

Abstract
Artificial target sequences for tissue-specific miRNAs have recently been introduced as a new means for altering the tissue tropism of viral replication. This approach can be used to improve the safety of oncolytic viruses for cancer virotherapy by restricting their replication in unwanted tissues, such as the liver. Semliki Forest virus (SFV) is a positive-strand RNA virus and, similar to the related alphaviruses, like Sindbis virus, has potential as a gene therapy vector and an oncolytic virotherapy agent, but this potential is limited by the neurovirulence of these alphaviruses. Here, we have generated a replicative SFV4 carrying six tandem targets for the neuron-specific miR124 between the viral nonstructural protein 3 and 4 (nsp3 and nsp4) genes. When administered intraperitoneally into adult BALB/c mice, SFV4-miRT124 displayed an attenuated spread into the central nervous system (CNS) and greatly increased survival. Peripheral replication was not affected, indicating neuron-specific attenuation. Moreover, a strong protective SFV immunity was elicited in these animals. Intracranial infection of adult mice with SFV4-miRT124 showed greatly reduced infection of neurons in the brain but led to the infection of oligodendrocytes in the corpus callosum. Taken together, our data show that miR124-mediated attenuation of neurovirulence is a feasible and promising strategy for generating safer oncolytic alphavirus virotherapy agents.
INTRODUCTION
MicroRNAs (miRNAs) are small noncoding RNA molecules that have important regulatory roles in gene expression by targeting mRNAs for cleavage or translational repression. Incomplete sequence complementarity between the miRNA and the target mRNA can induce translational repression without mRNA degradation, whereas a higher degree of sequence complementarity leads to catalytic degradation of the target mRNA (1). The human genome encodes more than 1,000 miRNAs, and over 50% of cellular mRNAs have been estimated to be under miRNA regulation (2). miRNAs have important regulatory roles in development, cell proliferation, differentiation, apoptosis, and stress responses, explaining why dysregulation of miRNA expression is associated with many diseases, most notably cancer (3).
miRNA targeting has recently been used to modify replicative tropism of both RNA and DNA viruses (4–10). By tissue-specific targeting the expression of viral genes that account for viral replication/pathogenicity, it is possible to generate rationally attenuated live vaccine viruses and safer oncolytic viruses for the treatment of cancer. For example, Edge et al. developed a tumor-specific vesicular stomatitis virus (VSV) by inserting 3 copies of Let7 miRNA target elements in the 3′ untranslated region of the VSV genomic RNA (5). Likewise, by inserting four copies of muscle-specific miRNA target elements into the coxsackievirus A21 genome, Kelly et al. could prevent lethal myositis developing in mice infected with the miRNA-targeted coxsackievirus A21 without compromising the tumor cell-killing potency of this virus (7).
Semliki Forest virus (SFV) is an enveloped positive-strand RNA virus of the family Togaviridae. The natural hosts of SFV include small wild animals, birds, and nonhuman primates, while the vectors are usually mosquitoes of the Aedes subspecies. Several strains of SFV exist. Strains such as SFV4 and L10 (the prototype SFV) are highly neurovirulent in mice of all ages, causing fatal encephalitis (11). In contrast, the avirulent strain A7(74) shows age-dependent neurovirulence, being lethal only to neonatal mice (12, 13). In the central nervous system (CNS) of older mice, the replication and spread of A7(74) are attenuated, and the infection remains asymptomatic. Human SFV infections are associated with a disease characterized by headache, fever, myalgia, and arthralgia of variable severity (14, 15). One fatal case of SFV encephalitis in a laboratory worker has been reported (16).
Replication-competent SFV vectors based on A7(74) have been considered promising tools for cancer virotherapy (17–19). Since SFV is neurotropic, infecting cells of the CNS, it has been of particular interest in virotherapy of brain tumors (see references 20 and 21). As an example of the potential of SFV vectors in the treatment of gliomas, Heikkilä et al. showed that systemically administered oncolytic SFV vector derived from the strain A7(74) could efficiently eradicate orthotopic human glioma xenografts in nude mice (20).
However, to further increase the safety of SFV-based virotherapy, additional measures to restrict SFV replication in the CNS are needed. One such measure is the protection of neurons. To address this issue, we have generated novel miRNA-targeted SFV vectors by inserting into their genomes multiple target elements for the neuron-specific miR124. SFV4 carrying liver-specific miR122 was used as a control. To summarize, we found that the novel miR124-targeted SFV4 virus showed attenuated neurovirulence in adult BALB/c mice, while retaining peripheral replication potency similar to that of the miR122 target-carrying control virus.
MATERIALS AND METHODS
Construction of miRNA-targeted replication competent SFVs.
pSP6-SFV4, a plasmid containing the genomic sequence of SFV4, was modified by creating a novel NdeI site (CATATG) upstream (starting at 5,514 nucleotides [nt]) of the sequence coding for the protease cleavage site (CTAGGCCGCGCGGGTGCA) located between the nonstructural protein 3 and 4 (nsp3 and nsp4) coding regions. A synthetic DNA sequence including duplicated sequence coding for the protease cleavage site and sequences for six target elements for miR122 or miR124 was inserted between the upstream naturally existing XhoI (CTCGAG; starting at 5,304 nt) site and the novel NdeI site, resulting in a plasmid carrying six miRNA-target elements flanking by sequences coding for the protease cleavage sites. Full sequences of the miRNA-targeted viruses are available upon request.
Cell cultures and viruses.
