. 1982. The interaction of antibody with the major surface glycoprotein of vesicular stomatitis virus. I. Analysis of neutralizing epitopes with monoclonal antibodies. Virology 121:157–167 [PubMed] [Google Scholar]
- 30. Chattopadhyay A, Rose JK. 2011. Complementing defective viruses that express separate paramyxovirus glycoproteins provide a new vaccine vector approach. J. Virol. 85:2004–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Beaty BJ, Calisher CH, Shope RE. 1995. Arboviruses, p 189–212 In Lennete ET, Lennete DA. (ed), Diagnostic procedures for viral, rickettsial and chlamydial infections, 7th ed American Public Health Association, Washington, DC [Google Scholar]
- 32. Schnell MJ, Buonocore L, Kretzschmar E, Johnson E, Rose JK. 1996. Foreign glycoproteins expressed from recombinant vesicular stomatitis viruses are incorporated efficiently into virus particles. Proc. Natl. Acad. Sci. U. S. A. 93:11359–11365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zavada J, Zavadova Z, Russ G, Polakova K, Rajcani J, Stencl J, Loksa J. 1983. Human cell surface proteins selectively assembled into vesicular stomatitis virus virions. Virology 127:345–360 [DOI] [PubMed] [Google Scholar]
- 34. Boritz E, Gerlach J, Johnson JE, Rose JK. 1999. Replication-competent rhabdoviruses with human immunodeficiency virus type 1 coats and green fluorescent protein: entry by a pH-independent pathway. J. Virol. 73:6937–6945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gardner J, Anraku I, Le TT, Larcher T, Major L, Roques P, Schroder WA, Higgs S, Suhrbier A. 2010. Chikungunya virus arthritis in adult wild-type mice. J. Virol. 84:8021–8032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rose JK, Whitt MA. 2001. Rhabdoviridae: the viruses and their replication. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 4th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 37. Zinkernagel RM, Adler B, Holland JJ. 1978. Cell-mediated immunity to vesicular stomatitis virus infections in mice. Exp. Cell Biol. 46:53–70 [DOI] [PubMed] [Google Scholar]
- 38. Muthumani K, Lankaraman KM, Laddy DJ, Sundaram SG, Chung CW, Sako E, Wu L, Khan A, Sardesai N, Kim JJ, Vijayachari P, Weiner DB. 2008. Immunogenicity of novel consensus-based DNA vaccines against Chikungunya virus. Vaccine 26:5128–5134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Couderc T, Khandoudi N, Grandadam M, Visse C, Gangneux N, Bagot S, Prost JF, Lecuit M. 2009. Prophylaxis and therapy for Chikungunya virus infection. J. Infect. Dis. 200:516–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hoarau JJ, Jaffar Bandjee MC, Krejbich Trotot P, Das T, Li-Pat-Yuen G, Dassa B, Denizot M, Guichard E, Ribera A, Henni T, Tallet F, Moiton MP, Gauzere BA, Bruniquet S, Jaffar Bandjee Z, Morbidelli P, Martigny G, Jolivet M, Gay F, Grandadam M, Tolou H, Vieillard V, Debre P, Autran B, Gasque P. 2010. Persistent chronic inflammation and infection by Chikungunya arthritogenic alphavirus in spite of a robust host immune response. J. Immunol. 184:5914–5927 [DOI] [PubMed] [Google Scholar]
- 41. Wauquier N, Becquart P, Nkoghe D, Padilla C, Ndjoyi-Mbiguino A, Leroy EM. 2011. The acute phase of Chikungunya virus infection in humans is associated with strong innate immunity and T CD8 cell activation. J. Infect. Dis. 204:115–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Johnson JE, Coleman JW, Kalyan NK, Calderon P, Wright KJ, Obregon J, Ogin-Wilson E, Natuk RJ, Clarke DK, Udem SA, Cooper D, Hendry RM. 2009. In vivo biodistribution of a highly attenuated recombinant vesicular stomatitis virus expressing HIV-1 Gag following intramuscular, intranasal, or intravenous inoculation. Vaccine 27:2930–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Simon ID, Publicover J, Rose JK. 2007. Replication and propagation of attenuated vesicular stomatitis virus vectors in vivo: vector spread correlates with induction of immune responses and persistence of genomic RNA. J. Virol. 81:2078–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Solignat M, Gay B, Higgs S, Briant L, Devaux C. 2009. Replication cycle of Chikungunya: a re-emerging arbovirus. Virology 393:183–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Partidos CD, Paykel J, Weger J, Borland EM, Powers AM, Seymour R, Weaver SC, Stinchcomb DT, Osorio JE. 2012. Cross-protective immunity against o'nyong-nyong virus afforded by a novel recombinant Chikungunya vaccine. Vaccine 30:4638–4643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Walton TE, Jochim MM, Barber TL, Thompson LH. 1989. Cross-protective immunity between equine encephalomyelitis viruses in equids. Am. J. Vet. Res. 50:1442–1446 [PubMed] [Google Scholar]
- 47. Wust CJ, Crombie R, Brown A. 1987. Passive protection across subgroups of alphaviruses by hyperimmune non-cross-neutralizing anti-Sindbis serum. Proc. Soc. Exp. Biol. Med. 184:56–63 [DOI] [PubMed] [Google Scholar]