

Abstract
The paramyxovirus entry machinery consists of two glycoproteins that tightly cooperate to achieve membrane fusion for cell entry: the tetrameric attachment protein (HN, H, or G, depending on the paramyxovirus genus) and the trimeric fusion protein (F). Here, we explore whether receptor-induced conformational changes within morbillivirus H proteins promote membrane fusion by a mechanism requiring the active destabilization of prefusion F or by the dissociation of prefusion F from intracellularly preformed glycoprotein complexes. To properly probe F conformations, we identified anti-F monoclonal antibodies (MAbs) that recognize conformation-dependent epitopes. Through heat treatment as a surrogate for H-mediated F triggering, we demonstrate with these MAbs that the morbillivirus F trimer contains a sufficiently high inherent activation energy barrier to maintain the metastable prefusion state even in the absence of H. This notion was further validated by exploring the conformational states of destabilized F mutants and stabilized soluble F variants combined with the use of a membrane fusion inhibitor (3g). Taken together, our findings reveal that the morbillivirus H protein must lower the activation energy barrier of metastable prefusion F for fusion triggering.
INTRODUCTION
Paramyxoviruses are enveloped, nonsegmented, negative-stranded RNA viruses which include pathogens of both humans and animals and collectively induce diseases with significant global health and economic impacts. For instance, respiratory syncytial virus (RSV), a pneumovirus, is a major cause of pneumonia in young children. Measles virus (MeV), a morbillivirus, still kills more than 120,000 people each year (1), whereas the closely related canine distemper virus (CDV) continues to rage among terrestrial and aquatic carnivores with high rates of morbidity and mortality. Emerging paramyxoviruses such as Hendra virus (HeV) and Nipah virus (NiV) cause zoonotic infections with high mortality rates (2, 3).
To initiate disease, paramyxoviruses enter host cells by using two envelope glycoproteins that tightly cooperate to mediate plasma membrane fusion at a neutral pH: the attachment (HN/H/G, depending on the genus) and the fusion (F) proteins (4). Functional paramyxovirus attachment proteins consist of a loosely associated pair of covalently linked dimers (5, 6); each monomer is composed of a short luminal tail, a single membrane-spanning region, and a large ectodomain. The extracellular region comprises a membrane-proximal stalk region supporting a membrane-distal globular head domain that contains the receptor-binding sites. While H/G proteins interact with specific proteinaceous receptors (7–12), HN proteins bind to sialic acid-containing molecules (4, 13). Partial crystal structures of several paramyxovirus attachment protein ectodomains have been solved, which revealed that the monomeric head domains invariably fold into a six-bladed beta-propeller conformation typical of sialidases (6, 14–16). However, substantial differences were observed with respect to their oligomeric organizations (6). Although these conformations were speculated to represent biologically relevant tetrameric conformations, the functional significance of individual tetramer arrangements remains to be demonstrated.
Like other class I viral fusion glycoproteins, the paramyxovirus F protein forms a homotrimer. The F protein is first synthesized as an inactive precursor (F0) that is proteolytically matured into two disulfide-linked subunits (F1 and F2). F1 contains a short luminal tail, a transmembrane domain, and a large ectodomain harboring conserved domains that are characteristic of class I viral fusion glycoproteins. These include a hydrophobic N-terminal fusion peptide (FP) and two heptad repeat regions (HRA and HRB), adjacent to the FP and the transmembrane domain, respectively (4, 17). The crystal structures of paramyxovirus F proteins in both the pre- and postfusion conformations have been determined and have considerably advanced our insight into F-protein structural rearrangements that occur upon the triggering of attachment-protein-mediated fusion. The X-ray structure of a soluble, prefusion-stabilized parainfluenza virus type 5 (PIV5) F trimer revealed a short three-helix bundle (3-HB) stalk region supporting a large globular head domain (18). This differs strikingly from the soluble, nonstabilized, Newcastle disease virus (NDV), hPIV3, and RSV F trimers, which featured a “golf tee-like” conformation that includes the six-helix bundle (6-HB) fusion core structure characteristic of class I viral fusion proteins in their postfusion state (19–21).
Receptor binding by attachment proteins is thought to activate F when a target membrane is present, which then undergoes extensive structural rearrangements leading to a membrane merger (4, 22–26). Whereas both structural and functional studies have strongly advanced our knowledge of receptor binding by paramyxovirus attachment proteins, it remains largely unknown how attachment proteins translate receptor binding to F triggering. It was speculated based on conformation-sensitive monoclonal antibody (MAb)-binding patterns that F triggering is due to receptor-induced conformational changes within the paramyxovirus attachment protein (27, 28). In the case of morbillivirus H proteins, both the head and the central stalk sections were furthermore shown to undergo oligomeric modifications as signals for F triggering (24, 29). However, the first proof of a direct link between H structural rearrangements and F triggering was obtained only very recently. In that study, native PAGE gel systems successfully illuminated discrete H migration profiles, which correlated with putative receptor-bound and -unbound conformational states (30).
Depending on the paramyxovirus type, the fusion and attachment proteins travel to the cell surface either as intracellularly preformed hetero-oligomeric complexes (31) or independently (32, 33), which implied different molecular mechanisms of attachment-protein-dependent F triggering. Two hypotheses emerged with respect to the energetic nature of F activation by paramyxovirus attachment proteins upon interactions with their receptors (34–38). According to the first model, the receptor-binding protein actively destabilizes the F protein to trigger the refolding cascade (referred to as the provocateur or association model). This notion was supported by data showing that the fusion protein maintains the prefusion state in the absence of the attachment protein (35) and that fusion activity is directly proportional to strength of the interaction between the two glycoproteins (model described for sialic acid-binding HN-carrying paramyxoviruses) (39, 40). In contrast, the second model predicts that prior to receptor binding, the attachment protein actively prevents the prefusion F trimer from undergoing irreversible structural rearrangements. Upon the interaction of the attachment protein with a host cell receptor, the F protein is released from the preassembled glycoprotein complex, which in turn spontaneously refolds (referred to as the clamp or dissociation model). The latter model takes into account a strong intracellular association observed between morbillivirus F and H proteins (31) and an inverse correlation between fusion activity and the strength of the physical interaction between the two glycoproteins (model described for proteinaceous receptor-binding H/G-carrying paramyxoviruses) (41–45). As recently documented, however, both models do not exclude any additional requirements for interactions of the fusion/attachment proteins which may occur beyond the initial F-triggering step (46, 47).
In the present study, we examined the intrinsic conformational stability of the morbillivirus F-protein trimer by monitoring the F conformation in the presence and absence of the attachment protein. Different conformational states of F were discriminated by employing newly identified pairs of conformation-sensitive anti-F MAbs. Based on this approach, we provide biochemical and functional evidence supporting the view that the morbillivirus F trimer inherently maintains a prefusion conformation regardless of the presence or absence of the H protein. This suggests that the H tetramer does not actively stabilize the prefusion F trimer. Rather, our findings demonstrate that H lowers the activation energy barrier of the self-stabilized morbillivirus prefusion F trimer, enabling the F-refolding cascade to proceed.
MATERIALS AND METHODS
Cell cultures and viruses.
293T cells, CHO cells, Vero cells, Vero cells expressing the SLAM receptor (Vero-SLAM cells), and Vero-SLAM cells additionally engineered to express Hwt (Vero-SLAM-H cells) were grown in Dulbecco's modified Eagle's medium (Gibco, Invitrogen) with 10% fetal calf serum at 37°C in the presence of 5% CO2. The modified vaccina Ankara (MVA)-T7 recombinant virus was used for a quantitative cell-cell fusion assay and was obtained from B. Moss, NIH, Bethesda, MD. The recombinant CDV A75/17 strain, containing an additional red fluorescent protein (RFP) gene (48, 49), was amplified in Vero-SLAM cells, whereas a previously described H-knockout derivative was amplified in Vero-SLAM-Hwt cells (50).
Construction of expression plasmids.
All single (and multiple) substitutions performed in pCI-CDV-F (derived from hemagglutinin [HA] of the CDV A75/17 strain [51]) or pCI-MeV Fedm (derived from the hemagglutinin of the MeV Edmonston strain) were obtained by using the QuikChange Lightning site-directed mutagenesis kit (Stratagene). FLAG tag insertions (DYKDDDK) within different locations of the morbillivirus F proteins were performed by site-directed mutagenesis as described above. Only the epitope tag insertion with the N-terminal region of F2 (between V216 and G217, according the CDV F-protein amino acid numbering) did not substantially alter F bioactivity. Soluble CDV F proteins were generated by removing the transmembrane and cytosolic tail of the membrane-embedded F-expressing plasmid (sF). To potentially stabilize F in the prefusion state, we fused the GCNt tag peptide (18) N terminally to the F HRB region (sF-GCNt). This was achieved by PCR amplification (High Fidelity PCR system; Roche) of the desired F fragments with RsrII-bearing primers. In addition, the reverse primer contained the GCNt nucleotide motif (encoding the sequence EDKIEEILSKIYHIENEIARIKKLIGEAPGGIEGR). PCR products were next digested with RsrII and subsequently cloned into the RsrII-cleaved pCI vector. Finally, both modified F genes were further tandemly tagged with hexahistidine (6×His) and FLAG epitopes by site-directed mutagenesis, using the same kit as that described above.
