

Abstract
The human genome contains approximately 50 copies of the replication-defective human endogenous retrovirus 9 (ERV-9) and thousands of copies of its solitary long term repeat (sLTR) element. While some sLTRs are located upstream of critical genes and have enhancer activity, other sLTRs are located within introns and may be transcribed as RNAs. We found that intronic RNAs arising from U3 sLTRs of ERV-9 were expressed as both sense (S) and antisense (AS) transcripts in all human cells tested but that expression levels differed in malignant versus nonmalignant cells. In nonmalignant cells, AS was expressed at higher levels than S and at higher levels than in malignant cells; in malignant cells, AS was expressed at amounts equivalent to those of S RNA. Critically, U3 AS RNA was found to physically bind to key transcription factors for cellular proliferation, including NF-Y, p53, and sp1, indicating that such RNA transcripts may function as decoy targets or traps for NF-Y and thus inhibit the growth of human cancer cells. Indeed, short U3 oligodeoxynucleotides (ODNs) based on these RNA sequences ably inhibited proliferation of cancer cell lines driven by cyclins B1/B2, the gene targets of NF-Y.
INTRODUCTION
Human endogenous retroviruses (HERVs) comprise approximately 8% of the human genome (1), and have physiological functions as well as a potential role in some human diseases (2). During primate evolution, thousands of copies of solitary long terminal repeats (sLTRs) were generated due to duplications of the founder provirus genome followed by recombinational deletion of most of the proviral genome, leaving intact sLTRs (3). Different from other known retroviral LTRs, endogenous retrovirus 9 (ERV-9) LTRs have a variable number of tandem repeats with multiple enhancer binding sites for NF-Y (CCAAT), MZF1 (GTGGGGA), and GATA-2 (GATA) in their U3 region (4, 5). Substantial progress has been made in uncovering the promoter activities of the ERV9 LTR U3 DNA (pertaining to its binding of transcription factors) in regulating several important genes, including p63 isoforms which protect the male germ line via tumor suppressor activity (6–8), and the globin gene (4, 5). However, little information has been published regarding the activities and functions of RNAs originating from the U3 region. Since some ERV-9 LTRs locate within introns of coding sequences of genes, such as the axin1 gene (in the reverse orientation) (9, 10) and the HLA-DRB gene (in the forward orientation) (11), both U3 S and AS RNAs should be transcribed in cells as the result of spliced by-products from mRNAs of these genes.
In this study, we evaluated the function not of the ERV-9 LTR U3 DNA but, rather, of the U3 RNA transcripts originating from the U3 repeat region.
MATERIALS AND METHODS
Cells.
Cell lines were maintained at 37°C in a humidified atmosphere of 5% CO2. Human cancer cells (MDA231, MCF-7, K562, LNcap, HepG2, HT1080, HTB77 and HTB78 cells) were cultured in either Dulbecco modified Eagle medium (DMEM) or RPMI medium. All of the media were purchased from Life Technologies, Inc.
ODN decoy treatment.
A total of 2.5 × 104 cells were treated with ERV-9 U3 sense (S) or antisense (AS) oligodeoxynucleotides (ODNs) at different concentrations for 72 h. Green fluorescent protein (GFP) ODN was used as a control to subtract the ∼3 to 6% nonspecific cytotoxicity of phosphorothioate-based ODN. G3139, GRN163, and MDM2AS were three positive controls. Cells were counted with a FACSCalibur (BD Company). Relative cell proliferation levels were expressed as percentages of control ODN treatment. ODNs and biotinylated ODNs were as follows: ERV-9LTR U3 S, CTCAAGGTTTGTAAACACACCAATCAG; ERV-9LTR U3 AS, CTGATTGGTGTGTTTACAAACCTTGAG (GenBank accession no. AF064190; 3,329 to 3,355 bp); U3 S mut, CTCAAGGTTTGTAAA CACAtgtgaCAG; U3 AS mut, CTGacactTGTGTTTACAAACCTTGAG; GFP, CATTATCAACAAAATACTCCAATTGGC; G3139, TCTCCCAGCGTGCGCCAT; GRN163, TAGGGTTAGACAA; and MDM2 AS, TGACACCTGTTCTCACTCAC, where lowercase letters represent mutated nucleotides.
Cell cycle analysis.
ODN-treated cells were suspended in phosphate buffer solution (PBS) and centrifuged (1,500 rpm for 8 min), and the supernatant was discarded. The suspension was fixed with precooled 75% ethanol overnight at −20°C and treated with RNase at 0.5 mg/ml at room temperature for 30 min and then with 10 μg/ml propidium iodide. After 30 min at room temperature, the percentage of the cells at different phases in the cell cycle and cell apoptosis was determined with a FACSCalibur (BD Company).
PCR primers.
For information about the PCR primers, see the supplementary material.
RNA extraction, reverse transcription, and real-time PCR quantification.
Total RNA was extracted from cells with a cell density of 75% confluence using TRIzol total RNA isolation reagent (Gibco BRL, Life Technologies, Gaithersburg, MD) as per the manufacturer's protocol and the RNeasy minikit (Qiagen; catalog number 74104). Total RNAs were treated by DNase I (Invitrogen; catalog number 18068015). There was no PCR amplification of ERV-9 LTR U3 products from DNase I-treated RNA samples (see Fig. S1a in the supplemental material). cDNA was synthesized from total RNA using gene-specific primers or random primers by using a high-capacity cDNA reverse transcription kit (Applied Biosystems [ABI]; part number 4368813). Real-time PCR was performed using an ABI 7300 sequence detection system. Real-time PCR was performed by using Integrated DNA Technologies (IDT)-designed or ABI probe and primers to detect the gene of interest with 18S rRNA or the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene as an internal control. Cellular and viral PCR primers for real-time evaluations are included in supporting information.
Primary ductal carcinomas.
RNAs of 4 matched pairs of human primary ductal carcinomas (BT1 to BT4) (TNM classification T2 [12]) and their respective adjacent normal mammary tissues (BN1 to BN4) designated K135 (BT1 and BN1), K165 (BT2 and BN2), K172 (BT3 and BN3), and K191 (BT4 and BN4) were purchased from Indivumed GmbH (RNA integrity number [RIN] > 7). K135 and K165 were estrogen receptor positive, while K172 and K191 were estrogen receptor negative. K135 and K172 were HER2 positive, while K165 and K191 were HER2 negative. All four were progesterone receptor negative.
Northern and Western blotting.
RNAs were separated on 12% denaturing urea-polyacryamide gels and transferred to a nylon membrane. S and AS ODN probes end labeled with 32P were used to detect AS and S endogenous U3 RNAs separately. RNAs of four matched pairs of human breast cancer and adjacent normal mammary tissues were obtained from Indivumed. The RNA of EL4, a mouse lymphoma cell line, was used as the negative control. NuPAGE 4 to 12% bis-tris gels and an LAS4000 biomolecular imager were used for Western blot analysis.
RNA-IP.
RNA immunoprecipitation (RNA-IP) was carried out using an RNA-IP assay kit (MBL; catalog number RN1001). Precleared cell lysates were incubated at 4°C overnight with 2 μg of antibodies to one of the following: NF-Y, p53, p21, p300, Est-1, sp1, histone deacetylase 1 (HDAC1), polypyrimidine tract-binding protein 1 (PTBP1), and mouse IgG. The RNA-protein immunocomplexes were brought down by protein A/G beads, and RNA was then purified from the complexes for real-time PCR (RT-PCR) as described previously. For in vitro competition, 100 μg of cellular lysate was incubated with 5 μg of different AS, S, and control ODNs and rocked at 4°C overnight. The same concept was used to design U3SL, U3ASL, U3SmL, U3ASmL, and KL. ODN sequences used in RNA-IP are included in the supplemental material. Antibodies to NFY-A/sc-10779, NFY-C/sc7715-R, NFY-B/sc-13045, NFKB-p65/sc372, HDAC1/sc-7872, Est-1/sc5558, sp1/sc-14027, p53/sc-6243, p21/DCS60, p300/sc-584, PTBP1/sc-133677, and biotin/sc-57636 were used for RNA-IP.
