

Abstract
There are no available vaccines for dengue, the most important mosquito-transmitted viral disease. Mechanistic studies with anti-dengue virus (DENV) human monoclonal antibodies (hMAbs) provide a rational approach to identify and characterize neutralizing epitopes on DENV structural proteins that can serve to inform vaccine strategies. Here, we report a class of hMAbs that is likely to be an important determinant in the human humoral response to DENV infection. In this study, we identified and characterized three broadly neutralizing anti-DENV hMAbs: 4.8A, D11C, and 1.6D. These antibodies were isolated from three different convalescent patients with distinct histories of DENV infection yet demonstrated remarkable similarities. All three hMAbs recognized the E glycoprotein with high affinity, neutralized all four serotypes of DENV, and mediated antibody-dependent enhancement of infection in Fc receptor-bearing cells at subneutralizing concentrations. The neutralization activities of these hMAbs correlated with a strong inhibition of virus-liposome and intracellular fusion, not virus-cell binding. We mapped epitopes of these antibodies to the highly conserved fusion loop region of E domain II. Mutations at fusion loop residues W101, L107, and/or G109 significantly reduced the binding of the hMAbs to E protein. The results show that hMAbs directed against the highly conserved E protein fusion loop block viral entry downstream of virus-cell binding by inhibiting E protein-mediated fusion. Characterization of hMAbs targeting this region may provide new insights into DENV vaccine and therapeutic strategies.
INTRODUCTION
Dengue imposes one of the largest socioeconomic burdens of any mosquito-borne human disease in the world, yet there is currently no available vaccine or specific treatment. Worldwide, there are an estimated 50 to 100 million cases of dengue infection per year, and 2.5 billion people living in regions where dengue is endemic are at risk of infection (1, 2). An estimated 500,000 people, many of them children, are hospitalized annually with severe dengue symptoms, including dengue hemorrhagic fever/dengue shock syndrome (DHF/DSS) (2, 3). Of note, after an extended absence, dengue has recently reemerged in south Florida (4, 5). Local transmission has also recently been reported in Southern France and Croatia (6, 7).
The four distinct serotypes of dengue virus (DENV) cocirculate in many areas of the world and give rise to sequential epidemic outbreaks when the number of susceptible individuals in the local population reaches a critical threshold and weather conditions favor reproduction of the mosquito vectors Aedes aegypti and Aedes albopictus. Initial infection with one DENV serotype usually generates a protective and long-lasting immune response against reinfection with the same serotype. While antibody cross-reactivity between serotypes is common, cross-serotype protection is only short-lived (8). Low levels of neutralizing antibodies, cross-reactive but nonneutralizing antibodies, or both from previous infections bind virions of other serotypes and target them to Fc receptors on macrophages and certain other cell types, enhancing infection of these cells (9). The presence of these cross-reactive and nonneutralizing antibodies also correlated with severe disease outcome (DHF/DSS) in several studies (10–12). Higher levels of viremia are associated with the development of DHF (12, 13). This antibody-dependent enhancement (ADE) effect may also explain the sequential nature of epidemic outbreaks, as well as the severe disease seen in infants as maternal antibodies wane (10, 11, 14).
Like other members of the genus Flavivirus, DENV has a lipid envelope and a positive-strand RNA genome that codes for a single large polyprotein. This polyprotein is cleaved into separate segments to form the capsid (C), premembrane (prM/M), and envelope (E) structural proteins and enzymatic components required for viral replication and transmission (15). The external E glycoprotein participates in cell recognition and cell entry and is physically arranged in a herringbone pattern as a series of 90 homodimers on the outer surface of the mature virus particle (16). On immature particles, the prM protein lies over the E protein and serves to protect the virus particle from undergoing premature fusion or inactivation within the secretory pathway of the host cell. prM is subsequently cleaved by a host protease to release the ectodomain and allow viral maturation (17). The E protein consists of three structural domains (D), DI, DII, and DIII (18, 19). At one end of the molecule is the fusion loop within DII, and at the other end is DIII, which is involved in host cell binding (20). Upon infection and entry of DENV into the acidic environment of the endosome, the E proteins undergo a conformational change and reassemble into 60 trimers with their fusion loops forming the tip of a trimeric spike oriented to insert into the endosomal membrane within the target cell. Subsequent reconfiguration of the E protein trimers results in fusion of the viral membrane and target cell endosomal membrane to facilitate release of the viral contents into the cytoplasm (21–23).
The nature of the human antibody response to DENV is likely to play a dominant role in defining the outcome of infection. A preponderance of antibodies that recognize neutralizing epitopes will lead to virus clearance and reduced symptoms, while an abundance of antibodies that recognize enhancing epitopes will lead to more severe disease. Multiple questions remain about the nature of the antibody balance, including which epitopes are most important for neutralization versus enhancement and whether these are distinct or overlapping epitopes.
Studies with sera from convalescent DENV patients have yielded conflicting information regarding the human antibody response and the epitopes that these antibodies target. Interestingly, while one of the predominant epitopes recognized by human serum antibodies appears to include the fusion loop and adjacent regions (24, 25), one study reported that these fusion loop antibodies are nonneutralizing (24). He et al. tested the ability of patient sera to block binding of DENV serotype 2 (DENV-2) to Vero cells and reported that neutralization occurred primarily by blocking cell attachment, suggestive of a major role for antibodies targeting DIII (26). In contrast, Wahala et al. subsequently reported that human antibodies directed toward epitopes other than DIII (presumably DI/II) are primarily responsible for neutralization (27).
Monoclonal antibodies (MAbs) have been used to further elucidate important epitopes. However, to date, most anti-DENV monoclonal antibodies are of murine origin (mMAbs), generated from mice (20, 28, 29). mMAbs may not accurately represent the human antibody response to DENV, as mice do not experience human disease other than a transitory viremia and produce an antibody response with more limited diversity and typically lower-affinity antibodies than humans.
Recent studies with human monoclonal anti-DENV antibodies (hMAbs) have highlighted both similarities and major differences between the behavior of sera from convalescent DENV patients and purified hMAbs. In the work of Schieffelin et al., three antibodies that targeted the E protein were isolated from a single donor (30). All three antibodies were cross-reactive with at least two DENV serotypes, one was neutralizing, and all were able to enhance DENV infection. Dejnirattisai et al. reported that in a panel of hMAbs from seven donors, the majority of the antibody response was against prM and was very poorly neutralizing but highly enhancing (31). Beltramello et al. described a wide variety of hMAbs from five DENV patients (32). They included hMAbs against prM, as well as E. However, in contrast to the findings of Dejnirattisai, et al., half of the prM hMAbs reported by Beltramello et al. showed substantial neutralization activity (32). Among the hMAbs recognizing E, Beltramello et al. described antibodies targeting DI/II and DIII. The DIII hMAbs were very highly neutralizing and included serotype-specific and cross-reactive examples. The neutralization activities of the DI/II hMAbs were more diverse and included nonneutralizing, serotype-specific neutralizing, and cross-neutralizing examples. Two of the cross-neutralizing DI/II hMAbs were mapped to the fusion loop using West Nile virus (WNV) E protein mutants.
de Alwis et al. reported that after primary infection most hMAbs were cross-reactive and weakly neutralizing and that many bound to prM (33). Using a modified screening procedure, they were able to detect rare DIII hMAbs that were serotype specific and strongly neutralizing. Recently, de Alwis et al. reported that the majority of antibodies in human sera bound to intact virions, not monomeric E (34). They found that though abundant in human sera, cross-reactive antibodies did not contribute to neutralization and that type-specific antibodies were responsible for potent neutralization. These findings were confirmed with 3 hMAbs that were isolated by first screening for antibodies that bound to intact virions and then screening for a subset of antibodies that were potently neutralizing. They generated escape mutants and mapped the mutations to the quaternary epitopes containing contacts on two different E proteins in the hinge region between DI and DII.
These studies with hMAbs emphasize the complexity of the human antibody response against DENV and highlight the importance of further examination of the roles of different epitopes in prM, in E protein DI/II (either the fusion loop or the hinge region), and in DIII and the mechanisms by which different antibodies neutralize DENV infection. For instance, an affected stage of viral entry—virus binding to the cell surface versus fusion between the viral envelope and endosomal membrane—has never been identified for any neutralizing hMAb.
In this work, we specifically screened for broadly cross-reactive and neutralizing hMAbs from three patients with distinct histories of DENV infection, and we identified three similar hMAbs that mapped to the conserved epitope containing the E protein DII fusion loop. These hMAbs were broadly reactive, high affinity, and conformationally sensitive. With some exceptions, they showed broad but intermediate neutralization activity against all four DENV serotypes and also enhanced all four serotypes. Using a novel assay, we confirmed that these hMAbs inhibited intracellular virus fusion during entry, rather than cell binding, and we provide a mechanistic characterization of these hMAbs.
(This study was presented in part at the 2nd Antivirals Congress, Cambridge, MA, 11 to 13 November 2012 [34a].)
MATERIALS AND METHODS
Cells, viruses, and recombinant E proteins.
The Macaca mulatta kidney epithelial cell line LLC-MK2 (American Type Culture Collection [ATCC], Manassas VA), used in neutralization assays and to propagate DENV, and the human embryonic kidney cell line HEK-293T (ATCC), used for cloning and expression of hMAbs, were grown in Dulbecco's modified Eagle medium (DMEM) containing 10% (vol/vol) fetal bovine serum (FBS), 2 mM Glutamax (Gibco, Carlsbad, CA), 100 U/ml penicillin G, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B at 37°C with 5% (vol/vol) CO2. K-562 human hematopoietic cells (ATCC), used for virus enhancement assays, were grown in RPMI 1640, 10% (vol/vol) FBS, 2 mM Glutamax, 100 U/ml penicillin G, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B at 37°C with 5% (vol/vol) CO2. The M. mulatta kidney epithelial cell line MA104 (ATCC) used in intracellular fusion and prefusion assays was grown in advanced DMEM reduced serum medium (ADMEM) (Gibco) supplemented with 10% fetal bovine serum, 25 mM HEPES, 292 μg/ml l-glutamine, 100 U/ml penicillin G, 100 μg/ml streptomycin at 37°C with 5% (vol/vol) CO2. The human embryonic kidney cell line HEK-293 (ATCC) used in epitope mapping to express prM/E mutants was grown in DMEM supplemented with 10% (vol/vol) FBS, 10 mM HEPES, 100 U/ml penicillin G, 100 μg/ml streptomycin, 1 mM sodium pyruvate, 2 mM l-glutamine (Mediatech), and 1× nonessential amino acid mixture (BioWhittaker, Lonza Walkersville, Inc., Walkersville, MD) at 37°C with 5% (vol/vol) CO2.
DENV-1 strain HI-1, DENV-2 strain NG-2, DENV-3 strain H-78, and DENV-4 strain H-42 were obtained from R. Tesh at the World Health Organization Arbovirus Reference Laboratory at the University of Texas at Galveston. Viruses were propagated in LLC-MK2 as previously described (30). LLC-MK2 cells were inoculated with DENV stock at 70% to 80% confluence and cultured in DMEM and 10% (vol/vol) FBS. Following the appropriate incubation period for the various DENV strains, cell culture supernatant was collected and used in neutralization and enhancement assays, or the culture medium was switched to Protein Free Hybridoma Medium (Gibco) prior to use in antibody detection enzyme-linked immunosorbent assays (ELISAs). Infected adherent cells, as well as supernatant fluids, were collected and treated with 1% (vol/vol) Triton X-100 to solubilize and inactivate virus, as described previously, and both were aliquoted and stored at −20°C for use in ELISAs (30).