Transfections and luciferase reporter gene content mix assay.
Vero cells, in 6-well plates at 90% confluence, were cotransfected with 2 μg of different pCI-F constructs (52), with 1 μg of the various pCI-H plasmids with 9 μl of Fugene HD (Roche), or with 1 μg of the various pCI-F plasmids in 24 wells with 3 μl of Fugene HD, according to the manufacturer's protocol. In some experiments, phase-contrast pictures were taken at 24 h posttransfection with a confocal microscope (Fluoroview FV1000; Olympus).
The quantitative fusion assay was performed as described previously (53, 54). Briefly, Vero cells were cotransfected with the F and H expression plasmids and 0.1 μg of pTM-Luc (kindly provided by Laurent Roux, University of Geneva). In parallel, separate 6-well plates of Vero-SLAM cells or keratinocytes at 30% confluence were infected with MVA-T7 (55) at a multiplicity of infection (MOI) of 1. After incubation overnight, both cell populations were mixed. After 2.5 h (Vero-SLAM cells), the cells were lysed by using Bright Glo lysis buffer (Promega), and the luciferase activity was determined by using a luminescence counter (PerkinElmer Life Sciences) and the Britelite reporter gene assay system (PerkinElmer Life Sciences).
Western blotting.
Western blots were performed as previously described (54, 56). Transfected cells were washed twice with cold phosphate-buffered saline (PBS) before the addition of 150 μl of lysis buffer (10 mM Tris [pH 7.4], 150 mM NaCl, 1% deoxycholate, 1% Triton X-100, 0.1% sodium dodecyl sulfate [SDS]) with a complete protease inhibitor (Roche Biochemicals). After incubation for 20 min at 4°C, the lysates were cleared by centrifugation at 5,000 × g for 15 min at 4°C, and the supernatant was mixed with an equal amount of 2× Laemmli sample buffer (Bio-Rad) containing 100 mM dithiothreitol, subsequently boiled at 95°C for 5 min, and fractionated on 8 or 10% SDS-polyacrylamide gels under denaturing conditions. Separated proteins were transferred onto nitrocellulose membranes by electroblotting. The membranes were then incubated with a polyclonal rabbit anti-CDV F antibody (57) or an anti-HA polyclonal antibody (Covance). Following incubation with a peroxidase-conjugated secondary antibody, the membranes were subjected to enhanced chemiluminescence (ECL) (Amersham Pharmacia Biotech), according to the manufacturer's instructions.
Soluble F-protein production and immunoprecipitation (IP).
293T cells were transfected with 10 μg of various soluble F-expressing DNA plasmids. At 3 days posttransfection, supernatants were harvested, and soluble F proteins were concentrated by using 30-kDa-cutoff filtration columns (Millipore). Subsequently, equal aliquots of supernatants were immunoprecipitated for 2 h with either anti-FLAG F3165 M2 (Sigma), anti-Pre (4941), or anti-Trig (4068) MAb (1:1,000 dilution) (58). This was followed by the addition of protein G-Sepharose beads (overnight), and the mixture was subsequently fractionated in 10% SDS-polyacrylamide gels under regular reducing conditions. Immunoprecipitated F proteins were finally revealed by Western blotting, as described above, using a polyclonal anti-F antibody.
Immunofluorescence (IF) staining and flow cytometry.
Vero or CHO cells were transfected with 1 μg of F-expressing DNA plasmids alone or combined with 1 μg H-expressing DNA plasmids. At 1 day posttransfection, unfixed and unpermeabilized cells were washed twice with ice-cold PBS and subsequently stained with one of the various MAbs (1:1,000 dilution) for 1 h at 4°C. In some experiments, brief heat shocks (10 min) were performed prior to the addition of first antibodies at the indicated temperatures. The anti-CDV F MAbs 3633, 4068, 4941, 4985, and G1 (58) and the anti-MeV F MAbs 186CB, 186CA, 19BG4, 16AG5, and 19GD6 (59, 60) were employed. We note that MAb 4941 was initially characterized as an anti-H antibody (58). However, by having expressed H and F separately in Vero cells, we clearly show in this study that MAb 4941 binds exclusively to F trimer. As a control, the anti-FLAG F3165 M2 MAb (Sigma) was employed. This was followed by washes with ice-cold PBS and incubation of the cells with Alexa Fluor 488-conjugated secondary antibody (1:500 dilution) for 1 h at 4°C. In some experiments, fluorescence emission was captured by confocal fluorescence microscopy (Fluoroview FV1000; Olympus). For flow cytometry analyses, cells were subsequently washed 2 times with ice-cold PBS and consequently detached from the wells by adding PBS-EDTA (50 μM) for 30 min at 37°C. The mean fluorescence intensity of 10,000 cells was then measured by using a BD LSRII flow cytometer (Becton Dickinson) or a FACSCanto II cytometer.
Virus neutralization assay.
A total of 100 50% tissue culture infective dose (TCID50) units of virus was incubated with the indicated dilutions of antibody for 1 h at 37°C. The virus-antibody mixture was then added to Vero cells and incubated for 72 h at 37°C. The cells were assayed for luciferase activity for MeV or the number of infectious units for CDV.
RESULTS
Ectodomain FLAG-tagged F-protein engineering.
To monitor F-protein expression in a conformation-independent manner, we first engineered ectodomain FLAG-tagged F proteins. Three candidate domains were selected for the insertion of a FLAG tag based on a structural model of prefusion CDV F: (i) a membrane-distal domain (C-terminal part of F2 [CDV FF2-FLAG]), (ii) a central domain (within the HRB-linker subdomain [CDV FHRB-link-FLAG]), and (iii) a membrane-proximal domain (within the 7 residues linking the transmembrane domain to the HRB subdomain [CDV FMPD-FLAG]) (Fig. 1A and B). While all three F mutants were properly expressed and proteolytically matured (Fig. 1C), bioactivities varied significantly: CDV FHRB-link-FLAG and CDV FMPD-FLAG were strongly impaired in fusion activity, whereas CDV FF2-FLAG induced fusion to an extent similar to that of untagged F (CDV F) (Fig. 1E). These results were fully confirmed by quantitative, reporter-based cell-to-cell fusion assays (Fig. 1D). Together, the results indicate that the insertion of an 8-amino-acid FLAG tag in the C-terminal region of F2 does not significantly alter the bioactivity of the fusion protein, suggesting minimal conformational modifications within this subdomain during F-trimer refolding into the postfusion conformation. CDV FF2-FLAG, renamed CDV FFLAG for simplicity, was thus selected for all subsequent experiments. An identical tagging strategy was applied for MeV Fedm (derived from the MeV Edmonston strain), which returned equivalent expression and bioactivity results (Fig. 1F and G); the F2 ectodomain-tagged MeV F variant, accordingly renamed MeV FedmFLAG, was used for all subsequent experiments with MeV F.
Fig 1.

Engineering of ectodomain FLAG-tagged morbillivirus F proteins. (A) Homology model of the prefusion CDV F trimer (52). Residues flanking the FLAG epitope insertion are highlighted in red. (B) Scheme of the morbillivirus F gene. Conserved regions among class I fusion proteins are shown. FP, fusion peptide; HRA and HRB, heptad repeat regions A and B, respectively; TM, transmembrane domain. The red boxes represent the positions along the gene selected for FLAG epitope insertions. (C and F) Expression and processing abilities of the different F variants. Total cell protein extraction from the various F mutants expressed in Vero cells was performed. Immunoblots were decorated with a polyclonal anti-HA (MeV F constructs) or polyclonal anti-F (CDV F constructs) antibody. (D) Quantitative fusion assay. Vero-SLAM cells (target cells) were infected with MVA-T7 (MOI of 1). In parallel, a population of Vero cells (effector cells) was transfected with the different F proteins, a plasmid encoding H, and a plasmid containing the luciferase reporter gene under the control of the T7 promoter. Twelve hours after transfection, effector cells were mixed with target cells and seeded into fresh plates. After 2.5 h at 37°C, fusion was indirectly quantified by using a commercial luciferase-measuring kit. For each experiment, the value obtained for the standard F/H combination was set to 100%. Means of data from three independent experiments in duplicate are shown. wt, wild type. (E and G) Syncytium formation assay. Shown are data for cell-cell fusion induction after the cotransfection of Vero-SLAM cells with plasmid DNA encoding various CDV F proteins and H. Representative fields of view were captured at 24 h posttransfection with a fluorescence confocal microscope (Fluoroview FV1000; Olympus).