Xenograft models.
BALB/c scid male mice (JAX mice), 4 to 6 weeks old, were used to make HT1080 and MDA231 xenografts. Cultured cells were washed with and resuspended in serum-free media. The suspension (2 × 106 cells in 0.2 ml per mouse) was then injected into the chest area of the mice. The mice were monitored by measuring tumor growth and body weight and by general clinical observation. Tumor-bearing mice were randomly divided into multiple treatment and control groups (8 mice per group) on day 7. All ODNs, dissolved in 0.2 ml of physiological saline (0.9% NaCl), were injected daily into the area adjacent to the tumor at doses of 50 μg/mouse for 7 days. The control group received saline only. Animals were sacrificed and tumors were isolated and weighed. ODN treatment was blinded and data were decoded.
PCR cloning and sequencing.
pCR@2.1-TOPO and pcDNA3.1/NT-GFP-TOPO vectors were used for cloning and sequencing.
Statistics.
A 2-tailed unpaired t test (GraphPad software) and sigma plot were used for analysis of variance (ANOVA). A P value of less than 0.05 was considered statistically significant. All real-time PCR and ODN growth inhibition data shown in this report are the means ± standard deviations (SDs) of 3 replicates. Each assay was repeated three times, with comparable results.
RESULTS
Expression of both S and AS RNAs of the ERV-9 LTR U3 repeat region in human tumor cell lines and in primary ductal carcinomas.
In initial experiments, we investigated the expression of ERV-9 LTR U3 RNAs in human cells by directional RT-PCR assays. A pair of primers was designed to specifically detect the first four repeat unit (0-1-2-3-4)1 sequences of the ERV-9 U3 LTR based on the sequence of the ERV-9 LTR located upstream of the human β-globin gene locus (4, 5, 9, 10) (Fig. 1a). Both U3 S and AS RNAs of the ERV-9 LTR (U3 S and U3 AS RNA, respectively) were detected as ∼140- and ∼200-nucleotide (nt) fragments in diverse human tumor cell lines (Fig. 1b). Cloning and sequencing of RT-PCR products from MDA231 revealed that the ∼140- and ∼200-nt amplicons are actually 155 nt and 196 nt, respectively. The 155-nt amplicon has repeat elements of sequence 0-1-3-4 (Fig. 2Ac; see also Fig. S2a in the supplemental material), while the 196-nt amplicon has repeat elements of sequence 0-1-2-3-4 (Fig. 2Ad; see also Fig. S2b in the supplemental material). The 155-nt amplicon has 2 and the 196-nt amplicon has 3 CCAAU motifs (AUUGG in AS). Real-time PCR primers were then used to compare the relative levels of AS and S within malignant and nonmalignant cells. The U3 AS RNA was found to be expressed at a 2-fold-higher level than the U3 S RNA in primary, nonmalignant, cells while U3 AS and S RNAs were expressed at equal levels in cancer cell lines (Fig. 1c).
Fig 1.

Detection of S and AS RNAs of the U3 ERV-9 LTR (1-2-3-4)1 repeat region by directional RT-PCR. (a) PCR primers used to detect the 200-bp S and AS transcripts from the U3 ERV-9 LTR. (b) Directional RT-PCR analysis of ERV-9 LTR U3 RNAs in nonmalignant primary human cells and in human cancer cell lines. P (positive control) is the U3 ERV-9 LTR directly amplified from K562 genomic DNA, and N (negative control) is the U3 ERV-9 LTR directly amplified from K562 RNA treated with DNase I. ERV-9 LTR U3 products could not be detected in DNase I-treated RNA samples in any cell line (see Fig. S1a in the supplemental material). (c) Real-time PCR analysis of U3 AS and S RNAs in nonmalignant and cancer cell lines. U3 AS RNA was expressed at a significantly higher level than U3 S RNA in normal cells (P < 0.05) but not in cancer cells (P > 0.05). The relative ratio of U3 RNAs between two samples was measured with GAPDH as a cDNA loading control, but the absolute copy numbers of U3 RNAs were not assessed. Relative fold was calculated by setting AS RNA expression level in each cell line as 1-fold. t test was used to evaluate significance.
Fig 2.

(A) ERV-9 LTR variants: a and b are LTRs at globin and axin1 genes, c and d are 196- and 155-nt LTRs cloned by a pair of PCR primers shown in Fig. 1, and e and f are 549 and 240 nt LTRs cloned by nested PCR primers (see supplemental material). Sequences were analyzed by NCBI/BLAST. The cDNA sequences of 155-, 196-, 240-, and 549-nt amplicons are shown in Fig. S1 in the supplemental material. (B) The locations of ERV-9 LTRs in introns of different coding genes: a is BRCAA1 transcript, b is a 549-nt LTR embedded in an alternative BRCAA1-012 transcript, c is a 196-nt LTR embedded in MAPK10-018 transcript, and d is a 196-nt LTR embedded in IL-15-008 transcript. Sequences were analyzed by ensemble/blast/blat.
Out of concern that cancer cell lines might not reflect the true expression of U3 RNAs in primary human cancers, we assessed expression of U3 RNAs in primary mammary ductal carcinomas (TNM classification T2 [12]) and in their donor-matched adjacent normal mammary tissues. Normal mammary glands expressed 2- to 3-fold-more U3 AS RNA and 2-fold-less U3 S RNA than did their adjacent primary breast tumor counterparts (Fig. 3a). High-resolution Northern blotting confirmed this differential expression of U3 RNAs: normal mammary tissues expressed higher levels of an ∼500-nt U3 AS RNA species and lower levels of an ∼200-nt U3 S RNA species than did primary ductal carcinomas (Fig. 3b). Thus, both PCR and Northern blot analyses indicate that U3 RNAs arising from the ERV-9 LTR are differentially expressed in nonmalignant and malignant cells, with a higher level of expression of AS in nonmalignant primary cells than in malignant tumor cells. The ∼200-nt S and ∼500-nt AS RNA species appear to be novel U3 RNA transcripts, since they have not hitherto been reported (13, 14) (Fig. 3b). PCR cloning and sequencing with nested U3 primers revealed that the ∼500-nt AS RNA is 549 nt (Fig. 2Ae) and that this 549-nt AS RNA originates in intron 10 of the BRCAA1-012 gene (AL732292.12) and contains six AUUGG motifs (see Fig. S2d in the supplemental material). BRCAA1-012 is a transcriptional variant of the BRCAA1 gene, sharing the last 6 exons with BRCAA1 (Fig. 2Ba and Bb). Moreover, the ∼200-nt S RNA was cloned and its actual size was found to be 240 nt (Fig. 2Bf). The transcriptional origin of the 240-nt U3 S RNA is not known as yet, since its sequence is found in regions of chromosome 5 (AC008885.5) and chromosome 12 (AC126564.7), from which no other transcripts have originated.
Fig 3.