Human hepatocellular carcinoma cell line Huh7, human cervical cancer cell line HeLa, human embryonal carcinoma cell line Tera-2, and human primary glioblastoma cell line U87 were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, St. Louis, MO) with 10% fetal calf serum (FCS; Life Technologies, Carlsbad, CA), 1% l-glutamine, and 1% penicillin-streptomycin at 37°C, 5% CO2. The Syrian hamster kidney fibroblast cell line BHK-21 was cultured in DMEM supplemented with 10% FCS (Autogen Bioclear, Witshire, United Kingdom), 1% l-glutamine (Sigma-Aldrich), 1% penicillin-streptomycin (P0781; Sigma-Aldrich), and 2.98 g/500 ml 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES; Sigma-Aldrich) at 37°C, 5% CO2. The African green monkey kidney epithelial cell line Vero(B) was cultured in DMEM supplemented with 5% FCS, 1% l-glutamine, 1% penicillin-streptomycin, and HEPES at 37°C, 5% CO2.
Preparation of replication competent SFVs has been described previously (22). Briefly, wild-type SFV4 or viruses carrying miRNA target elements were produced from in vitro transcribed RNA by transfection into the BHK-21 cell line. Quantification of viruses was done by plaque titration in the Vero(B) cell line.
For virus titration of mouse tissues, one half of each brain and a part of each liver were snap-frozen in liquid nitrogen and stored at −70°C until homogenized in 1% bovine serum albumin (BSA)–phosphate-buffered saline (PBS) for virus titration. The homogenate was centrifuged at 4,500 × g at +4°C for 5 min, and the supernatant was used for titration. Blood samples were drawn from mice immediately after euthanasia, and the serum was separated using Microvette 500 Z-gel tubes (Sarstedt, Nümbrecht, Germany). Titration was done as described above with a plaque analysis in Vero(B) cells.
Cell viability assays.
All cell lines tested were infected with SFV4, SFV4-miRT122, or SFV4-miRT124 at a multiplicity of infection (MOI) of 0.1. Cell viability was measured 24 h postinfection (p.i.) (Tera-2), 48 h postinfection (Huh7, BHK-21, and HeLa), or 72 h postinfection (U87) using the CellTiter 96 AQueous one solution cell proliferation assay (Promega, Fitchburg, WI), and a multiwell plate reader (Multiskan EX; Thermo Fisher Scientific, Waltham, MA) was used to determine the optical density of the reactions at 492 nm.
Western blot analysis.
Cells were lysed with 1% NP-40 lysis buffer [150 mM NaCl, 50 mM Tris-HCl (pH 7.9), 1% Nonidet P-40) in the presence of protease inhibitors. Protein concentrations were determined by the Bio-Rad protein assay (Bio-Rad, Hercules, CA), and 10 μg of each sample was separated by SDS-PAGE and then blotted onto a nitrocellulose membrane (Bio-Rad). After blocking overnight at 4°C with PBS plus 0.05% Tween 20 containing 5% nonfat dry milk, the membranes were treated with rabbit polyclonal antibodies against nsp3 and nsp4 (kindly donated by Tero Ahola, University of Helsinki) or rabbit anti-SFV against envelope and capsid proteins as primary antibodies and then with IRDye 800 secondary antibody against rabbit immunoglobulins (LI-COR, Lincoln, NE). The Odyssey infrared imaging system (LI-COR) was used for the detection and quantification of SFV proteins.
Quantitative RT-PCR.
Total RNA was extracted from infected cells using the GenElute mammalian total RNA miniprep kit (Sigma-Aldrich). The concentration of the purified RNA was measured, and 100 ng of total RNA was used for cDNA synthesis with Fermentas RevertAid Moloney murine leukemia virus (M-MuLV) reverse transcriptase (RT) (Thermo Fisher Scientific). The oligonucleotide sequences for the amplification of the SFV positive-strand genome were as follows: forward primer, 5′-TGGAGCTGACCACAGACTTG-3′, and reverse primer, 5′-GGCCACAACGTCAGTATCTC-3′. PCR was run using an Mx3005P quantitative PCR System (Stratagene) with Power SYBR green PCR master mix (Applied Biosystems, Foster City, CA). After an initial incubation at 95°C for 10 min, 40 amplification cycles were conducted as follows: denaturation at 95°C for 15 s, annealing, and extension at 60°C for 1 min.
Animal experiments.
Adult (5- to 7-week-old) female BALB/c-mice (BALB/cAnNTac; Taconic) were infected with SFV4-miRT122, SFV4-miRT124, or SFV4 by injecting 1 × 106 PFU (in 100 μl of PBS) intraperitoneally (i.p.) (n = 8 per group), or 100 PFU to 1 × 105 PFU (in 10 μl of PBS) intracranially (i.c.) (n = 5 or 6 per group). For intracranial injections, mice were sedated with ketamine and medetomidine (75 mg/kg and 1 mg/kg of body weight, respectively, i.p.) and kept under 2% isoflurane gas anesthesia. Injection was done into the caudate putamen of mice, 2 mm right and 1 mm anterior from bregma in 3 mm depth. The antisedative drug atipamezole (1 mg/kg, i.p.) and the analgesic carprofen (5 mg/kg, subcutaneous [s.c.]) were administered after the operation.
After the virus injection, mice were monitored for neurological symptoms and distress. Symptoms were graded as follows: 0, no symptoms; 1, mild symptoms (ruffled fur, hunched back, weakness of limbs, and optic neuritis); 2, partial paralysis of hind limbs; 3, paralysis of hind or front limbs; 4, severe paralysis, tetraplegia; and 5, moribund/dead. In the case of severe neurological symptoms, paralyzed (grade 3 or higher) or distressed mice were sacrificed and tissue samples (brain, liver, and blood) were collected for immunohistochemical (IHC) analysis and titration. The animal experiments were performed at biosafety level 2 containment, obeying the guidelines of the local committee of animal welfare.
Measurements from tissue samples.
Beta interferon (IFN-β) was detected from serum samples or homogenized brain tissue samples prepared as described above. An analysis was done using a VeriKine mouse IFN-β enzyme-linked immunosorbent assay (ELISA) kit (PBL Interferon Source, Piscataway, NJ) according to the manufacturer's instructions, with samples diluted 1:2 or 1:10 in a buffer provided with the kit.