Purified DENV-2 strain NG-2 virions used in SDS-PAGE were prepared from large-scale cultures of infected LLC-MK2 cells as follows. Virus particles in cell culture supernatant were precipitated in 8% (wt/vol) polyethylene glycol (PEG) 8000 in an SLA-3000 rotor at 9,300 rpm for 51 min at 4°C, pelleted in a 24% (wt/vol) sucrose cushion using an SW-41ti rotor at 32,000 rpm for 90 min at 4°C, equilibrium banded using a 10 to 35% potassium sodium tartrate step density gradient in a SW-41ti rotor at 32,000 rpm for 2 h at 4°C, and dialyzed and concentrated using an Amicon Ultra-4 centrifugal filter unit (Millipore, Billerica, MA) in NTE buffer (120 mM NaCl, 12 mM Tris, 1 mM EDTA, pH 8.0).
Recombinant DENV-1, -2, -3, and -4 soluble E protein (sE) containing the N-terminal 80% of E protein expressed in Drosophila melanogaster strain Schneider 2 cells and purified by affinity chromatography were obtained from Hawaii Biotech Inc. (Aiea, HI) (18, 35). Recombinant DENV-2 N-terminal E protein containing domains I and II (sDI/II) and DENV-2 E protein domain III (sDIII) expressed in Escherichia coli were obtained from Meridian Life Science (Saco, ME).
Patient samples.
The collection and use of human blood samples for this project were reviewed and approved by the institutional review boards of Florida Gulf Coast University, Tulane University School of Medicine, and Tan Tock Seng Hospital. Informed written consent was obtained for all patients. Three patients were identified as having recovered from DENV infection. Patient 7B had acquired DENV while traveling in Myanmar. Blood was drawn from this patient 2 years posthospitalization, as previously described (30). Patient DA003 was hospitalized in Singapore and had blood drawn approximately 4 weeks postrecovery. As DENV IgG antibodies were detected, in addition to IgM antibodies, the patient was diagnosed with secondary dengue infection with low disease severity, since no hemoconcentration or bleeding was present. Patient 8C contracted DENV in Jamaica and had blood drawn approximately 3 months postrecovery. The patient had fever for 12 days, headache, retro-orbital pain, and blood in sputum on day 10. No information on the type of DENV antibodies present was available from this patient. For all patients, blood was drawn after informed written consent was obtained, and peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Hypaque gradient centrifugation and viably frozen in liquid nitrogen. The patient sera were tested by ELISA and neutralization assays to positively determine infection with DENV.
Epstein-Barr virus transformation.
The production of hMAbs by Epstein-Barr virus (EBV) transformation of B cells has been described previously (30, 36–38). Using this method, transformed clonal B cell lines were produced for hMAbs 4.8A (30) and D11C. Briefly, cryopreserved PBMCs were thawed, placed in culture, and inoculated with EBV (strain B95-8). The cells were resuspended in RPMI containing 20% (vol/vol) FBS, Primacin (InVivoGen, San Diego, CA), and 2 μg/ml CpG 2006 (Midland Certified Reagent Co., Midland, TX) and plated at 104 cells per well in 96-well tissue culture plates. These plates had been previously seeded with approximately 50,000 irradiated mature macrophages per well derived from PBMCs of healthy blood donors that served as feeder cultures to promote outgrowth of transformed B cells. Antibody-positive wells containing viable cells were subcultured at several dilutions and rescreened by ELISA. Cell lines that continued to grow and produce antibody during several low-cell-density passages were finally cloned at limiting dilution. Definitively cloned cell lines were expanded to grow as suspension cultures in stationary 490-cm2 roller bottle cultures (Corning, Corning, NY) from which cell culture fluid was harvested weekly. hMAbs were purified from 1 to 2 liters of culture supernatant by protein A affinity chromatography. The IgG subclass and light-chain type of each antibody was determined by reactivity with mMAbs to the four heavy- chain subclasses (Southern Biotech, Birmingham, AL) and polyclonal goat antibodies to kappa and lambda chains by ELISA using established methods.
Cloning and expression of human monoclonal antibodies.
To generate hMAbs, transient stimulation of memory B cells was used as an alternative approach to EBV transformation (39). hMAb 1.6D was isolated using this method. Cryopreserved PBMCs were thawed and washed as described above and then resuspended in RPMI containing 20% (vol/vol) FBS, Primacin, 2.5 μg/ml R848, Toll-like receptor 7 (TLR7) and TLR8 agonist (InvivoGen, San Diego, CA), and 50 U/ml recombinant human interleukin-2 (Roche Diagnostics Corporation, Indianapolis, IN). After incubating for 3 days at 37°C and 5% (vol/vol) CO2, the cells were recounted and plated at 500 to 104 cells per well in 96-well tissue culture plates containing feeder cells.
To generate molecular clones of hMAbs, we constructed linear full-length Ig heavy- and light-chain gene expression cassettes as described previously (40). Molecular clones were constructed for hMAbs 1.6D, from stimulated PBMCs, and D11C, from the transformed B-cell line. Briefly, RNA extracted from hMAb-positive B cells was reverse transcribed and cloned into gene expression cassettes. Purified PCR products of the paired Ig heavy- and light-chain gene expression cassettes were cotransfected into 80 to 90% confluent HEK-293T cells grown in 48-well tissue culture plates (300 ng of each chain per well) using Fugene Transfection Reagent (Roche Diagnostics Corporation, Indianapolis, IN) following the manufacturer's instructions. Transfection supernatants were tested for hMAb production against all four DENV serotypes by concanavalin A (ConA) ELISA. Positive cultures were seeded into 96-well plates in DMEM with 10% (vol/vol) FBS and 20 μg/ml Blasticidin S (InvivoGen) to ensure formation of stable hMAb-producing cell lines. Cultures were cloned by serial subculture at progressively lower cell densities in 96-well plates, with repeated antibody screening at each step.
Antibody detection with enzyme-linked immunosorbent assay.
B-cell cultures were screened for antibody production using ELISA as described previously (30, 36). Briefly, 96-well plates (Costar; Corning, Corning, NY) were coated with ConA at 25 μg/ml in 0.01 M HEPES (Gibco) for 1 h. The wells were washed (PBS containing 0.1% [vol/vol] Tween 20), and Triton X-100-solubilized DENV produced in serum-free medium was incubated for 1 h. All steps in this ELISA were performed at room temperature. After a wash step, unreacted ConA binding sites in the wells were blocked with RPMI 1640 medium and 10% (vol/vol) FBS for 30 min. Samples from B-cell cultures were transferred to assay plates and incubated for 1 h. The wells were again washed and incubated with peroxidase-conjugated goat anti-human IgG-gamma (Zymed, San Francisco, CA) or peroxidase-conjugated affinity-purified anti-mouse IgG (Rockland, Gilbertsville, PA) diluted 1:2,000 in PBS containing 0.5% (vol/vol) Tween 20, 10% (wt/vol) whey (BiPro, Le Sueur, MN), and 10% (vol/vol) FBS for 1 h. After a final wash step, color was developed with tetramethylbenzidine-peroxide (TMB)-H2O2 as the substrate for peroxidase. The reaction was stopped after 4 min by adding 1% (vol/vol) phosphoric acid, and color was read as the optical density (OD) at 450 nm.
Confocal microscopy.
DENV-infected cells were immunostained with hMAbs and imaged using confocal microscopy. LLC-MK2 cells were grown on no. 1.5 Gold Seal coverglass coverslips (Erie Scientific Company, Portsmouth, NH) placed in each well of a 6-well plate overnight to 80% confluence. Wells containing coverslips were infected with DENV in serum-free medium at a multiplicity of infection (MOI) of 0.002 for 1 h at 37°C and aspirated; fresh culture medium was added, and the coverslips were incubated for 3 days at 37°C. Infected cultures were fixed with 10% (wt/vol) formalin overnight at 4°C, permeabilized with 70% (vol/vol) ethanol for 20 min, and rinsed with PBS prior to immunostaining. Virus proteins were detected using 1 μg/ml hMAb 4.8A, D11C, or 1.6D overnight at 4°C, followed by 2 μg/ml goat anti-human Alexa Fluor 488 (Invitrogen Corporation, Carlsbad, CA) for 4 h at room temperature. The cells were then counterstained with 2 μg/ml Hoechst stain (Cambrex, Walkersville, MD) for 10 min at room temperature and washed with PBS. The coverslips were mounted on Fisherbrand Superfrost microscope slides (Fisher Scientific, Pittsburgh, PA) using Fluormount-G (Southern Biotech, Birmingham, AL) and visualized on an Olympus FV1000 Confocal Microscope System.
Western blotting.
Purified DENV-2; DENV-2 sE, produced as described previously (18, 35) (Hawaii Biotech Inc., Aiea, HI); DENV-2 E sDI/II; and DENV-2 E sDIII (Meridian Life Science, Saco, ME) were subjected to SDS-PAGE using 4 to 15% (wt/vol) or 15% (wt/vol) Tris-HCl polyacrylamide preparative gels for purified DENV-2 and soluble recombinant proteins, respectively (Bio-Rad, Hercules CA). Unless otherwise specified, samples were electrophoresed under nonreducing conditions in 25 mM Tris, 192 mM glycine, 3.5 mM SDS (Sigma-Aldrich, St. Louis, MO) and loaded in a buffer containing 0.7% (wt/vol) SDS. The reduced samples were loaded in a buffer containing 0.005% (wt/vol) SDS and 40 mM dithiothreitol (DTT). A Precision Plus protein Kaleidoscope ladder was used as a standard (Bio-Rad, Hercules, CA). Proteins were transferred to Amersham Hybond-LFP polyvinylidene difluoride (PVDF) membranes (GE Healthcare, Piscataway NJ) in 25 mM Tris, 192 mM glycine, and 20% (vol/vol) methanol (Fisher, Pittsburgh PA), and membrane strips were blocked in 3% (wt/vol) bovine serum albumin (BSA) (Sigma-Aldrich, St. Louis, MO), 0.1% (vol/vol) Tween 20 in PBS and then probed overnight at 4°C with 5 μg/ml of hMAbs 4.8A, D11C, and 1.6D; mMAbs 3H5.1 (Millipore, Billerica MA) specific for DENV-2 E DIII and 4G2 specific for DENV E DI/II; or 30% (vol/vol) cell culture supernatant mMAb D2-C2 specific for DENV-2 and -4 capsid protein (41) diluted in blocking buffer. The membrane strips were then incubated with Alexa Fluor 488-conjugated goat anti-human or anti-mouse antibody (Invitrogen, Carlsbad, CA) diluted in 0.1% (vol/vol) Tween 20 in PBS for 4 h at room temperature and rinsed in 0.1% (vol/vol) Tween 20 in PBS prior to scanning with a Typhoon Trio Variable Mode Imager (GE Healthcare, Piscataway NJ). Photomultiplier tube (PMT) voltage settings used for detecting antibody binding on blot strips ranged from 220 V to 562 V depending on the primary-antibody–secondary-antibody combination. Increasing the PMT voltage increases the signal level, but not the signal-to-noise ratio. The PMT voltage values used for visualizing the individual blots are indicated in the figure legends.
Biolayer interferometry binding assay.
Real-time binding assays between purified hMAbs and DENV-1, -2, -3, and -4 sE proteins (Hawaii Biotech Inc.) were performed using biolayer interferometry with an Octet QK system (Fortebio, Menlo Park, CA) as previously described but with human IgG high-binding sensors instead of streptavidin sensors (30, 42). Briefly, hMAbs 4.8A, D11C, and 1.6D were coupled to kinetics grade anti-human IgG Fc capture (AHC) biosensors (Fortebio, Menlo Park, CA). hMAb binding concentrations that gave a signal between 0.8 and 1.2 nm binding to the sensor surfaces within 200 s were used for sE binding studies. Unbound hMAbs were removed from the surfaces of the sensors by incubation in kinetics buffer containing 1 mM phosphate, 15 mM NaCl, 0.002% (vol/vol) Tween 20, 0.005% (wt/vol) sodium azide, 0.1 mg/ml (wt/vol) BSA, pH 7.4, in PBS. Probes coupled to hMAbs were allowed to bind to sE at several different concentrations, and the binding kinetics were calculated using the Octet QK software package, which fit the observed binding curves to a 1:1 binding model to calculate the association rate constants. DENV-1, -2, -3, and -4 sE proteins were allowed to dissociate by incubation of the sensors in kinetics buffer. Dissociation kinetics were calculated using the Octet QK software package, which fit the observed dissociation curves to a 1:1 model to calculate the dissociation rate constants. Association and dissociation rate constants were calculated using at least two different concentrations of sE. Equilibrium dissociation constants were calculated as the kinetic dissociation rate constant divided by the kinetic association rate constant.