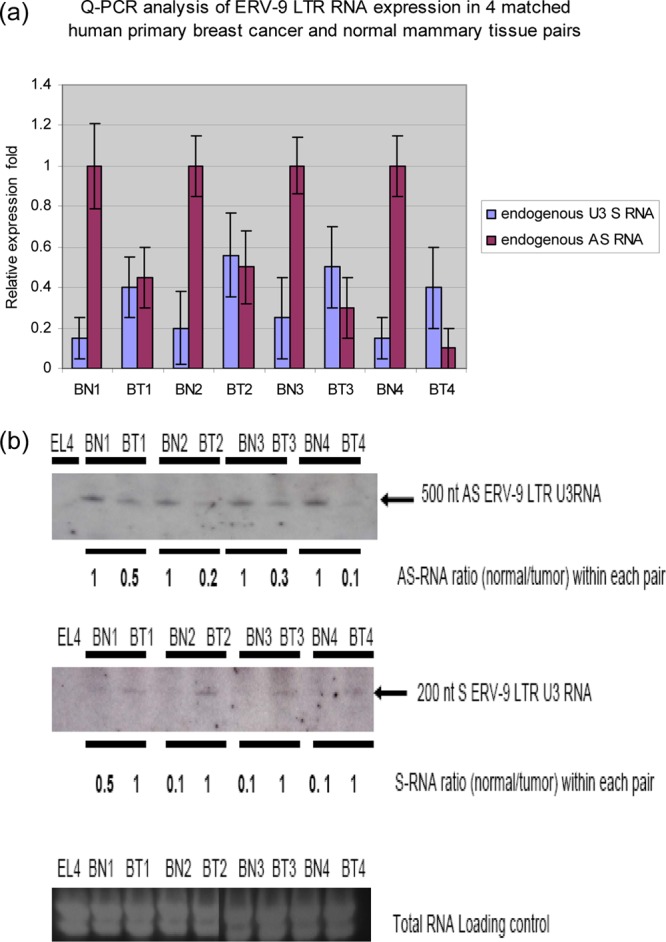
(a) Real-time PCR analysis of U3 AS and S RNAs in matched human primary ductal carcinomas (BT) and adjacent normal mammary tissue pairs (BN). U3 AS RNA was expressed at a significantly higher level than U3 S RNA in normal tissues (P < 0.05) but not in cancer tissues (P > 0.05) within each sample pair. Relative fold was calculated by setting AS RNA expression level in each corresponding normal tissue as 1-fold. ANOVA was used to evaluate significance. (b) High-resolution Northern blot analysis of U3 AS and S RNAs in matched pairs of human ductal carcinomas and adjacent normal mammary tissue. Ductal carcinomas expressed less U3 AS and more U3 S RNAs than adjacent normal tissues. The ∼500-nt AS RNA was detected by nested U3 (1-2-3-4)1 S ODN probes, and the ∼200-nt S RNA was detected by nested U3 (1-2-3-4)1 AS ODN probes. The nested probes were then used as PCR primers to clone these two novel RNAs (Fig. 2).
ERV-9 LTR U3 AS RNA binds to NF-Y, p53, and sp1 in vivo.
U3 RNAs of the ERV-9 LTR contain multiple CCAAT motifs (see Fig. S1b in the supplemental material) which are known binding sites for NF-Y, a transcription factor associated with cellular proliferation, principally through its binding to the CCAAT motifs of cyclin B1 and B2 genes (15). Moreover, NF-Y itself binds to additional factors important in cellular proliferation, including p53, sp1, p300, Ets-1, and HDAC1 (16), Thus, it was plausible that modulating levels of free NF-Y via binding to U3 RNAs of ERV-9 LTR could potentially modulate cellular proliferation by serving as a “decoy” target or a “trap” in competing with binding of free NF-Y and its associated factors to genes mediating proliferation such as those for cyclins B1 and B2, CDC25C, CDC2, and topoisomerase (15). Therefore, we investigated whether endogenous U3 RNAs bound to NF-Y and its associated factors in vivo by using antibodies to specifically immunoprecipitate NF-Y and other transcription factors in the cytoplasmic fraction of cells, followed by PCR to detect NF-Y-bound U3 RNAs. In malignant cell lines (HT1080 and MDA231) and in quiescent and proliferating T cells, the endogenous ∼140- and ∼200-bp U3 AS RNAs bound to NF-Y, p53, and sp1 but not to p300, Ets-1, or HDAC1 (Fig. 4). Cloning and sequencing of AS ∼140- and ∼200-nt U3 amplicons from such immunoprecipitations (Fig. 4) confirmed that they are the same 155- and 196-nt U3 amplicons as identified previously (Fig. 2Ac and Ad). The binding of NF-Y to U3 AS RNAs of ERV-9 LTR was relatively specific, since NF-Y did not bind to the AS RNA of the ERV-K111 LTR (see Fig S2a in the supplemental material). As the U3 AS 196-nt transcript originates from the mitogen-activated protein kinase 10 (MAPK-10) and/or interleukin 15 (IL-15) gene loci, and the transcription factor CREB regulates the expression of MAPK-10 (Genes-to-Systems Breast Cancer Database [http://www.itb.cnr.it/breastcancer/php/array.php?id=5602]) and transcription factors GR, ISGF-3, STAT3, GR-alpha, STAT5A, and AP-2 gamma regulate the expression of IL-15 (17; GeneCards compendium [http://www.genecards.org/cgi-bin/carddisp.pl?gene=IL15]), a higher expression level of MAPK-10 and IL-15, as has been reported previously for normal mammary glands versus those with mammary tumors (17; see also Genes-to-Systems Breast Cancer Database and GeneCards compendium above), may correlate with the higher expression level of the AS 196-nt decoy in normal cells. However, the transcriptional origin of the AS 155-nt amplicon is uncertain, since its sequence was not found within other identified transcripts, and therefore will require additional study. To our knowledge, this is the first report of an endogenous RNA molecule specifically binding to an important transcription factor in eukaryotic cells. Thus, the low expression level of U3 AS RNAs in breast cancer cells may facilitate the high-level binding of NF-Y to the cyclin B1 and B2 genes. Indeed, we found that all primary breast tumors had heightened expression of cyclins B1 and B2 RNAs without increased NF-Y mRNA levels (see Fig. S3a in the supplemental material). Although the expression of cyclin B1 and B2 mRNAs can be upregulated by NF-Y protein (15) and downregulated by p53 as well as by p21/WAF1 (18–21), the factors driving the maintenance of high expression levels of cyclins B1 and B2 in primary breast cancers (22) have not been fully elucidated. Indeed, we found reduced levels of message for cell cycle inhibitors p21/WAF1 and p15INK4b in primary breast tumors (see Fig. S3a in the supplemental material).
Fig 4.

Analysis of transcription factors assembled with ERV-9 LTR U3 RNAs by RNA-IP. A total of 5 × 107 cells from tumor cell lines and stimulated or unstimulated primary cells were used for RNA-IP (RIP) analysis. The cell lysates were treated with antibodies to each of the following: NF-Y, p53, p300, Ets-1, Sp1, HDAC1, IgG (nonspecific negative control shown in Fig. S2 in the supplemental material), and p21/WAF1 (transcription factor-specific negative control), as well as antibody to PTBP1 as a loading control. The RT-PCR detection of ERV-9 LTR U3 region is described in the legend to Fig. 1b. NF-Y, p53, and sp1 assemble with U3 AS RNAs in HT1080 and MDA231 cells and in quiescent and stimulated human primary T cells.
In vitro and in vivo inhibitory effects of U3 S and AS ODNs in growth of human cancer cell lines.
