

Abstract
Japanese encephalitis virus (JEV) is a mosquito-borne pathogenic flavivirus responsible for acute viral encephalitis in humans. The cellular entry of JEV is poorly characterized in terms of molecular requirements and pathways. Here we present a systematic study of the internalization mechanism of JEV in fibroblasts and neuroblastoma cells. To verify the roles of distinct pathways of cell entry, we used fluorescently labeled virus particles, a combination of pharmacological inhibitors, RNA interference (RNAi), and dominant-negative (DN) mutants of regulatory proteins involved in endocytosis. Our study demonstrates that JEV infects fibroblasts in a clathrin-dependent manner, but it deploys a clathrin-independent mechanism to infect neuronal cells. The clathrin-independent pathway requires dynamin and plasma membrane cholesterol. Virus binding to neuronal cells leads to rapid actin rearrangements and an intact and dynamic actin cytoskeleton, and the small GTPase RhoA plays an important role in viral entry. Immunofluorescence analysis of viral colocalization with endocytic markers showed that JEV traffics through Rab5-positive early endosomes and that release of the viral nucleocapsid occurs at the level of the early and not the late endosomes.
INTRODUCTION
Japanese encephalitis virus (JEV) belongs to the genus Flavivirus in the family Flaviviridae. Other members of the genus include dengue virus (DENV), West Nile virus (WNV), yellow fever virus (YFV), and tick-borne encephalitis virus (TBEV). Most flaviviruses are transmitted by mosquito or tick vectors and cause serious human and animal disease (1). JEV is a major cause of epidemic encephalitis worldwide, with a potential to cause permanent neuropsychiatric sequelae, and is sometimes fatal in children living in areas of endemicity such as Southeast Asia. JEV transmission has also been observed in the Southern Hemisphere and has the potential to become a worldwide public health threat (2, 3).
JEV particles are small particles (∼50 nm), in which a glycoprotein-containing lipid envelope surrounds the capsid, which has a single-stranded positive-sense 11-kb RNA genome. The viral RNA carries a single open reading frame with genes for three structural proteins, i.e., capsid (C), premembrane (prM), and envelope (E), and seven nonstructural (NS) proteins, i.e., NS1, NS2a, NS2b, NS3, NS4a, NS4b, and NS5 (1). The E glycoprotein is the major antigenic determinant on flavivirus particles and mediates binding and fusion during virus entry (4, 5). After internalization, flaviviruses are trafficked to an endosomal compartment where low pH induces conformational changes necessary for virus uncoating and capsid disassembly (6).
Clathrin-mediated endocytosis (CME) is believed to be the major route of flavivirus cell entry. The earliest evidence for this was obtained from ultrastructural studies showing the presence of Kunjin virus and YFV in coated pits (7, 8). Subsequently, DENV and WNV have also been shown to infect mosquito cells via CME (9–11). Single-particle tracking of dengue virus in live cells has demonstrated virus movement along the cell surface in a diffusive manner before it is captured by a preexisting clathrin-coated pit (12). To date only one study has reported an alternate clathrin-independent infectious entry pathway for DENV-2 in mammalian cells (13). Using biochemical inhibitors and dominant-negative (DN) mutants, entry of DENV-1 was demonstrated to be clathrin dependent, while that of DENV-2 was clathrin, cholesterol, and caveolin independent in Vero cells. In contrast, DENV-2 entry in A549 cells was clathrin dependent, as previously reported for HeLa, C6/36, and BS-C-1 cells (14). It is possible that the entry pathway(s) utilized by the virus may be cell type dependent. Two recent RNA interference studies for dengue virus entry and multiplication into monocytes and HepG2 cells show dependence on clathrin heavy chain (CHC) and dyamin-2 (15, 16).
The cell biology of JEV entry remains relatively unexplored. An electron microscopy study showed that JEV is transported across cerebral blood vessels and breaches the blood brain barrier, and both coated and uncoated vesicles could be seen in the capillary endothelium (17). Data in support of a clathrin-mediated pathway for JEV internalization are limited mainly to pharmacological studies using the inhibitor chlorpromazine (18) and to the use of dominant-negative constructs of Eps15, a key protein involved in CME (19). It has also been shown that bafilomycin, a specific inhibitor of vacuolar-type H+-ATPase, interfered with JEV infection of Vero cells (20).
In addition to CME, several endocytic pathways that do not use clathrin have also been described (21, 22). At least three clathrin-independent internalization pathways have been reported in mammalian cells, but they are not yet completely characterized (23, 24). These pathways vary in the cargoes they transport and in the protein machinery that facilitates the endocytic process. Some of these pathways are constitutive, whereas others are triggered by specific signals or are hijacked by viruses (25). Considerable plasticity exists in these endocytic mechanisms, and certain components, such as dynamin-2, Rho GTPases, and actin, can participate in more than one pathway. The large GTPase dynamin-2 was originally noted for its role in severing clathrin-coated vesicles from the plasma membrane and was subsequently found to be involved in a clathrin-independent pathway mediated by caveolae (22). In addition, some members of the ADP-ribosylation factor (Arf) and Rho subfamilies of small GTPases were recently suggested to play key roles in regulating clathrin-independent endocytic pathways (26). Rho GTPases are also known to be involved in the control of actin dynamics (27). Dynamin-2 and F-actin are crucial to most endocytic processes that coexist within the cell. These common factors must be tightly controlled and perhaps differentially regulated according to the endocytic mechanism.
It is now being increasingly appreciated that many viruses can utilize more than one entry pathway to infect cells (25, 28). There have been many recent studies showing that viruses can induce macropinocytosis for productive entry and infection (29–31). Here, we address the roles of different endocytic molecules and pathways involved in JEV internalization in Vero (green monkey kidney fibroblast), Neuro2a (mouse neuroblastoma), and SH-SY5Y (human neuroblastoma) cells. Using a combination of pharmacological and molecular approaches, we show that a clathrin-independent pathway operates in neuronal cells for JEV infection. Dynamin-2, membrane cholesterol, and a dynamic actin cytoskeleton are specifically required for neuronal cell entry and infection of JEV. In addition, the small GTPase RhoA and myosin II motors aid JEV internalization. Immunofluorescence analysis of viral colocalization with endocytic markers showed that JEV is trafficked to Rab5-positive early endosomes, where membrane fusion occurs. The infection process in both fibroblasts and neuronal cells requires acid-dependent fusion at the level of the early endosomes.
MATERIALS AND METHODS
Cells, antibodies, inhibitors, and plasmids.
Mouse neuroblastoma (Neuro2a) and human neuroblastoma (SH-SY5Y) cells were grown in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (HyClone). Porcine stable kidney (PS) cells (National Center for Cell Sciences, Pune, India) and Vero (African green monkey kidney) cells were grown in Eagle's minimal essential medium (MEM) with 10% FBS. All media was additionally supplemented with 100 μg/ml penicillin-streptomycin and 2 mM l-glutamine. Antibodies to JEV E protein, actin (loading control), and myc were from Abcam. CHC antibody was from Cell Signaling Technology and CLC antibody from Santa Cruz. All inhibitors, i.e., chlorpromazine hydrochloride, dynasore, 5-N-ethyl-N-isoproamiloride (EIPA), methyl-β-cyclodextrin (MβCD), filipin, cytochalasin D (CytoD), latrunculin A, jasplakinolide, blebbistatin, and bafilomycin, were purchased from Sigma. The chemical inhibitor for RhoA (CT04) and the Rho, Rac, and Cdc42 G-LISA Activation Assay Biochem kits were purchased from Cytoskeleton Inc. (Denver, CO). All chemical inhibitor stock solutions were prepared according to the manufacturer's directions. Fluorescent dye DiD and fluorophore-coupled transferrin (Tf), phalloidin, secondary anti-mouse, and anti-rabbit antibodies and ProLong Gold antifade reagent with DAPI (4′,6′-diamidino-2-phenylindole), were obtained from Invitrogen Corporation. Horseradish peroxidase (HRP)-coupled secondary antibodies were obtained from Jackson Immunochemicals. The Eps15 mutants DIIIδ2(control) and DIII, subcloned in pEGFP-C2, were kind gifts from Alexandre Benmerah (Universite Paris, Paris, France) (32). Plasmids for wild-type (wt), dominant-active (DA), and dominant-negative (DN) Rho, Rac, Cdc42, Rab5, Rab7, and dyn-2K44A were obtained from Addgene: plasmids 15899, 15900, 15901, 15903, 15904, 15905, 15906, and 15907 deposited by Alan Hall (33); plasmids 12660 and 12605 deposited by Richard Pagano (34); plasmid 14437 deposited by Ari Helenius (35); plasmid 28045 deposited by Qing Zhong (36); and plasmid 34687 deposited by Sandra Schmid.
Virus generation, purification, and fluorescent labeling.
For all studies, JEV isolate Vellore P20778 generated in PS cells was used. The culture supernatant was harvested when 75% of the cells showed a cytopathic effect, usually 36 to 48 h after infection, and was clarified by centrifugation at 1,000 × g for 30 min at 4°C. Virus titers were determined using monolayers of PS cells as described earlier (37). Virus was further purified over a 20% sucrose cushion in a Beckman Coulter ultracentrifuge (Optima L-100K) at 80,000 × g for 4 h at 4°C. Purified virus was exchanged into phosphate-buffered saline (PBS) through cycles of concentration by centrifugation (800 × g) and dilution with PBS, using 50-ml ultrafiltration tubes (10 kDa; Amicon). Virus was labeled with DiD by injecting 2 nmol of dye into virus stock under intensive vortexing for 10 min at room temperature. Excess dye was removed by purification through Micro Spin G-25 columns (GE Healthcare). Labeling did not abolish viral infectivity. Labeled virus was used immediately for experiments.