IHC analysis.
Brain tissue samples for IHC analysis were collected from mice suffering from neurological symptoms as follows. Mice were euthanized with CO2 and perfused with cold PBS through the left ventricle. After perfusion, a part of the liver and the right hemisphere of the brain were dissected and placed into 4% paraformaldehyde (4% PFA in PBS) solution overnight (+4°C). Next-day tissues were transferred from PFA to 70% ethyl alcohol (EtOH) and stored at +4°C until embedded into paraffin blocks. Paraffin-embedded tissue samples were cut into 7-μm sections using a microtome, and an IHC analysis was done using a Vectastain ABC kit (Vector Laboratories, Burlingame, CA) and polyclonal anti-SFV antibody (prepared in rabbit).
RT-PCR from mouse brain tissue.
Total RNA was extracted from the brain tissue of the mouse displaying more severe neurological symptoms when infected i.p. with SFV4-miRT124 using the GenElute mammalian total RNA miniprep kit (Sigma-Aldrich). cDNA was generated with SFV4-specific primer (5′-AATTTTGCGGCCGCTAGCACGTTCTG-3′) using Fermentas RevertAid M-MuLV reverse transcriptase (Thermo Fisher Scientific). For PCR-based amplification of the target element region, the following primers were used: forward primer, 5′-AATTTTCTCGAGAACCCGATTCCTCC-3′, and reverse primer, 5′-AGATTGGAATTCCTAACGGATTTTTGTTGT-3′. The resulting PCR product was then cloned into the pJET1.2 vector using the Fermentas CloneJET PCR cloning kit (Thermo Fisher Scientific), and 18 individual clones were verified by sequencing.
RESULTS
Construction and in vitro characterization of novel miRNA-targeted SFVs.
In order to attenuate neurovirulence of SFV, we inserted perfectly complementary target elements for the neuronally expressed miR124 into the genome of SFV4. miR124 was chosen for this purpose because it has been shown to be expressed in high levels in the central nervous system (CNS) of mice and reported to be useful for suppressing poliovirus replication in the brain (4, 23). However, due to the shortage of well-validated cell line models expressing miR124, we first tested the feasibility of miRNA targeting to suppress SFV replication using the liver-specific miR122 and the Huh7 hepatocellular carcinoma cell line as an experimental system. Huh7 cells express large amounts of miR122 (24) and have been successfully used as an in vitro model to demonstrate the capacity of miR122 target elements to suppress adenovirus replication in the liver (10, 25).
Three different strategies to generate miRNA-regulated SFV4 were tested. Target elements for miR122 were inserted into the 5′ and 3′ nontranslated regions (NTR) or between the nsp3 and nsp4 genes in the SFV4 genome. In the latter case, an additional protease cleavage site was introduced between nsp3 and nsp4, followed by insertion of miR122 target elements between the additional and original cleavage sites. Preliminary characterization of these modified viruses indicated that miRNA targeting was most effective when the target elements were inserted between nsp3 and nsp4 (data not shown), and this strategy was chosen for all subsequent experiments. This intergenic strategy for the insertion of six copies of miR122 (SFV4-miRT122) or miR124 (SFV4-miRT124) target elements is schematically presented in Fig. 1.
Fig 1.

Schematic presentation of the SFV4 vector carrying tissue-specific miRNA target sequences. (A) Target elements contained 6 copies of miR122 (liver-specific) or miR124 (neuron-specific) target sequences (bold and underlined) with the nsp3/nsp4 protease cleavage site sequences at both ends of the element (white on black background). (B) Element was inserted between the SFV4 nsp3 and nsp4 genes with the nsp3/nsp4 protease cleavage sites (black boxes) at both ends of miRNA target element (gray box). (C) Presentation of the whole SFV4 genome with miRNA target element location is marked with a black arrow.
To confirm the correct proteolytic processing of nsp3 and nsp4 in cells infected with these viruses, BHK-21 cells were infected with SFV4, SFV4-miRT122, or SFV4-miRT124 at a multiplicity of infection (MOI) of 1. At 12 h postinfection (p.i.), the processing of nsp3 and nsp4 was analyzed by Western blotting. As shown in Fig. 2A, SFV4-miRT122 and SFV4-miRT124 viruses showed protein bands of similar size compared to SFV4 for both nsp3 and nsp4, indicating the correct processing of both proteins from the polyprotein.
Fig 2.
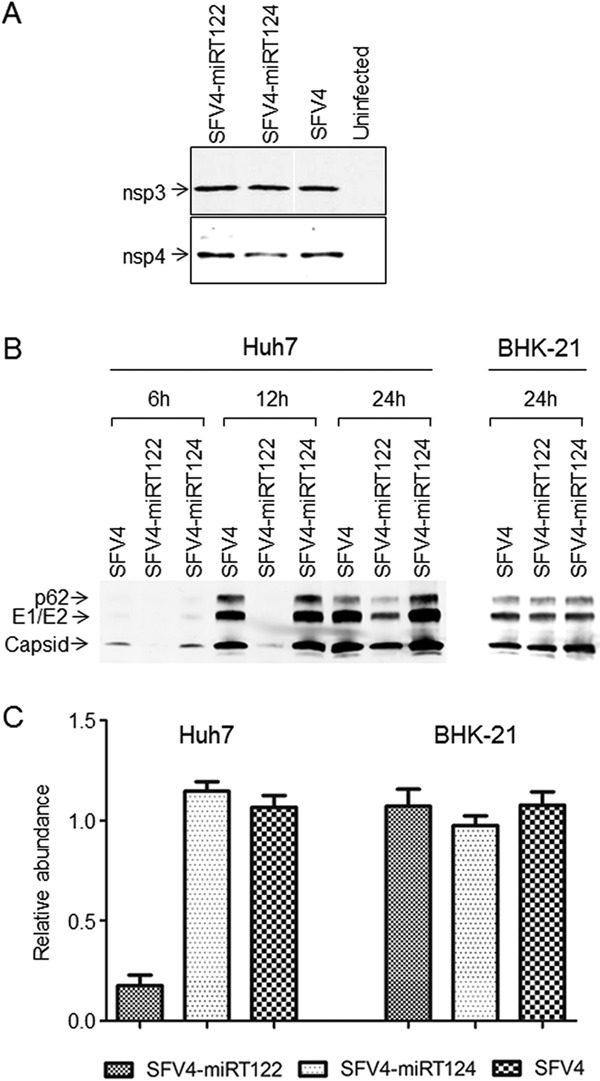
In vitro characterization of miRNA-targeted SFV4 viruses. (A) Western blot analysis of viral nsp3 and nsp4 proteins. Samples collected from BHK-21 cells infected with SFV4-miRT124, SFV4-miRT122, and SFV4 or left uninfected. (B) Viral proteins detected with Western blotting from infected Huh7 or BHK-21 cells collected at 6 h, 12 h, or 24 h postinfection. (C) The amount of viral genomic RNA measured with quantitative PCR from infected Huh7 and BHK-21 cells at 24 h postinfection.