Identification of conformation-sensitive anti-F monoclonal antibodies.
Having thus established the conformation-independent immunodetection of the F ectodomain (Fig. 2A), we next screened previously reported anti-CDV F and anti-MeV F MAbs (58–60) in search of specific antibodies that differentiate between distinct F conformations. For identification, CDV F was coexpressed with CDV H in CDV receptor-negative Vero cells (Fig. 2B), and MeV F was coexpressed with MeV H in MeV receptor-negative CHO cells (Fig. 2E). The CDV FFLAG and H variants used in this study originated from the CDV A75/17 strain, which poorly replicates in Vero cells. We therefore expected that F would maintain a prefusion conformation in this system.
Fig 2.

Identification of conformational anti-F monoclonal antibodies (MAbs). (A) Scheme of the morbillivirus F gene. Conserved regions among class I fusion proteins are shown. FP, fusion peptide; HRA and HRB, heptad repeat regions A and B, respectively; TM, transmembrane domain. The red box represents the position at the C-terminal part of F2 used for the FLAG epitope insertion. (B and E) Vero (for CDV) or CHO (for MeV) cells were cotransfected with F and H expression plasmids. For IF analysis, cells were stained with the different anti-F MAbs (FLAG, 4941, and 4068 for CDV and FLAG, 186CA, and 19GD6 for MeV) at 24 h posttransfection at 4°C. In some experiments, brief heat shocks (10 min at the indicated temperatures) were performed prior to IF. Alexa Fluor 488-conjugated secondary antibody was then added, and images were captured with a fluorescence confocal microscope (Fluoroview FV1000; Olympus). (C and F) Reactivity of the pair of conformational MAbs with F trimers. Cells were transfected and stained as described in above for panels B and E. Stained cells were then subjected to flow cytometry to record mean fluorescence intensities (MFI). In some experiments, brief heat shocks (10 min at the indicated temperatures) were performed prior to flow cytometry. Means of data from three independent experiments performed in duplicates are shown. (D and G) Virus neutralization assay. A total of 100 TCID50 units of virus was incubated with the indicated dilution of antibody for 1 h at 37°C. The virus-antibody mixture were then added to Vero cells, and the cells were incubated for 72 h at 37°C. The cells were assayed for luciferase activity for MeV or the number of infectious units for CDV.
To trigger F refolding and drive the trimers into a postfusion conformation, we subjected the transfected cells to a brief (10-min) heat shock of 40°C, 50°C, or 60°C. The anti-CDV or MeV F MAb reactivity was then determined by immunofluorescence analysis performed at 4°C. Of the different anti-CDV F MAbs tested, only one (anti-F MAb 4941) efficiently stained the cells in the absence of heat treatment (Fig. 2B). All other MAbs similarly showed poor reactivity in the absence of heat shock or after exposure to lower temperatures (37°C, 40°C, and 50°C), whereas after exposure to a temperature of 60°C, transfected cells were stained strongly by these MAbs (Fig. 2B). Thus, the reactivity of these MAbs with F shows a direct correlation with increasing heat shock temperatures, while MAb 4941 shows an inverse correlation (Fig. 2B). Likewise, a pair of conformation-sensitive anti-MeV F MAbs with inverse and direct correlations of reactivity to increasing heat shock temperatures was identified (186CA and 19GD6) (Fig. 2E). For the control, cells were stained with the anti-FLAG MAb M2, which recognizes a linear epitope and returned strong staining under all conditions. This underscores that F surface steady-state levels remained unchanged during the experiment.
In conclusion, these data suggest that anti-CDV F MAb 4941 and anti-MeV F MAbs 186CA and 186CB bind specifically to an epitope present in a prefusion-like F trimer, whereas all other MAbs may recognize epitopes present only in triggered F structures. Based on their high reactivity with prefusion and triggered F proteins, anti-CDV F MAbs 4941 and 4068 and anti-MeV F MAbs 186CA and 19GD6, respectively, were selected for further experiments and are referred to as “anti-Pre” and “anti-Trig” MAbs, accordingly.
Flow cytometry conducted after immunofluorescence staining confirmed the above-described findings for both pairs of MAbs and glycoproteins: at 37°C, each F protein was strongly recognized by the respective anti-Pre MAbs, whereas after heat shock, the F proteins became increasingly reactive with the virus-specific anti-Trig MAb (Fig. 2C and F).
To further characterize the bioactivity of these conformation-sensitive MAbs, virus neutralization assays were conducted. For MeV, the anti-Pre MAb efficiently inhibited viral-cell entry, while in contrast, the anti-Trig MAbs did not substantially neutralize the virus (Fig. 2D and G). As controls, an anti-H MAb (B5, kindly provided by Makato Takeda, National Institute of Infectious Disease, Japan) with documented neutralizing ability was included. Unexpectedly, none of the anti-CDV F antibodies neutralized CDV, whereas the commercially available anti-H MAb (1C42H11) strongly inhibited virus entry (Fig. 2D). The efficient neutralization activity of the MeV F anti-Pre MAb corroborates binding to an epitope present in prefusion F. Despite the exclusive reactivity of the CDV F anti-Pre antibody for an F conformational state assumed (i) in receptor-negative cells, (ii) at 37°C, and (iii) in the presence of H, we surprisingly found that this MAb did not neutralize virus infectivity. A low affinity or avidity of this antibody for the F trimer may, however, impede efficient neutralization. Alternatively, a distinct lipidic membrane curvature and/or density of glycoprotein complexes on the envelope of viral particles may prevent the proper binding of MAbs to F trimers.
Taken together, we have successfully identified pairs of anti-F MAbs that allow the differentiation of morbillivirus (MeV and CDV) F conformations. Our findings are consistent with the notion that the anti-Pre MAbs recognize prefusion conformations of the respective F trimers, while the anti-Trig MAbs bind to refolding intermediates or postfusion F.
Anti-Trig MAbs recognize F trimers triggered under physiological conditions.
To assess whether the conformation of F proteins achieved upon heat shock treatment is consistent with that of F trimers that is triggered by H upon receptor engagement, we developed a slightly different flow cytometry-based F-triggering assay. In the latter, F and H proteins were expressed in receptor-negative Vero cells, and at 1 day posttransfection, cells were (i) cocultured with receptor-positive Vero-SLAM cells, (ii) cocultured with receptor-negative Vero cells, or (iii) left untreated for 1 h at 4°C. Anti-Trig MAbs were added during the coculture (receptor-binding) step. The cells were then switched (or not) to 37°C for 1.5 h and returned to 4°C. Secondary antibodies were finally added at 4°C, and the cells were subjected to flow cytometry to record quantitative values. Because we noted a significantly smaller amount of total membrane-bound F proteins under conditions resulting in membrane fusion activity, the binding efficiencies of the anti-Trig MAbs are shown normalized to the total amount of F trimers exposed on the plasma membrane (monitored by anti-FLAG MAb).
The results presented in Fig. 3 indicate that only when F/H-expressing Vero cells were overlaid with Vero-SLAM cells followed by incubation at 37°C were substantially larger amounts of F trimers detected by the anti-Trig antibodies. In contrast, when no or receptor-negative cells were added to the system and switched to a higher temperature, anti-Trig antibodies exhibited reactivities very similar to those in F/H-expressing cells that did not receive heat shock treatment. This assay thus provided clear evidence that the conformational state of F trimers achieved through heat treatments is similar to the conformation of F proteins triggered by receptor-induced H conformational changes. Taken together, these and previously reported results are in perfect agreement with the notion that anti-Trig MAbs bind to physiologically relevant triggered F conformations.
Fig 3.