Cell transfection and transient expression.
Cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocols. For transfection, cells were grown to 60 to 70% confluence on 18-mm coverslips or 35-mm coverslip dishes and transfected with 1 μg DNA. Transfections were typically allowed to proceed for 18 to 24 h before infection with JEV.
Small interfering RNA (siRNA) depletion experiments.
Mouse-specific clathrin heavy chain (CHC)-targeting short hairpin RNA (shRNA) plasmid DNAs (SHCLND_NM_001003908) were purchased from Sigma. The best silencing efficiency was observed with clone NM_001003908.1-5688s1c1 containing the sequence CCGGGCCGACAAAGACAACACTAATCTCGAGATTAGTGTTGTCTTTGTCGGCTTTTTG. Cells were transfected with shRNA plasmid and harvested at 24, 48, and 72 h to check for protein knockdown. Significant depletion of CHC was observed at 72 h. For clathrin light chain (CLC) depletion, an shRNA construct was generated by cloning appropriate oligonucleotides containing the targeting sequence in the pSIREN-RetoQZsGreen plasmid (Clontech). The sequence of the forward primer oligonucleotide designed for the CLC shRNA was GATCCGGAGCCTGAAAGTATCCGTATTCAAGAGATACGGATACTTTCAGGCTCTTTTTTGTCGACG. This sequence of the clathrin light chain targeted in the shRNA construct is evolutionarily conserved from rodents to primates. The green fluorescent protein (GFP)-CLCshRNA clone was used for gene silencing in both Neuro2a and Vero cells. This construct confers the additional advantage that cells receiving the shRNA also express GFP. A time course of CLC depletion was done, and maximal protein knockdown was observed at 72 h. Cell lysates were run on SDS-PAGE, and Western blotting was done for CLC, CHC, and actin. For infection experiments, cells were transfected with shRNA plasmids for either CHC or CLC for 72 h, following which they were infected with JEV.
Virus infection and cell treatment.
Neuro2a cells seeded on 35-mm coverslip dishes or 18-mm coverslips were either transfected as described above or treated with inhibitors prior to infection. The incubation time, dosage, and cell viability upon treatment were determined for each inhibitor. For all inhibitors the final dimethyl sulfoxide (DMSO) concentration never exceeded 0.2% of the total culture medium. Cells were infected with JEV at a multiplicity of infection (MOI) of 0.4 for 1 h at 37°C with or without inhibitors. For experiments involving dynasore, cells were grown in 10% Nuserum (BD Biosciences)-containing medium. For cholesterol depletion, cells were washed four or five times with serum-free medium before addition of methyl-β-cyclodextrin (MβCD) or filipin. For RhoA inhibition, cells were serum starved for 2 h, and inhibitor CT04 was added to serum-free medium and left for 2 h. For inhibitors requiring serum-free conditions, infections were done using purified virus in PBS with 0.1% glucose. For experiments involving transfections, cells were infected with JEV at an MOI of 1 (which results in about 30% infection in GFP-transfected cells) for 1 h at 37°C. Following infection, cells were washed twice with PBS and complete medium was added. Cells were fixed at 24 h postinfection (hpi) in 2% paraformaldehyde and permeabilized using 0.4% Triton X-100 or 0.04% saponin in PBS for 20 min at room temperature. Blocking was done with 2 mg/ml bovine serum albumin (BSA) in PBS for 1 h prior to incubation with anti-JEV E antibody, followed by Alexa 488/568 anti-mouse secondary antibody. For transfection experiments using overexpression constructs of myc-tagged Rho, Rac, and Cdc42, cells were processed for double immunostaining with antibodies against the myc epitope tag and JEV E antigen.
Virus colocalization studies.
For transferrin and JEV colocalization studies, labeled virus (MOI, 10) was allowed to bind cells on ice for 1 h. After virus binding, cells were incubated with labeled Tf for 5, 10, and 15 min at 37°C. For colocalization with dsRed-Rab5, and GFP-Rab7, binding of labeled virus to cells was done on ice for 1 h, after which cells were warmed to 37°C for the specified times. Cells were given a low-pH wash (0.1 M sodium acetate, 0.05 M NaCl, pH 5.5; 5 min) to remove virus particles sticking to cells, fixed in 2% paraformaldehyde, and imaged.
Immunofluorescence microscopy and image processing.
Confocal microscopy was performed using an Olympus FV1000 confocal microscope. For quantification of entry, images were acquired with a 20× Plan Apo objective with a numerical aperture (NA) of 1.20. Images were acquired for 8 to 10 fields of view per coverslip. Quantification of JEV infection was done by counting cells that were immune stained versus nonstained for virus envelope (E) antigen. For experiments involving transfections, quantitation was done by counting cells that were both transfected and infected versus those that were transfected but remained uninfected. The infection studies were normalized to either solvent-treated or GFP expressing controls. All experiments were done in duplicate or triplicate. Results are expressed as mean ± standard deviation (SD). Significance was determined using a Student t test. For colocalization experiments, images were acquired with a 60× PlanApo objective lens (NA, 1.4). Z stacks were acquired at 0.25 μm per slice by sequential scanning. FluoView software (Olympus) was used to generate cross-sectional and maximum-intensity projection images.
qRT-PCR.
Neuro2a cells were plated in 35-mm dishes at a density of 0.5 × 106 cells/dish and were pretreated with inhibitors. Viral adsorption (MOI, 10) to cells was performed at 4°C for 1 h, followed by one wash with cold PBS and a shift to 37°C for 1 h in the presence of inhibitor. After incubation, cells were washed with chilled PBS and low-pH buffer and lysed in TRIzol reagent (Invitrogen). Quantitative real-time PCR (qRT-PCR) primers were procured from Sigma. JEV positive-strand cDNA was generated using the primer AATAAGTTGTAGTTGGGCACTCTG. JEV was amplified using the following probes: TaqMan probe, CCACGCCACTCGACCCATAGACTG (5′ end, 6-carboxyfluorescein [FAM]; 3′ end, 6-carboxytetramethylrhodamine [TAMRA]); 5′ primer, AGAGCACCAAGGGAATGAAATAGT; 3′ primer, AATAAGTTGTAGTTGGGCACTCTG. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as an internal control with the following probes: TaqMan probe, ACAACCTGGTCCTCAGTGTAGC (5′ end, FAM; 3′ end, TAMRA); 5′ primer, CCTGCCAAGTATGATGAC; 3′ primer, GGAGTTGCTGTTGAAGTC. The PCR conditions were as follows: 94°C for 2 min (1 cycle) and 94°C for 15 s, 55°C for 30 s, and 72°C for 1 min (40 cycles). qPCR was done on Applied Biosystems ABI 7500 instrument.
Quantification of transferrin uptake by flow cytometry and microscopy.
Transferrin internalization by cells after treatment with chlorpromazine was measured by flow cytometry. Cells pretreated with 25 μM or 50 μM chlorpromazine were given a 10-min pulse of Alexa 488-Tf in labeling medium (DMEM or MEM with 10% serum). After incubation, excess label was washed off with chilled PBS and low-pH buffer to remove surface-bound Tf. Cells were fixed and detached, and Alexa 488 fluorescence was analyzed using a Becton Dickinson (BD) FACSCantoII flow cytometer. The average of measured geometric means of internalized Tf in control and inhibitor-treated cells was calculated. For quantification of transferrin uptake in cells transfected with different endocytic mutants/CLC shRNA, a pulse of Alexa 568/647 Tf was given for 10 min in labeling medium. Cells were processed as described above, fixed, and imaged at ×20. Total fluorescence intensity per cell was calculated using Olympus FV1000 analysis software. In each experiment, fluorescence was calculated from 10 to 12 fields of view from duplicate slides for each transfection condition. Integrated values of cell fluorescence were corrected for background autofluorescence. Tf uptake is represented as mean and standard error of the mean of integrated fluorescence intensity from two independent experiments.
Rho GTPase activation assays.
Neuro2a cells were transfected with wt, DA, and DN plasmids of Rho, Rac, and Cdc42. Rho and Cdc42 activation was measured at 24 h posttransfection with a G-LISA activation kit (kit BK124 or kit BK 127; Cytoskeleton Inc.). To measure Rac activation, mock-, Rac DA-, and Rac DN-transfected cells were serum starved for 24 h, followed by treatment with complete medium for 10 min before washing with cold PBS and lysis (kit BK125). To measure Rho activation in response to virus binding, JEV (MOI, 10) was adsorbed to cells at 4°C for 1 h, followed by one wash with cold PBS and a shift to 37°C to allow infection for the indicated times. Cells were washed with cold PBS, lysed, and processed for enzyme-linked immunosorbent assay (ELISA). The activated form of the G protein was detected by incubation with specific primary antibody followed by a secondary antibody conjugated to HRP and a detection reagent. The signal was read by measuring absorbance at 490 nm using a microplate reader.
RESULTS
JEV internalization is dynamin-2 dependent.