Focus-forming-unit reduction neutralization assay.
LLC-MK2 target cells were seeded at a density of approximately 500,000 cells in each well of a 12-well plate 24 h prior to DENV inoculation. Approximately 100 focus-forming units (FFU) of DENV were incubated with dilutions of heat-inactivated patient serum or purified hMAb (0.4, 2, 4, 20, and 40 μg/ml final) in serum-free DMEM for 1 h at room temperature. DENV mixtures were allowed to infect confluent target cell monolayers for 1 h at 37°C, with rocking every 15 min, after which the inoculum was aspirated and overlaid with fresh MEM-10% (vol/vol) FBS containing 1.2% (wt/vol) microcrystalline cellulose (Avicel; FMC, Newark, DE). The infected cells were then incubated at 37°C with 5% (vol/vol) CO2 for two (DENV-4) or three (DENV-1, -2, and -3) days. The cells were fixed, and foci were visualized as for confocal microscopy, as described above, except a horseradish peroxidase-conjugated goat anti-mouse immunoglobulin (Pierce, Rockford, IL) was used as the detection antibody and developed using 3,3′-diaminobenzidine tetrahydrochloride (Sigma-Aldrich, St. Louis, MO). Fifty percent inhibitory concentrations (IC50s) (in μg/ml) were determined graphically from the percent neutralization plots. A sigmoidal curve fit program was not used, since several of the DENV serotype-antibody combinations did not reach 100% inhibition and thus fit poorly to sigmoidal curves, skewing the calculations. Results are expressed as pooled data from two independent experiments with three replicates each.
Antibody-dependent enhancement assay.
Antibody-dependent enhancement assays were performed as previously described (43). Briefly, 250 FFU of DENV was incubated with various concentrations of hMAbs for 1 h at 37°C. Each DENV-antibody mixture was added to 300 μl of K562 cells (cell density = 2.7 × 105/ml) and incubated for 3 days at 37°C, 5% (vol/vol) CO2. The final hMAb concentrations were 0.04, 0.4, 2, 4, 20, and 40 μg/ml. Afterward, cells were collected and total RNA was isolated using an RNeasy Mini-kit (Qiagen, Valencia, CA) following the manufacturer's protocol. A quantitative reverse transcription (qRT)-PCR was performed on isolated RNA using a universal DENV primer pair (44). The amplification conditions were 95°C for 5 s, 61°C for 20 s, and 72°C for 30 s.
Virus-liposome fusion assay.
The fusogenic activity of dengue virions toward liposomes was characterized using a novel high-throughput plate reader assay, a version of an assay described previously (45). Viral particles were labeled with the fluorescent lipid DiD (Vybrant cell-labeling kit; Molecular Probes, Eugene, OR) in a self-quenching concentration, as described in reference 45. Large unilamellar liposomes 100 nm in diameter were formed by an extrusion technique from the 1:1 (mol/mol) mixture of 1,2-dioleoyl-sn-glycero-3-phosphocholine (PC) and 1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (PG) (Avanti Polar Lipids, Alabaster, AL). DiD-labeled viral particles (∼105 infectious units) in PBS without calcium and magnesium, pH 7.5, were incubated with different concentrations of the antibodies for 1 h at room temperature in a total volume of 50 μl. Virions preincubated with antibodies were then mixed in the wells of 96-well plates (3 wells for each condition) with acidified liposome-containing buffer (final concentration of PC and PG, 30 μM, pH 5.5). After 10 min of coincubation of virions and liposomes at acidic pH, the fluorescence was recorded at excitation and emission wavelengths of 630 and 665 nm. At the end of each recording (10 min of incubation at 22°C), Triton X-100 was added to a final concentration of 0.1% (vol/vol) to fully dequench the DiD. The efficiency of fusion is presented as the difference between fluorescence intensities measured after 10 min of coincubation of labeled virions with liposomes at pH 5.5 and at pH 7.5 normalized to the difference between fluorescence intensities measured for fully dequenched DiD and at pH 7.5. In control experiments, we used dengue virions inactivated by an application of a histidine-modifying reagent, diethylpyrocarbonate (DEPC) (Sigma, St. Louis, MO) (2 mM; 15 min; room temperature).
Virus intracellular fusion and precellular fusion assays.
DENV-2 virions were labeled with DiD as described above. Virus-endosome fusion events were detected as an increase in cell fluorescence upon DiD dilution (45). MA104 cells (ATCC; ∼103 cells/well) were grown overnight in 96-well microtiter plates (Ibidi,Verona, WI). The cells were then incubated for 30 min at 11°C, followed by 5 min at 37°C with 104 DiD-labeled infectious DENV-2 particles that had been preincubated with hMAbs in 100 μl of serum-free ADMEM for 1 h at room temperature. Unbound DENV-2 and hMAbs were removed by washing twice with 400 μl of serum-free ADMEM, and the cells were incubated for an additional 25 min at 37°C. For each well, we captured images of 5 randomly chosen fields of view using a Zeiss Observer Z1 (oil immersion objective; 40×; Carl Zeiss Microscopy, LLC, Thornwood, NY) and generated maximum-intensity z projections based on 15 z-slices of 0.5 μm each for the subsequent analysis. The projections of the cells were analyzed using ImageJ software to subtract the background and threshold using the software's Triangle algorithm. For each condition, we averaged fluorescence intensities in 15 fields (5 fields for each of 3 wells). The data are presented as the mean and standard deviation of the mean for the averaged intensities (n = 3) normalized to the averaged intensities measured for the cells incubated with DENV-2 in the absence of hMAbs.
After taking the images for the above-mentioned analysis, we examined the effects of the hMAbs on the total number of cell-associated virions using a novel assay that measured dequenching of DiD incorporated into unfused viral envelopes. MA104 cells incubated without DENV-2, with DENV-2 and 10 μg/ml of heparan sulfate, or with DENV-2 and 100 μg/ml of hMAb 4.8A, D11C, or 1.6D were lysed by a 15-min incubation with 0.1% (vol/vol) Triton X-100 at 37°C. The lysates were cleared by a 5-min centrifugation at 14,000 × g, and 80 μl of each supernatant was mixed with 1,920 μl of a 20 mM HEPES, 150 mM NaCl, pH 7.5, buffer. Using a Fluoromax 4 Horiba Jobin Yvon spectrophotometer (Horiba Scientific, Edison, NJ), we measured the emission fluorescence at 665 nm using an excitation wavelength of 600 nm. The data are presented as the mean and standard deviation of the mean of three independent experiments normalized to the fluorescence intensity measured for DENV-2-infected cells in the absence of hMAbs.
Antibody binding competition enzyme-linked immunosorbent assay.
HMAbs 4.8A, D11C, and 1.6D were tested for cross-competition with each other to determine whether they recognized overlapping or nonoverlapping sites on DENV-1 E protein using an enzyme-linked immunosorbent assay (30, 46, 47). Solubilized dengue E protein in detergent-treated, serum-free culture fluid was immobilized in ConA-coated wells. The plates were washed and blocked for 30 min at room temperature. Purified hMAbs or dilution buffer was incubated in the wells for 30 min at room temperature. Biotinylated hMAbs were then added to the wells at dilutions that gave less than maximal binding and incubated for 1 h at room temperature. Bound biotinylated hMAb was detected with horseradish peroxidase-streptavidin (Vector, Burlingame, CA). After a wash step, color was developed with TMB-H2O2 as the substrate for peroxidase. The reaction was stopped after 4 min by adding 1% (vol/vol) phosphoric acid, and the color was read as the OD at 450 nm.
Antibody binding competition biolayer interferometry assay.
Real-time competition assays between purified hMAb 1.6D and purified DENV-2 sE were performed using biolayer interferometry with an Octet QK system (Fortebio, Menlo Park, CA). To determine whether the hMAbs recognized overlapping or nonoverlapping sites, we analyzed hMAb 1.6D for competition with itself, as well as with mMAbs 4G2 and 3H5.1. Anti-HIV hMAb 1.7B was used as a negative control. Twenty-five micrograms per milliliter of hMAb 1.6D diluted in kinetics buffer containing 1 mM phosphate, 15 mM NaCl, 0.002% (vol/vol) Tween 20, 0.005% (wt/vol) sodium azide, 0.1 mg/ml (wt/vol) BSA, pH 7.4, in PBS was coupled with AHC biosensors (Fortebio, Menlo Park, CA). Unbound hMAb 1.6D was removed from the surfaces of the sensors by incubation in kinetics buffer. sE was preincubated with hMAb or mMAbs at a 1:1 molar ratio. hMAb 1.6D-coupled AHC sensors were then incubated with 50 nM sE, either prebound to antibodies or in kinetics buffer only. Association of sE with the hMAb 1.6D-coupled sensor was measured by light interference.
Epitope mapping using prM/E mutants.
Mutations were introduced into the prM/E polyprotein of DENV-3 (strain CH53489) by PCR using a Diversity Mutagenesis kit (Clontech Laboratories, Inc., Mountain View, CA), sequenced, and selected to test for hMAb reactivity from a larger library of mutations. Expression plasmids encoding each mutant were transfected into HEK-293 cells, fixed in 4% (vol/vol) paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) 18 h posttransfection, and permeabilized for 45 min with 0.1% (wt/vol) saponin (Sigma-Aldrich) in PBS plus calcium and magnesium (PBS++). Cells were stained for 1 h with hMAbs 4.8A, D11C, 1.6D (0.11 μg/ml in 10% normal goat serum [NGS] [Sigma]-0.1% [wt/vol] saponin), a human polyclonal serum (1:1,000), or the anti-DENV E mMAb 1A1D-2, (1:10,000 mouse ascites fluid, kindly provided by John Roehrig, CDC) (48). Cells were washed three times with PBS++ containing 0.1 (wt/vol) saponin, followed by the addition of 0.4 μg/ml horseradish peroxidase (HRP)-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 h. Following washes, Femto Substrate (Pierce) was added to each well, and luminescence values were measured after 5 min (Wallac Victor 2; PerkinElmer, Waltham, MA). All incubations were performed at room temperature. Antibody reactivities against each mutant E protein clone were calculated relative to wild-type E protein reactivity by subtracting the signal from mock-transfected controls and normalizing to the signal from wild-type E-transfected controls. Mutations within critical clones were identified as critical to the hMAb epitope if they did not support reactivity of the test hMAb but supported reactivity of human polyclonal serum and the conformation-dependent mMAb 1A1D-2. The critical residue within critical clones that contained more than one mutation was identified by assessing other clones containing each of those mutations.
RESULTS
Broadly reactive anti-dengue virus antibodies were isolated from three different patients.