To further study the biological functions of ERV-9 LTR U3 S and AS RNAs in cell growth, we administered short U 3S and U3 AS ODN sequences, based on sequences in these LTR elements, to cancer cells. Phosphorothioate ODNs (27 nt) corresponding to subunit 2 of the (1-2-3-4)1 tandem repeat in S and AS orientations were constructed (S ODN and AS ODN) (Fig. 1a). These two ODNs contain one NF-Y binding motif (CCAAT in sense and ATTGG in antisense) and, critically, lack CpG sequences which are known to induce nonspecific inflammatory or interferon (IFN) responses that could diminish cell proliferation. Both U3 S and AS ODNs reduced the growth of HT1080 and MDA231 2- to 3-fold but did not inhibit the growth of normal primary keratinocytes stimulated by EGF, quiescent or proliferating peripheral blood lymphocytes (PBL), or monocytes stimulated with pooled human serum (see Fig. S3 in the supplemental material). Control U3 S and AS ODNs (U3Sm and U3ASm) which were mutated so that they lacked the NF-Y binding motif showed significant restoration of cellular proliferation, indicating the criticality of the NF-Y binding motifs (see Fig. S4 in the supplemental material). We then tested these ODNs in six diverse human cancer cell lines and found that both ODNs reduced cell growth 1.5- to 2-fold greater than did G3139 (23), GRN163 (24), and MDM2 AS (25) ODNs (Fig. 5a). G3139, GRN163, and MDM2 AS ODNs are known to inhibit growth of malignant cells by targeting Bcl2, telomerase, and MDM2, respectively. In a chemosensitivity assay, S and AS ODNs sensitized all six cell lines to etoposide phosphate (VP16) 1.5- to 2-fold greater than did the three positive controls (see Fig. S5 in the supplemental material). To assess whether such effects on in vitro tumor cell growth would be realized in vivo, we investigated the effects of ERV-9 LTR U3 S and AS ODNs on tumor growth in a xenotransplant model, using HT1080 and MDA231 tumors in SCID mice. We found that U3 S and AS ODNs potently inhibited in vivo tumor growth (Fig. 5b).
Fig 5.

(a) Treatment of human cancer cell lines with ERV-9 LTR U3 S and AS ODNs in vitro. A total of 2.5 × 104 cells was treated with ODNs at 10, 20, or 30 μg/ml for 72 h. The inhibition of proliferation mediated by both U3 S and AS ODNs exceeded those of G3139, GRN163, and MDM2 AS ODNs at all three concentration levels (P < 0.05) in all six cell lines. ANOVA was used to evaluate significance. Relative fold was calculated by setting each cell number in the GFP-treated sample as 1. (b) Treatment of human tumor xenografts with ERV-9 LTR U3 S and AS ODNs in vivo. Animals were inoculated with 2 × 106 tumor cells on day 0. Tumor bearing mice were randomly sorted into 5 groups (n = 8) on day 7. Injection of the tumor site with ODNs was initiated on day 7 and continued daily for 7 days. U3 S and AS ODNs diminished HT1080 and MDA 231 tumor growth to a greater extent than did PBS and GFP controls (P < 0.05). ANOVA was used to evaluate significance. G3139 diminished HT1080 and MDA 231 tumor growth, but the value was not statistically significantly different from that of the negative control by ANOVA.
ERV-9 LTR U3 ODNs regulate the cell cycle by acting as NF-Y traps.
Since U3 S and AS ODNs contain NF-Y binding motifs and NF-Y has been shown to regulate cell cycle progression (15), we performed cell cycle analyses following treatment of cell lines with S and AS ODNs. Compared to the GFP ODN negative control, U3 S and AS ODNs increased the percentage of cells in G0/G1 phases from 80% to 90% in HT1080 and from 64% to 77% in MDA231, while the three positive controls increased the percentage of cells in the G0/G1 phases from 80% to 82% (HT1080) and from 64% to 71% (MDA231) (Fig. 6). These results indicate that one of the mechanisms by which U3 S and AS ODNs of ERV-9 LTRs inhibit human cancer cell growth is by causing arrest in G0/G1. To uncover the molecular basis by which ERV-9 LTR ODNs caused G0/G1 phase arrest, we assessed cell cycle genes related to the NF-Y pathway by real-time PCR in ODN-treated cells. Both U3 S and AS ODNs downregulated mRNAs of cyclins B1 and B2, CDC2, and CDC25C 2- to 3-fold and upregulated mRNA of the cell cycle inhibitor p21/WAF1 3-fold in HT1080 and MDA231cells (Fig. 7). Additionally, U3 AS ODN upregulated mRNA for IL-24, a cytokine known to inhibit cell proliferation, 7-fold in MDA231 cells (Fig. 7c). These ODNs failed to downregulate mRNAs of cyclins D1 and D3, which are not strongly regulated by NF-Y (Fig. 7a and b), and, critically, neither U3 S nor AS ODNs altered the mRNA levels of critical transcription factors themselves, including NF-Y, sp1, and p53 (see Fig. S6 in the supplemental material). Because some ODNs induce type I interferons which are known to cause G0/G1-phase arrest (26), we evaluated whether type I interferon was involved in U3 ODN-mediated G1 arrest. Thus, we cotreated HT1080 and MDA231 cells with U3 ODNs and a soluble interferon receptor, B18R, which has high affinity for type I interferons and blocks both autocrine and paracrine functions of type I interferons. We found that B18R did not significantly reduce the antiproliferative activity of U3 ODNs but ably blocked the antiproliferative activity of the IFN-β control, indicating that type I interferons did not play a major role in G1-phase arrest by U3 ODNs (see Fig. S7 in the supplemental material). We also failed to find a role for enhanced apoptosis as an explanation for diminished cell growth in ODN-treated cells. Moreover, the nonspecific cytotoxicity of the ODNs themselves was less than 6% in all tested samples.
Fig 6.

Cell cycle analysis of ODN-treated HT1080 and MDA231 cells. A total of 2.5 × 104 cells was treated with ODNs at 30 μg/ml for 72 h. G3139, GRN163, and MDM2 AS ODNs were positive controls, and GFP ODN was the universal phosphorothioate ODN control. ODN-treated cells were then subjected to flow cytometric cycle analysis. Both U3S and AS ODNs increased the percentage of cells in G0/G1 and reduced the percentage of cells in S/G2 phase.
Fig 7.

Real-time PCR analysis of cell cycle genes in U3 ODN-treated HT1080 and MDA231 cells. (a and b) U3 ODNs downregulated the mRNA of cyclins B1 and B2, CDC2, and CDC25C (P < 0.05) but not of cyclins D1 and D3 in both cell lines. (c) U3 ODNs upregulated mRNA of p21/WAF1 (P < 0.05). U3 AS ODN also upregulated mRNA of IL-24 (P < 0.05) in MDA231 cells. ANOVA was used to evaluate significance. Relative fold was calculated by setting gene expression levels in GFP ODN-treated cells as 1-fold and defining significance by ANOVA. GFP, U3 Sm, and U3 ASm ODN treatments were used as controls (a).
Since NF-Y has been reported to have a minimal impact on the p16/cyclin D1/CDK4/pRB proliferation pathway (15), U3 ODNs theoretically should have less inhibitory effect in cancer cell lines whose proliferation is largely mediated by activation of the p16/cyclin D pathway. To test whether this was indeed the case, we evaluated two ovarian cancer cell lines that differed with respect to dependence on cyclins D and B. In comparing the expression of 15 cell cycle-related genes, we found that HTB77 had lost p16 expression but had no significant differences in the remaining 14 genes compared to HTB78 (see Fig. S8a and b in the supplemental material) (27). Since p16 inhibits the cyclinD/CDK4/6 pathway, the loss of p16 allows constitutive activation of the cyclin D/CDK4/pRB pathway. In treating these two cell lines with U3 ODNs, we found that while U3 AS ODN inhibited the growth of HTB78 cells, it did not significantly inhibit the growth of the cyclin D-driven HTB77 cells (see Fig. S8c). The HTB77 cell line was, however, inhibited by NFKB AS ODN, which targets the cyclin D pathway (see Fig. S8c). This result further supports the specificity of U3 AS ODN for the cyclin B1 and B2 proliferation pathway.