Dynamin-2 is a large GTPase which acts by mediating release of newly formed endocytic vesicles from the plasma membrane. It thus plays a critical role in CME as well as in some of the non-clathrin-dependent pathways (38). To examine the role of dynamin-2 in JEV entry in different cell lines, the effect of dynasore, a potent and specific dynamin-2 inhibitor, was tested (39). Neuro2a, SH-SY5Y, and Vero cells treated with 80 μM dynasore showed close to a 90% block in JEV infection (Fig. 1A, C, and D). At 80 μM, dynasore specifically blocks dynamin-2 function, which was confirmed via a significant inhibition in internalization of transferrin (Tf), a cargo that becomes internalized via the dynamin- and clathrin-dependent endocytic mechanism. To confirm that dynasore was blocking JEV endocytosis and not inhibiting any downstream event necessary for infection, qRT-PCR of JEV positive-strand RNA was performed at 1 hpi to estimate viral entry in untreated and dynasore-treated Neuro2acells. There was a 70% decrease in the viral load in dynasore-treated cells relative to the control (Fig. 1B), indicating that JEV endocytosis is likely to require functional dynamin.
Fig 1.

JEV internalization is dynamin dependent. (A) Neuro2a cells grown on coverslips were either untreated or treated with 80 μM dynasore for 1 h, following which they were infected with JEV (MOI, 0.4) in the presence of the inhibitor. At 24 hpi cells were fixed and stained with anti-JEV E primary and Alexa 488 anti-mouse IgG secondary antibodies. Cells were mounted in DAPI containing antifade reagent and imaged at ×20 on a confocal microscope. Cell nuclei can be seen in blue (DAPI) and JEV E in green. Bar, 20 μm. (B) Neuro2a cells were infected with JEV (MOI, 10) in the presence of 80 μM dynasore for 1 h, following which the endocytosed viral load was estimated by qRT-PCR of JEV positive-strand RNA. (C) Quantitation of JEV infection (MOI, 0.4) in Neuro2a, SH-SY5Y, and Vero cell lines in the presence of 80 μM dynasore was done by microscopy and image analysis as described in Materials and Methods. The results are normalized to solvent-treated control cells. (D) After 24 hpi (MOI, 0.4), the virus titer (mean ± SD) in culture supernatants was calculated by plaque assay. (E) Neuro2a cells transfected with GFP or GFP-dyn2-K44A were infected with JEV (MOI, 1; ∼30% infection in GFP-transfected cells). Cells were fixed at 24 hpi, stained with anti-JEV E primary and Alexa 568 anti-mouse IgG secondary antibody, and imaged at ×60 on a confocal microscope. Bar, 10 μm. (F) JEV infection in Neuro2a, SH-SY5Y, and Vero cells expressing GFP and GFPdyn2K44A was quantitated as described above. Infection was normalized to GFP-transfected cells. For panels B, C, and F, Student's t test was used to generate P values. **, P < 0.01; *, P < 0.05.
We next tested the ability of a plasmid expressing the dominant-negative K44A mutant of dynamin-2 (dyn2K44A) (40) to block JEV infection. Neuro2a cells were transfected with either a control GFP plasmid (Fig. 1E, left panel) or GFPdyn2K44A (Fig. 1E, right panel) and 24 h later were infected with JEV. Infected cells were immunostained for JEV E protein. Whereas cells expressing GFP alone were infected with JEV (Fig. 1E, left panel), those expressing the dyn2K44A mutant showed nearly a 70% block in infection (Fig. 1E, right panel, and F). This observation was also confirmed in SH-SY5Y and Vero cells (Fig. 1F). These experiments thus suggest that JEV internalization in cells is dynamin-2 dependent.
JEV internalization is independent of clathrin-mediated endocytic cargo.
Since JEV infection was dynamin dependent, we examined the role of clathrin in the internalization process. Transferrin, which binds to the transferrin receptor, is a well-characterized cargo of clathrin-coated pits and served as a control in these studies.
As a first approach, we used fluorescently labeled virus particles to follow endocytic internalization of JEV. To detect single JEV particle entry events in Neuro2a cells, we labeled virions with the membrane-permeative lipophilic dye DiD. This approach has been used successfully with dengue, influenza, and hepatitis C viruses (12, 41, 42). A homogeneously labeled particle suspension was obtained, as indicated by confocal microscopy of labeled particles attached to glass coverslips (Fig. 2A, left panel). A majority of the DiD-labeled viral particles have uniform fluorescence emissions. To confirm that the DiD signal is specific to labeled JEV particles, we immunostained DiD-labeled JEV with JEV envelope antibody (Fig. 2A, middle and left panels, insets). There was no significant loss of infectivity of DiD-labeled virus particles as tested by plaque assays (Fig. 2B).
Fig 2.

DiD-labeled JEV is internalized independently of clathrin-mediated endocytosis cargo transferrin. (A) DiD-labeled JEV particles were spotted onto a poly-d-lysine-coated coverslip, immunostained with JEV E antibody, and imaged on a 60× objective. Images are presented as gray scales for individual colors and were pseudocolored as indicated prior to being merged. Insets show magnified views of the region corresponding to the asterisk. Bar, 10 μm. (B) DiD-labeled JEV and unlabeled JEV were processed identically, and infectious viral titers were calculated by plaque assays. (C) DiD-labeled JEV (pseudocolored in green) was allowed to bind to Neuro2a cells on ice for 1 h, washed with chilled PBS, and then incubated at 37°C for different times in the presence of Alexa 568/Alexa 647-Tf (pseudocolored in red). Cells were imaged at ×60. The upper right panel represents a differential interference contrast (DIC) image (superimposed with fluorescence from the DiD channel, pseudocolored in green) of the fluorescent image in the upper left panel. Note that DiD-labeled JEV endosomes remain segregated from Tf endosomes until 15 min (arrowheads), and some overlap is detected only by 30 min (arrows). Bar, 5 μm.
DiD-labeled JEV was allowed to bind to Neuro2a cells on ice for 1 h, followed by a pulse with labeled Tf for different times. By 10 min, virus particles can be seen associated with the cell body, with several virus particles attached to filopodia (Fig. 2C, upper right panel, arrowheads). At this time point, no colocalization of the internalized virus particles is seen with Tf (Fig. 2C, upper left panel, arrowheads). Virus internalization was a slow process, and the virus-containing vesicles remained segregated from Tf endosomes until 15 min postentry (Fig. 2B, lower left panel, arrowheads). After 30 min of internalization, there was about 50% colocalization between virus and Tf endosomes, and this could imply presence of the virus in Rab5 early endosomes (Fig. 2B, lower right panel, arrows). Virus entry in Neuro2a cells appeared to be independent of Tf endocytosis, since most of the internalized virus was seen in vesicles that did not colocalize with Tf endosomes at early time points.
JEV entry in Neuro2a cells is clathrin independent.
The role of clathrin-mediated endocytosis in JEV infection was further tested by employing different strategies to disrupt the pathway. Chlorpromazine is commonly used to inhibit clathrin-mediated endocytosis; however, this drug also elicits other secondary effects on the cell, including inhibition of voltage-gated potassium channels in neuronal cells and inhibition of lysosomal and cytosolic phospholipases (43, 44). Studies have shown that chlorpromazine can inhibit JEV infection of Vero cells (18). We tested this effect of chlorpromazine on Neuro2a, SH-SY5Y, and Vero cells. A concentration of 50 μM was chosen for infection studies, as this dosage showed maximum inhibition in Tf uptake in all three cell lines (nearly 50% or more for Neuro2a and SH-SY5Y cells and 35% for Vero cells) (Fig. 3A), and exposure to higher drug concentrations led to significant cell death. While there was a greater than 60% block in JEV infection of Vero cells, there was no significant inhibition of infection of Neuro2a and SH-SY5Y cells (Fig. 3B and D). Treatment of Neuro2a cells with chlorpromazine also did not have any effect on the amount of JEV endocytosed as calculated by qRT-PCR (Fig. 3C). These data suggest that JEV entry is likely to be cell type dependent.
Fig 3.

JEV entry and infection in neuronal cells are not inhibited by chlorpromazine treatment. (A) Neuro2a, SH-SY5Y, and Vero cells were pretreated for 30 min with chlorpromazine at the indicated concentrations and given a pulse of Alexa 488-Tf for 10 min. Cells were fixed, and transferrin uptake was quantified by flow cytometry. The averages ± SDs of measured geometric means of internalized Tf in control and chlorpromazine-treated cells are shown. (B) Neuro2a, SH-SY5Y, and Vero cells were pretreated for 30 min with 50 μM chlorpromazine and infected with JEV (MOI, 0.4) in the presence of inhibitor. Infection was scored as described in Materials and Methods. Student's t test was used to generate P values. *, P < 0.05. (C) Neuro2a cells were infected with JEV (MOI, 10) in the presence of 50 μM chlorpromazine for 1 h, and the amount of virus endocytosed was estimated by qRT-PCR of JEV positive-strand RNA. (D) Virus titers (mean ± SD) in culture supernatants at 24 hpi (MOI, 0.4) were calculated by plaque assay.