To evaluate the capacity of miRNA targeting to restrict SFV replication, we infected miR122-expressing Huh7 cells (24) and miR122-negative BHK-21 cells (26) with SFV4-miRT122, SFV4-miRT124, or unmodified SFV4. After 6 h, 12 h, or 24 h of infection of Huh7 cells or after 24 h of infection of BHK-21 cells with an MOI of 0.1, the levels of SFV envelope and capsid proteins were analyzed by Western blotting and detected by infrared imaging (Fig. 2B). In the BHK-21 cell line, SFV4-miRT122 and SFV4-miRT124 produced similar amounts of proteins compared to SFV4. In the Huh7 cell line, SFV4-miRT124 and SFV4 expressed similar amounts of viral proteins at all time points analyzed, whereas viral protein production of SFV4-miRT122 was strongly reduced, especially at the early time points (less than 5% of the amount measured from SFV4- or SFV4-miRT124-infected Huh7 cells). At 24 h p.i., this difference was less pronounced, but viral protein expression in SFV4-miRT122-infected Huh7 cells was still clearly decreased compared to SFV4- or SFV4-miRT124-infected cultures, where further accumulation of SFV proteins was probably already limited by virus-induced cytotoxicity.
To test whether the reduced levels of viral proteins in the Huh7 cell line infected with SFV4-miRT122 were due to miRNA-mediated catalytic cleavage of SFV RNA or resulted from translational repression, we infected the Huh7 and BHK-21 cell lines with SFV4, SFV4-miRT122, or SFV4-miRT124 at an MOI of 0.1 and measured the amount of SFV genomic RNA by quantitative PCR at 24 h p.i. (Fig. 2C). In BHK-21 cells infected with SFV4-miRT122 or SFV4-miRT124, the levels of genomic RNA were similar to those in cells infected with SFV4, whereas in the Huh7 cell line, the amount of genomic RNA in cells infected with SFV4-miRT122 was strongly (over 83%) reduced compared to cells infected with SFV4-miRT124 or SFV4. As could be expected, based on the perfect homology of the miR122 target elements, these results indicated that the miRNA-specific targeting indeed occurred via RNA destruction rather than inhibition of protein translation.
To evaluate the oncolytic potency of the miRNA-targeted SFV4 viruses in vitro, we infected a panel of tumor cell lines with SFV4, SFV4-miRT122, or SFV4-miRT124 at an MOI of 0.1 and monitored the lytic cell killing and the spread of infection using a colorimetric cell viability assay. Infection with SFV4 or SFV4-miRT124 led to efficient destruction of all the tested cell lines (Fig. 3). Infection with SFV4-miRT122 led to the efficient destruction of all other cell lines except the miR122-expressing Huh7 cell line. These results suggest that the replication of SFV4-miRT122 was specifically suppressed by miR122. In addition, the insertion of miRNA-target elements into the SFV4 genome did not seem to diminish the oncolytic potency of these viruses in vitro.
Fig 3.

miRNA-targeted SFV4 viruses show oncolytic potency in vitro. Five cell lines of the miR122-deficient (BHK-21, HeLa, Tera-2, and U87) or miR122-expressing cell line Huh7 were infected with SFV4, SFV4-miRT122, or SFV4-miRT124 at an MOI of 0.1. Cell survival in the infected cells was measured at 24 h postinfection (Tera-2), 48 h postinfection (Huh7, BHK-21, and HeLa), or 72 h postinfection (U87) using an ATP-based cell viability assay and plotted on the y axis as the percentage of the control values measured from uninfected cultures. The data are presented as the means of four repetitions ± standard deviations.
SFV4-miR124 shows attenuated spread in the brains of mice.
To evaluate the infection and replication characteristics of the miRNA-targeted SFVs in vivo, we infected adult 5- to 7-week-old BALB/c mice intraperitoneally with 1 × 106 PFU of SFV4 or SFV4-miRT124 in 100 μl PBS. Since miR122 is not expressed in the CNS, we used SFV4-miRT122 as a control virus to rule out the nonspecific effects on viral replication due to the insertion of miRNA target elements into the SFV4 genome. As shown in Fig. 4A, mice infected with SFV4 started to show neurological symptoms expressed as limb weakness and paralysis from day 4 p.i., leading to severe symptoms or death in 4 out of 8 mice within 10 days, whereas only 2 mice remained asymptomatic. Infection of mice with SFV4-miRT122 produced a similar phenotype, leading to severe CNS-derived symptoms or death in 6 out of 8 mice, and no statistical survival difference between the SFV4 and SFV4-miRT122 groups could be observed. In contrast, in the group of mice infected with SFV4-miRT124, severe neurologic symptoms could be witnessed only in 1 out of 8 mice and a significant survival benefit was obtained over the SFV4-miRT122 control group (P = 0.0106, log-rank test), indicating that SFV4-miRT124 replication was severely attenuated in the CNS.