With the goal of understanding the human antibody response in naturally occurring DENV infections, we isolated hMAbs from peripheral blood B cells obtained from patients with distinct histories of DENV infection. These three patients, 7B, 8C, and DA003, contracted DENV in geographically distinct regions, Myanmar, Jamaica, and Singapore, respectively. Cryopreserved PBMC samples were collected at different times postrecovery (approximately 2 years for 7B, 3 months for 8C, and 4 weeks for DA003). Patient DA003 was diagnosed with secondary dengue infection. All three patients were confirmed seropositive to DENV antigens, as shown in Fig. 1A and reported previously for patient 7B (30). From each patient, several hMAbs were produced, either by EBV transformation of B cells (7B and DA003) (30) or by memory B-cell stimulation, followed by molecular cloning (8C) (39, 40). To screen for hMAbs binding to glycosylated DENV proteins, we used a previously described ELISA in which Triton X-100-solubilized DENV proteins were captured in ConA-coated wells of ELISA plates (30). This selection procedure likely biased identification toward cross-reactive hMAbs recognizing the E and prM proteins. Initial selection was done using DENV-2 (7B) or DENV-1 and -3 (8C and DA003). Based on ELISA reactivity to E proteins from all four DENV serotypes (for neutralizing activity, see below), we selected 4.8A, D11C, and 1.6D from patients 7B, DA003, and 8C, respectively. The broad reactivity is illustrated in Fig. 1B for hMAbs D11C and 1.6D and in Schieffelin et al. (30) for hMAb 4.8A. Since we used a number of different methods to isolate these antibodies, we cannot determine what percentage of the total repertoire the antibodies represent. However, since we isolated the antibodies from three out of three patients with different infection histories, we can conclude that they are not uncommon. The three hMAbs were composed of IgG1 heavy chains and kappa light chains.
Fig 1.

Broadly reactive patient-derived monoclonal antibodies. (A) DENV-1, -2, -3, and -4 glycosylated antigens were captured on ConA-coated plates and probed with dilutions of patient 8C and DA003 sera. The data points show the means of one experiment with three replicates. The error bars show standard deviations. (B) DENV-1, -2, -3, and -4 glycosylated antigens were captured on ConA-coated plates and probed with dilutions of hMAbs D11C and 1.6D. Representative data show the means of one experiment with three replicates. The error bars show standard deviations. (C) LLC-MK2 cells infected with DENV-1, -2, -3, and -4 at an MOI of 0.002 were probed with hMAbs 4.8A, D11C, and 1.6D and imaged by confocal microscopy. The nuclei were counterstained with Hoechst stain.
We further showed that the hMAbs 4.8A, D11C, and 1.6D could bind to DENV antigens expressed in DENV-1-, -2-, -3-, or -4-infected monkey epithelial LLC-MK2 cells, using immunofluorescence assays (Fig. 1C). All three hMAbs exhibited a characteristic crescent-shaped perinuclear staining against all four DENV serotypes under fluorescence confocal microscopy. No staining was observed in uninfected cells. The low virus MOI (0.002) used allowed a clear distinction between staining of virus-infected versus noninfected cells.
Recognition of DENV E protein by human monoclonal anti-dengue virus antibodies.
To confirm that the hMAbs recognize DENV E protein, we prepared Western blots using gradient-purified DENV-2 particles under reducing and nonreducing conditions and probed the blot strips with equal amounts (5 μg/ml) of hMAbs 4.8A, D11C, and 1.6D. As shown in Fig. 2A, all three hMAbs recognized a 52-kDa band consistent with the size of DENV-2 E protein in nonreduced samples. No other bands were observed. The 52-kDa band was not present in reduced samples, indicating that all three hMAbs bound to epitopes dependent on disulfide bonds. As a control, an anti-DENV capsid mMAb, D2-C2 (41), recognized bands consistent with the size of DENV-2 capsid protein in both reduced and nonreduced samples. To confirm that the hMAbs bound specifically to E protein, we also prepared Western blots under nonreducing conditions using recombinant DENV-2 sE, which contains the ectodomain of the E protein, and reacted blot strips with hMAbs 4.8A, D11C, and 1.6D, along with mMAbs 4G2 (49, 50) and 3H5.1 (51) as controls. As shown in Fig. 2B, a band consistent with the size of sE was observed for all hMAbs that was identical to the size of the bands recognized by the two control mMAbs specific for DENV E protein.
Fig 2.
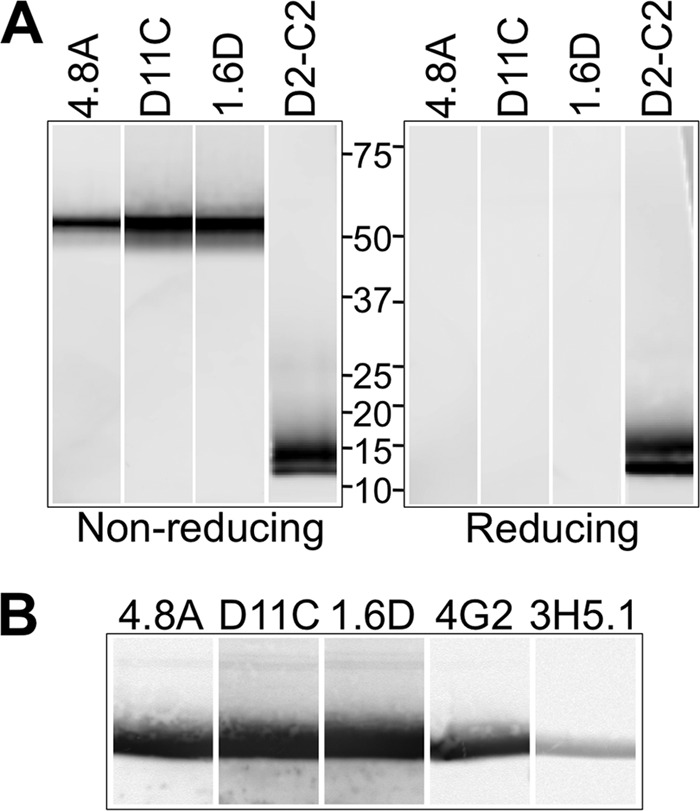
Recognition of the E protein. (A) Western blots were prepared with gradient-purified DENV-2 particles, and blot strips were probed with hMAbs 4.8A, D11C, and 1.6D or anti-DENV capsid mMAb D2-C2 (41) under reducing and nonreducing conditions. Binding of hMAbs to DENV-2 proteins on the blot strips was detected at a PMT voltage of 400 V. Protein standards are indicated in kilodaltons. (B) Western blots were prepared with DENV-2 sE, and blot strips were probed with hMAbs 4.8A, D11C, 1.6D, and control mMAbs 4G2 and 3H5.1 under nonreducing conditions. Binding of hMAbs and mMAbs to DENV-2 sE on the blot strips was detected at a PMT voltage of 220 V.
To determine how tightly hMAbs 4.8A, D11C, and 1.6D bound to sE, we performed biolayer interferometry binding assays with hMAbs coupled to IgG binding sensors. After removing unbound hMAbs, we incubated them with different concentrations of DENV-1, -2, -3, or -4 sE. Binding of the sE proteins to the hMAbs on the surfaces of the probes was measured by the change in interference from light reflected from the surface of the probe. After binding, the probes were placed in a solution without sE protein to similarly measure sE-hMAb dissociation. Antibody on and off rates and equilibrium dissociation constants were calculated assuming a 1:1 binding ratio. As expected from patient serum and the hMAb ELISA results, all three of the hMAbs bound to all four serotypes of DENV equally well, with equilibrium dissociation constants (KDs) in the 10−9 to 10−10 M range (Table 1).
Table 1.
Equilibrium dissociation constants of hMAbs 4.8A, D11C, and 1.6D bound to DENV-1, -2, -3, and -4 sE
| DENV sE bound | Equilibrium KD (M) (mean ± SD) of hMAb: |
||
|---|---|---|---|
| 4.8A | D11C | 1.6D | |
| DENV-1 sE | 1.2 × 10−9 ± 1.6 × 10−9 | 1.4 × 10−10 ± 1.2 × 10−10 | 1.5 × 10−10 ± 5.0 × 10−11 |
| DENV-2 sE | 1.3 × 10−9 ± 1.1 × 10−9 | 1.2 × 10−10 ± 9.4 × 10−11 | 3.5 × 10−10 ± 4.5 × 10−10 |
| DENV-3 sE | 7.4 × 10−10 ± 7.7 × 10−10 | 6.2 × 10−10 ± 3.2 × 10−10 | 1.8 × 10−10 ± 8.0 × 10−11 |
| DENV-4 sE | 7.6 × 10−10 ± 5.4 × 10−10 | 2.9 × 10−10 ± 1.5 × 10−10 | 2.4 × 10−10 ± 5.6 × 10−11 |
Broadly neutralizing activities of human monoclonal anti-dengue virus antibodies.
We analyzed neutralizing antibodies in patient sera using focus-forming-unit reduction neutralization assays in monkey epithelial LLC-MK2 cells in which serial dilutions of patient sera were incubated with DENV-1, -2, -3, or -4. Sera from patients 8C and DA003 neutralized all four serotypes (Fig. 3A). Serum from patient 7B was previously reported to strongly neutralize DENV-1 and -3 and weakly neutralize DENV-2 and -4 (30). To characterize the neutralizing activities of the hMAbs derived from the subjects, we performed neutralization assays with each hMAb (Fig. 3B to D). All three hMAbs neutralized DENV-1 through -4 to some extent in a dose-dependent manner. Some of the hMAbs were stronger neutralizers than others, whereas some neutralized specific serotypes more strongly than others. For example, the IC50s of hMAbs D11C and 1.6D were 1 μg/ml or below (Fig. 3C and D), whereas hMAb 4.8A did not reach 50% inhibition of infectivity against DENV-2 or DENV-4 over the hMAb concentrations tested (Fig. 3B). The observed neutralization activity of hMAb 4.8A was consistent with patient 7B serum activity (30). Additionally, D11C neutralized DENV-1, -2, and -4 more strongly than DENV-3 (Fig. 3C). HMAb 1.6D neutralized DENV-1 through -4 with similar activity (Fig. 3D).
Fig 3.

Broad neutralizing activity. Focus-forming-unit reduction neutralization assays were performed by incubating DENV-1, -2, -3, and -4 with serial dilutions of sera from patients 8C and DA003 (A), hMAb 4.8A (B), hMAb D11C (C), and hMAb 1.6D (D) prior to infecting monolayers of LLC-MK2 cells. IC50s (in μg/ml) were determined graphically and were as follows: for hMAb 4.8A with DENV-1, 2.1 ± 1.1, DENV-2, >40, DENV-3, 2.4 ± 0.1, and DENV-4, >40; for hMAb D11C with DENV-1, 1.5 ± 0.1, DENV-2, 1.0 ± 0.4, DENV-3, 10.2 ± 0.8, and DENV-4, 1.6 ± 0.6; and for hMAb 1.6D with DENV-1, 1.5 ± 1.1, DENV-2, 0.2 ± 0.0, DENV-3, 0.5 ± 0.1, and DENV-4, 2.7 ± 0.8. The pooled data points show the means of at least two independent experiments with three replicates each. The error bars indicate standard deviations.
To determine the neutralization potential of the hMAbs against other flaviviruses, we performed neutralization assays using yellow fever virus (YF-17D) and YF-17D pseudotyped with West Nile virus E glycoprotein (Fig. 3B to D). The hMAbs neutralized WNV to some extent but did not appreciably neutralize yellow fever virus.
Antibody-dependent enhancement mediated by human monoclonal anti-dengue virus antibodies.
At certain concentrations and with the proper Fc domain, all anti-DENV antibodies have the potential to mediate antibody-dependent enhancement in Fc receptor-bearing cells in vitro. For neutralizing antibodies, this enhancement effect decreases as the antibody concentration increases due to the antibody's ability to completely coat the virus and effectively neutralize it. However, for nonneutralizing antibodies, the enhancement potential remains high even at high antibody concentrations (30). To determine the antibody-dependent enhancement potential of the three hMAbs, we incubated each of the four DENV serotypes with increasing concentrations of hMAbs 4.8A, D11C, and 1.6D and infected the Fc receptor II-bearing human macrophage-like cell line K562. Subsequent viral replication was measured by DENV-specific qRT-PCR. In the absence of antibodies that could serve to mediate DENV infection, K562 cells were more permissive to DENV-2 infection than to DENV-1, -3, and -4. As a result, normalized enhancements were typically lower for DENV-2 than for the other 3 serotypes. As presented in Fig. 4A to D, each antibody displayed a similar general trend, with a peak enhancement of infection at antibody concentrations of 0.4 to 4 μg/ml, followed by neutralization, resulting in reduced infection at increasing antibody concentrations.