The role of clathrin-mediated endocytosis was further tested by the expression of GFP-tagged dominant-negative (DN) Eps15 mutants. Eps15 is a crucial component of clathrin-coated pits, where it interacts with adaptor protein 2 (AP-2), the major clathrin adaptor complex. Clathrin-mediated endocytosis can be blocked by overexpression of the DN Eps15 mutant DIII, which has a large N-terminal deletion, leaving only its C-terminal DIII domain intact (32). The DIII mutant with an additional deletion of its AP-2 binding sites (DIIIδ2), was used as a control to monitor overexpression effects. Overexpression of the DN Eps15 decreased transferrin internalization in Neuro2a cells by around 50% (Fig. 4A versus B; quantitation in shown in Fig. 4C). However, when cells overexpressing the DN Eps15 mutant and control plasmids were infected with JEV, there was a significant block in infection in Vero cells, whereas Neuro2a and SH-SY5Y cells showed infection comparable to that of the control (Fig. 4D and E). These results imply that JEV internalization in neuronal cells can occur independently of the clathrin-mediated endocytic pathway.
Fig 4.

JEV infection in neuronal cells is clathrin independent. (A and B) Neuro2a cells were transfected with plasmids for GFP-Eps15-DIIIδ2 (A) and GFP-Eps15-DIII (B) and 24 h later were given a pulse of Alexa 568-Tf for 10 min. Images are presented as gray scales for individual colors and were pseudocolored as indicated prior to being merged. Note that the majority of cells transfected with Eps15-DIII (arrows) show a block in Tf uptake, whereas cells transfected with control Eps15-DIIIδ2 (arrows) show Tf uptake. Bar, 20 μm. (C) Quantitation of Tf internalization in Neuro2a cells transfected with GFP-Eps15 constructs DIIIδ2 and DIII was done as described in Materials and Methods. (D) Neuro2a, SH-SY5Y, and Vero cells transfected with GFP-Eps15-DIIIδ2 and GFP-Eps15 DIII were infected with JEV (MOI, 1). At 24 hpi, cells were fixed and stained for JEV E antigen. Infection was scored by counting the number of GFP-transfected cells stained versus unstained for JEV E and normalized to GFP-expressing infected cells. (E) Virus titers (mean ± SD) in culture supernatants of transfected cells at 24 hpi (MOI, 1) were calculated by plaque assay. (C and D) Student's t test was used to generate P values; **, P < 0.01; *, P < 0.05.
To further dissect the clathrin-independent endocytic mechanism in neuronal cells, we focused on the cell line Neuro2a along with Vero fibroblasts. The main components of clathrin-coated pits and vesicles are clathrin triskelions, consisting of three heavy and three light chains. To specifically inhibit clathrin-mediated endocytosis, we used siRNA to knock down the expression of both the clathrin light chain (CLC) and clathrin heavy chain (CHC) in independent experiments. Both siRNAs have been previously used to inhibit clathrin-mediated endocytosis (45). CLC was depleted by use of a GFP-tagged shRNA construct that targets an evolutionarily conserved region of the gene. Overexpression of this construct in Neuro2a cells showed a significant inhibition (∼60%) of Tf internalization in GFP-positive cells by 72 h (Fig. 5A, compare left panel [mock transfected] versus right panel [CLC shRNA transfected]; quantitation is shown in Fig. 5B), and nearly 90% of CLC was depleted from cells as tested by Western blotting in both Neuro2a and Vero cells (Fig. 5C). Neuro2a cells depleted of CLC did not show any reduction in JEV infection compared to cells transfected with shRNA vector alone, whereas Vero cells showed close to 50% inhibition of infection (Fig. 5D and E). Depletion of CHC in Neuro2a cells (Fig. 5F) also did not show any significant decrease in JEV infection compared to that in mock-transfected cells (Fig. 5G and H). These experiments strongly suggest that JEV internalization in mouse neuroblastoma cells can occur efficiently via a clathrin-independent mechanism.
Fig 5.

Depletion of clathrin light-chain and clathrin heavy-chain proteins has no effect on JEV infection of neuronal cells. (A) Neuro2a cells were transfected with either GFP-Retro Q shRNA plasmid vector alone (left panel) or GFP-CLC shRNA (right panel) and 72 h later were given a 10-min pulse of Alexa 647-Tf (pseudocolored in red). Note that cells expressing vector alone show efficient Tf uptake, while cells expressing CLC shRNA show a block in Tf endocytosis. Bar, 5 μm. (B) Quantitation of Tf uptake was done as described in Materials and Methods. (C) Western blots showing depletion of CLC in Neuro2a and Vero cells transfected with CLCshRNA plasmid for 72 h and loading control actin. (D) Neuro2a and Vero cells were transfected with shRNA plasmids directed against clathrin light chain (CLC) or control mock plasmid. After 72 h, cells were infected with JEV (MOI, 1). Cells were scored at 24 hpi by immunofluorescence staining of JEV E antigen. Infection was quantified as described above. (E) JEV titers in Neuro2a and Vero cells transfected with control and CLCshRNA plasmids, estimated at 24 hpi (MOI, 1). (F) Western blot showing depletion of CHC in Neuro2a cells transfected with shRNA plasmids for 72 h and loading control actin. (G) JEV infection in Neuro2a cells in the background of CHC depletion was scored as described above. (H) Virus titers (mean ± SD) in culture supernatants of Neuro2a cells depleted of CHC, infected at an MOI of 1, were calculated at 24 hpi by plaque assays. Student's t test was used to generate P values. **, P < 0.01; *, P < 0.05.
Cholesterol is required for JEV infection.
Since a number of viral entry pathways depend on cholesterol, we next examined whether JEV internalization was sensitive to lowering of membrane cholesterol levels, which is a major determinant of many endocytic processes (46). Though several studies imply the requirement of cholesterol to be an indicator of caveolar/lipid raft endocytosis, cholesterol dependence of cell entry is an operational definition and does not imply a specific pathway of entry (24). Most endocytic pathways are sensitive to cholesterol perturbation, with both clathrin-dependent (47, 48) and clathrin-independent (26, 49, 50) pathways being inhibited by removal of cholesterol. Also, cargo such as simian virus 40 (SV40) and cholera toxin B, which were earlier believed to be markers for caveolar endocytosis, can be internalized via caveolin-independent pathways (51, 52). Thus, the definition of a caveolar endocytic pathway in terms of its specific cargo is now redundant.
For the dengue and West Nile flaviviruses, cholesterol was shown to be required for infection (53–55). Mouse neural stem/progenitor cells depleted of cholesterol before JEV infection also showed a reduction in viral load and infective virus particle production (19). In contrast, another study showed an increase in JEV infection upon treatment of Huh-7 cells with methyl-β-cyclodextrin (56).
Our experiments demonstrated that membrane cholesterol was required for JEV infection of both neuronal cells and fibroblasts. Cholesterol depletion was done by treating cells with methyl-β-cyclodextrin, a drug that selectively extracts cholesterol from the plasma membrane (57), or with filipin, a compound that binds selectively to cholesterol, forming complexes in the plasma membrane that sequester cholesterol and induce structural disorder (58). Cells were treated with 5 mM and 7.5 mM methyl-β-cyclodextrin or with 0.5 μM and 1 μM filipin before being infected with JEV. Both treatments resulted in a marked inhibition (60% or greater) of JEV infection, highlighting an essential role for cholesterol in both clathrin-dependent and clathrin-independent endocytosis of JEV in different cell lines (Fig. 6A). Quantitative RT-PCR also showed an inhibition of JEV endocytosis in cholesterol-depleted Neuro2a cells (Fig. 6B).
Fig 6.

Cholesterol is required for JEV infection. (A) Neuro2a and Vero cells were treated with the indicated concentrations of MβCD or filipin for 1 h and infected with JEV (MOI, 0.4) in the presence of inhibitor. At 24 hpi, cells were fixed and infection was quantified as described in Materials and Methods. (B) Neuro2a cells were treated with the indicated concentrations of MβCD or filipin for 1 h and infected with JEV (MOI, 10) in the presence of inhibitor. The endocytosed viral load was estimated by qRT-PCR of JEV positive-strand RNA. Student's t test was used to generate P values. **, P < 0.01; *, P < 0.05.
JEV binding on cells leads to active actin rearrangements.
The actin cytoskeleton is an important regulator of all membrane processes. Studies have shown that upon interaction with filopodia, several viruses, such as HIV, murine leukemia virus (MLV), and vesicular stomatitis virus (VSV), undergo rapid actin-and myosin II-driven transport, “surfing” to entry sites at the cell body (59). To test whether JEV binding and infection events lead to any global actin rearrangements, virus was added to Neuro2a cells and left for different times and actin was observed via phalloidin staining. Uninfected cell surfaces were smooth and showed a clear margin with peripheral F-actin staining and random filopodial projections (Fig. 7A, upper left panel). As early as 3 min postinfection (mpi), several cells showed increased intensity of cortical actin structures and filopodial projections (Fig. 7A, upper right panel), which increased with time (Fig. 7A, lower panels). By 5 min postinfection, several cells showed a dramatic increase in the number of filopodia that showed a regular arrangement around the cell periphery (Fig. 7A, lower left panel). This effect peaked at 10 min postinfection (Fig. 7A, lower right panel). This increased distribution of filopodia on cells is likely to represent induction of an efficient infectious pathway brought about by some signaling event initiated by virus binding.
Fig 7.

Role of actin and myosin motors in JEV entry in neuronal cells. (A) JEV (MOI, 10) was added to Neuro2a cells and left for the indicated times at 37°C, following which cells were washed with chilled PBS, fixed, and stained with Alexa 546-phalloidin. Images were taken at ×60. Bar, 10 μm. (B) Neuro2a and Vero cells were treated with inhibitors cytochalasin D, latrunculin, jasplakinolide, and blebbistatin at the indicated concentrations. Cells were infected with JEV (MOI, 0.4) in the presence of inhibitor, and infection was scored as described in Materials and Methods. (C) Neuro2a cells with treated with inhibitors at the indicated concentrations and infected with JEV at an MOI of 10 in the presence of the inhibitor. Virus entry was quantified by qRT-PCR. Student's t test was used to generate P values. **, P < 0.01; *, P < 0.05.