Fig 4.

SFV4-miRT124 shows an attenuated spread into the CNS following intraperitoneal injection in mice. (A) Survival and symptoms witnessed following intraperitoneal injection of BALB/c mice (n = 8) with 1 × 106 PFU of the virus and graded as follows: 0, no symptoms; 1, ruffled fur, hunched back, weakness of limbs, and optic neuritis; 2, partial paralysis of hind limbs; 3, paralysis of hind or front limbs; 4, severe paralysis, tetraplegia; and 5, moribund/dead. (B) Virus titers measured from serum samples collected from groups of mice (n = 3) 1, 2, and 3 days postinfection (p.i.). (C) Virus titers measured from brain tissue samples collected from groups of mice (n = 3) 2, 3, and 4 days p.i. nd, not detectable.
To examine if the course of infection in the single mouse that developed severe neurologic symptoms upon SFV4-miRT124 administration might have been due to mutational loss of the miR124 target elements, we isolated RNA from the brain of this animal. The relevant region of the viral genome was amplified by RT-PCR, and 18 individual clones containing these PCR fragments were sequenced. Sixteen out of 18 clones showed no mutations in the miRNA target region. In 1 clone, 2 target elements had been deleted, while the remaining 4 target elements were intact. Only 1 clone out of 18 had lost all the six target elements and had reverted to the wild-type-like virus. While these results showed that mutations in the miR124 target elements do occur, the late emergence and low frequency of escape mutants suggested that this was a relatively uncommon event and unlikely to explain the severe outcome of the infection in this animal.
Virus titration from the sera of infected mice revealed a high level of viremia at day 1 p.i., with a subsequent rapid drop between days 2 and 3 (Fig. 4B). Importantly, all viruses showed comparable serum titers, indicating that the miRNA-targeted viruses had retained wild-type-like replication capacity in the periphery. Titers obtained from homogenized brain tissues showed an increasing CNS virus load between days 2 and 4 p.i. (Fig. 4C). However, the average brain titer for SFV4-miRT124 virus remained at 103 PFU/g, while infection with SFV-miRT122 and SFV4 reached 105 to 106 PFU/g (100- to 1,000-fold higher).
To evaluate the antiviral interferon responses in the CNS of these mice, we measured the levels of beta interferon (IFN-β) in brain homogenates prepared from these animals. While high levels of IFN-β were found in the brain tissue samples collected from SFV4- and SFV4-miRT122-infected mice, no IFN-β could be detected in the brains of SFV4-miRT124-infected mice (Fig. 5). No IFN-β was detected in the serum samples (data not shown). Thus, the IFN-β levels in the CNS of these animals correlated very well with our data on the differential capacity of these viruses to replicate in the brain.
Fig 5.

Beta interferon measurement from mouse brain tissue. Samples collected from mice infected intraperitoneally with 1 × 106 PFU of virus 4 days postinfection (p.i.). IFN-β measured using ELISA (n = 3). Detection limit 31 pg/ml (dashed line). nd, not detectable.
Interestingly, an immunohistochemical (IHC) analysis of the brain sample obtained from the SFV4-miRT124-infected mouse suffering from severe symptoms revealed only a limited focal infection of the brain (Fig. 6A). In contrast, analysis of the brain samples from the SFV4-miRT122- and SFV4-infected groups demonstrated a wide-spread neuronal infection of the midbrain, thalamus, and cerebellum (Purkinje neurons) in those mice suffering from neurological symptoms (Fig. 6B and C).
Fig 6.

Immunohistochemical analysis of mouse brain tissue collected following intraperitoneal infection. Mice infected with (A) SFV4-miRT124, (B) SFV4-miRT122, or (C) wild-type SFV4 (1 × 106 PFU of virus). Samples were collected 6 days (A and B) or 5 days (C) postinfection. Seven-micrometer sagittal brain sections stained with anti-SFV antibody (left). Magnifications taken from areas indicated with black boxes (right). Scale bar, 200 μm.
Taken together, these results suggested that insertion of miR124 target elements into the virus genome strongly attenuates the neurovirulence of SFV4 when administered via a peripheral route.
SFV4-miRT124 infection elicits protective immunity against lethal SFV challenge.
To determine whether the SFV4-miRT124-infected mice that survived i.p. virus administration had developed immunity against SFV, we infected 5 such asymptomatic mice i.p. with 1 × 106 PFU of the lethal L10 strain. As shown in Fig. 7, all the mice previously infected with SFV4-miRT124 survived the L10 infection and remained asymptomatic for the 20-day follow-up period, while the mice without the preceding SFV infection died within 4 days. Thus, these results showed that efficient anti-SFV immunity had developed in SFV4-miRT124-infected mice, suggesting a potential for the use of miR124-attenuated viruses as live attenuated alphavirus vaccines.
Fig 7.

SFV4-miRT124 elicits antiviral immunity against SFV strain L10 in mice. Kaplan-Meier survival curves of mice infected intraperitoneally with a lethal dose of SFV strain L10 with (white squares) or without (filled circles) a preceding intraperitoneal infection with SFV4-miR124T.
Intracranial infection with SFV4-miRT124 leads to restricted infection of the corpus callosum.
We next wanted to examine the capacity of SFV4-miRT124 to replicate in neurons when introduced directly into the brains of mice. To this end, 100, 10,000, or 100,000 PFU of SFV4, SFV4-miRT122, or SFV4-miRT124 were administered into groups of BALB/c mice (n = 5 to 6) via an intracranial injection. Although a significant survival benefit (P ≤ 0.0009, log-rank test) of SFV4-miRT124-infected mice was obtained compared to SFV4- or SFV4-miRT122-infected animals, all mice eventually developed lethal neurological symptoms (Fig. 8).