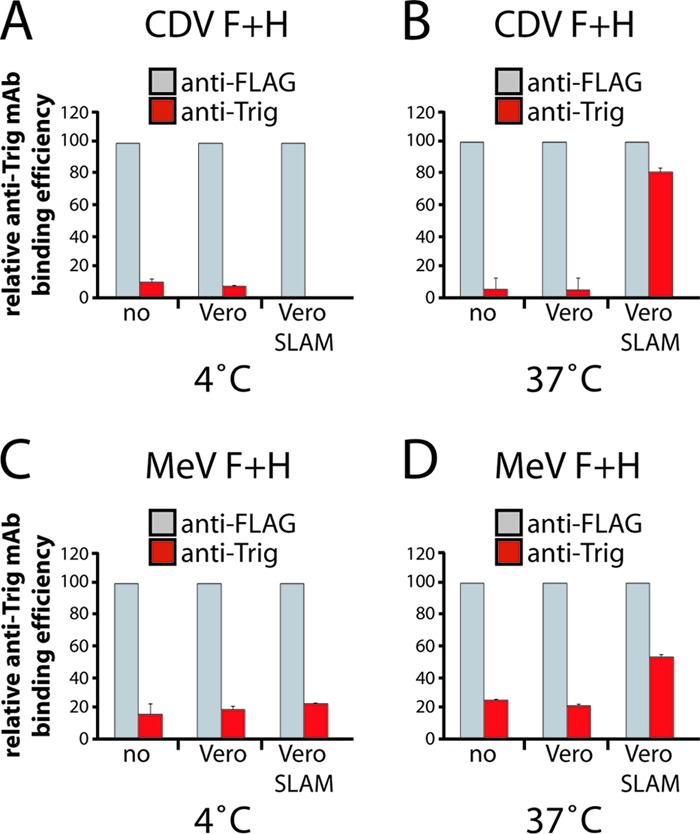
Conformation of heated F trimers correlates with triggered F proteins under physiological conditions. (A and B) Reactivity of the anti-Trig conformational MAbs with CDV F trimers. Vero cells were transfected with standard CDV F- and H-expressing plasmids. At 1 day posttransfection, cells were overlaid with receptor-negative Vero cells, with receptor-positive Vero-SLAM cells, or without cells for 1 h at 4°C together with the anti-Trig MAb. Both cell populations were then switched (or not) to 37°C for 1.5 h and were then returned to 4°C. Secondary antibodies were added at 4°C, and to record quantitative values, mean fluorescence intensities were monitored by flow cytometry. Means of data from three independent experiments performed in duplicates are shown. (C and D) Reactivity of the anti-Trig conformational MAbs with MeV F trimers. Experimental settings identical to those described above for panels A and B were used but with MeV F- and H-expressing Vero cells. In this series of experiments, to allow MeV H (Edmonston) to trigger fusion exclusively through interactions with SLAM, a CD46-binding-deficient mutant was used (70). Means of data from three independent experiments performed in duplicates are shown.
High reactivity of destabilized F mutants with MAbs binding to triggered F proteins.
F mutants harboring conformation-destabilizing mutations are anticipated to undergo heat shock-induced refolding at temperatures lower than those observed for standard F. A previously described destabilized CDV F mutant (L372G) (52) was assessed accordingly using our newly identified pair of conformation-sensitive MAbs. In parallel, two residues in MeV FedmFLAG were replaced with alanine (L457A and V459A), with the aim to destabilize the prefusion state. These residues were selected due to their location at the interface between the F-stalk and head domains, which was previously shown to be involved in maintaining the conformational stability of prefusion MeV F (61).
Both F mutants were coexpressed with their homotypic H protein in receptor-negative cells (Vero cells for CDV and CHO cells for MeV), followed by immunofluorescence analysis with the appropriate pair of MAbs. In the absence of any additional temperature stimulation, both the CDV F-L372G and MeV F-L457A/V459A mutants were strongly reactive with the anti-Trig MAbs (Fig. 4A and B). As controls, unmodified CDV and MeV F proteins were included, which exhibited the expected conformation-sensitive MAb staining profile (Fig. 4A and B). A quantitative assessment of MAb-binding activity by flow cytometry confirmed the microscopy results (Fig. 4C and D). In summary, destabilized CDV and MeV F mutants are recognized by anti-Trig MAbs that usually react with F only after heat stimulation in a receptor-negative host cell system. These results underscore the conformational specificity of the different MAbs and highlight a strong inherent conformational stability of wild-type morbillivirus F proteins under physiological conditions.
Fig 4.

Prefusion conformation-destabilized F variants are reactive with MAbs recognizing triggered F structures. (A and B) Localization of the substituted residues within the structural homology model of the prefusion CDV F trimer (left and top). Each F monomer is color coded for clarification. IF was performed as described in the legend of Fig. 2B and E (right bottom). In some experiments, brief heat shocks (10 min at the indicated temperatures) were performed prior to IF. Monoclonal antibodies (MAbs) 4941 (CDV) and 186CA (MeV) were named “anti-Pre” MAbs, and MAbs 4068 (CDV) and 19GD6 (MeV) were named “anti-Trig” antibodies, due to their specificity in the binding of either the prefusion or triggered F structures, respectively.
Self-stabilization of the morbillivirus prefusion F conformation.
We next determined the conformation of morbillivirus F expressed in the absence of the homotypic H protein. Plasmid-encoded CDV and MeV F proteins were expressed in Vero cells, followed by qualitative (immunofluorescence staining) and quantitative (flow cytometry) conformational analysis using the two pairs of conformation-sensitive MAbs. Closely resembling the binding profile observed in the presence of H, both morbillivirus F proteins remained reactive with the anti-Pre MAbs and showed only little reactivity with the anti-Trig antibodies (Fig. 5A and C). Again, destabilized CDV and MeV F mutants were substantially more reactive with the anti-Trig MAbs when expressed in the absence of H. We noticed, however, that the CDV F-L372G mutant was more resistant to spontaneous refolding than the destabilized MeV F variant, since part of the membrane-bound F population was still partially recognized by the anti-Pre MAb (Fig. 5A and C). As expected, heat shock (60°C for 10 min) prior to IF and flow cytometry analyses significantly triggered the refolding of the unmodified F proteins. CDV and MeV F proteins were then efficiently bound by anti-Trig MAbs (Fig. 5B and D). These results suggest that the prefusion state of the CDV F protein is thermodynamically more stable than that of MeV F, since a substantial portion of the total CDV F population expressed at the cell surface was recognized by the anti-Pre MAb even after heat shock treatment. As expected, destabilized F mutants (CDV F-L372G and MeV F-V457A/L459A) were fully triggered by heat exposure and then reacted exclusively with the anti-Trig MAbs (Fig. 5B and D). Interestingly, we noted that after heat shock, CDV F trimers were more reactive to the anti-Trig MAb when expressed with H than when expressed alone (CDV FFLAG at 60°C) (compare Fig. 4C and 5B). Although clearly nonproductive under physiological conditions, these data are in agreement with the notion that wild-type H proteins may improperly bind to an unknown receptor expressed in Vero cells. It is therefore conceivable that under nonphysiological conditions (after heat shock), the putative suboptimal H-receptor interaction leads to slight but appreciable H-mediated F triggering. In sum, we hypothesize that the treatment of H/F-expressing Vero cells at 60°C results in F triggering through heat- and receptor-induced H conformational changes.
Fig 5.
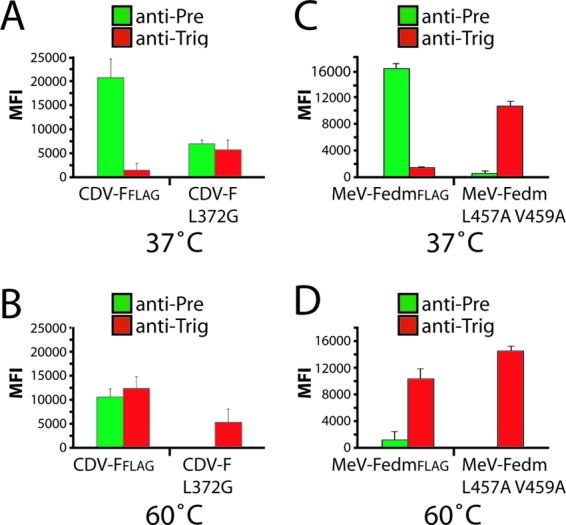
Self-stabilization of morbillivirus prefusion F trimers. Shown are the reactivities of the pair of conformational MAbs with F trimers. Vero cells were transfected with standard CDV or MeV F-expressing plasmids. Cells were then stained, as described in the legend of Fig. 2B and E, with the pairs of conformation-sensitive anti-F MAbs. To record quantitative values, mean fluorescence intensities were monitored by flow cytometry. In some experiments, brief heat shocks (10 min at 60°C) were performed prior to IF. Means of data from three independent experiments performed in duplicates are shown.
Taken together, these data demonstrate that morbillivirus F proteins do not require the presence of the H protein to maintain a metastable prefusion conformation.
A prefusion conformation of destabilized F mutants is conserved by a fusion-inhibitory compound.
A previously developed small-molecule antiviral compound (AS-48) potently blocks MeV glycoprotein-mediated virus-to-cell and cell-to-cell fusion activity (62, 63). Moreover, this inhibitor was demonstrated previously to efficiently restore the intracellular transport competence of hyperfusogenic, structurally destabilized MeV F mutants in a fusion-competent conformation (64). We have furthermore shown that the inhibitory activity of a close chemical analog of AS-48 with comparable bioactivity, 3g, extends to CDV (65). Using the destabilized CDV and MeV F variants and the conformation-sensitive anti-F MAbs, we next directly examined the effect of the compound on the F-trimer conformation. When the F mutants (CDV F-L372G and MeV F-L457A/V459A) were expressed in the presence of 3g, we first found that, consistent with the original observations by Doyle and colleagues (64), the cell surface transport competence of the CDV F-L372G mutant was fully restored in the presence of the compound (Fig. 6A). In contrast, the MeV F-L457A/V459A variant remained improperly surface expressed even in the presence of the antiviral compound, which suggested a strong destabilization phenotype of this specific mutant (Fig. 6D). Strikingly, however, IF and fluorescence-activated cell sorter (FACS) analyses indicated that 3g stabilized a prefusion conformation of both the CDV F-L372G (completely) and MeV F-L457A/V459A (partially) mutants, since both proteins regained reactivity with the anti-Pre MAbs in the presence of the compound (Fig. 6B and E). In the context of the unmodified CDV F protein, 3g likewise exerted a stabilizing effect on prefusion F, since even the small population reactive with the anti-Trig MAb disappeared under these conditions (Fig. 6C and F). This effect was not as pronounced for unmodified MeV FedmFLAG, possibly reflecting that different epitopes are recognized by the individual anti-CDV and MeV MAbs, which may be differentially affected by the presence of the compound. In summary, consistent with previously reported speculations, these data now clearly indicate that this fusion-inhibitory compound class binds and stabilizes a transport-competent, metastable conformation of the F trimer, very likely by increasing the energy barrier required for F triggering.