JEV entry in neuronal cells is actin and myosin II dependent.
The induction of actin-rich filopodia on cells after virus binding implies that viral entry is likely to be an actin- and motor-driven process. We further examined the role of the actin cytoskeleton in JEV entry by using chemical inhibitors of actin polymerization and depolymerization. Cytochalasin D (CytoD) reversibly targets barbed ends of F-actin, inducing depolymerization of existing filaments and increasing the cellular pool of ADP-bound actin monomers (60). Latrunculin A reversibly disrupts actin dynamics by targeting monomeric G-actin and preventing actin polymerization (61). Treatment of cells with CytoD at concentrations of 5 μM and 2.5 μM showed a 70% block in JEV infection in Neuro2a cells and a 20% decrease in Vero cells (Fig. 7B). Similarly, treatment with latrunculin at concentrations of 5 μM and 2.5 μM significantly reduced JEV infection in Neuro2a cells while having a marginal effect in Vero cells (Fig. 7B). To block turnover of actin microfilaments, we used a cell-permeative drug, jasplakinolide, that selectively binds to F-actin and dramatically decreases rate of actin depolymerization (62). Treatment with jasplakinolide at concentrations ranging from 0.5 to 2 μM resulted in about 80% inhibition of JEV infection in Neuro2a cells (Fig. 7B). We also confirmed that these drugs block JEV entry inside cells by qPCR (Fig. 7C). Collectively, our pharmacological and immunofluorescence labeling data strongly suggest that the dynamic reorganization (depolymerization/reploymerization) of actin microfilaments is critical for JEV entry in neuronal cells.
We further examined whether myosin-driven contractions would provide the mechanical force for JEV infection process. It has been shown that myosin motors are involved in movement of actin filaments in cell surface protrusions (63), a process known as rearward or retrograde F-actin flow. Nonmuscle myosin II is a plus-end motor that localizes to the lamellum and retraction fibers and is involved in viral surfing on filopodia. Since myosin II is an important contributor to the cytoskeleton of neuronal cells and may influence the trafficking of JEV, we tested its involvement in viral entry by using its specific inhibitor blebbistatin (64). Blebbistatin interferes neither with binding of myosin to actin nor with ATP-induced actomyosin dissociation, but it blocks myosin II in an actin-detached state. Treatment of cells with blebbistatin (50 μM) resulted in about a 50% block in JEV infection in Neuro2a cells and a 30% block in Vero cells (Fig. 7B and C). These results suggest that myosin is essential for infection and that JEV entry may involve virus movement along filopodia to the cell body utilizing the underlying actin-myosin II machinery.
Role of the small GTPases in JEV infection.
We next examined the role of the Rho family of small GTPases, i.e., Rho, Rac, and Cdc42, that are involved in spatiotemporal control of actin polymerization and also play a regulatory role in endocytic processes (65). To dissect the effects of individual Rho family GTPases, we expressed wild-type, dominant-negative, and dominant-active isoforms of RhoA, Rac1, and Cdc42 and tested their activity in cells (Fig. 8A, B, and C). While the dominant-active isoforms showed severalfold-higher GTPase activity than the wild type, the dominant-negative isoforms showed close to a 50% decrease in GTPase activity when overexpressed. These constructs were expressed in Neuro2a cells and checked for their effects in the JEV infection process. Cells expressing myc-tagged isoforms of wt RhoA, wt Cdc42, Cdc42N17 (DN), Cdc42L61 (DA), Rac1L61 (DA), and Rac1N17 (DN) had roughly equal efficiency of JEV infection. Only the RhoAN19 (DN) isoform showed about a 50% reduction in JEV infection (Fig. 8D). The dominant-active RhoA (L63) had a minor effect and reduced JEV infection by about 20%. This could be because of decreased GTP-GDP turnover rates of endogenous wt Rho protein in the background of overexpression of dominant-active RhoA. As an alternative, we also tested the effect of CT04, a specific RhoA inhibitor (66, 67). The active site of CT04 is the exoenzyme C3 transferase from Clostridium botulinum. CT04 specifically inhibits RhoA, -B, and -C proteins by ADP-ribosylation on asparagine 41 in the effector binding domain of the GTPase (68). Rho inhibition by CT04 was tested in Neuro2a cells by cell morphology phenotypes and phalloidin staining. Cells treated with CT04 at a concentration of 2 μg/ml showed close to a 60% block in JEV infection and a 50% block in JEV entry, strongly suggesting the involvement of RhoA in the JEV endocytic internalization process in neuronal cells (Fig. 8F and G).
Fig 8.

The GTPase Rho A is required for JEV infection of neuronal cells. (A to C) Neuro2a cells were transfected with myc-tagged constructs of wt RhoA, RhoA L63, RhoA N19, Rac1 L61, Rac1 N17, wt Cdc42, Cdc42 L61, and Cdc42 N17. The GTPase activity of the overexpressed constructs was quantified using GTPase-specific ELISA as described in Materials and Methods. (D) Neuro2a cells transfected with the wt, DA, and DN constructs of Rho, Rac, and Cdc42 were infected with JEV (MOI, 1). At 24 hpi, double immunofluorescence staining was done for myc and JEV E. Cells staining positive for both myc and JEV E were scored and normalized to wt RhoA-expressing infected cells. (E) Time course of Rho activation in response to JEV binding using Rho GTPase-specific ELISA as described in Materials and Methods. (F) Neuro2a cells were serum starved for 2 h before treatment with CT04 inhibitor for another 2 h. Cells were infected with JEV (MOI, 0.4) in the presence of inhibitor. Infection was quantified as described in Materials and Methods. (G) Virus endocytosis (MOI, 10) was quantified in control and CT04-treated cells by qRT-PCR. Student's t test was used to generate P values, **, P < 0.01; *, P < 0.05.
Since RhoA appeared to play a critical role in viral infection, we investigated the activation status of RhoA during the initial stages of JEV entry in Neuro2a cells. JEV (MOI, 10) was used to infect cells, and Rho activation was measured with the G-LISA Rho activation kit following the manufacturer's instructions. The results showed that Rho activation is an immediate and robust event during JEV entry, reaching a maximum (close to 2-fold) at 10 mpi compared to that in mock-infected cells (Fig. 8E). This result further corroborates that JEV entry induces RhoA activation in neuronal cells.
JEV infection requires passage through Rab5-positive early endosomes but is independent of Rab7.
It is established that all flaviviruses require acidification in the endosome for uncoating and release of the viral nucleic acid for replication (11, 69). We confirmed that JEV entry in Neuro2a cells was acid dependent, as pretreatment of cells with 100 to 200 nM bafilomycin A1, a drug that is a potent inhibitor of the vacuolar ATPase and specifically prevents acidification of endosomal vesicles (70), led to about a 95% decrease in JEV infection in both Neuro2a and Vero cells (Fig. 9B).
Fig 9.
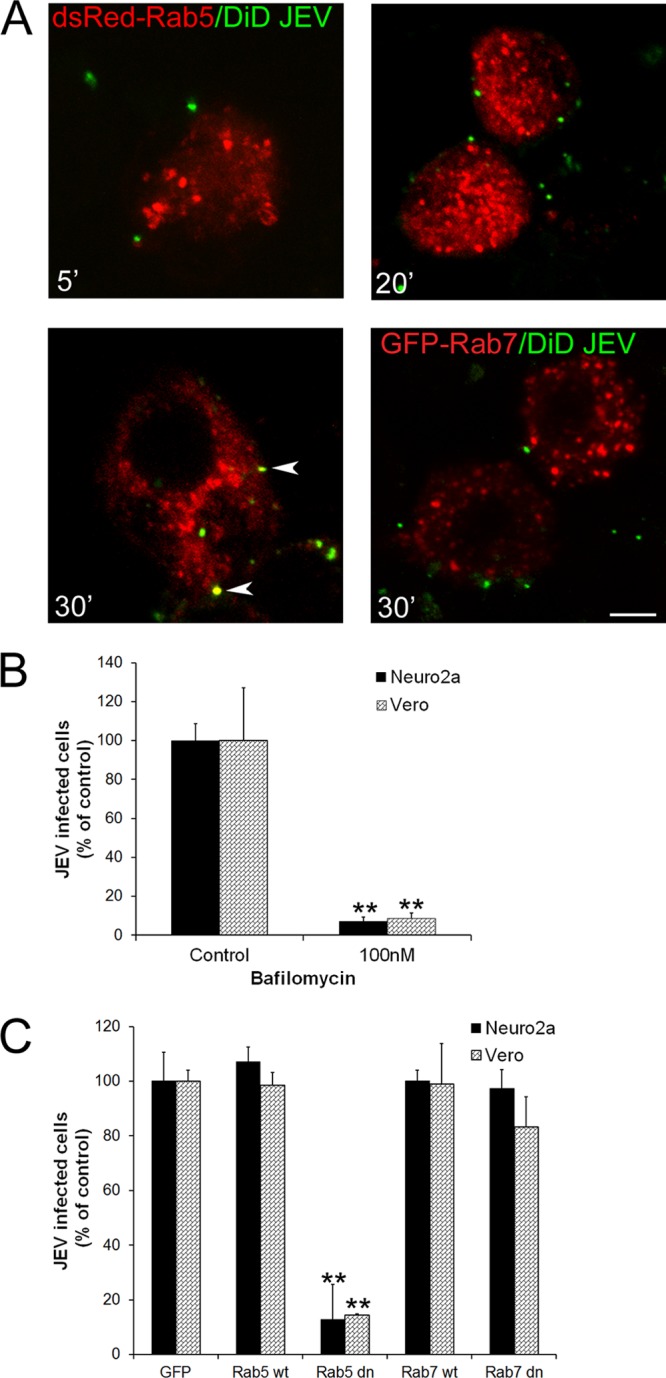
JEV infection requires trafficking through Rab5-positive compartments but is independent of Rab7. (A) DiD-labeled JEV (pseudocolored in green) was added to Neuro2a cells transfected with dsRed-Rab5 or GFP-Rab7 (pseudocolored in red) on ice for 1 h. Cells were warmed to 37°C for the indicated times, washed with low-pH buffer, fixed, and imaged. DiD-labeled JEV colocalization with Rab5 is seen only by 30 min postinternalization (lower left panel, arrowheads). Bar, 10 μm. (B) Neuro2a and Vero cells were pretreated with 100 nM bafilomycin before infection with JEV. Infection was quantified as described in Materials and Methods. (C) Neuro2a and Vero cells transfected with GFP, GFP-wt Rab5, GFP-Rab5dn, GFP-wt Rab7, or GFP-Rab7dn were infected with JEV and processed at 24 hpi as described above. Infection was normalized to cells transfected with GFP alone. Student's t test was used to generate P values. **, P < 0.01.