Fig 8.

Mice infected intracranially with SFV4-miRT124 show increased survival compared to SFV4-miRT122 or wild-type SFV4. (A) Survival and symptoms witnessed in groups of BALB/c mice infected intracranially with 100 (n = 6), 10,000 (n = 5), or 100,000 (n = 6) PFU of SFV4-miRT122 (A), 100 (n = 6), 10,000 (n = 6), or 100,000 (n = 6) PFU of SFV4-miRT124 (B), and 100 (n = 6) or 10,000 (n = 5) PFU of SFV4 (C) and graded as follows: 0, no symptoms; 1, ruffled fur, hunched back, weakness of limbs, and optic neuritis; 2, partial paralysis of hind limbs; 3, paralysis of hind or front limbs; 4, severe paralysis, tetraplegia; and 5, moribund/dead.
Interestingly, an IHC analysis of the brains of these mice showed that infection with SFV4-miRT124 was associated with a dramatically restricted virus spread in the neurons. Only a few affected neurons could be seen, while the virus was predominantly detected in oligodendrocytes of the white matter tract of the corpus callosum (Fig. 9A). In contrast, the control miRNA target bearing SFV4-miRT122 caused a global infection of the brain involving also the Purkinje neurons of the cerebellar cortex (Fig. 9B).
Fig 9.

Immunohistochemical analysis of mouse brain tissue collected following intracranial infection. Samples collected from sacrificed mice following a 1 × 105 PFU intracranial injection of SFV4-miRT124 (A) or SFV4-miRT122 (B). Samples were collected 3 days (A) or 2 days postinfection. (B). Magnified pictures taken from the areas indicated with black boxes. Staining is done from paraffin-embedded 7-μm tissue slices using a polyclonal anti-SFV antibody. Virus antigen in infected cells is shown in brown. Background stained with hematoxylin. Scale bar, 200 μm.
DISCUSSION
Here we show that the neuropathogenicity of Semliki Forest virus can be selectively attenuated by inserting target elements for the neuron-specific microRNA miR124 between nsp3 and nsp4 in the viral genome. This genomic modification (SFV4-miRT124) did not affect normal nsp polyprotein processing, as both nsp3 and nsp4 were produced identically to wild-type SFV4. Using a well-validated miR122-based cell line model system, the capacity of cell type-specific microRNAs to control SFV replication was first demonstrated in vitro. These studies also confirmed that the mechanism of SFV attenuation exercised by perfectly complementary microRNAs target elements placed between nsp3 and nsp4 involved viral RNA degradation rather than translational suppression. Our in vivo results in BALB/c mice showed that neuronal detargeting by miR124 was effective as demonstrated by strongly reduced neurovirulence and pathology. Virus titers in serum samples collected from intraperitoneally infected mice showed similar peripheral replication kinetics for SFV4-miRT124 compared to the nonattenuated control virus (SFV4-miRT122), indicating that the reduced neuropathology observed after SFV4-miRT124 infection was not due to peripheral attenuation of the virus.
The initial peripheral replication of our viruses did not induce detectable levels of IFN-β in the serum. Instead, IFN-β was detected in the brain tissue following intraperitoneal infection with SFV4 or SFV4-miRT122. Levels of IFN-β in the brain correlated with virus loads, and consequently, no IFN-β was detected in SFV4-miRT124-infected mouse brain samples. These results are in line with our previous finding that, in the mouse, CNS expression of cytokines correlates with the rate of SFV replication (27).
Unlike the mice infected with SFV4-miRT122 or wild-type SFV4, only 1 out of 8 animals infected i.p. with SFV4-miRT124 displayed severe neurological symptoms. To study if this severe course of the disease could be explained by a loss of the miR124 target elements, we sequenced several independent viral genomes from brain tissue RNA prepared from this animal. While some mutations could be found, the great majority of the viruses (89%) contained the original sequence introduced between nsp3 and nsp4, arguing against a role of escape mutants in this case. Moreover, these studies suggested that the mutational inactivation of microRNA target elements placed in this position of an alphavirus is a relatively infrequent and slow process and does not pose a major threat to the utility of this approach in general.
Of note, we found that a prior infection of mice with SFV4-miRT124 could provide antiviral immunity and complete protection against an otherwise lethal challenge with a highly virulent wild-type virus (L10), suggesting that, besides applications in the area of cancer virotherapy, cell type-specific miRNA targeting could also be exploited in the development of more potent and safer alphaviral vaccine vectors. In a related approach, Kamrud et al. utilized target elements for ubiquitously expressed miRNAs introduced into the helper RNAs of a vector system based on Venezuelan equine encephalitis virus (VEEV) to build an additional safety measure into the production of virus-like replicon particles (VRP) for VEEV vaccine development (28).
To further characterize the basis of the reduced neuropathogenicity associated with systemic infection with an miR124-controlled virus, we also tested the intracranial administration of SFV4-miRT124. This resulted in a restricted infection pattern where a predominant infection of oligodendrocytes of corpus callosum was observed. This infection pattern correlates well with the known expression of miR124 in different parts of the mouse brain (29, 30). Interestingly, a similar region-limited infection result was reported in a recent study where avirulent SFV and SFV replicons were used (31). The observed infection limited to corpus callosum might explain the attenuated but nevertheless lethal phenotype of SFV4-miRT124 following i.c. infection. However, a significant survival benefit was achieved in mice infected with SFV4-miRT124. It is likely that the infectious doses used for i.c. injections exceeded the amount of virus that generally enters the brain following systemic infection. Thus, despite the fatal outcome by the i.c. route, we conclude that the SFV4-miRT124 virus is strongly attenuated in its neurovirulence and shows diminished pathogenicity.