Finally, to evaluate whether such exogenous U3 ODNs functioned as traps to bind NF-Y, we performed a competition assay in which U3 ODNs competed for NF-Y binding to its endogenous ligand, U3 AS RNA. AS ODN diminished binding of NF-Y, p53, and sp1 to U3 AS RNA 5-fold compared to its U3 AS mut ODN counterpart lacking an NF-Y binding site, while U3 S ODN diminished binding of NF-Y, p53, and sp1 to U3 AS RNA 2-fold compared to its mutant control counterpart (Fig. 8a). To more directly assess the physical binding of NF-Y to the ODNs, we designed a pair of long (SL and ASL) ODNs (158 nt) with 5 tandem repeats of the original 27-nt S and AS sequences containing 5 NF-Y binding motifs (Fig. 8b) flanked by a 12-nt murine leukemia virus (MLV) PCR linker on both ends to distinguish the exogenous ODN from the endogenous ERV-9 LTR U3 DNA. Our data demonstrated that ASL ODNs strongly bound to NF-Y but not directly to p53 or sp1, while SL ODNs more weakly bound to NF-Y but not directly to p53 or sp1 (Fig. 8c) compared to the long U3 mutant and ERV-K controls lacking such NF-Y binding sites.
Fig 8.

(a) RNA-IP and real-time PCR analysis of transcription factors assembled with ERV-9 LTR U3 RNAs in MDA231 cell lysates from cells treated with U3 ODNs. U3 ODNs diminished binding of NF-Y, p53, and sp1 with endogenous U3 AS RNAs (P < 0.05). PTBP1 primers were added with ERV-9 LTR primers to amplify PTBP1 RNA as a loading control. Relative fold was calculated by setting the gene expression level in control GFP ODN as 1-fold. ANOVA was used to evaluate significance. (b) Design of a pair of longer U 3 SL and ASL ODNs (158 nt) with 5 tandem repeats of the original 27-nt S or AS RNA in the middle and flanked by a12-nt murine leukemia virus (MLV) PCR linker on both ends. SmL, ASmL, and KL (ERV-K111) are control ODNs which also have 5 tandem repeats of their corresponding sequences (lacking NF-Y binding motifs) in the middle and MLV PCR linker on both ends. (c) Both SL and ASL ODNs bound to NF-Y but not to p53 or sp1, with ASL ODN demonstrating more avid binding to NF-Y. MLV PCR primers detected all constructs with MLV linkers. (d) A total of 2.5 × 104 cells was treated with biotinylated ODNs at 30 μg/ml for 12 h, washed, and lysed. The lysates from each treatment were immunoprecipitated separately with antibodies to NF-Y and biotin. The precipitated pellets were analyzed by Western blotting. IgG and NFKB antibodies were used as negative controls for immunoprecipitation. ERV-K, U3 Sm, and U3 ASm ODNs were used as negative controls for ODN treatment. The ratio of U3 ODN-biotin antibody/NF-Y antibody is the percentage of cellular NF-Y bound by U3 ODNs. The U3 S and AS ODNs bound approximately 10% and 30% total NF-Y, respectively. DR, density ration of bands.
Although we could not directly address what fraction of NF-Y was bound to endogenous U3 RNAs, because endogenous U3 RNAs cannot specifically be isolated by current immunoprecipitation methods, we used biotinylated U3 ODNs as a surrogate. We treated unstimulated PBL and MDA231 with biotinylated ODNs and then used an antibody against biotin to specifically pull down NF-Y protein complexed with the U3 ODN. Our data demonstrated that U3 S ODN bound to ∼10% total NF-Y and U3 AS ODN bound to ∼30% total NF-Y protein (Fig. 8d) compared to ERV-K and both U3 mutant ODNs (lacking NF-Y binding sites). Taken together, these results indicate that the CCAAT motif plays an essential role in NF-Y binding to U3 ODNs and suggest that the mechanism by which exogenous U3 ODNs inhibit cell cycle progression is by outcompeting the binding of NF-Y to both endogenous U3 AS RNA and to the CCAAT motifs of the cyclin B1 and B2, CDC2, and CDC25C genes. These findings also indicate that endogenous U3 S RNA binds to NF-Y. However, its relatively weak affinity likely explains why we could not detect the binding of U3 S RNA to NF-Y previously.
As regards the lack of inhibitory effect of U3 ODNs on the growth of normal cell populations such as PBL, two factors may be important. First, PBL and many other cell types proliferate predominantly through a cyclin D-mediated pathway (28–30). Second, nonmalignant cells such as PBL express 2-fold-more AS U3 RNA and 1.5- to 3-fold-less NF-Y mRNA than do HT1080 and MDA231 cells (see Fig. S9 in the supplemental material), suggesting that higher, perhaps plateau, levels of endogenous antiproliferative U3 AS RNA and lower levels of NF-Y provide relative resistance to effects of exogenous U3 ODNs. That stimulated PBL maintain high levels of U3 AS RNA also indicates that a loss of high levels of expression of U3 AS RNA may be unique to the malignant state. Studies are under way to examine the basis for loss of high-level expression of U3 AS RNA in tumor cells.
DISCUSSION
Our data regarding the pattern of expression of ERV9-LTR U3 RNA transcripts in primary human cells and in their malignant primary cancer and cancer cell line counterparts suggest that U3 AS RNAs with AUUGG motifs function as decoy receptors or traps for NF-Y that diverts it from binding to cyclin B genes and thus keeps cell proliferation in check. This hypothesis was supported by the finding that U3 AS RNAs of ERV-9 LTRs do indeed form RNA-protein complexes with NF-Y, p53, and sp1. That U3 S RNAs were not found to bind such factors, despite containing CCAAU motifs, further supports the known complexity regarding binding of transcription factors to their ligands (see description of methods used to detect protein-RNA interactions on the Pierce Protein Biology Products website [http://www.piercenet.com/browse.cfm?fldID=B7A23C70-5056-8A76-4E39-B7DB24C3742C]) as well as the limit of sensitivity of our assay, as our long U3S ODN construct, containing multiple NF-Y binding sites, did show binding. The difference in NF-Y binding to U3 S and AS RNAs may thus be significantly influenced by tertiary structure, abundance, and stability of U3 S and AS RNAs (see Pierce web page above). Our decoy hypothesis is further supported by a recent study in which it was proposed that the 4,000 DNA copies of the sLTR in the human genome may competitively bind NY-F, present in limiting amounts, and selectively transfer NF-Y to the promoters of cis-linked genes (5). Indeed, our findings provide evidence that the noncoding AS RNA of ERV-9 LTR U3 may actually serve a novel function as a natural regulatory reservoir (Fig. 9a) for cell cycle transcription factors such as NF-Y, p53, and sp1 in human cells, as distinct from previously elucidated functions of noncoding RNAs, including the direct targeting of RNA polymerases, X-inactivating-specific RNA transcripts, and histone-modifying enzymes (31). A similar small noncoding U3 RNA (104 nt) derived from the feline leukemia retrovirus was also recently documented (33).
Fig 9.