Rab proteins are known to orchestrate membrane traffic in the cell and show distinct localizations to specific subcellular compartments. Cargo from CME is sorted into early endosomes that are marked by Rab5 and EEA1. From the early endosomes, molecules are either sorted for recycling or directed to late endosomes that are Rab7 and LAMP1 positive. Endosomes carrying cargo internalized via clathrin-independent pathways also fuse with Rab5-positive early endosomes (71, 72). Studies with Dengue virus have demonstrated that the virus moves from Rab5-positive early endosomes to Rab7-positive late endosomes before viral RNA is released in the cytoplasm (11, 12). West Nile virus was also shown to traffic from EEA1-positive early endosomes to LAMP1-positive late endosomes, where virus uncoating finally occurred (10, 73). In contrast, another study based on RNA interference showed entry of dengue and West Nile viruses to be dependent on Rab5 but not on Rab7, indicating that virus uncoating occurs at the level of early endosomes (74).
To investigate the endocytic trail followed by JEV, DiD-labeled virus was allowed to bind to Neuro2a cells transfected with either dsRed-Rab5 or GFP-Rab7 on ice for 1 h and then incubated at 37°C for different times to follow virus internalization in the cell. Virus internalization was a slow process, and virus particles were seen in endosomes distinct from Rab5 until 20 min of internalization (Fig. 9A, upper panels). After 30 min of uptake, virus-containing vesicles had acquired/fused with Rab5 endosomes (Fig. 9A, lower left panel, arrowheads). By 1 h of internalization, the virus signal significantly decreased and was no longer detectable inside the cell. Labeled virus particles were not detected in GFP-Rab7-positive late endosomes at any time point, indicating that virus fusion is happening at a level upstream of Rab7 late endosomes (Fig. 9A, lower right panel).
To further confirm this observation, Neuro2a and Vero cells were transfected with GFP-dnRab5 or GFP-dnRab7 for 24 h and virus infection was monitored. Whereas cells transfected with dnRab5 showed about a 90% block in JEV infection, cells transfected with dnRab7 showed no significant inhibition. This indicates that JEV fusion occurs at the level of the early endosome and not the late endosome (Fig. 9C).
DISCUSSION
Current knowledge indicates that a complex network of diverse, continuing, and triggered pathways operate at the eukaryotic plasma membrane. Cargo specificity, coat proteins, and scission molecules are used to define most of these mechanisms. The orderly transport of endocytic cargo is tightly regulated and requires the participation of numerous lipids and accessory proteins. Endocytosis also requires alterations of fine cellular structures and mechanical force to internalize a vesicle. Viruses are adept at exploiting these mechanisms for gaining entry (25). Nearly all viruses utilize an endocytic mechanism to gain entry into a permissive cell and establish infection. These pathways serve to deliver viruses to vesicles and compartments conducive to viral membrane fusion and release of the core into the cell cytoplasm at a site permissive to replication. The route of virus entry can differ between cell types. In addition to utilizing the already-operational endocytic pathways, in several cases viruses can induce pathways conducive to entry by receptor binding and signaling events.
The results presented here indicate that entry of JEV in neuronal cells involves strategies different from those described for many other flaviviruses. JEV internalization occurs through a clathrin-independent mechanism, and the cellular factors needed for entry are dynamin, cholesterol, dynamic actin cytoskeleton, the small GTPase RhoA, and myosin II motors. After endocytosis, the virus traffics through Rab5-positive early endosomes, which are possibly the sites for viral uncoating and genome release.
The information currently available suggests that flavivirus internalization is clathrin dependent. Studies have shown that JEV infection of Vero and neural stem/progenitor cells can be inhibited by chlorpromazine (18, 19). We also observed a clathrin-dependent pathway for JEV infection in Vero cells. By depleting the key coat proteins clathrin heavy chain and clathrin light chain, our study clearly demonstrates that a clathrin-independent pathway is operational for JEV entry in neuronal cells. Neuronal cells are highly relevant for investigating JEV infection, since JEV is primarily a neurotropic virus. It is likely that the endocytic route followed by the virus is cell type dependent.
Cholesterol dependence is often considered to be a marker of the caveolar/lipid raft-mediated pathway (75). However, cholesterol is essential for most membrane processes. For flaviviruses as well, membrane cholesterol is a requirement for entry (19, 54). Our study with different cell types shows that membrane cholesterol is an absolute requirement for JEV internalization.
JEV was seen associated with filopodia and the cell body. Virus binding induced dynamic rearrangements of the actin cytoskeleton at the early stages of infection. Several viruses activate small GTPases to form lamellipodia and filopodia in cells (76, 77). Our data from experiments with drugs that alter actin organization show that both actin polymerization and depolymerization are required during the virus infection cycle. Filopodia are connected to the cortical actin network, which, in association with myosin II, controls their contractility at their base, and the motor activity of myosin II could be required for JEV infection. Our results point to a greater role for the actin cytoskeleton in early viral entry events in neuronal cells.
After receptor binding, viruses undergo a period of diffusive or directed motion on the plasma membrane until they become confined (78). Some viruses can move along filopodia to internalization sites by receptor interaction with the actin cytoskeleton and retrograde flow within filopodia (59, 79). The actin cytoskeleton plays a structural role in endocytosis. Its dynamic nature is essential for its function. Existing actin filaments undergo severing and depolymerization in response to cellular requirements and stimuli, while new actin filaments are polymerized from monomeric actin subunits and by branching off from existing filaments. These processes are regulated by the Rho GTPases. Studies on herpesviruses have shown that RhoA can induce filopodia in infected cells (76). Entry of Kaposi's sarcoma-associated herpesvirus (KSHV) induces RhoA GTPase and rearrangements of both microtubules and the actin cytoskeleton in fibroblasts (80), and RhoA GTPase is also important for virus entry in HEK293 cells (81). Our study also points to an important role for RhoA in virus entry. The binding of JEV to its specific receptor is likely to activate signaling of cells via RhoA activation.
Several viruses induce macropinocytosis and use it as a route for internalization and infection (29, 30, 82, 83). JEV binding and uptake in Neuro2a cells did not show characteristics of macropinocytic uptake. Further JEV internalization was not affected by treatment of cells with amiloride, an inhibitor that specifically blocks Na+/H+ exchange and macropinocytois (data not shown).
The present findings provide the first evidence for a clathrin-independent, actin-dependent pathway utilized by JEV for infection of neuronal cells. At least three clathrin-independent mechanisms at the plasma membrane have been described, primarily on the basis of cargo, dynamin dependence/independence, the role of cellular factors such as Arf and Rho GTPases, and actin and cholesterol requirements (23, 24). It is still unclear whether JEV utilizes a preexisting clathrin-independent endocytic pathway or whether a specific pathway is induced in response to virus binding and downstream signaling. The receptor for JEV on neuronal cells is still uncharacterized. It is possible that binding to a specific receptor and entry through a clathrin-independent pathway confer on the virus a greater advantage to establish infection in neuronal cells. Like all viruses, JEV relies on host cell factors and physiological processes for key steps of its replication cycle. Identification of these processes and factors not only will allow a better insight into pathogenic mechanism but may identify novel targets for future therapeutic development.
ACKNOWLEDGMENTS
This work was supported by the Department of Biotechnology (DBT), Government of India (grant BT/MB/01/VIDRC/08 to S.V.).
We thank Alexandre Benmerah for the Eps15 constructs. M.K. is grateful to Shahid Jameel, Sankar Bhattacharya, and Arup Banerjee for critical input on the manuscript. We thank Manpreet Kaur, Vikas Sood, and Taranjeet Kaur for assistance with flow cytometry, RT-PCR, and virus generation.