Fig 6.

The antiviral compound 3g stabilizes spontaneously refolding F mutants into the metastable, prefusion state. (A and D) Vero cells were transfected with standard F- or variant-expressing plasmids in the presence or absence of 3g. IF was performed, as described in the legend of Fig. 2B and E, with the FLAG MAb. Cell surface expression (CSE) was then recorded by flow cytometry analysis. (B, C, E and F) Vero cells were transfected with standard F- or variant-expressing plasmids in the presence or absence of 3g. IF was performed, as described in the legend of Fig. 2B and E, with the pairs of conformation-sensitive anti-F MAbs. To determine the MAb-binding abilities with the indicated F trimers, mean fluorescence intensities were recorded by flow cytometry. (G) Scheme of engineered soluble F proteins. Conserved regions among class I fusion proteins are shown. FP, fusion peptide; HRA and HRB, heptad repeat regions A and B, respectively; TM, transmembrane domain; green box, trimerizing peptide (GCNt); red box, FLAG epitope; blue box, 6×His epitope. (H) Immunoprecipitation (IP) of soluble F variants with the conformation-insensitive (FLAG) or -sensitive (anti-Pre and anti-Trig) MAbs. Immunoblots were decorated with a polyclonal anti-F antibody. In some experiments, brief heat shocks (10 min at 60°C) were performed prior to IP. (I) Experiments identical to those described above for panel H were performed but in the presence and absence of 3g.
Compound 3g stabilizes soluble F protein in a prefusion state.
We next asked the question of whether the 3g fusion blocker is also capable of stabilizing soluble F proteins in a prefusion conformation and consequently generated soluble F (sF) expression constructs lacking the transmembrane domain and cytosolic tail (Fig. 6G). Equivalent paramyxovirus sF proteins were previously shown to fold into a highly stable postfusion-like conformation featuring the 6-HB fusion core (19–21), presumably reflecting that the transmembrane and cytosolic domains contribute to stabilize a metastable prefusion form of the F trimer. This effect was successfully mimicked for soluble PIV5 F by fusing a GCNt trimerization domain to the HRB domain, which allowed the crystallization of the protein in a prefusion state (18). We generated an equivalently stabilized CDV sF protein by fusing the GCNt motif to the CDV sF HRB domain (sF-GCNt) (Fig. 6G). For immunoprecipitation and purification, both soluble F proteins contained tandem FLAG and hexahistidine (6×His) tags at their C termini (Fig. 6G).
We first probed the conformation assumed by these sF variants in the absence of compound. Concentrated supernatants of transfected 293T cells were subjected to immunoprecipitations (IPs) with the following MAbs: conformation-insensitive anti-FLAG and conformation-sensitive anti-Pre and anti-Trig F MAbs. Immunoprecipitates were gel fractionated, and F antigenic material was detected by using a polyclonal anti-F antiserum. The results shown in Fig. 6H (top) indicate that both sF forms are efficiently immunoprecipitated by the anti-FLAG MAb. However, only the anti-Trig MAb efficiently precipitated sF, while the anti-Pre antibody reacted with this F construct very weakly. In contrast, the precipitation of sF-GCNt with these MAbs returned a reversed reactivity profile: strong reactivity with anti-Pre and limited binding of the anti-Trig antibody. Both sF variants were next subjected to heat shock prior to IP. As expected, this resulted in predominant F precipitation by the anti-Trig MAb (Fig. 6H, bottom). Equivalent to data from previous studies with PIV5 F, these findings confirm that CDV sF-GCNt is expressed in a prefusion conformation, but refolding can be triggered through heat stress. In contrast, sF spontaneously assumes a postfusion-like conformation at a physiological temperature. Thus, we successfully engineered soluble CDV F variants that either are stabilized in a prefusion form (sF-GCNt) or fold spontaneously into a postfusion-like conformation (sF). When we repeated these experiments after sF expression in the presence of 3g, we observed a nearly exclusive reactivity of sF-GCNt with the anti-Pre MAb (Fig. 6I, bottom right). Remarkably, the conformation of sF was also stabilized by 3g, since we again detected a predominant precipitation of this construct by the anti-Pre antibody (Fig. 6I, top right). These results indicate that sF, which spontaneously assumes a postfusion conformation when expressed in the absence of compound, is efficiently stabilized in a prefusion form by the antiviral compound.
Significant external energy is required to initiate morbillivirus F refolding.
To better appreciate the energy barrier controlling morbillivirus F refolding, we next applied the MAb-based assay to map the temperature at which CDV and MeV F refolding occurs. For these experiments, heat shock was performed prior to the addition of the MAbs at different temperatures (55°C, 60°C, and 65°C for CDV F and 52°C, 54°C, 58°C, and 60°C for MeV Fedm). This revealed a higher intrinsic stability of the CDV F isolate than of MeV Fedm, since the former required exposure to 60°C before binding of the anti-Trig antibody was observed, while MeV Fedm had already gained reactivity with its specific anti-Trig MAb at 54°C (Fig. 7A and C). Consistent with the antiviral compound elevating the prefusion F activation energy barrier, a significant increase in the CDV F reactivity with the anti-Trig MAb was noted only at 65°C (Fig. 7B). Likewise, the refolding of MeV Fedm was initiated at 58°C instead of at 54°C in the presence of 3g (Fig. 7C). In addition, and confirming these observations, it appeared that in the absence of the antiviral compound, the anti-Pre MAbs started to exhibit a clear loss of F-trimer reactivity at lower temperatures than in the presence of the 3g compound (Fig. 7A and B [for CDV F] and C and D [for MeV F]).
Fig 7.

Substantial external energy is required to trigger morbillivirus prefusion F trimers. (A to D) Vero cells were transfected with standard F- or variant-expressing plasmid DNA in the presence or absence of 3g. IF was performed, as described in the legend of Fig. 2B and E, with the pairs of conformation-sensitive anti-F MAbs. To determine the MAb-binding ability with the indicated F trimers, mean fluorescence intensities were recorded by flow cytometry. In some experiments, brief heat shocks (10 min at the indicated temperatures) were performed prior to IF. (E and F) Cell-cell fusion induction after cotransfection of Vero-SLAM (for CDV) or Vero (for MeV) cells with plasmid DNA encoding CDV or MeV F and H proteins in the presence of increasing concentrations of the fusion inhibitor 3g. At 24 h posttransfection, cells were incubated at 42°C for 5 h (for CDV) or 8 h (for MeV) in the presence of 3g. Representative fields of view were captured at 24 h posttransfection with a fluorescence confocal microscope (Fluoroview FV1000; Olympus).
By combining the fusion inhibitor 3g with H/F coexpression in a receptor-positive host system, we observed that H-fusion promotion and external thermal energy have an additive effect on F triggering. When the cell-to-cell fusion of Vero-SLAM cells cotransfected with CDV H and F in the presence of increasing 3g concentrations at different incubation temperatures was monitored, we found that the fusion activity was efficiently restored after incubation at 42°C, instead of at 37°C, for 5 h (Fig. 7E). While longer incubation periods at elevated temperatures were required, an equivalent effect was observed with 3g and the MeV F/H glycoprotein system (Fig. 7F).
Thus, the fusion support provided by receptor-bound H and external thermal energy have an additive effect on the triggering of the refolding of 3g-stabilized prefusion F. Taken together, these results confirm that considerable external energy is required to initiate morbillivirus F refolding and suggest that H functions by lowering the activation energy barrier that keeps F in a metastable prefusion conformation.
DISCUSSION
To enter target cells and initiate disease, paramyxoviruses contain two glycoproteins that tightly cooperate to fuse the viral envelope with host cell membranes (4). It has been hypothesized that upon receptor binding, the paramyxovirus attachment protein undergoes structural rearrangements that trigger the fusion protein to refold from a metastable, prefusion state into a highly stable postfusion conformation, which ultimately leads to fusion pore formation (4, 23). However, how precisely the H protein translates receptor binding to F triggering remains largely unexplored.