(a) Model depicting the interactions of the U3 AS RNA of ERV-9 LTR with NF-Y, p53, and sp1 as they bear on inhibition of NF-Y binding to promoters of cyclin B1 and B2 in normal cells. (b) Cancer cells express less U3 AS RNA and more NF-Y transcripts, favoring binding to and upregulation of cyclins B1 and B2. Novel U3 miRNA may be present in tumor cells. (c) Exogenous U3 S and AS ODN compete for binding to NF-Y with both endogenous U3 AS RNA of ERV-9 LTR and the promoters of cyclins B1 and B2, thereby inhibiting progression through the cell cycle.
That the origin of the AS RNA decoys described in these studies is from ERV9 LTR sequences is best supported by the homologies of such RNA transcripts to the sequences and order of the repeat elements in the ERV9 LTR associated with the β-globin gene. Thus, the 196-nt amplicon contains four identical subunits (subunits 1, 2, 3, and 4) and the identical repeat alignment 0-1-2-3-4 (Fig. 2Aa and Ad) and is ∼99% homologous to the β-globin's LTR sequence, while the 155-nt amplicon contains three identical subunits (subunits1, 3, and 4) and a similar, but not identical, repeat alignment (0-1-3-4) (Fig. 2Aa and Ac) and is ∼75% homologous to the β-globin LTR sequence (due to missing subunit 2) (Fig. 2Aa and Ac). The 155- and 196-nt amplicons do not appear to be processed from the 240- or 549-nt species, since the alignments of the repeat elements differ among these species (Fig. 2Ac, Ad, Ae, and Af). Therefore, it is likely that they arose independently from different chromosomal loci (Fig. 2Ac, Ad, Ae, and Af). Additional efforts will be made to evaluate the evolutionary origins of these sLTR elements. Also, additional ∼140- and 200-nt amplicons will be sequenced to look for processed transcriptional variants other than the identified 155- and 196-nt RNAs.
Our studies further identified a 549-nt AS RNA transcript expressed at a higher level in primary nonmalignant cells than in malignant cells and, reciprocally, a 240-nt S RNA transcript expressed at a higher level in malignant breast tumors than in primary cells. These U3 AS (549-nt) and U3 S (240-nt) RNAs appear to be novel species, since they are much smaller than the previously reported 1,350-nt RNA of the ERV-9 LTR (11, 13) and are different in the alignment of their repeat elements from ERV-9 LTRs at the globin and axin 1 gene loci (Fig. 2Aa, Ab, Ae, and Af). Moreover, as the 549-nt AS originates from sequences in intron 10 of the BRCAA1-012 gene (AL732292.12), a transcriptional variant of the BRCAA1 gene, transcription factors such as HOXA9B, HOXA9, CULT1, LCR-F1, YY1, FOX04, Sox9, IRF-7A, Meis-1a, and Meis have the potential to regulate the expression of this transcript via their known regulation of BRCAA1 (see ARIB4D in the GeneCard compendium [http://www.genecards.org/cgi-bin/carddisp.pl?gene=ARID4B]). The relationship between these 10 transcription factors and the expression of the 549-nt RNA will be studied by using small interfering RNAs (siRNAs) to individually knock down these transcription factors. BRCAA1 is a known tumor suppressor gene, since its deficiency in mice leads to leukemia (32), and mutations in this gene contribute to development of human diseases and cancer through epigenetic mechanisms (32). However, given that the 549-nt AS RNA is embedded in BRCAA1 intronic elements, it is tempting to speculate that one potential mechanism of the tumor suppression mediated by BRCAA1 gene pertains to potential antiproliferative activity of this AS species, which contains NF-Y binding sites. Thus, higher expression of the tumor suppressor gene BRCAA1 RNA in normal mammary glands compared to breast cancers (see ARIB4D in the GeneCard compendium) may correlate with the higher expression of the ∼500-nt (549-nt) AS RNA in normal versus malignant tissues in our Northern blot. But why levels of BRCAA1 and the 549-nt AS RNA are diminished in tumor cells is not yet clear. The expression pattern of this 549-nt AS, whether it binds NF-Y protein, and whether it diminishes cell proliferation will be examined in multiple cancer cell lines. The 240-nt sequence is not found embedded in other identified transcripts, and thus, its origin and the molecular basis of the differential expression of U3 S RNA between normal and tumors require further study. Further characterization of both U3 RNA species as regards their origin, function, and splicing fates are under way. There is, thus far, no evidence to support the possibility that intronic ERV-9 LTRs can also act as promoter or enhancer elements to regulate their own, or cis-linked, genes (4, 5, 9, 10). Genomic knockout studies are being considered to further address this point. Additional contributions of ERV9 U3 RNAs to tumor cell proliferation may arise from microRNAs (miRNAs) associated with such transcripts. Thus, given the equal levels of expression of S and AS RNA transcripts in cancer cells, we evaluated whether such S and AS hybridized with each other to form stem structures (Fig. 9b). Three ERV-9 U3 RNAs cloned from a primary breast tumor (BT1 in Fig. 3) appear to be microRNAs in preliminary studies. These U3 RNAs have CCAAU motifs in the stem structure and may also have a role in NF-Y regulation.
ERV9 U3 ODNs may have potential as therapeutic agents. Indeed, they induced G1-phase arrest in the cell cycle, decreased the expression of proproliferative cyclins B1 and B2, CDC2, and CDC25C, and increased the expression of antiproliferative p21/WAF1 and IL-24, but they did not affect the RNA expression levels of NF-Y, p53, or sp1. Their induction of G0/G1-phase arrest is highly similar to the cell cycle arrest mediated by other antitumor ODNs which induce cell cycle arrest in ∼7 to 10% of cells (34, 35). Important differences in the decoy mechanism of the U3 ODNs, as opposed to the endogenous U3 AS, include the fact that p53 and sp1 did not form protein-protein complexes with NF-Y bound to ODN, perhaps due to structural constraints. This failure to bind p53 and sp1 may well be critical to the potency of AS ODN activity, as increased free levels of p53 and sp1 have been reported to enhance expression of p21/WAF1 (19, 36–39) and IL-24 (40), both of which mediate antiproliferative activity via G0/G1-phase arrest (41, 42). Indeed, the high level of induction of IL-24 mRNA in AS ODN-treated MDA1 cells may pertain, at least in part, to the activity of free sp1 (Fig. 8a and c). This hypothesis will be evaluated in future studies. The complexity of IL-24 mRNA induction is further highlighted by the fact that its induction was observed only in U3 AS ODN-treated MDA231 cells and not in HT1080 cells, indicating that there may also be tissue-specific factors involved. That the antiproliferative activity of U3 AS ODN depended on its binding to NF-Y, and thus affected the cyclin B1 and B2 proliferative pathway, was shown not only by specific controls lacking NF-Y binding sites but also by the lack of effect of U3AS ODN on cells whose proliferation is predominantly driven by the cyclin D pathway (see Fig. S8 in the supplemental material). Finally, there remains the question of the mechanism by which S RNA and ODNs mediated antiproliferative activity, as we failed to document binding of NF-Y to endogenous S RNA. Nonetheless, we did find that our extended S ODN bound weakly to NF-Y, and therefore, it is possible that both U3 S and AS ODNs have similar mechanisms of action in competing for binding of NF-Y with cellular CCAAT motifs in normal cells, suggesting that the failure to observe binding of endogenous U3 S to NF-Y, using our current RT-PCR protocol, may be related to the lack of sensitivity of the assay. Based on our data, we postulate that dysregulation of expression of ERV9 LTR AS RNA and NF-Y may be important in inducing and/or maintaining a malignant phenotype. Since ODNs were used at a relatively high dosage in vitro as well as being delivered directly into the tumor bed in vivo, it is uncertain whether such U3 ODN would have inhibitory function at a lower level or delivered by a less direct route. Therefore, methods to increase bioavailability and cell delivery of ODN are critical challenges (43).