Fig 4.

Antibody-dependent enhancement. Enhanced infection of Fc receptor-bearing K562 cells was measured by DENV-specific qRT-PCR following infection with DENV-1 (A), DENV-2 (B), DENV-3 (C), and DENV-4 (D) in the presence of hMAbs 4.8A, D11C, and 1.6D. Each data point is the mean of three replicates. The error bars indicate standard deviations.
Neutralizing activities of human monoclonal anti-dengue virus antibodies correlate with inhibition of fusion, not binding.
Antibodies directed against virus surface proteins are predicted to inhibit an early entry step into target cells. DENV enters through receptor-mediated endocytosis, where the E glycoprotein binds to a cellular receptor on the plasma membrane, followed by endocytosis and fusion of the viral and cellular membranes in the low-pH environment of endocytic vesicles, allowing the viral genome to enter target cells. To determine the details of the mechanism of neutralization, we explored the effects of our antibodies on different stages of viral entry.
To investigate whether hMAbs could inhibit DENV-2 fusion, we used an assay that measures fusogenic activity of DENV particles toward liposomes (45, 52). DENV-2 particles labeled with a self-quenching concentration of a fluorescent lipid, DiD, were pretreated with hMAbs prior to coincubation with liposomes at acidic pH. Lipid mixing between labeled viral and unlabeled liposomal membranes was monitored as an increase in fluorescence, reflecting DiD dilution. As expected, no increase in the fluorescence, and thus no lipid mixing, was observed for virions inactivated by a histidine-modifying reagent, diethylpyrocarbonate (45). In contrast to the negative-control anti-HIV gp120 hMAb EH21, all three anti-DENV E hMAbs strongly inhibited virus-liposome fusion in a dose-dependent manner (Fig. 5A). The relative fusion-inhibiting activities of the hMAbs, with 1.6D being the most potent and 4.8A the least potent, corresponded to their relative neutralization activities (Fig. 3B to D).
Fig 5.

Mechanism of neutralization. (A) Low-pH-activated virus-liposome fusion was measured using fluorescently labeled DENV-2 incubated with hMAbs 4.8A, D11C, and 1.6D. The fluorescence signal was normalized to the signal generated in the absence of hMAbs to calculate percent liposome fusion. EH21 is an irrelevant anti-HIV hMAb. (B) Intracellular fusion of DiD-labeled DENV-2 within endosomes leads to dequenching of DiD. Confluent monolayers of MA104 cells were infected with equivalent amounts of DENV-2 preincubated with or without 100 μg/ml hMAbs, as indicated. Intracellular structures at the site of fusion events fluoresce red. Cells were counterstained with DAPI to visualize nuclei. (C) Intracellular fusion levels were quantified after incubation of DENV-2 with different concentrations of hMAbs. Fluorescence levels were normalized to those of virus-only controls. (D) Total fluorescence of all bound DENV-2 was quantified by fully dequenching the cells. DENV-2 was incubated with 100 μg/ml of each hMAb. Fluorescence levels were normalized to those of virus-only controls. Heparan sulfate at 10 μg/ml, a known inhibitor of DENV binding, was used as a positive control for binding inhibition. For panels A, C, and D, each data point is the mean of three replicates. The error bars indicate standard deviations.
Since virus-liposome fusion relies on random collisions between virions and liposomes rather than on E-mediated virion-liposome binding, the ability of hMAbs 4.8A, D11C and 1.6D to inhibit fusion between virions and liposomes suggested that viral entry in vivo might also be inhibited at the fusion stage of the entry. To test this hypothesis, we directly examined the effects of the antibodies on intracellular fusion of DENV-2 and on the prefusion stages of viral entry into rhesus macaque kidney epithelial (MA104) cells. For DENV-2 labeled with DiD at a self-quenching concentration, fusion events along the endocytic pathway dilute DiD and thus lead to an increase in fluorescence signal (45, 53, 54). We quantified the efficiency of intracellular fusion by measuring cell fluorescence with a novel microtiter plate version of the assay described previously (45). Virions were preincubated with the antibodies and then applied to the cells at 11°C for 30 min to permit binding while holding the virions in a temperature-arrested state. The temperature was then raised to 37°C to allow uptake and fusion of the virions. After the first 5 min of incubation at 37°C, we removed unbound virions and antibodies by rinsing and, after 25 additional minutes, assayed intracellular fusion by fluorescence microscopy (Fig. 5B). Fusion of DiD-labeled virus within endosomes leads to dequenching of DiD and the appearance of brightly fluorescent intracellular structures. Cells were counterstained with DAPI (4′,6-diamidino-2-phenylindole) to visualize the nuclei. All three anti-DENV hMAbs inhibited intracellular fusion in a dose-dependent manner (Fig. 5C) corresponding to their relative inhibiting activities in viral neutralization and virus-liposome fusion assays (1.6D was the most potent and 4.8A the least potent) (Fig. 3B to D and 5A). In contrast, a control anti-HIV gp120 hMAb, EH21, did not inhibit intracellular fusion. These results suggest that 4.8A, D11C, and 1.6D directly interfere with the structural transitions required for the virus to fuse to the endosomal membrane.
For virions to reach endosomes and fuse, they must first bind to the cell surface and undergo internalization. In order to test whether our hMAbs inhibited virus-cell binding, the total number of virions associated with cells must be evaluated, including (i) cell surface-bound virions, (ii) internalized but yet unfused virions, and, finally, (iii) fused virions. Note that when we measured fusion after a 30-min incubation at 37°C, fused virions represented only a small fraction of all cell-associated virions (54) and only fused virions were dequenched. After measuring the intracellular fusion efficiency, we lysed the cells and fully dequenched the DiD probe in all unfused virions, using Triton X-100 to disrupt the viral membranes. The level of unquenched DiD fluorescence was therefore proportional to the total number of cell-associated virions and thus can be used to evaluate the effects of different reagents on virus-cell binding (Fig. 5D). As expected, heparan sulfate (10 μg/ml), which inhibits DENV binding to cells (55), dramatically lowered the numbers of cell-associated virions, and consequently, the DiD fluorescence of cell lysates. Preincubation of virions with high concentrations of our hMAbs (100 μg/ml, sufficient to profoundly inhibit intracellular fusion) had no effect on cell lysate DiD fluorescence intensity, indicating that these antibodies do not appreciably affect virus-cell binding. These findings demonstrate that hMAbs 4.8A, D11C, and 1.6D block viral infection downstream of virus-cell binding at the stage of virus-endosome fusion.
Interestingly, hMAb 4.8A did not completely suppress DENV-2 fusion even at very high concentrations, correlating with the observed neutralization activity of this hMAb against DENV-2. The inability of some antibodies to completely neutralize infection and fusion has been previously reported (52, 56–58), suggesting that even at saturation these antibodies only partially neutralize the fusogenic activity of each E protein. Alternatively, the epitopes at some of the viral surface E proteins may be inaccessible, reflecting the heterogeneity of virions and/or E protein chemical environments. For all three hMAbs, inhibition of lipid mixing required somewhat higher concentrations of hMAbs than virus neutralization. This could reflect different conditions (in the neutralization assay, we used 102 infectious units versus 105 and 104 infectious units in liposome and intracellular fusion assays, respectively). This difference may also indicate that for DENV, as for several other viruses (59, 60), early stages of viral fusion (detected as lipid mixing in our assay) require fewer functional fusion proteins and thus are more difficult to inhibit than opening of a fusion pore large enough to release viral RNA, a prerequisite for viral infection. As a result, at neutralizing concentrations of the antibodies, virions may still have enough functional (i.e., not antibody bound) fusion proteins to mediate lipid mixing.
Taken together, our results show that hMAbs 4.8A, D11C, and 1.6D neutralize infection by inhibiting E protein-mediated membrane fusion rather than prefusion stages of viral entry.
Targeting of distinct but overlapping fusion loop epitopes.
To determine which E protein domain(s) hMAbs 4.8A, D11C, and 1.6D interacted with, we subjected recombinant DENV-2 E proteins sDI/II and sDIII to SDS-PAGE under nonreducing conditions and probed with equal amounts of hMAbs 4.8A, D11C, and 1.6D and control mMAbs 3H5.1 (specific for DENV-2 E DIII) or 4G2 (specific for DENV E DII fusion loop). As illustrated in Fig. 6A, hMAbs 4.8A, D11C, and 1.6D interacted specifically with sDI/II and not with sDIII.
Fig 6.

Coarse-level epitope mapping. (A) Western blots were prepared with DENV-2 sDI/II and sDIII, and blot strips were probed with 5 μg/ml of hMAbs 4.8A, D11C, 1.6D, and control mMAbs 4G2 and 3H5.1 under nonreducing conditions. Binding of antibodies to sDI/II on the blot strips was detected at a PMT voltage of 475 V or 562 V for hMAbs and mMAbs, respectively, whereas binding of both hMAbs and mMAbs to sDIII on blot strips was detected at a PMT voltage of 420 V. (B) A competition ELISA was used to determine whether hMAbs 4.8A, D11C, and 1.6D and mMAb 4G2 recognized overlapping epitopes on DENV-1 E protein. HMAb EH21 against HIV-1 ENV was used as a negative control. Unlabeled antibodies (shown on the x axis) were added to DENV-1 E protein-coated wells. Upon removal of unbound antibodies, the wells were probed with biotinylated antibodies as shown. (C) Antibody binding competition was measured using biolayer interferometry. Biosensor probes were coupled to hMAb 1.6D and subsequently incubated with either DENV-2 sE alone or sE complexed with hMAb 1.6D or control anti-HIV 1.7B or with mMAb 4G2 or 3H5.1.
To determine whether hMAbs 4.8A, D11C, and 1.6D bound to overlapping epitopes on E protein, we used an ELISA binding competition assay, as previously reported (30). Unlabeled hMAbs 4.8A, D11C, 1.6D, and EH21 (an anti-HIV-1 gp120 antibody) and mMAb 4G2 (which binds to the DENV fusion loop) were incubated with DENV-1 antigen bound in ConA-coated plates. Biotinylated antibodies were then added to wells containing prebound unlabeled antibodies. If the labeled and unlabeled antibodies bound to the same epitope or overlapping epitopes, then the labeled antibody would not bind, resulting in a low signal upon development with streptavidin-conjugated enzyme. As shown in Fig. 6B, biotinylated hMAbs 4.8A, D11C, and 1.6D were unable to bind to wells containing any of their unlabeled counterparts. In addition, mMAb 4G2, which binds to the fusion loop, was unable to bind in the presence of hMAb 4.8A, D11C, or 1.6D. These results suggest that the three hMAbs and 4G2 share overlapping epitopes. Of note, when wells preincubated with unlabeled mMAb 4G2 were incubated with labeled hMAbs, the hMAbs were able to displace mMAb 4G2 to some extent. This result could suggest that the hMAbs and mMAb 4G2 bind to different epitopes. However, the results could also arise if hMAbs 4.8A, D11C, and 1.6D bind to the same E protein epitope as mMAb 4G2 but with higher affinities (as is further suggested by experiments described below). As a validation of the competition assay, the negative-control hMAb EH21 did not compete for binding with either the hMAbs or mMAb 4G2.