Two fundamental models emerged from previous studies of paramyxovirus entry (34, 36–38). In the association or “provocateur” model, it has been speculated that sialic acid-binding HN proteins associate with F upon receptor binding, destabilizing the trimer for the triggering of refolding and membrane fusion. In the dissociation or “clamp” model, it has been hypothesized that paramyxoviruses that bind proteinaceous receptors express an attachment protein that serves as a molecular scaffold preventing premature prefusion F structural rearrangements. Upon an H-protein interaction with the receptor, F spontaneously refolds as a result of the dissociation of preassembled fusion complexes.
To address the question of whether morbillivirus F proteins require the attachment protein tetramer to maintain a prefusion fold, we examined the conformations of the MeV and CDV F proteins in the presence and absence of the homotypic attachment proteins. Toward this goal, we screened panels of anti-F MAbs (58–60) in an attempt to identify individual antibodies that recognize conformational epitopes. Although further work is required to characterize the molecular nature of the different epitopes, we found in either case discrete MAbs that specifically interacted with nontriggered prefusion F (anti-Pre MAbs) or triggered F (anti-Trig MAbs). By combining these pairs of conformation-sensitive MAbs with biochemical and functional assays, we obtained several lines of evidence demonstrating that morbillivirus H is not necessary to stabilize prefusion F, indicating that the active dissociation of F from H is required to trigger F refolding. First, we demonstrate that both the MeV and CDV F proteins are strongly reactive with anti-F MAbs specifically recognizing a prefusion state when expressed in the absence of their homotypic attachment proteins. This provides the first clear evidence that the prefusion conformation of the morbillivirus F trimer is intrinsically controlled by a sufficiently high activation energy barrier. Thus, an intracellular association of H with F is not required to stabilize F in the prefusion state. Rather, our data indicate that morbillivirus fusion proteins are efficiently self-stabilized.
Second, we demonstrate that F triggering can be efficiently induced by external energy. This finding is consistent with results obtained with PIV5 F (35, 71) and also indicates that morbillivirus H proteins must destabilize prefusion F for triggering the refolding cascade. Interestingly, in the case of PIV5 F, elevating the temperature coincided with an enhanced membrane fusion activity of F in the absence of HN (66). Although we found that both MeV and CDV F trimers efficiently refold when heat treated, fusion pore formation was not observed under these conditions or during incubation at elevated temperatures. We hypothesize that a sustained engagement of the H protein with its membrane-embedded surface receptor promotes a “fusion-permissive” environment required for morbillivirus F-mediated fusion (30). We note that PIV5 F is one of the rare fusion proteins among the Paramyxovirinae subfamily that can naturally achieve slight but significant fusion activity in the absence of the attachment protein. Thus, we cannot exclude that PIV5 F evolved to directly bind a host cell surface receptor and that the MeV and CDV F proteins did not, thereby explaining the inability of morbillivirus F trimers to induce membrane fusion even when triggered by heat. However, in favor of the model that a continuous engagement of the attachment protein with its receptor is needed to achieve an effective F-dependent induction of membrane fusion, Porotto and colleagues recently obtained very similar conclusions using different paramyxovirus glycoprotein complexes. In that study, although direct biochemical evidences were not provided, it was further proposed that paramyxovirus F and attachment proteins may exhibit productive interactions even beyond the initial F activation step (46).
Third, consistent with previously reported results (64), our data demonstrate that a small fusion inhibitor (3g) (63, 65, 67) efficiently stabilizes the prefusion conformation of intrinsically destabilized, spontaneously refolding, morbillivirus F variants. In agreement with this mechanism of action, more external energy is required for both the unmodified CDV and MeV F proteins to switch conformations in the presence of 3g, since refolding occurred only at higher heat shock temperatures. Remarkably, a transmembrane- and cytosolic tail-deleted F variant, which spontaneously assumes a triggered conformational state, was substantially stabilized in the prefusion conformation by the antiviral compound. Taken together, our findings clearly demonstrate that the 3g class of morbillivirus fusion inhibitors enhances the energy barrier required for F-trimer activation, in turn counteracting H-mediated F triggering and the ensuing membrane fusion activity.
We also noted that the temperature-induced conformational changes of F proteins isolated from a highly neurovirulent CDV strain required more external energy than the MeV Fedm trimer (60°C versus 54°C). This may reflect an inherently higher intrinsic stability of prefusion CDV F than MeV F or may highlight a gradual destabilization of MeV Fedm during tissue culture adaptation. In addition, data presented in a previous study of an attenuated PIV5 strain demonstrated that this F protein switched conformations at around 50°C (35), thus demonstrating an even lower intrinsic energy. These findings underscore that the energetic costs to initiate paramyxovirus F refolding differ wildly, at least among laboratory-adapted viruses. It is tempting to speculate that the stability of the prefusion F trimer may be more tightly controlled in clinical isolates, since it will likely directly affect viral pathogenesis by modulating the efficiency of virus-to-cell and cell-to-cell fusion in vivo.
Finally, we show that increasing the amount of external energy added to the system restores F/H-mediated membrane fusion in the presence of the fusion inhibitor compound. These data are consistent with 3g stabilizing a prefusion F conformation by raising the F-refolding activation energy barrier. The reactivation of fusion activity at higher temperatures under these conditions likely results from temperature-mediated increased molecular energy present in the prefusion F trimer, combined with a lowering of the activation energy barrier through H.
Taken together, in this study, we provide a large body of evidence supporting the conclusion that both the MeV and CDV H proteins trigger F refolding by lowering the activation energy barrier that controls the metastable, prefusion conformation of the F trimer. This clearly illuminates a common mechanism of morbillivirus membrane fusion triggering, a concept that may even extend to other members of the Paramyxovirinae subfamily, since very similar conclusions were obtained with PIV5 HN-tetramer-mediated F triggering (35).
How can the attachment protein affect the energy barrier for triggering F? The hypothesis of conformational changes within the attachment protein induced by receptor interactions is, to date, the most relevant hypothesis (4, 27, 28). With regard to the nature of these structural modifications, our recently reported results (29) combined with the present findings prompt us to speculate two hypotheses for morbillivirus H-mediated membrane fusion triggering. In the first model, upon receptor engagement by H present in preassembled H and F fusion complexes, the putative metastable tetrameric supercoiled lower section of the attachment protein stalk may realign with the upper, membrane-distal, straight 4-HB section. Subsequent oligomeric rearrangements of the immediate upper region may result in the activation of the F trimer, thereby increasing the intrinsic thermal energy level of F. This will result in a lowering of the relative activation energy barrier and, ultimately, will drive F trimers to irreversible structural rearrangements. Alternatively, H-stalk conformational changes may result directly in the release of the associated F trimer, and the ensuing change in the microenvironment at the former protein-protein interface may transiently destabilize the prefusion trimer, resulting in a lowering of the relative energy barrier and F refolding.
There is now compelling structural and functional evidence indicating that paramyxoviruses adhere to their target cells differently (6, 25, 68), which infers discrete mechanisms of receptor-induced conformational changes in the attachment proteins leading to F triggering. While our model does not exclude any different structural rearrangements of the attachment proteins' head domain resulting in stalk activation (69), we suggest that the conformational changes occurring in the central section of the receptor-binding protein stalk domain are the ultimate active signals required to trigger the paramyxovirus fusion protein.
In summary, we postulate that the lowering of the prefusion F activation energy barrier by the action of the attachment protein stalk domain emerges as a common denominator in the initiation of paramyxovirus infection. This principle would be independent of whether attachment and F proteins preassemble or not prior to receptor engagement.
ACKNOWLEDGMENTS
We thank M. Ehnlund for generously providing MAbs directed against the MeV F protein and Makoto Takeda for the anti-MeV H MAb B5.
This work was supported by the Swiss National Science Foundation (reference no. 310030_132887 to P.P.) and, in part, by U.S. Public Health Service grant AI083402 (to R.K.P.).