In conclusion, this is the first report that describes the differential expression of RNAs of the ERV-9 LTR U3 region in normal human primary and cancer cells, the capacity of ERV-9 LTR U3 AS RNAs to assemble a protein-RNA complex with transcription factors, and the ability of U3 ODNs to diminish proliferation of some tumor cells. Since U3 ODNs are equally as or more potent than some known antitumor ODN agents, we believe that targeting the NF-Y pathway with ERV-9 LTR U3 ODN decoys could be considered a potential therapeutic approach with appropriate development, but one unlikely to mediate sustained tumor regression in the absence of agents that also target the cyclin D proliferation pathway. Since thousands of copies of sLTRs of ERV-9 have been inherited by successive generations and are widely expressed in multiple tissues, it is possible that there may be additional beneficial activities not yet uncovered (1). Thus, sLTRs of ERV-9 in both DNA and RNA forms may have important epigenetic activity in multiple critical cellular functions, potentially explaining their evolutionary preservation and offering the possibility of therapeutics based on their sequences. Overall, the studies of the interaction between hosts and endogenous retroviruses as well as retrotransposons may lead to novel therapeutic molecular arsenals in the future (44–46).
Supplementary Material
ACKNOWLEDGMENTS
We thank Yaqin Zhang (Division of Therapeutic Proteins, FDA) for MDA231 and MCF-7 cells, the Division of Monoclonal Antibodies (FDA) for human PBL and monocytes, and Carole Yee (Laboratory of Tumor Virus Biology and the Laboratory of Pathology, NCI) for human keratinocytes.
This work was supported by NHGRI intramural program funds (to Fabio Candotti).
Footnotes
Published ahead of print 24 October 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01648-12.
REFERENCES
- 1. Subramanian R, Wildschutte J, Russo C, Coffin J. 2011. Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology 8:90 doi:10.1186/1742-4690-8-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stengel A, Roos C, Hunsmann G, Seifarth W, Leib-Mösch C, Greenwood AD. 2006. Expression profiles of endogenous retroviruses in Old World monkeys. J. Virol. 80:4415–4421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bannert N, Kurth R. 2006. The evolutionary dynamics of human endogenous retroviral families. Annu. Rev. Genomics Hum. Genet. 7:149–173 [DOI] [PubMed] [Google Scholar]
- 4. Pi W, Yang Z, Wang J, Ruan L, Yu X, Ling J, Krantz S, Isales C, Conway SJ, Lin S, Tuan D. 2004. The LTR enhancer of ERV-9 human endogenous retrovirus is active in oocytes and progenitor cells in transgenic zebrafish and humans. Proc. Natl. Acad. Sci. U. S. A. 101:805–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pi W, Yang Z, Wu M, Wang Y, Fulzele S, Eroglu A, Ling J, Tuan D. 2010. 2010. Long-range function of an intergenic retrotransposon. Proc. Natl. Acad. Sci. U. S. A. 107:12992–12997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beyer U, Moll-Rocek J, Moll UM, Dobbelstein M. 2011. Endogenous retrovirus drives hitherto unknown proapoptotic p63 isoforms in the male germ line of humans and great apes. Proc. Natl. Acad. Sci. U. S. A. 108:3624–3629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu M, Eiden MV. 2011. Role of human endogenous retroviral long terminal repeats (LTRs) in maintaining the integrity of the human germ line. Viruses 3:901–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Signoretti S, Waltregny D, Dilks J, Isaac B, Lin D, Garraway L, Yang A, Montironi R, McKeon F, Loda M. 2000. p63 is a prostate basal cell marker and is required for prostate development. Am. J. Pathol. 157:1769–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ling J, Pi W, Bollag R, Zeng S, Keskintepe M, Saliman H, Krantz S, Whitney B, Tuan D. 2002. The solitary long terminal repeats of ERV-9 endogenous retrovirus are conserved during primate evolution and possess enhancer activities in embryonic and hematopoietic cells. J. Virol. 76:2410–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Long Q, Bengra C, Li C, Kutlar F, Tuan D. 1998. A long terminal repeat of the human endogenous retrovirus ERV-9 is located in the 5′ boundary area of the human β-globin locus control region. Genomics 54:542–555 [DOI] [PubMed] [Google Scholar]
- 11. Svensson AC, Setterblad N, Sigurdardottir S, Rask L, Andersson G. 1995. Primate DRB genes from the DR3 and DR8 haplotypes contain ERV9 LTR elements at identical positions. Immunogenetics 92:74–82 [DOI] [PubMed] [Google Scholar]
- 12. Sobin LH, Wittekind C. (ed). 2002. TNM classification of tumour viruses, 6th ed International Union against Cancer, Geneva, Switzerland [Google Scholar]
- 13. Lania L, Di Cristofano A, Strazzullo M, Pengue G, Majello B, La Mantia G. 1992. Structural and functional organization of the human endogenous retroviral ERV9 sequences. Virology 191:464–468 [DOI] [PubMed] [Google Scholar]
- 14. Svensson AC, Raudsepp T, Larsson C, Di Cristofano A, Chowdhary B, La Mantia G, Rask L, Andersson G. 2001. Chromosomal distribution, localization and expression of the human endogenous retrovirus ERV9. Cytogenet. Cell Genet. 92:89–96 [DOI] [PubMed] [Google Scholar]
- 15. Benatti P, Basile V, Merico D, Fantoni LI, Taqliafico E, Imbriano C. 2008. A balance between NF-Y and p53 governs the pro- and anti-apoptotic transcriptional response. Nucleic Acids Res. 36:1415–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peart MJ, Prives C. 2006. Mutant p53 gain of function: the NF-Y connection. Cancer Cell 10:173–174 [DOI] [PubMed] [Google Scholar]
- 17. Sanders AJ, Ye L, Wei XQXQ, Mansel RE, Jiang WG. 2011. Expression of interleukin-15 (IL-15) and the IL-15 receptor in human breast cancer. Cancer Res. 71(24 Suppl):P1-01-08 doi:10.1158/0008-5472.SABCS11-P1-01-.08 [Google Scholar]
- 18. Archer SY, Johnson J, Kim HJ, Ma Q, Mou H, Daesety V, Meng S, Hodin RA. 2005. The histone deacetylase inhibitor butyrate downregulates cyclin B1 gene expression via a p21/WAF-1-dependent mechanism in human colon cancer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 289:G696–G703 [DOI] [PubMed] [Google Scholar]
- 19. Han JW, Ahn SH, Kim YK, Bae GU, Yoon JW, Hong S, Lee HY, Lee YW, Lee HW. 2001. Activation of p21(WAF1/Cip1) transcription through Sp1 sites by histone deacetylase inhibitor apicidin: involvement of protein kinase C. J. Biol. Chem. 276:42084–42090 [DOI] [PubMed] [Google Scholar]
- 20. Ho J, Benchimol S. 2003. Transcriptional repression mediated by the p53 tumour suppressor. Cell Death Differ. 10:404–408 [DOI] [PubMed] [Google Scholar]