To further investigate the relative binding affinities of hMAbs to their epitopes, we chose hMAb 1.6D for additional studies in an antibody binding competition biolayer interferometry assay. hMAb 1.6D was coupled to human IgG binding sensors. After removing unbound hMAb 1.6D, we applied DENV-2 sE that had been preincubated at a 1:1 molar ratio with hMAb 1.6D, mMAb 4G2 or 3H5.1, or medium only to the sensors. As before, binding of the sE protein to the hMAbs on the surfaces of the probes was measured by the change in interference from light reflected from the surface of the probe. The magnitude of the signal was indicative of the thickness of the antibody-sE complexes. It was anticipated that if an antibody effectively competed for binding to the hMAb 1.6D epitope, sE precomplexed with that particular antibody would not be able to bind to the hMAb 1.6D-coated sensor. In contrast, if a particular antibody bound to a different epitope on sE, the sE-antibody complex would be able to bind to the hMAb 1.6D-coated sensor. Figure 6C illustrates the individual binding curves. As expected, DENV-2 sE bound to the hMAb 1.6D-coated sensor, generating a signal proportional to the thickness of the antibody on the sensor plus the sE protein. When sE was precomplexed with hMAb 1.6D prior to addition (sE plus 1.6D), sE binding to hMAb 1.6D captured on the sensor was profoundly reduced, indicating that hMAb 1.6D can compete very effectively with itself for binding. When mMAb 3H5.1, which binds to DIII, was precomplexed with sE, the sE-3H5.1 complex bound to the hMAb 1.6D-coated sensor, resulting in an increased signal due to the increased thickness of the probe-coupled complex, which consisted of sE plus two antibodies. As a control, when an irrelevant anti-HIV hMAb (1.7B) was added to sE, the binding signal was equivalent to that of sE alone. When mMAb 4G2 was precomplexed with sE, sE bound to the hMAb 1.6D-coated sensor; however, the thickness of the complex was indicative of only sE binding to the sensor with no additional antibody. This result is likely due to effective competition of hMAb 1.6D with the mMAb 4G2 binding epitope, consistent with competition by ELISA (Fig. 6B). The antibody binding competition biolayer interferometry assay further established that hMAbs bound to epitopes on the E protein fusion loop and suggested that these hMAbs may have higher affinities than similar mMAbs.
To more precisely define the epitopes for hMAbs 4.8A, D11C, and 1.6D, we screened a library of DENV-3 E point mutants to identify mutations that reduce hMAb binding. Three residues, W101, L107, and G109, that when mutated significantly reduced 4.8A, D11C, or 1.6D binding compared to wild-type E protein were identified (Fig. 7A). These residues were located directly within the fusion loop (residues 98 to 109) and mapped in close proximity on the structure of the E protein (Fig. 7B). Each E protein mutant reacted to a human polyclonal serum and the conformation-dependent mMAb 1A1D-2 that targets a different epitope (61), confirming that each clone was expressed and was not simply globally misfolded. hMAb 4.8A binding was reduced by mutations at any of the three positions, while D11C and 1.6D binding was reduced by mutations at only W101 or G109. These data suggest that 4.8A, D11C, and 1.6D have overlapping but distinct epitopes in the fusion loop, consistent with their ability to compete with each other and with a fusion loop mMAb.
Fig 7.

Molecular-level epitope mapping. (A) Cells expressing DENV E mutants were fixed and immunostained with the indicated antibodies. Clones with reactivities of ≤25% relative to wild-type (WT) DENV-3 E were identified as critical for hMAb binding. The reactivities of mutant clones containing each critical residue with hMAbs 4.8A, D11C, and 1.6D and the control mMAb 1A1D-2 and human polyclonal serum (hPAb) are shown. The experiments were repeated three times, and standard deviations of quadruplicate wells are shown. (B) Critical residues for hMAbs 4.8A (W101, L107, and G109), D11C (W101 and G109), and 1.6D (W101 and G109) were visualized on a structure of DENV-3 E protein (Protein Data Bank accession code 1uzg [ 87]). DI, DII, and DIII are depicted in red, yellow, and blue, respectively, and the fusion loop (residues 98 to 109) is circled.
DISCUSSION
This study focused on the portion of the human antibody response that is broadly neutralizing and potentially protective against all DENV serotypes. Several other classes of DENV-neutralizing hMAbs are primarily serotype specific, including hMAbs that target E protein DIII (31–33) and hMAbs that recognize quaternary epitopes between two E proteins (34, 62). We established the mechanism of action of broadly neutralizing antibodies produced in three human dengue patients. Though the hMAbs were isolated from patients from different countries and diverse ethnic backgrounds, with different infecting viruses, and at different times postrecovery, similar broadly neutralizing hMAbs were produced, suggesting that the target of these hMAbs is a common epitope that plays an important role in DENV infectivity. With the goal of determining the mechanism of neutralization, using a novel assay, we uncoupled DENV binding to target cells from fusion and found that the neutralization activity of the hMAbs correlated with inhibition of fusion rather than virus-cell binding. We further mapped the binding of the hMAbs to the highly conserved fusion loop region in DII of the E glycoprotein.
A common theme among different structural classes of enveloped virus fusion proteins is the existence of an internal or N-terminal hydrophobic fusion loop or fusion peptide. Neutralizing antibodies directed against these fusion regions have been well described in other virus systems, including closely related flaviviruses (63), more distantly related alphaviruses (64), and unrelated orthomyxoviruses (65). Our results are consistent with those of a recent study that identified two other broadly neutralizing hMAbs from a single patient that target DENV DI/II and whose binding to WNV DI/II was ablated when residues in the fusion loop were altered, suggesting that these antibodies may also bind to the DENV fusion loop (32). Importantly, reports focusing on polyclonal antibody fractions from DENV patient-derived serum have shown that the predominant fraction of the broadly neutralizing activity targets DI/II and specifically the fusion loop, consistent with our hMAb results (24, 25, 27). Broadly neutralizing chimpanzee MAbs and mMAbs targeting the DENV fusion loop have been described previously (49, 50, 66). Our finding that hMAbs recognizing the fusion loop inhibit the fusion stage of DENV entry into mammalian cells is consistent with earlier reports that a chimpanzee MAb against the fusion loop inhibits fusion between mosquito cells mediated by cell surface-bound dengue virions (67). The hMAbs reported here can individually block the binding of an mMAb recognizing the fusion loop, confirming that they share overlapping epitopes. However, mMAb prebound to E could not block binding by the hMAbs, indicating that the particular mMAb used either has a lower affinity than the hMAbs or that hMAbs bind to the fusion loop differently, in a manner that allows the hMAbs to displace the mMAb.
The DENV fusion loop is highly conserved, so it is not clear why hMAb 4.8A inhibited DENV-2 and -4 less strongly than DENV-1 and -3 nor why hMAb D11C inhibited DENV-3 less strongly. Additionally, other flaviviruses with nearly identical fusion loop sequences are not inhibited effectively, with hMAbs 4.8A, D11C, and 1.6D achieving only an intermediate level of neutralization against WNV and very poor neutralization against yellow fever virus. It is possible that the fusion loop region may be oriented differently or have altered accessibility in different viruses (56). These hMAbs bind to E under native conditions but do not bind denatured and reduced E protein (Fig. 2A), suggesting that disulfide bridges preserve a structural conformation of the epitopes. Additional nonconserved, fusion loop-adjacent residues may also contribute to antibody binding. Such residues could have a cumulative effect on binding energetics that is not detected when individual residues are mutated in isolation. These potential additional contact residues might be on the same E protein or part of an adjacent E protein on the virus surface. Binding to recombinant sE monomers and dissociated E protein in ELISAs and Western blots is not identical to binding the E proteins as they are arranged on the surface of a virion. E protein dimers are located in distinct symmetry positions on assembled viruses, and steric hindrance may alter the binding of antibodies to these positions, similar to observations with binding to WNV (68).
Both antibody-virus binding (56) and virus-cell binding (45) can be ineffective at 4°C, the temperature that is often used in binding assays (54, 69). Thus, while our virus-cell binding assay based on measuring DiD fluorescence can be used to quantify binding at different temperatures, we measured the efficiency of virus binding at physiological temperature. Note that, like other virus-cell binding assays (for instance, references 69 and 70), our assay does not distinguish between potential nonproductive, nonspecific binding to the cell surface and specific, productive binding between virions and a yet unidentified specific cell surface receptor for DENV, the identity of which is under debate (reviewed in reference 71). We therefore questioned whether our hMAbs might neutralize virus by inhibiting specific virus-receptor binding. We estimated that to be undetectable within the margin of error of our data, any putative specific binding would represent only a small fraction of total binding. Additionally, the hMAbs would need to inhibit only this specific binding and not the nonspecific binding, and only this specific binding would allow productive viral entry and infection. While we cannot exclude the possibility that our hMAbs block viral entry and infection by blocking unknown specific virus-receptor binding yet causing no changes in total virus-cell binding, we consider the conclusion that hMAbs block entry at the fusion stage rather than at the virus-cell binding stage to be the most likely interpretation of our data. The liposome fusion assay results also support this conclusion.
Broadly neutralizing hMAbs can potentially be used in the development of therapeutic treatments. Most previous work in this area has focused on the use of mMAbs (29). The present study has shown that, for binding to E, an anti-DENV hMAb can outcompete an mMAb with a similar epitope. This observation is not surprising, since human antibodies tend to have longer variable regions than mouse antibodies (72, 73). When used therapeutically in patients, hMAbs are also much less likely to provoke an immune response, which can even be directed against the antigenically distinct variable regions in humanized mMAbs, where the heavy- and light-chain constant regions have been replaced with human sequences (74). A recent study using a mouse model of lethal DENV infection showed that hMAbs protected mice after exposure to DENV, highlighting the important role that hMAbs can play in the development of DENV therapeutics (32).
While the neutralization activities reported here are lower than those of some recently described hMAbs (31–33), it is difficult to compare neutralization potencies between assay systems in different laboratories, as the potency can vary depending on the specific assay used, the serotype and strain of virus, the target cell line, and the incubation conditions of the assay (48, 75). Despite difficulties comparing methodologies, neutralization potency alone offers an incomplete view of the human antibody response. Given our study and the work of others, there appears to be a wide spectrum of hMAb responses directed against the DENV surface proteins, ranging from potently neutralizing, serotype-specific antibodies to nonneutralizing, cross-reactive antibodies, and many hMAbs falling between these two extremes (30–34). Using vesicular stomatitis virus mMAbs, Bachmann et al. demonstrated that in vivo protection was independent of immunoglobulin subclass, avidity, and in vitro neutralization activity and that above a minimal avidity threshold (>2 × 107 M−1), protection depended simply on a minimum serum concentration (76). For therapeutic or protective purposes, whether it would be preferable to have multiple serotype-specific, highly neutralizing anti-DENV hMAbs or a single cross-reactive and moderately neutralizing hMAb is currently unknown.
We have not characterized the extent to which the virus preparations we used for our neutralization assays contain mature, immature, or partially mature particles. Thus, we do not know if hMAbs 4.8A, D11C, and 1.6D neutralize infectivity by preferentially binding to completely mature, partially mature, or completely immature virions. A previous study suggested a structural basis for the preferential binding of fusion loop antibodies to the partially exposed fusion loop region on immature flaviviruses (63), but differences between the mMAb used in that study, which bound to the bc loop in addition to the fusion loop, and our hMAbs make it difficult to speculate on the role of mature versus immature virion structure in our results.
One of the most striking outcomes of other recent studies of hMAbs against DENV is the discovery that the response is dominated by broadly reactive but nonneutralizing antibodies directed against prM and E that serve only to enhance DENV infection in macrophages and other Fc receptor-bearing cells (30–33). The majority, if not all, of DENV vaccine candidates approaching or in clinical trials contain full-length DENV prM and E proteins (77–86). Full-length DENV prM and E proteins, whether expressed as part of an attenuated DENV strain or expressed in another manner, may induce a broadly reactive and primarily nonneutralizing antibody response. Although both neutralizing and nonneutralizing antibodies can enhance infection, large numbers of broadly reactive nonneutralizing antibodies could shift the response in favor of enhancement, which may result in an increased risk of severe disease in vaccine recipients. However, if immunogens that present the fusion loop in the proper context can be developed, a broadly reactive neutralizing response might be possible for a DENV vaccine. The enhancing activity induced by such an immunogen might be reduced compared to full-length prM and E.