Footnotes
Published ahead of print 17 October 2012
REFERENCES
- 1. Chen SY, Anderson S, Kutty PK, Lugo F, McDonald M, Rota PA, Ortega-Sanchez IR, Komatsu K, Armstrong GL, Sunenshine R, Seward JF. 2011. Health care-associated measles outbreak in the United States after an importation: challenges and economic impact. J. Infect. Dis. 203:1517–1525 [DOI] [PubMed] [Google Scholar]
- 2. Chua KB, Bellini WJ, Rota PA, Harcourt BH, Tamin A, Lam SK, Ksiazek TG, Rollin PE, Zaki SR, Shieh W, Goldsmith CS, Gubler DJ, Roehrig JT, Eaton B, Gould AR, Olson J, Field H, Daniels P, Ling AE, Peters CJ, Anderson LJ, Mahy BW. 2000. Nipah virus: a recently emergent deadly paramyxovirus. Science 288:1432–1435 [DOI] [PubMed] [Google Scholar]
- 3. Murray K, Selleck P, Hooper P, Hyatt A, Gould A, Gleeson L, Westbury H, Hiley L, Selvey L, Rodwell B, Ketterer P. 1995. A morbillivirus that caused fatal disease in horses and humans. Science 268:94–97 [DOI] [PubMed] [Google Scholar]
- 4. Lamb RA, Parks GD. 2007. Paramyxoviridae: the viruses and their replication, p 1449–1496 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 4th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 5. Brindley MA, Plemper RK. 2010. Blue native PAGE and biomolecular complementation reveal a tetrameric or higher-order oligomer organization of the physiological measles virus attachment protein H. J. Virol. 84:12174–12184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hashiguchi T, Ose T, Kubota M, Maita N, Kamishikiryo J, Maenaka K, Yanagi Y. 2011. Structure of the measles virus hemagglutinin bound to its cellular receptor SLAM. Nat. Struct. Mol. Biol. 18:135–141 [DOI] [PubMed] [Google Scholar]
- 7. Bonaparte MI, Dimitrov AS, Bossart KN, Crameri G, Mungall BA, Bishop KA, Choudhry V, Dimitrov DS, Wang LF, Eaton BT, Broder CC. 2005. Ephrin-B2 ligand is a functional receptor for Hendra virus and Nipah virus. Proc. Natl. Acad. Sci. U. S. A. 102:10652–10657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dorig RE, Marcil A, Chopra A, Richardson CD. 1993. The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell 75:295–305 [DOI] [PubMed] [Google Scholar]
- 9. Negrete OA, Levroney EL, Aguilar HC, Bertolotti-Ciarlet A, Nazarian R, Tajyar S, Lee B. 2005. EphrinB2 is the entry receptor for Nipah virus, an emergent deadly paramyxovirus. Nature 436:401–405 [DOI] [PubMed] [Google Scholar]
- 10. Negrete OA, Wolf MC, Aguilar HC, Enterlein S, Wang W, Muhlberger E, Su SV, Bertolotti-Ciarlet A, Flick R, Lee B. 2006. Two key residues in ephrinB3 are critical for its use as an alternative receptor for Nipah virus. PLoS Pathog. 2:e7 doi:10.1371/journal.ppat.0020007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tatsuo H, Ono N, Tanaka K, Yanagi Y. 2000. SLAM (CDw150) is a cellular receptor for measles virus. Nature 406:893–897 [DOI] [PubMed] [Google Scholar]
- 12. Tatsuo H, Ono N, Yanagi Y. 2001. Morbilliviruses use signaling lymphocyte activation molecules (CD150) as cellular receptors. J. Virol. 75:5842–5850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Villar E, Barroso IM. 2006. Role of sialic acid-containing molecules in paramyxovirus entry into the host cell: a minireview. Glycoconj. J. 23:5–17 [DOI] [PubMed] [Google Scholar]
- 14. Colf LA, Juo ZS, Garcia KC. 2007. Structure of the measles virus hemagglutinin. Nat. Struct. Mol. Biol. 14:1227–1228 [DOI] [PubMed] [Google Scholar]
- 15. Hashiguchi T, Kajikawa M, Maita N, Takeda M, Kuroki K, Sasaki K, Kohda D, Yanagi Y, Maenaka K. 2007. Crystal structure of measles virus hemagglutinin provides insight into effective vaccines. Proc. Natl. Acad. Sci. U. S. A. 104:19535–19540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Santiago C, Celma ML, Stehle T, Casasnovas JM. 2010. Structure of the measles virus hemagglutinin bound to the CD46 receptor. Nat. Struct. Mol. Biol. 17:124–129 [DOI] [PubMed] [Google Scholar]
- 17. Smith EC, Popa A, Chang A, Masante C, Dutch RE. 2009. Viral entry mechanisms: the increasing diversity of paramyxovirus entry. FEBS J. 276:7217–7227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McLellan JS, Yang Y, Graham BS, Kwong PD. 2011. Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J. Virol. 85:7788–7796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Swanson K, Wen X, Leser GP, Paterson RG, Lamb RA, Jardetzky TS. 2010. Structure of the Newcastle disease virus F protein in the post-fusion conformation. Virology 402:372–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yin HS, Paterson RG, Wen X, Lamb RA, Jardetzky TS. 2005. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. U. S. A. 102:9288–9293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Crennell S, Takimoto T, Portner A, Taylor G. 2000. Crystal structure of the multifunctional paramyxovirus hemagglutinin-neuraminidase. Nat. Struct. Biol. 7:1068–1074 [DOI] [PubMed] [Google Scholar]
- 23. Lamb RA. 1993. Paramyxovirus fusion: a hypothesis for changes. Virology 197:1–11 [DOI] [PubMed] [Google Scholar]
- 24. Navaratnarajah CK, Oezguen N, Rupp L, Kay L, Leonard VH, Braun W, Cattaneo R. 2011. The heads of the measles virus attachment protein move to transmit the fusion-triggering signal. Nat. Struct. Mol. Biol. 18:128–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu K, Rajashankar KR, Chan YP, Himanen JP, Broder CC, Nikolov DB. 2008. Host cell recognition by the henipaviruses: crystal structures of the Nipah G attachment glycoprotein and its complex with ephrin-B3. Proc. Natl. Acad. Sci. U. S. A. 105:9953–9958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zaitsev V, von Itzstein M, Groves D, Kiefel M, Takimoto T, Portner A, Taylor G. 2004. Second sialic acid binding site in Newcastle disease virus hemagglutinin-neuraminidase: implications for fusion. J. Virol. 78:3733–3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aguilar HC, Ataman ZA, Aspericueta V, Fang AQ, Stroud M, Negrete OA, Kammerer RA, Lee B. 2009. A novel receptor-induced activation site in the Nipah virus attachment glycoprotein (G) involved in triggering the fusion glycoprotein (F). J. Biol. Chem. 284:1628–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bishop KA, Hickey AC, Khetawat D, Patch JR, Bossart KN, Zhu Z, Wang LF, Dimitrov DS, Broder CC. 2008. Residues in the stalk domain of the Hendra virus G glycoprotein modulate conformational changes associated with receptor binding. J. Virol. 82:11398–11409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ader N, Brindley MA, Avila M, Origgi FC, Langedijk JP, Orvell C, Vandevelde M, Zurbriggen A, Plemper RK, Plattet P. 2012. Structural rearrangements of the central region of the morbillivirus attachment protein stalk domain trigger F protein refolding for membrane fusion. J. Biol. Chem. 287:16324–16334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brindley M, Takeda M, Plattet P, Plemper RK. Proc. Natl. Acad. Sci. U.S.A., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Plemper RK, Hammond AL, Cattaneo R. 2001. Measles virus envelope glycoproteins hetero-oligomerize in the endoplasmic reticulum. J. Biol. Chem. 276:44239–44246 [DOI] [PubMed] [Google Scholar]
- 32. Li J, Quinlan E, Mirza A, Iorio RM. 2004. Mutated form of the Newcastle disease virus hemagglutinin-neuraminidase interacts with the homologous fusion protein despite deficiencies in both receptor recognition and fusion promotion. J. Virol. 78:5299–5310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Paterson RG, Johnson ML, Lamb RA. 1997. Paramyxovirus fusion (F) protein and hemagglutinin-neuraminidase (HN) protein interactions: intracellular retention of F and HN does not affect transport of the homotypic HN or F protein. Virology 237:1–9 [DOI] [PubMed] [Google Scholar]