Footnotes
Published ahead of print 10 October 2012
REFERENCES
- 1. Lindenbach BD, Thiel Heinz-Jurgen Rice CM. 2007. Flaviviridae: the viruses and their replication, p 1101–1152 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed, vol 1 Lippincott-Raven Publishers, Philadelphia, PA [Google Scholar]
- 2. Mackenzie JS, Gubler DJ, Petersen LR. 2004. Emerging flaviviruses: the spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat. Med. 10:S98–109 [DOI] [PubMed] [Google Scholar]
- 3. Pyke AT, Williams DT, Nisbet DJ, van den Hurk AF, Taylor CT, Johansen CA, Macdonald J, Hall RA, Simmons RJ, Mason RJ, Lee JM, Ritchie SA, Smith GA, Mackenzie JS. 2001. The appearance of a second genotype of Japanese encephalitis virus in the Australasian region. Am. J. Trop. Med. Hyg. 65:747–753 [DOI] [PubMed] [Google Scholar]
- 4. Allison SL, Schalich J, Stiasny K, Mandl CW, Heinz FX. 2001. Mutational evidence for an internal fusion peptide in flavivirus envelope protein E. J. Virol. 75:4268–4275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kuhn RJ, Zhang W, Rossmann MG, Pletnev SV, Corver J, Lenches E, Jones CT, Mukhopadhyay S, Chipman PR, Strauss EG, Baker TS, Strauss JH. 2002. Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell 108:717–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gollins SW, Porterfield JS. 1986. The uncoating and infectivity of the flavivirus West Nile on interaction with cells: effects of pH and ammonium chloride. J. Gen. Virol. 67:1941–1950 [DOI] [PubMed] [Google Scholar]
- 7. Ishak R, Tovey DG, Howard CR. 1988. Morphogenesis of yellow fever virus 17D in infected cell cultures. J. Gen. Virol. 69:325–335 [DOI] [PubMed] [Google Scholar]
- 8. Ng ML, Lau LC. 1988. Possible involvement of receptors in the entry of Kunjin virus into Vero cells. Arch. Virol. 100:199–211 [DOI] [PubMed] [Google Scholar]
- 9. Acosta EG, Castilla V, Damonte EB. 2011. Infectious dengue-1 virus entry into mosquito C6/36 cells. Virus Res. 160:173–179 [DOI] [PubMed] [Google Scholar]
- 10. Chu JJ, Leong PW, Ng ML. 2006. Analysis of the endocytic pathway mediating the infectious entry of mosquito-borne flavivirus West Nile into Aedes albopictus mosquito (C6/36) cells. Virology 349:463–475 [DOI] [PubMed] [Google Scholar]
- 11. Mosso C, Galvan-Mendoza IJ, Ludert JE, del Angel RM. 2008. Endocytic pathway followed by dengue virus to infect the mosquito cell line C6/36 HT. Virology 378:193–199 [DOI] [PubMed] [Google Scholar]
- 12. van der Schaar HM, Rust MJ, Chen C, van der Ende-Metselaar H, Wilschut J, Zhuang X, Smit JM. 2008. Dissecting the cell entry pathway of dengue virus by single-particle tracking in living cells. PLoS Pathog. 4:e1000244 doi:10.1371/journal.ppat.1000244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Acosta EG, Castilla V, Damonte EB. 2009. Alternative infectious entry pathways for dengue virus serotypes into mammalian cells. Cell. Microbiol. 11:1533–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Acosta EG, Castilla V, Damonte EB. 2008. Functional entry of dengue virus into Aedes albopictus mosquito cells is dependent on clathrin-mediated endocytosis. J. Gen. Virol. 89:474–484 [DOI] [PubMed] [Google Scholar]
- 15. Alhoot MA, Wang SM, Sekaran SD. 2011. Inhibition of dengue virus entry and multiplication into monocytes using RNA interference. PLoS Negl. Trop. Dis. 5:e1410 doi:10.1371/journal.pntd.0001410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alhoot MA, Wang SM, Sekaran SD. 2012. RNA interference mediated inhibition of dengue virus multiplication and entry in HepG2 cells. PLoS One 7:e34060 doi:10.1371/journal.pone.0034060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liou ML, Hsu CY. 1998. Japanese encephalitis virus is transported across the cerebral blood vessels by endocytosis in mouse brain. Cell Tissue Res. 293:389–394 [DOI] [PubMed] [Google Scholar]
- 18. Nawa M, Takasaki T, Yamada K, Kurane I, Akatsuka T. 2003. Interference in Japanese encephalitis virus infection of Vero cells by a cationic amphiphilic drug, chlorpromazine. J. Gen. Virol. 84:1737–1741 [DOI] [PubMed] [Google Scholar]
- 19. Das S, Chakraborty S, Basu A. 2010. Critical role of lipid rafts in virus entry and activation of phosphoinositide 3′ kinase/Akt signaling during early stages of Japanese encephalitis virus infection in neural stem/progenitor cells. J. Neurochem. 115:537–549 [DOI] [PubMed] [Google Scholar]
- 20. Nawa M. 1997. Japanese encephalitis virus infection in Vero cells: the involvement of intracellular acidic vesicles in the early phase of viral infection was observed with the treatment of a specific vacuolar type H+-ATPase inhibitor, bafilomycin A1. Microbiol. Immunol. 41:537–543 [DOI] [PubMed] [Google Scholar]
- 21. Lamaze C, Dujeancourt A, Baba T, Lo CG, Benmerah A, Dautry-Varsat A. 2001. Interleukin 2 receptors and detergent-resistant membrane domains define a clathrin-independent endocytic pathway. Mol. Cell 7:661–671 [DOI] [PubMed] [Google Scholar]
- 22. Pelkmans L, Helenius A. 2002. Endocytosis via caveolae. Traffic 3:311–320 [DOI] [PubMed] [Google Scholar]
- 23. Kirkham M, Parton RG. 2005. Clathrin-independent endocytosis: new insights into caveolae and non-caveolar lipid raft carriers. Biochim. Biophys. Acta 1746:349–363 [DOI] [PubMed] [Google Scholar]
- 24. Mayor S, Pagano RE. 2007. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 8:603–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marsh M, Helenius A. 2006. Virus entry: open sesame. Cell 124:729–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Naslavsky N, Weigert R, Donaldson JG. 2004. Characterization of a nonclathrin endocytic pathway: membrane cargo and lipid requirements. Mol. Biol. Cell 15:3542–3552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jaffe AB, Hall A. 2005. Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21:247–269 [DOI] [PubMed] [Google Scholar]
- 28. Kalia M, Jameel S. 2011. Virus entry paradigms. Amino acids 41:1147–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. de Vries E, Tscherne DM, Wienholts MJ, Cobos-Jimenez V, Scholte F, Garcia-Sastre A, Rottier PJ, de Haan CA. 2011. Dissection of the influenza A virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog. 7:e1001329 doi:10.1371/journal.ppat.1001329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mercer J, Helenius A. 2008. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 320:531–535 [DOI] [PubMed] [Google Scholar]
- 31. Saeed MF, Kolokoltsov AA, Albrecht T, Davey RA. 2010. Cellular entry of Ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathog. 6:e1001110 doi:10.1371/journal.ppat.1001110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Benmerah A, Bayrou M, Cerf-Bensussan N, Dautry-Varsat A. 1999. Inhibition of clathrin-coated pit assembly by an Eps15 mutant. J. Cell Sci. 112:1303–1311 [DOI] [PubMed] [Google Scholar]
- 33. Nobes CD, Hall A. 1999. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J. Cell Biol. 144:1235–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Choudhury A, Dominguez M, Puri V, Sharma DK, Narita K, Wheatley CL, Marks DL, Pagano RE. 2002. Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. J. Clin. Invest. 109:1541–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vonderheit A, Helenius A. 2005. Rab7 associates with early endosomes to mediate sorting and transport of Semliki forest virus to late endosomes. PLoS Biol. 3:e233 doi:10.1371/journal.pbio.0030233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sun Q, Westphal W, Wong KN, Tan I, Zhong Q. 2010. Rubicon controls endosome maturation as a Rab7 effector. Proc. Natl. Acad. Sci. U. S. A. 107:19338–19343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vrati S, Agarwal V, Malik P, Wani SA, Saini M. 1999. Molecular characterization of an Indian isolate of Japanese encephalitis virus that shows an extended lag phase during growth. J. Gen. Virol. 80:1665–1671 [DOI] [PubMed] [Google Scholar]
- 38. Doherty GJ, McMahon HT. 2009. Mechanisms of endocytosis. Annu. Rev. Biochem. 78:857–902 [DOI] [PubMed] [Google Scholar]
- 39. Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. 2006. Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 10:839–850 [DOI] [PubMed] [Google Scholar]
- 40. Damke H, Baba T, Warnock DE, Schmid SL. 1994. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J. Cell Biol. 127:915–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Coller KE, Berger KL, Heaton NS, Cooper JD, Yoon R, Randall G. 2009. RNA interference and single particle tracking analysis of hepatitis C virus endocytosis. PLoS Pathog. 5:e1000702 doi:10.1371/journal.ppat.1000702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lakadamyali M, Rust MJ, Babcock HP, Zhuang X. 2003. Visualizing infection of individual influenza viruses. Proc. Natl. Acad. Sci. U. S. A. 100:9280–9285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mathie A, Wooltorton JR, Watkins CS. 1998. Voltage-activated potassium channels in mammalian neurons and their block by novel pharmacological agents. Gen. Pharmacol. 30:13–24 [DOI] [PubMed] [Google Scholar]