ACKNOWLEDGMENTS
This work was supported by the Defense Threat Reduction Agency under award numbers HDTRA1-08-1-0003, HDTRA1-09-1-0004, and HDTRA1-10-1-0009 and the National Science Foundation under grant number CBET-0923030 to S.I. and S.F.M.; the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health (NIH), and by a National Institute of Allergy and Infectious Diseases (NIAID) NIH Intramural Biodefense Research grant and the Intramural AIDS Targeted Antiviral Program to L.V.C.; by NIAID contract HHSN272200900055C to B.J.D.; and by NIH Centers of Biomedical Research Excellence award P20RR021970-06 to J.S.S. and J.E.R.
J.M.C., E.Z., J.E.R., B.J.D., L.V.C., S.F.M., J.S.S., and S.I. conceived and designed experiments. J.M.C., E.Z., K.M.K., C.O.N., D.K.R., A.S.G., L.E.B., G.H., M.F.S., R.H.F., S.-T.Y., J.S.S., and S.I. performed the experiments. L.L. contributed reagents, materials, and analysis tools. J.M.C., E.Z., K.M.K., B.J.D., L.V.C., S.F.M., J.S.S., and S.I. analyzed the data. S.F.M. and S.I. wrote the paper.
B.J.D. is a shareholder in Integral Molecular, Inc.
Footnotes
Published ahead of print 21 October 2012
REFERENCES
- 1. Mackenzie JS, Gubler DJ, Petersen LR. 2004. Emerging flaviviruses: the spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat. Med. 10:S98–S109 [DOI] [PubMed] [Google Scholar]
- 2. WHO 2012. Dengue and severe dengue. Fact sheet no. 117. http://www.who.int/mediacentre/factsheets/fs117/en/
- 3. Gubler DJ. 2002. Epidemic dengue/dengue hemorrhagic fever as a public health, social and economic problem in the 21st century. Trends Microbiol. 10:100–103 [DOI] [PubMed] [Google Scholar]
- 4. CDC 2010. Locally acquired dengue—Key West, Florida, 2009–2010. MMWR Morb. Mortal. Wkly. Rep. 59:577–581 [PubMed] [Google Scholar]
- 5. Graham AS, Pruszynski CA, Hribar LJ, DeMay DJ, Tambasco AN, Hartley AE, Fussell EM, Michael SF, Isern S. 2011. Mosquito-associated dengue virus, Key West, Florida, USA, 2010. Emerg. Infect. Dis. 17:2074–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. La Ruche G, Souares Y, Armengaud A, Peloux-Petiot F, Delaunay P, Despres P, Lenglet A, Jourdain F, Leparc-Goffart I, Charlet F, Ollier L, Mantey K, Mollet T, Fournier JP, Torrents R, Leitmeyer K, Hilairet P, Zeller H, Van Bortel W, Dejour-Salamanca D, Grandadam M, Gastellu-Etchegorry M. 2010. First two autochthonous dengue virus infections in metropolitan France, September 2010. Euro Surveill. 15:19676. [PubMed] [Google Scholar]
- 7. Schmidt-Chanasit J, Haditsch M, Schoneberg I, Gunther S, Stark K, Frank C. 2010. Dengue virus infection in a traveller returning from Croatia to Germany. Euro Surveill. 15:pii=19677. http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=19677 [DOI] [PubMed] [Google Scholar]
- 8. Sabin AB. 1952. Research on dengue during World War II. Am. J. Trop. Med. Hyg. 1:30–50 [DOI] [PubMed] [Google Scholar]
- 9. Halstead SB, O'Rourke EJ. 1977. Dengue viruses and mononuclear phagocytes. I. Infection enhancement by non-neutralizing antibody. J. Exp. Med. 146:201–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Halstead SB. 1988. Pathogenesis of dengue: challenges to molecular biology. Science 239:476–481 [DOI] [PubMed] [Google Scholar]
- 11. Kliks SC, Nimmanitya S, Nisalak A, Burke DS. 1988. Evidence that maternal dengue antibodies are important in the development of dengue hemorrhagic fever in infants. Am. J. Trop. Med. Hyg. 38:411–419 [DOI] [PubMed] [Google Scholar]
- 12. Vaughn DW, Green S, Kalayanarooj S, Innis BL, Nimmannitya S, Suntayakorn S, Endy TP, Raengsakulrach B, Rothman AL, Ennis FA, Nisalak A. 2000. Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J. Infect. Dis. 181:2–9 [DOI] [PubMed] [Google Scholar]
- 13. Vaughn DW, Green S, Kalayanarooj S, Innis BL, Nimmannitya S, Suntayakorn S, Rothman AL, Ennis FA, Nisalak A. 1997. Dengue in the early febrile phase: viremia and antibody responses. J. Infect. Dis. 176:322–330 [DOI] [PubMed] [Google Scholar]
- 14. Simmons CP, Chau TN, Thuy TT, Tuan NM, Hoang DM, Thien NT, Lien le B, Quy NT, Hieu NT, Hien TT, McElnea C, Young P, Whitehead S, Hung NT, Farrar J. 2007. Maternal antibody and viral factors in the pathogenesis of dengue virus in infants. J. Infect. Dis. 196:416–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chang G-JJ. 1997. Molecular biology of dengue viruses, p 175–198 In Gubler DJ, Kuno G. (ed), Dengue and dengue hemorrhagic fever. CABI, Wallingford, United Kingdom [Google Scholar]
- 16. Kuhn RJ, Zhang W, Rossmann MG, Pletnev SV, Corver J, Lenches E, Jones CT, Mukhopadhyay S, Chipman PR, Strauss EG, Baker TS, Strauss JH. 2002. Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell 108:717–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yu IM, Zhang W, Holdaway HA, Li L, Kostyuchenko VA, Chipman PR, Kuhn RJ, Rossmann MG, Chen J. 2008. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science 319:1834–1837 [DOI] [PubMed] [Google Scholar]
- 18. Modis Y, Ogata S, Clements D, Harrison SC. 2003. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 100:6986–6991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rey FA, Heinz FX, Mandl C, Kunz C, Harrison SC. 1995. The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature 375:291–298 [DOI] [PubMed] [Google Scholar]
- 20. Crill WD, Roehrig JT. 2001. Monoclonal antibodies that bind to domain III of dengue virus E glycoprotein are the most efficient blockers of virus adsorption to Vero cells. J. Virol. 75:7769–7773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Harrison SC. 2008. Viral membrane fusion. Nat. Struct. Mol. Biol. 15:690–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heinz FX, Allison SL. 2003. Flavivirus structure and membrane fusion. Adv. Virus Res. 59:63–97 [DOI] [PubMed] [Google Scholar]
- 23. Zhang W, Chipman PR, Corver J, Johnson PR, Zhang Y, Mukhopadhyay S, Baker TS, Strauss JH, Rossmann MG, Kuhn RJ. 2003. Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat. Struct. Biol. 10:907–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lai CY, Tsai WY, Lin SR, Kao CL, Hu HP, King CC, Wu HC, Chang GJ, Wang WK. 2008. Antibodies to envelope glycoprotein of dengue virus during the natural course of infection are predominantly cross-reactive and recognize epitopes containing highly conserved residues at the fusion loop of domain II. J. Virol. 82:6631–6643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lin HE, Tsai WY, Liu IJ, Li PC, Liao MY, Tsai JJ, Wu YC, Lai CY, Lu CH, Huang JH, Chang GJ, Wu HC, Wang WK. 2012. Analysis of epitopes on dengue virus envelope protein recognized by monoclonal antibodies and polyclonal human sera by a high throughput assay. PLoS Negl. Trop. Dis. 6:e1447 doi:10.1371/journal.pntd.0001447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. He RT, Innis BL, Nisalak A, Usawattanakul W, Wang S, Kalayanarooj S, Anderson R. 1995. Antibodies that block virus attachment to Vero cells are a major component of the human neutralizing antibody response against dengue virus type 2. J. Med. Virol. 45:451–461 [DOI] [PubMed] [Google Scholar]
- 27. Wahala WM, Kraus AA, Haymore LB, Accavitti-Loper MA, de Silva AM. 2009. Dengue virus neutralization by human immune sera: role of envelope protein domain III-reactive antibody. Virology 392:103–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Halstead SB. 2003. Neutralization and antibody-dependent enhancement of dengue viruses. Adv. Virus Res. 60:421–467 [DOI] [PubMed] [Google Scholar]
- 29. Sukupolvi-Petty S, Austin SK, Engle M, Brien JD, Dowd KA, Williams KL, Johnson S, Rico-Hesse R, Harris E, Pierson TC, Fremont DH, Diamond MS. 2010. Structure and function analysis of therapeutic monoclonal antibodies against dengue virus type 2. J. Virol. 84:9227–9239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schieffelin JS, Costin JM, Nicholson CO, Orgeron NM, Fontaine KA, Isern S, Michael SF, Robinson JE. 2010. Neutralizing and non-neutralizing monoclonal antibodies against dengue virus E protein derived from a naturally infected patient. Virol. J. 7:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dejnirattisai W, Jumnainsong A, Onsirisakul N, Fitton P, Vasanawathana S, Limpitikul W, Puttikhunt C, Edwards C, Duangchinda T, Supasa S, Chawansuntati K, Malasit P, Mongkolsapaya J, Screaton G. 2010. Cross-reacting antibodies enhance dengue virus infection in humans. Science 328:745–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Beltramello M, Williams KL, Simmons CP, Macagno A, Simonelli L, Quyen NT, Sukupolvi-Petty S, Navarro-Sanchez E, Young PR, de Silva AM, Rey FA, Varani L, Whitehead SS, Diamond MS, Harris E, Lanzavecchia A, Sallusto F. 2010. The human immune response to Dengue virus is dominated by highly cross-reactive antibodies endowed with neutralizing and enhancing activity. Cell Host Microbe 8:271–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. de Alwis R, Beltramello M, Messer WB, Sukupolvi-Petty S, Wahala WM, Kraus A, Olivarez NP, Pham Q, Brien JD, Tsai WY, Wang WK, Halstead S, Kliks S, Diamond MS, Baric R, Lanzavecchia A, Sallusto F, de Silva AM. 2011. In-depth analysis of the antibody response of individuals exposed to primary dengue virus infection. PLoS Negl. Trop. Dis. 5:e1188 doi:10.1371/journal.pntd.0001188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. de Alwis R, Smith SA, Olivarez NP, Messer WB, Huynh JP, Wahala WM, White LJ, Diamond MS, Baric RS, Crowe JE, Jr, de Silva AM. 2012. Identification of human neutralizing antibodies that bind to complex epitopes on dengue virions. Proc. Natl. Acad. Sci. U. S. A. 109:7439–7444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34a. Isern S, Costin JM, Zaitseva E, Kahle KM, Nicholson CO, Rowe DK, Graham AS, Bazzone LE, Hogancamp G, Figueroa Sierra M, Fong RH, Yang S-T, Lin L, Robinson JE, Doranz BJ, Chernomordik LV, Michael SF, Schieffelin JS. 2012. Abstr 2nd Antivirals Congress, Cambridge, MA, abstr P-64 [Google Scholar]
- 35. Cuzzubbo AJ, Endy TP, Nisalak A, Kalayanarooj S, Vaughn DW, Ogata SA, Clements DE, Devine PL. 2001. Use of recombinant envelope proteins for serological diagnosis of Dengue virus infection in an immunochromatographic assay. Clin. Diagn. Lab. Immunol. 8:1150–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Robinson JE, Holton D, Liu J, McMurdo H, Murciano A, Gohd R. 1990. A novel enzyme-linked immunosorbent assay (ELISA) for the detection of antibodies to HIV-1 envelope glycoproteins based on immobilization of viral glycoproteins in microtiter wells coated with concanavalin A. J. Immunol. Methods 132:63–71 [DOI] [PubMed] [Google Scholar]