The severe neurovirulence of SFV4 has limited its utility as a potential cancer virotherapy agent. The selectively attenuated replication of SFV4-miRT124 in the CNS now helps to overcome this problem and opens new possibilities for using SFV as an oncolytic virus in preclinical mouse cancer models. In this regard, the rationally and selectively attenuated SFV4-miRT124 offers distinct advantages compared to the naturally attenuated strains. Wild-type strains like SFV4 have been reported to be more potent in their ability to inhibit type I interferon responses in target cells compared to the attenuated strains (32). Indeed, type I interferons produced by the infected tumor cells have been found to play a key role in limiting oncolysis by the avirulent SFV vector VA7 (33). It seems reasonable to expect that SFV4 attenuated by microRNA targeting might be less susceptible to type I interferon-mediated antiviral immunity.
On the other hand, Ruotsalainen et al. also showed that treatment of mice with cyclophosphamide led to enhanced VA7 replication in healthy brain tissue, including neurons (33). Since this and other immunosuppressive treatments are increasingly being tested to enhance the oncolytic potential of cancer virotherapy agents, additional safety measures are clearly needed. In the case of SFV, an attractive approach for increasing safety would be to combine the features of SFV4-miRT124 and VA7 by engineering the miR124 target elements into the VA7 genome.
Taken together, our results show for the first time that the tissue tropism of a fully replicative and neurovirulent alphavirus can be altered by the insertion of tissue-specific miRNA target elements into the viral genome. By engineering SFV replication sensitive to neuron-specific miR124, we were able to strongly attenuate the neurovirulence of SFV4, thus providing new opportunities for studies on SFV in preclinical mice models for cancer virotherapy and in alphavirus vaccine development.
ACKNOWLEDGMENTS
This study was supported by grants from the Academy of Finland (K.S. and A.H.), the Cancer Centre of Eastern Finland (A.H.), the Oskar Öflund Foundation (A.H.), the Kuopio University Hospital (EVO) (A.H.), Understödsföreningen Liv och Hälsa (A.H.), Helsinki University Central Hospital Research Council (K.S.), the Sigrid Juselius Foundation (K.S.), and the Jenny and Antti Wihuri Foundation (E.Y.). E.Y. was supported in part by Helsinki Biomedical Graduate School.
We thank the personnel of the Experimental Animal Center of UEF for help in maintaining good animal care, Virpi Syvälahti for technical assistance, and Kristina Zumer for help and discussion.
The permissions for animal work were obtained from the National Committee for animal experiments.
Footnotes
Published ahead of print 17 October 2012
REFERENCES
- 1. Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297 [DOI] [PubMed] [Google Scholar]
- 2. Cullen BR. 2011. Viruses and microRNAs: RISCy interactions with serious consequences. Genes Dev. 25:1881–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Croce CM. 2009. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 10:704–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barnes D, Kunitomi M, Vignuzzi M, Saksela K, Andino R. 2008. Harnessing endogenous miRNAs to control virus tissue tropism as a strategy for developing attenuated virus vaccines. Cell Host Microbe 4:239–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Edge RE, Falls TJ, Brown CW, Lichty BD, Atkins H, Bell JC. 2008. A let-7 MicroRNA-sensitive vesicular stomatitis virus demonstrates tumor-specific replication. Mol. Ther. 16:1437–1443 [DOI] [PubMed] [Google Scholar]
- 6. Hikichi M, Kidokoro M, Haraguchi T, Iba H, Shida H, Tahara H, Nakamura T. 2011. MicroRNA regulation of glycoprotein B5R in oncolytic vaccinia virus reduces viral pathogenicity without impairing its antitumor efficacy. Mol. Ther. 19:1107–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kelly EJ, Hadac EM, Greiner S, Russell SJ. 2008. Engineering microRNA responsiveness to decrease virus pathogenicity. Nat. Med. 14:1278–1283 [DOI] [PubMed] [Google Scholar]
- 8. Leber MF, Bossow S, Leonard VH, Zaoui K, Grossardt C, Frenzke M, Miest T, Sawall S, Cattaneo R, von Kalle C, Ungerechts G. 2011. MicroRNA-sensitive oncolytic measles viruses for cancer-specific vector tropism. Mol. Ther. 19:1097–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee CY, Rennie PS, Jia WW. 2009. MicroRNA regulation of oncolytic herpes simplex virus-1 for selective killing of prostate cancer cells. Clin. Cancer Res. 15:5126–5135 [DOI] [PubMed] [Google Scholar]
- 10. Ylösmäki E, Hakkarainen T, Hemminki A, Visakorpi T, Andino R, Saksela K. 2008. Generation of a conditionally replicating adenovirus based on targeted destruction of E1A mRNA by a cell type-specific MicroRNA. J. Virol. 82:11009–11015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Santagati MG, Määttä JA, Itäranta PV, Salmi AA, Hinkkanen AE. 1995. The Semliki Forest virus E2 gene as a virulence determinant. J. Gen. Virol. 76(Pt 1):47–52 [DOI] [PubMed] [Google Scholar]
- 12. Bradish CJ, Allner K, Maber HB. 1971. The virulence of original and derived strains of Semliki Forest virus for mice, guinea-pigs and rabbits. J. Gen. Virol. 12:141–160 [DOI] [PubMed] [Google Scholar]