- 34. Chang A, Dutch RE. 2012. Paramyxovirus fusion and entry: multiple paths to a common end. Viruses 4:613–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Connolly SA, Leser GP, Jardetzky TS, Lamb RA. 2009. Bimolecular complementation of paramyxovirus fusion and hemagglutinin-neuraminidase proteins enhances fusion: implications for the mechanism of fusion triggering. J. Virol. 83:10857–10868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Iorio RM, Mahon PJ. 2008. Paramyxoviruses: different receptors—different mechanisms of fusion. Trends Microbiol. 16:135–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee B, Ataman ZA. 2011. Modes of paramyxovirus fusion: a Henipavirus perspective. Trends Microbiol. 19:389–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Plemper RK, Brindley MA, Iorio RM. 2011. Structural and mechanistic studies of measles virus illuminate paramyxovirus entry. PLoS Pathog. 7:e1002058 doi:10.1371/journal.ppat.1002058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Corey EA, Mirza AM, Levandowsky E, Iorio RM. 2003. Fusion deficiency induced by mutations at the dimer interface in the Newcastle disease virus hemagglutinin-neuraminidase is due to a temperature-dependent defect in receptor binding. J. Virol. 77:6913–6922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Melanson VR, Iorio RM. 2004. Amino acid substitutions in the F-specific domain in the stalk of the Newcastle disease virus HN protein modulate fusion and interfere with its interaction with the F protein. J. Virol. 78:13053–13061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aguilar HC, Matreyek KA, Choi DY, Filone CM, Young S, Lee B. 2007. Polybasic KKR motif in the cytoplasmic tail of Nipah virus fusion protein modulates membrane fusion by inside-out signaling. J. Virol. 81:4520–4532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aguilar HC, Matreyek KA, Filone CM, Hashimi ST, Levroney EL, Negrete OA, Bertolotti-Ciarlet A, Choi DY, McHardy I, Fulcher JA, Su SV, Wolf MC, Kohatsu L, Baum LG, Lee B. 2006. N-glycans on Nipah virus fusion protein protect against neutralization but reduce membrane fusion and viral entry. J. Virol. 80:4878–4889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bishop KA, Stantchev TS, Hickey AC, Khetawat D, Bossart KN, Krasnoperov V, Gill P, Feng YR, Wang L, Eaton BT, Wang LF, Broder CC. 2007. Identification of Hendra virus G glycoprotein residues that are critical for receptor binding. J. Virol. 81:5893–5901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Corey EA, Iorio RM. 2007. Mutations in the stalk of the measles virus hemagglutinin protein decrease fusion but do not interfere with virus-specific interaction with the homologous fusion protein. J. Virol. 81:9900–9910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Plemper RK, Hammond AL, Gerlier D, Fielding AK, Cattaneo R. 2002. Strength of envelope protein interaction modulates cytopathicity of measles virus. J. Virol. 76:5051–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Porotto M, DeVito I, Palmer SG, Jurgens EM, Yee JL, Yokoyama CC, Pessi A, Moscona A. 2011. Spring-loaded model revisited: paramyxovirus fusion requires engagement of a receptor binding protein beyond initial triggering of the fusion protein. J. Virol. 85:12867–12880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Porotto M, Palmer SG, Palermo LM, Moscona A. 2012. Mechanism of fusion triggering by human parainfluenza virus type III: communication between viral glycoproteins during entry. J. Biol. Chem. 287:778–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rothlisberger A, Wiener D, Schweizer M, Peterhans E, Zurbriggen A, Plattet P. 2010. Two domains of the V protein of virulent canine distemper virus selectively inhibit STAT1 and STAT2 nuclear import. J. Virol. 84:6328–6343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wiener D, Vandevelde M, Zurbriggen A, Plattet P. 2010. Investigation of a unique short open reading frame within the 3′ untranslated region of the canine distemper virus matrix messenger RNA. Virus Res. 153:234–243 [DOI] [PubMed] [Google Scholar]
- 50. Wyss-Fluehmann G, Zurbriggen A, Vandevelde M, Plattet P. 2010. Canine distemper virus persistence in demyelinating encephalitis by swift intracellular cell-to-cell spread in astrocytes is controlled by the viral attachment protein. Acta Neuropathol. 119:617–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zipperle L, Langedijk JP, Orvell C, Vandevelde M, Zurbriggen A, Plattet P. 2010. Identification of key residues in virulent canine distemper virus hemagglutinin that control CD150/SLAM-binding activity. J. Virol. 84:9618–9624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Plattet P, Langedijk JP, Zipperle L, Vandevelde M, Orvell C, Zurbriggen A. 2009. Conserved leucine residue in the head region of morbillivirus fusion protein regulates the large conformational change during fusion activity. Biochemistry 48:9112–9121 [DOI] [PubMed] [Google Scholar]
- 53. Nussbaum O, Broder CC, Berger EA. 1994. Fusogenic mechanisms of enveloped-virus glycoproteins analyzed by a novel recombinant vaccinia virus-based assay quantitating cell fusion-dependent reporter gene activation. J. Virol. 68:5411–5422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Plattet P, Cherpillod P, Wiener D, Zipperle L, Vandevelde M, Wittek R, Zurbriggen A. 2007. Signal peptide and helical bundle domains of virulent canine distemper virus fusion protein restrict fusogenicity. J. Virol. 81:11413–11425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sutter G, Ohlmann M, Erfle V. 1995. Non-replicating vaccinia vector efficiently expresses bacteriophage T7 RNA polymerase. FEBS Lett. 371:9–12 [DOI] [PubMed] [Google Scholar]
- 56. Wiener D, Plattet P, Cherpillod P, Zipperle L, Doherr MG, Vandevelde M, Zurbriggen A. 2007. Synergistic inhibition in cell-cell fusion mediated by the matrix and nucleocapsid protein of canine distemper virus. Virus Res. 129:145–154 [DOI] [PubMed] [Google Scholar]
- 57. Cherpillod P, Beck K, Zurbriggen A, Wittek R. 1999. Sequence analysis and expression of the attachment and fusion proteins of canine distemper virus wild-type strain A75/17. J. Virol. 73:2263–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Orvell C, Sheshberadaran H, Norrby E. 1985. Preparation and characterization of monoclonal antibodies directed against four structural components of canine distemper virus. J. Gen. Virol. 66(Pt 3):443–456 [DOI] [PubMed] [Google Scholar]
- 59. Malvoisin E, Wild F. 1990. Contribution of measles virus fusion protein in protective immunity: anti-F monoclonal antibodies neutralize virus infectivity and protect mice against challenge. J. Virol. 64:5160–5162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sheshberadaran H, Chen SN, Norrby E. 1983. Monoclonal antibodies against five structural components of measles virus. I. Characterization of antigenic determinants on nine strains of measles virus. Virology 128:341–353 [DOI] [PubMed] [Google Scholar]
- 61. Lee JK, Prussia A, Snyder JP, Plemper RK. 2007. Reversible inhibition of the fusion activity of measles virus F protein by an engineered intersubunit disulfide bridge. J. Virol. 81:8821–8826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Plemper RK, Erlandson KJ, Lakdawala AS, Sun A, Prussia A, Boonsombat J, Aki-Sener E, Yalcin I, Yildiz I, Temiz-Arpaci O, Tekiner B, Liotta DC, Snyder JP, Compans RW. 2004. A target site for template-based design of measles virus entry inhibitors. Proc. Natl. Acad. Sci. U. S. A. 101:5628–5633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sun A, Prussia A, Zhan W, Murray EE, Doyle J, Cheng LT, Yoon JJ, Radchenko EV, Palyulin VA, Compans RW, Liotta DC, Plemper RK, Snyder JP. 2006. Nonpeptide inhibitors of measles virus entry. J. Med. Chem. 49:5080–5092 [DOI] [PubMed] [Google Scholar]
- 64. Doyle J, Prussia A, White LK, Sun A, Liotta DC, Snyder JP, Compans RW, Plemper RK. 2006. Two domains that control prefusion stability and transport competence of the measles virus fusion protein. J. Virol. 80:1524–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Singethan K, Hiltensperger G, Kendl S, Wohlfahrt J, Plattet P, Holzgrabe U, Schneider-Schaulies J. 2010. N-(3-Cyanophenyl)-2-phenylacetamide, an effective inhibitor of morbillivirus-induced membrane fusion with low cytotoxicity. J. Gen. Virol. 91:2762–2772 [DOI] [PubMed] [Google Scholar]
- 66. Paterson RG, Russell CJ, Lamb RA. 2000. Fusion protein of the paramyxovirus SV5: destabilizing and stabilizing mutants of fusion activation. Virology 270:17–30 [DOI] [PubMed] [Google Scholar]
- 67. Plemper RK, Doyle J, Sun A, Prussia A, Cheng LT, Rota PA, Liotta DC, Snyder JP, Compans RW. 2005. Design of a small-molecule entry inhibitor with activity against primary measles virus strains. Antimicrob. Agents Chemother. 49:3755–3761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yuan P, Thompson TB, Wurzburg BA, Paterson RG, Lamb RA, Jardetzky TS. 2005. Structural studies of the parainfluenza virus 5 hemagglutinin-neuraminidase tetramer in complex with its receptor, sialyllactose. Structure 13:803–815 [DOI] [PubMed] [Google Scholar]
- 69. Mirza AM, Aguilar HC, Zhu Q, Mahon PJ, Rota PA, Lee B, Iorio RM. 2011. Triggering of the Newcastle disease virus fusion protein by a chimeric attachment protein that binds to Nipah virus receptors. J. Biol. Chem. 286:17851–17860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhou Q, Schneider IC, Gallet M, Kneissl S, Buchholz CJ. 2011. Resting lymphocyte transduction with measles virus glycoprotein pseudotyped lentiviral vectors relies on CD46 and SLAM. Virology 413:149–152 [DOI] [PubMed] [Google Scholar]
- 71. Connolly SA, Leser GP, Yin HS, Jardetzky TS, Lamb RA. 2006. Refolding of a paramyxovirus F protein from prefusion to postfusion conformations observed by liposome binding and electron microscopy. Proc. Natl. Acad. Sci. U. S. A. 103:17903–17908 [DOI] [PMC free article] [PubMed] [Google Scholar]