- 21. Innocente S, Abrahamson J, Cogswell J, Lee JM. 1999. p53 regulates a G2 checkpoint through cyclin B1. Proc. Natl. Acad. Sci. U. S. A. 96:2147–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Suzuki T, Urano T, Miki Y, Moriya T, Akahira J, Ishida T, Horie K, Inoue S, Sasano H. 2007. Nuclear cyclin B1 in human breast carcinoma as a potent prognostic factor. Cancer Sci. 98:644–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wacheck V, Krepler C, Strommer S, Heere-Ress E, Klem R, Pehamberger H, Eichler HG, Jansen B. 2002. Antitumor effect of G3139 Bcl-2 antisense oligonucleotide is independent of its immune stimulation by CpG. Antisense Nucleic Acid Drug Dev. 12:359–367 [DOI] [PubMed] [Google Scholar]
- 24. Asai A, Oshima Y, Yamamoto Y, Uochi TA, Kusaka H, Akinaga S, Yamashita Y, Pongracz K, Pruzan R, Wunder E, Piatyszek M, Li S, Chin AC, Harley CB, Gryaznov S. 2003. A novel telomerase template antagonist (GRN163) as a potential anticancer agent. Cancer Res. 63:3931–3939 [PubMed] [Google Scholar]
- 25. Zhang Z, Li M, Wang H, Agrawal S, Zhang RW. 2003. Antisense therapy targeting MDM2 oncogene in prostate cancer: effects on proliferation, apoptosis, multiple gene expression, and chemotherapy. Proc. Natl. Acad. Sci. U. S. A. 100:11636–11641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sangfelt O, Erickson S, Castro J, Heiden T, Gustafsson A, Einhorn S, Grandér D. 1999. Molecular mechanisms underlying interferon-alpha-induced G0/G1 arrest: CKI-mediated regulation of G1 Cdk-complexes and activation of pocket proteins. Oncogene 18:2798–2810 [DOI] [PubMed] [Google Scholar]
- 27. Schuyer M, van Staveren IL, Klijn JG, vd Burg ME, Stoter G, Henzen-Logmans SC, Foekens JA, Berns EM. 1996. Sporadic CDKN2 (MTS1/p16ink4) gene alterations in human ovarian tumours. Br. J. Cancer 74:1069–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Caetano MS, Vieira-de-Abreu A, Teixeira LK, Werneck MB, Barcinski MA, Viola JPB. 2002. NFATC2 transcription factor regulates cell cycle progression during lymphocyte activation: evidence of its involvement in the control of cyclin gene expression. FASEB J. 16:1940–1942 [DOI] [PubMed] [Google Scholar]
- 29. Mori N, Fujii M, Hinz M, Nakayama K, Yamada Y, Ikeda S, Yamasaki Y, Kashanchi F, Tanaka Y, Tomonaga M, Yamamoto N. 2002. Activation of cyclin D1 and D2 promoters by human T-cell leukemia virus type I tax protein is associated with IL-2-independent growth of T cells. Int. J. Cancer 99:378–385 [DOI] [PubMed] [Google Scholar]
- 30. White PC, Shore AM, Clement M, Mclaren J, Soeiro I, Lam EW, Brennan P. 2006. Regulation of cyclin D2 and the cyclin D2 promoter by protein kinase A and CREB in lymphocytes. Oncogene 25:2170–2180 [DOI] [PubMed] [Google Scholar]
- 31. Barrandon C, Spiluttini B, Barrandon O. 2008. Non-coding RNAs regulating the transcriptional machinery. Biol. Cell 100:83–95 [DOI] [PubMed] [Google Scholar]
- 32. Wu MY, Eldin KW, Beaudet AL. 2008. Identification of chromatin remodeling genes Arid4a and Arid4b as leukemia suppressor genes. J. Natl. Cancer Inst. 3:1247–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Forman LW, Pal-Ghosh R, Spanjaard RA, Faller DV, Ghosh SK. 2009. Identification of LTR-specific small non-coding RNA in FeLV infected cells. FEBS Lett. 583:1386–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Akiyama M, Hideshima T, Shammas MA, Hayashi T, Hamasaki M, Tai YT, Richardson P, Gryaznov S, Munshi NC, Anderson KC. 2003. Effects of oligonucleotide N3′→P5′ thio-phosphoramidate (GRN163) targeting telomerase RNA in human multiple myeloma cells. Cancer Res. 63:6187–6194 [PubMed] [Google Scholar]
- 35. Meye A, Würl P, Bache M, Bartel F, Grünbaum U, Mansa-ard J, Schmidt H, Taubert H. 2000. Colony formation of soft tissue sarcoma cells is inhibited by lipid-mediated antisense oligodeoxynucleotides targeting the human mdm2 oncogene. Cancer Lett. 28:181–188 [DOI] [PubMed] [Google Scholar]
- 36. Elbendary AA, Cirisano FD, Evans AC, JR, Davis PL, Iglehart JD, Marks JR, Berchuck A. 1996. Relationship between p21 expression and mutation of the p53 tumor suppressor gene in normal and malignant ovarian epithelial cells. Clin. Cancer Res. 2:1571–1575 [PubMed] [Google Scholar]
- 37. Gartel AL, Goufman E, Najmabadi F, Tyner AL. 2000. Sp1 and Sp3 activate p21 (WAF1/CIP1) gene transcription in the Caco-2 colon adenocarcinoma cell line. Oncogene 19:5182–5188 [DOI] [PubMed] [Google Scholar]
- 38. Saramäki A, Banwell CM, Campbell MJ, Carlberg C. 2006. Regulation of the human p21(waf1/cip1) gene promoter via multiple binding sites for p53 and the vitamin D3 receptor. Nucleic Acids Res. 34:543–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xiao H, Hasegawa T, Isobe K. 2000. p300 collaborates with Sp1 and Sp3 in p21(waf1/cip1) promoter activation induced by histone deacetylase inhibitor. J. Biol. Chem. 275:1371–1376 [DOI] [PubMed] [Google Scholar]
- 40. Pan L, Pan H, Jiang H, Du J, Wang X, Huang B, Lu J. 2010. HDAC4 inhibits the transcriptional activation of mda-7/IL-24 induced by Sp1. Cell. Mol. Immunol. 7:221–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lepley DM, Pelling JC. 1997. Induction of p21/WAF1 and G1 cell-cycle arrest by the chemopreventive agent apigenin. Mol. Carcinog. 19:74–82 [DOI] [PubMed] [Google Scholar]
- 42. Sainz-Perez A, Gary-Gouy H, Gaudin F, Maarof G, Marfaing-Koka A, de Revel T, Dalloul A. 2008. IL-24 induces apoptosis of chronic lymphocytic leukemia B cells engaged into the cell cycle through dephosphorylation of STAT3 and stabilization of p53 expression. J. Immunol. 181:6051–6060 [DOI] [PubMed] [Google Scholar]
- 43. Zhao Q, Matson S, Herrera CJ, Fisher Yu E H, Krieg AM. 1993. Comparison of cellular binding and uptake of antisense phosphodiester, phosphorothioate, and mixed phosphorothioate and methylphosphonate oligonucleotides. Antisense Res. Dev. 3:53–66 [DOI] [PubMed] [Google Scholar]
- 44. Bowen NJ, Jordan K, Epstein J, Wood V, Levin H. 2003. Retrotransposons and their recognition of pol II promoters: a comprehensive survey of the transposable elements from the complete genome sequence of Schizosaccharomyces pombe. Genome Res. 13:1984–1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Feschotte C, Gilbert C. 2012. Endogenous viruses: insights into viral evolution and impact on host biology Nat. Rev. Genet. 13:283–296 [DOI] [PubMed] [Google Scholar]
- 46. Mills RE, Bennett EA, Iskow RC, Luttig CT, Tsui C, Pittard WS, Devine SE. 2006. Recently mobilized transposons in the human and chimpanzee genomes. Am. J. Hum. Genet. 78:671–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.