- 44. Sathasivam N, Brammah S, Wright LC, Delikatny EJ. 2003. Inhibition of tetraphenylphosphonium-induced NMR-visible lipid accumulation in human breast cells by chlorpromazine. Biochim. Biophys. Acta 1633:149–160 [DOI] [PubMed] [Google Scholar]
- 45. Motley A, Bright NA, Seaman MN, Robinson MS. 2003. Clathrin-mediated endocytosis in AP-2-depleted cells. J. Cell Biol. 162:909–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Parton RG, Richards AA. 2003. Lipid rafts and caveolae as portals for endocytosis: new insights and common mechanisms. Traffic 4:724–738 [DOI] [PubMed] [Google Scholar]
- 47. Rodal SK, Skretting G, Garred O, Vilhardt F, van Deurs B, Sandvig K. 1999. Extraction of cholesterol with methyl-beta-cyclodextrin perturbs formation of clathrin-coated endocytic vesicles. Mol. Biol. Cell 10:961–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Subtil A, Gaidarov I, Kobylarz K, Lampson MA, Keen JH, McGraw TE. 1999. Acute cholesterol depletion inhibits clathrin-coated pit budding. Proc. Natl. Acad. Sci. U. S. A. 96:6775–6780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chadda R, Howes MT, Plowman SJ, Hancock JF, Parton RG, Mayor S. 2007. Cholesterol-sensitive Cdc42 activation regulates actin polymerization for endocytosis via the GEEC pathway. Traffic 8:702–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cheng ZJ, Singh RD, Sharma DK, Holicky EL, Hanada K, Marks DL, Pagano RE. 2006. Distinct mechanisms of clathrin-independent endocytosis have unique sphingolipid requirements. Mol. Biol. Cell 17:3197–3210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Damm EM, Pelkmans L, Kartenbeck J, Mezzacasa A, Kurzchalia T, Helenius A. 2005. Clathrin- and caveolin-1-independent endocytosis: entry of simian virus 40 into cells devoid of caveolae. J. Cell Biol. 168:477–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kirkham M, Fujita A, Chadda R, Nixon SJ, Kurzchalia TV, Sharma DK, Pagano RE, Hancock JF, Mayor S, Parton RG. 2005. Ultrastructural identification of uncoated caveolin-independent early endocytic vehicles. J. Cell Biol. 168:465–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee CJ, Lin HR, Liao CL, Lin YL. 2008. Cholesterol effectively blocks entry of flavivirus. J. Virol. 82:6470–6480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Medigeshi GR, Hirsch AJ, Streblow DN, Nikolich-Zugich J, Nelson JA. 2008. West Nile virus entry requires cholesterol-rich membrane microdomains and is independent of alphavbeta3 integrin. J. Virol. 82:5212–5219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Puerta-Guardo H, Mosso C, Medina F, Liprandi F, Ludert JE, del Angel RM. 2010. Antibody-dependent enhancement of dengue virus infection in U937 cells requires cholesterol-rich membrane microdomains. J. Gen. Virol. 91:394–403 [DOI] [PubMed] [Google Scholar]
- 56. Tani H, Shiokawa M, Kaname Y, Kambara H, Mori Y, Abe T, Moriishi K, Matsuura Y. 2010. Involvement of ceramide in the propagation of Japanese encephalitis virus. J. Virol. 84:2798–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kilsdonk EP, Yancey PG, Stoudt GW, Bangerter FW, Johnson WJ, Phillips MC, Rothblat GH. 1995. Cellular cholesterol efflux mediated by cyclodextrins. J. Biol. Chem. 270:17250–17256 [DOI] [PubMed] [Google Scholar]
- 58. Bolard J. 1986. How do the polyene macrolide antibiotics affect the cellular membrane properties? Biochim. Biophys. Acta 864:257–304 [DOI] [PubMed] [Google Scholar]
- 59. Lehmann MJ, Sherer NM, Marks CB, Pypaert M, Mothes W. 2005. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J. Cell Biol. 170:317–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Goddette DW, Frieden C. 1986. Actin polymerization. The mechanism of action of cytochalasin D. J. Biol. Chem. 261:15974–15980 [PubMed] [Google Scholar]
- 61. Spector I, Shochet NR, Kashman Y, Groweiss A. 1983. Latrunculins: novel marine toxins that disrupt microfilament organization in cultured cells. Science 219:493–495 [DOI] [PubMed] [Google Scholar]
- 62. Bubb MR, Senderowicz AM, Sausville EA, Duncan KL, Korn ED. 1994. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J. Biol. Chem. 269:14869–14871 [PubMed] [Google Scholar]
- 63. Ponti A, Machacek M, Gupton SL, Waterman-Storer CM, Danuser G. 2004. Two distinct actin networks drive the protrusion of migrating cells. Science 305:1782–1786 [DOI] [PubMed] [Google Scholar]
- 64. Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. 2003. Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science 299:1743–1747 [DOI] [PubMed] [Google Scholar]
- 65. Ellis S, Mellor H. 2000. Regulation of endocytic traffic by rho family GTPases. Trends Cell Biol. 10:85–88 [DOI] [PubMed] [Google Scholar]
- 66. Benink HA, Bement WM. 2005. Concentric zones of active RhoA and Cdc42 around single cell wounds. J. Cell Biol. 168:429–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Burakov A, Nadezhdina E, Slepchenko B, Rodionov V. 2003. Centrosome positioning in interphase cells. J. Cell Biol. 162:963–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Aktories K, Wilde C, Vogelsgesang M. 2004. Rho-modifying C3-like ADP-ribosyltransferases. Rev. Physiol. Biochem. Pharmacol. 152:1–22 [DOI] [PubMed] [Google Scholar]
- 69. Nawa M. 1998. Effects of bafilomycin A1 on Japanese encephalitis virus in C6/36 mosquito cells. Arch. Virol. 143:1555–1568 [DOI] [PubMed] [Google Scholar]
- 70. Bowman EJ, Siebers A, Altendorf K. 1988. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. U. S. A. 85:7972–7976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kalia M, Kumari S, Chadda R, Hill MM, Parton RG, Mayor S. 2006. Arf6-independent GPI-anchored protein-enriched early endosomal compartments fuse with sorting endosomes via a Rab5/phosphatidylinositol-3′-kinase-dependent machinery. Mol. Biol. Cell 17:3689–3704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Naslavsky N, Weigert R, Donaldson JG. 2003. Convergence of non-clathrin- and clathrin-derived endosomes involves Arf6 inactivation and changes in phosphoinositides. Mol. Biol. Cell 14:417–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chu JJ, Ng ML. 2004. Infectious entry of West Nile virus occurs through a clathrin-mediated endocytic pathway. J. Virol. 78:10543–10555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Krishnan MN, Sukumaran B, Pal U, Agaisse H, Murray JL, Hodge TW, Fikrig E. 2007. Rab 5 is required for the cellular entry of dengue and West Nile viruses. J. Virol. 81:4881–4885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Fielding CJ, Fielding PE. 2003. Relationship between cholesterol trafficking and signaling in rafts and caveolae. Biochim. Biophys. Acta 1610:219–228 [DOI] [PubMed] [Google Scholar]
- 76. Clement C, Tiwari V, Scanlan PM, Valyi-Nagy T, Yue BY, Shukla D. 2006. A novel role for phagocytosis-like uptake in herpes simplex virus entry. J. Cell Biol. 174:1009–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Favoreel HW, Enquist LW, Feierbach B. 2007. Actin and Rho GTPases in herpesvirus biology. Trends Microbiol. 15:426–433 [DOI] [PubMed] [Google Scholar]
- 78. Burckhardt CJ, Greber UF. 2009. Virus movements on the plasma membrane support infection and transmission between cells. PLoS Pathog. 5:e1000621 doi:10.1371/journal.ppat.1000621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Schelhaas M, Ewers H, Rajamaki ML, Day PM, Schiller JT, Helenius A. 2008. Human papillomavirus type 16 entry: retrograde cell surface transport along actin-rich protrusions. PLoS Pathog. 4:e1000148 doi:10.1371/journal.ppat.1000148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sharma-Walia N, Naranatt PP, Krishnan HH, Zeng L, Chandran B. 2004. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 envelope glycoprotein gB induces the integrin-dependent focal adhesion kinase-Src-phosphatidylinositol 3-kinase-rho GTPase signal pathways and cytoskeletal rearrangements. J. Virol. 78:4207–4223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Veettil MV, Sharma-Walia N, Sadagopan S, Raghu H, Sivakumar R, Naranatt PP, Chandran B. 2006. RhoA-GTPase facilitates entry of Kaposi's sarcoma-associated herpesvirus into adherent target cells in a Src-dependent manner. J. Virol. 80:11432–11446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kalin S, Amstutz B, Gastaldelli M, Wolfrum N, Boucke K, Havenga M, DiGennaro F, Liska N, Hemmi S, Greber UF. 2010. Macropinocytotic uptake and infection of human epithelial cells with species B2 adenovirus type 35. J. Virol. 84:5336–5350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nanbo A, Imai M, Watanabe S, Noda T, Takahashi K, Neumann G, Halfmann P, Kawaoka Y. 2010. Ebola virus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog. 6:e1001121 doi:10.1371/journal.ppat.1001121 [DOI] [PMC free article] [PubMed] [Google Scholar]