- 13. Fazakerley JK, Pathak S, Scallan M, Amor S, Dyson H. 1993. Replication of the A7(74) strain of Semliki Forest virus is restricted in neurons. Virology 195:627–637 [DOI] [PubMed] [Google Scholar]
- 14. Mathiot CC, Grimaud G, Garry P, Bouquety JC, Mada A, Daguisy AM, Georges AJ. 1990. An outbreak of human Semliki Forest virus infections in Central African Republic. Am. J. Trop. Med. Hyg. 42:386–393 [DOI] [PubMed] [Google Scholar]
- 15. Strauss JH, Strauss EG. 1994. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 58:491–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Willems WR, Kaluza G, Boschek CB, Bauer H, Hager H, Schutz HJ, Feistner H. 1979. Semliki Forest virus: cause of a fatal case of human encephalitis. Science 203:1127–1129 [DOI] [PubMed] [Google Scholar]
- 17. Ketola A, Hinkkanen A, Yongabi F, Furu P, Määttä AM, Liimatainen T, Pirinen R, Björn M, Hakkarainen T, Mäkinen K, Wahlfors J, Pellinen R. 2008. Oncolytic Semliki Forest virus vector as a novel candidate against unresectable osteosarcoma. Cancer Res. 68:8342–8350 [DOI] [PubMed] [Google Scholar]
- 18. Määttä AM, Mäkinen K, Ketola A, Liimatainen T, Yongabi FN, Vähä-Koskela M, Pirinen R, Rautsi O, Pellinen R, Hinkkanen A, Wahlfors J. 2008. Replication competent Semliki Forest virus prolongs survival in experimental lung cancer. Int. J. Cancer 123:1704–1711 [DOI] [PubMed] [Google Scholar]
- 19. Vähä-Koskela MJ, Kallio JP, Jansson LC, Heikkilä JE, Zakhartchenko VA, Kallajoki MA, Kähäri VM, Hinkkanen AE. 2006. Oncolytic capacity of attenuated replicative Semliki Forest virus in human melanoma xenografts in severe combined immunodeficient mice. Cancer Res. 66:7185–7194 [DOI] [PubMed] [Google Scholar]
- 20. Heikkilä JE, Vähä-Koskela MJ, Ruotsalainen JJ, Martikainen MW, Stanford MM, McCart JA, Bell JC, Hinkkanen AE. 2010. Intravenously administered alphavirus vector VA7 eradicates orthotopic human glioma xenografts in nude mice. PLoS One 5:e8603 doi:10.1371/journal.pone.0008603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vähä-Koskela MJ, Tuittila MT, Nygårdas PT, Nyman JK, Ehrengruber MU, Renggli M, Hinkkanen AE. 2003. A novel neurotropic expression vector based on the avirulent A7(74) strain of Semliki Forest virus. J. Neurovirol. 9:1–15 [DOI] [PubMed] [Google Scholar]
- 22. Sedensky MM, Hudson SJ, Everson B, Morgan PG. 1994. Identification of a mariner-like repetitive sequence in C. elegans. Nucleic Acids Res. 22:1719–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. 2002. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 12:735–739 [DOI] [PubMed] [Google Scholar]
- 24. Chang J, Nicolas E, Marks D, Sander C, Lerro A, Buendia MA, Xu C, Mason WS, Moloshok T, Bort R, Zaret KS, Taylor JM. 2004. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 1:106–113 [DOI] [PubMed] [Google Scholar]
- 25. Cawood R, Chen HH, Carroll F, Bazan-Peregrino M, van Rooijen N, Seymour LW. 2009. Use of tissue-specific microRNA to control pathology of wild-type adenovirus without attenuation of its ability to kill cancer cells. PLoS Pathog. 5:e1000440 doi:10.1371/journal.ppat.1000440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tian W, Dong X, Liu X, Wang G, Dong Z, Shen W, Zheng G, Lu J, Chen J, Wang Y, Wu Z, Wu X. 2012. High-throughput functional microRNAs profiling by recombinant AAV-based microRNA sensor arrays. PLoS One 7:e29551 doi:10.1371/journal.pone.0029551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tuittila M, Nygårdas, Hinkkanen A. 2004. mRNA expression of proinflammatory cytokines in mouse CNS correlates with replication rate of Semliki Forest virus but not with the strain of viral proteins. Viral Immunol. 17:287–297 [DOI] [PubMed] [Google Scholar]
- 28. Kamrud KI, Coffield VM, Owens G, Goodman C, Alterson K, Custer M, Murphy MA, Lewis W, Timberlake S, Wansley EK, Berglund P, Smith J. 2010. In vitro and in vivo characterization of microRNA-targeted alphavirus replicon and helper RNAs. J. Virol. 84:7713–7725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deo M, Yu JY, Chung KH, Tippens M, Turner DL. 2006. Detection of mammalian microRNA expression by in situ hybridization with RNA oligonucleotides. Dev. Dyn. 235:2538–2548 [DOI] [PubMed] [Google Scholar]
- 30. Pena JT, Sohn-Lee C, Rouhanifard SH, Ludwig J, Hafner M, Mihailovic A, Lim C, Holoch D, Berninger P, Zavolan M, Tuschl T. 2009. miRNA in situ hybridization in formaldehyde and EDC-fixed tissues. Nat. Methods 6:139–141 doi:10.1038/nmeth.1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fazakerley JK, Cotterill CL, Lee G, Graham A. 2006. Virus tropism, distribution, persistence and pathology in the corpus callosum of the Semliki Forest virus-infected mouse brain: a novel system to study virus-oligodendrocyte interactions. Neuropathol. Appl. Neurobiol. 32:397–409 [DOI] [PubMed] [Google Scholar]
- 32. Deuber SA, Pavlovic J. 2007. Virulence of a mouse-adapted Semliki Forest virus strain is associated with reduced susceptibility to interferon. J. Gen. Virol. 88:1952–1959 [DOI] [PubMed] [Google Scholar]
- 33. Ruotsalainen J, Martikainen M, Niittykoski M, Huhtala T, Aaltonen T, Heikkilä J, Bell J, Vähä-Koskela M, Hinkkanen A. 2012. Interferon-beta sensitivity of tumor cells correlates with poor response to VA7 virotherapy in mouse glioma models. Mol. Ther. doi:10.1038/mt.2012.53 [DOI] [PMC free article] [PubMed] [Google Scholar]