- 37. Robinson JE, Holton D, Pacheco-Morell S, Liu J, McMurdo H. 1990. Identification of conserved and variant epitopes of human immunodeficiency virus type 1 (HIV-1) gp120 by human monoclonal antibodies produced by EBV-transformed cell lines. AIDS Res. Hum. Retrovir. 6:567–579 [DOI] [PubMed] [Google Scholar]
- 38. Xiang SH, Doka N, Choudhary RK, Sodroski J, Robinson JE. 2002. Characterization of CD4-induced epitopes on the HIV type 1 gp120 envelope glycoprotein recognized by neutralizing human monoclonal antibodies. AIDS Res. Hum. Retrovir. 18:1207–1217 [DOI] [PubMed] [Google Scholar]
- 39. Pinna D, Corti D, Jarrossay D, Sallusto F, Lanzavecchia A. 2009. Clonal dissection of the human memory B-cell repertoire following infection and vaccination. Eur. J. Immunol. 39:1260–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liao HX, Levesque MC, Nagel A, Dixon A, Zhang R, Walter E, Parks R, Whitesides J, Marshall DJ, Hwang KK, Yang Y, Chen X, Gao F, Munshaw S, Kepler TB, Denny T, Moody MA, Haynes BF. 2009. High-throughput isolation of immunoglobulin genes from single human B cells and expression as monoclonal antibodies. J. Virol. Methods 158:171–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Puttikhunt C, Ong-Ajchaowlerd P, Prommool T, Sangiambut S, Netsawang J, Limjindaporn T, Malasit P, Kasinrerk W. 2009. Production and characterization of anti-dengue capsid antibodies suggesting the N terminus region covering the first 20 amino acids of dengue virus capsid protein is predominantly immunogenic in mice. Arch. Virol. 154:1211–1221 [DOI] [PubMed] [Google Scholar]
- 42. Costin JM, Jenwitheesuk E, Lok SM, Hunsperger E, Conrads KA, Fontaine KA, Rees CR, Rossmann MG, Isern S, Samudrala R, Michael SF. 2010. Structural optimization and de novo design of dengue virus entry inhibitory peptides. PLoS Negl. Trop. Dis. 4:e721 doi:10.1371/journal.pntd.0000721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nicholson CO, Costin JM, Rowe DK, Lin L, Jenwitheesuk E, Samudrala R, Isern S, Michael SF. 2011. Viral entry inhibitors block dengue antibody-dependent enhancement in vitro. Antivir. Res. 89:71–74 [DOI] [PubMed] [Google Scholar]
- 44. Chutinimitkul S, Payungporn S, Theamboonlers A, Poovorawan Y. 2005. Dengue typing assay based on real-time PCR using SYBR Green I. J. Virol. Methods 129:8–15 [DOI] [PubMed] [Google Scholar]
- 45. Zaitseva E, Yang ST, Melikov K, Pourmal S, Chernomordik LV. 2010. Dengue virus ensures its fusion in late endosomes using compartment-specific lipids. PLoS Pathog. 6:e1001131 doi:10.1371/journal.ppat.1001131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cole KS, Alvarez M, Elliott DH, Lam H, Martin E, Chau T, Micken K, Rowles JL, Clements JE, Murphey-Corb M, Montelaro RC, Robinson JE. 2001. Characterization of neutralization epitopes of simian immunodeficiency virus (SIV) recognized by rhesus monoclonal antibodies derived from monkeys infected with an attenuated SIV strain. Virology 290:59–73 [DOI] [PubMed] [Google Scholar]
- 47. Robinson JE, Cole KS, Elliott DH, Lam H, Amedee AM, Means R, Desrosiers RC, Clements J, Montelaro RC, Murphey-Corb M. 1998. Production and characterization of SIV envelope-specific rhesus monoclonal antibodies from a macaque asymptomatically infected with a live SIV vaccine. AIDS Res. Hum. Retrovir. 14:1253–1262 [DOI] [PubMed] [Google Scholar]
- 48. Roehrig JT, Hombach J, Barrett AD. 2008. Guidelines for plaque-reduction neutralization testing of human antibodies to dengue viruses. Viral Immunol. 21:123–132 [DOI] [PubMed] [Google Scholar]
- 49. Gentry MK, Henchal EA, McCown JM, Brandt WE, Dalrymple JM. 1982. Identification of distinct antigenic determinants on dengue-2 virus using monoclonal antibodies. Am. J. Trop. Med. Hyg. 31:548–555 [DOI] [PubMed] [Google Scholar]
- 50. Henchal EA, McCown JM, Burke DS, Seguin MC, Brandt WE. 1985. Epitopic analysis of antigenic determinants on the surface of dengue-2 virions using monoclonal antibodies. Am. J. Trop. Med. Hyg. 34:162–169 [DOI] [PubMed] [Google Scholar]
- 51. Sukupolvi-Petty S, Austin SK, Purtha WE, Oliphant T, Nybakken GE, Schlesinger JJ, Roehrig JT, Gromowski GD, Barrett AD, Fremont DH, Diamond MS. 2007. Type- and subcomplex-specific neutralizing antibodies against domain III of dengue virus type 2 envelope protein recognize adjacent epitopes. J. Virol. 81:12816–12826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cockburn JJ, Navarro Sanchez ME, Goncalvez AP, Zaitseva E, Stura EA, Kikuti CM, Duquerroy S, Dussart P, Chernomordik LV, Lai CJ, Rey FA. 2012. Structural insights into the neutralization mechanism of a higher primate antibody against dengue virus. EMBO J. 31:767–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ayala-Nunez NV, Wilschut J, Smit JM. 2011. Monitoring virus entry into living cells using DiD-labeled dengue virus particles. Methods 55:137–143 [DOI] [PubMed] [Google Scholar]
- 54. van der Schaar HM, Rust MJ, Waarts BL, van der Ende-Metselaar H, Kuhn RJ, Wilschut J, Zhuang X, Smit JM. 2007. Characterization of the early events in dengue virus cell entry by biochemical assays and single-virus tracking. J. Virol. 81:12019–12028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen Y, Maguire T, Hileman RE, Fromm JR, Esko JD, Linhardt RJ, Marks RM. 1997. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat. Med. 3:866–871 [DOI] [PubMed] [Google Scholar]
- 56. Dowd KA, Jost CA, Durbin AP, Whitehead SS, Pierson TC. 2011. A dynamic landscape for antibody binding modulates antibody-mediated neutralization of West Nile virus. PLoS Pathog. 7:e1002111 doi:10.1371/journal.ppat.1002111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Thompson BS, Moesker B, Smit JM, Wilschut J, Diamond MS, Fremont DH. 2009. A therapeutic antibody against West Nile virus neutralizes infection by blocking fusion within endosomes. PLoS Pathog. 5:e1000453 doi:10.1371/journal.ppat.1000453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vogt MR, Moesker B, Goudsmit J, Jongeneelen M, Austin SK, Oliphant T, Nelson S, Pierson TC, Wilschut J, Throsby M, Diamond MS. 2009. Human monoclonal antibodies against West Nile virus induced by natural infection neutralize at a postattachment step. J. Virol. 83:6494–6507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chernomordik LV, Kozlov MM. 2003. Protein-lipid interplay in fusion and fission of biological membranes. Annu. Rev. Biochem. 72:175–207 [DOI] [PubMed] [Google Scholar]
- 60. Cohen FS, Melikyan GB. 2004. The energetics of membrane fusion from binding, through hemifusion, pore formation, and pore enlargement. J. Membr. Biol. 199:1–14 [DOI] [PubMed] [Google Scholar]
- 61. Lok SM, Kostyuchenko V, Nybakken GE, Holdaway HA, Battisti AJ, Sukupolvi-Petty S, Sedlak D, Fremont DH, Chipman PR, Roehrig JT, Diamond MS, Kuhn RJ, Rossmann MG. 2008. Binding of a neutralizing antibody to dengue virus alters the arrangement of surface glycoproteins. Nat. Struct. Mol. Biol. 15:312–317 [DOI] [PubMed] [Google Scholar]
- 62. Teoh EP, Kukkaro P, Teo EW, Lim AP, Tan TT, Yip A, Schul W, Aung M, Kostyuchenko VA, Leo YS, Chan SH, Smith KG, Chan AH, Zou G, Ooi EE, Kemeny DM, Tan GK, Ng JK, Ng ML, Alonso S, Fisher D, Shi PY, Hanson BJ, Lok SM, MacAry PA. 2012. The structural basis for serotype-specific neutralization of dengue virus by a human antibody. Sci. Transl. Med. 4:139ra183 doi:10.1126/scitranslmed.3003888 [DOI] [PubMed] [Google Scholar]
- 63. Cherrier MV, Kaufmann B, Nybakken GE, Lok SM, Warren JT, Chen BR, Nelson CA, Kostyuchenko VA, Holdaway HA, Chipman PR, Kuhn RJ, Diamond MS, Rossmann MG, Fremont DH. 2009. Structural basis for the preferential recognition of immature flaviviruses by a fusion-loop antibody. EMBO J. 28:3269–3276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hammar L, Markarian S, Haag L, Lankinen H, Salmi A, Cheng RH. 2003. Prefusion rearrangements resulting in fusion peptide exposure in Semliki forest virus. J. Biol. Chem. 278:7189–7198 [DOI] [PubMed] [Google Scholar]
- 65. Hashem AM, Van Domselaar G, Li C, Wang J, She YM, Cyr TD, Sui J, He R, Marasco WA, Li X. 2010. Universal antibodies against the highly conserved influenza fusion peptide cross-neutralize several subtypes of influenza A virus. Biochem. Biophys. Res. Commun. 403:247–251 [DOI] [PubMed] [Google Scholar]
- 66. Goncalvez AP, Engle RE, St Claire M, Purcell RH, Lai CJ. 2007. Monoclonal antibody-mediated enhancement of dengue virus infection in vitro and in vivo and strategies for prevention. Proc. Natl. Acad. Sci. U. S. A. 104:9422–9427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Goncalvez AP, Purcell RH, Lai CJ. 2004. Epitope determinants of a chimpanzee Fab antibody that efficiently cross-neutralizes dengue type 1 and type 2 viruses map to inside and in close proximity to fusion loop of the dengue type 2 virus envelope glycoprotein. J. Virol. 78:12919–12928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pierson TC, Xu Q, Nelson S, Oliphant T, Nybakken GE, Fremont DH, Diamond MS. 2007. The stoichiometry of antibody-mediated neutralization and enhancement of West Nile virus infection. Cell Host Microbe 1:135–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Thaisomboonsuk BK, Clayson ET, Pantuwatana S, Vaughn DW, Endy TP. 2005. Characterization of dengue-2 virus binding to surfaces of mammalian and insect cells. Am. J. Trop. Med. Hyg. 72:375–383 [PubMed] [Google Scholar]
- 70. Watterson D, Kobe B, Young PR. 2012. Residues in domain III of the dengue virus envelope glycoprotein involved in cell-surface glycosaminoglycan binding. J. Gen. Virol. 93:72–82 [DOI] [PubMed] [Google Scholar]
- 71. Hidari KI, Suzuki T. 2011. Dengue virus receptor. Trop. Med. Health 39:37–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Johnson G, Wu TT. 1998. Preferred CDRH3 lengths for antibodies with defined specificities. Int. Immunol. 10:1801–1805 [DOI] [PubMed] [Google Scholar]
- 73. Schroeder HW., Jr 2006. Similarity and divergence in the development and expression of the mouse and human antibody repertoires. Dev. Comp. Immunol. 30:119–135 [DOI] [PubMed] [Google Scholar]
- 74. Hwang WY, Foote J. 2005. Immunogenicity of engineered antibodies. Methods 36:3–10 [DOI] [PubMed] [Google Scholar]
- 75. Thomas SJ, Nisalak A, Anderson KB, Libraty DH, Kalayanarooj S, Vaughn DW, Putnak R, Gibbons RV, Jarman R, Endy TP. 2009. Dengue plaque reduction neutralization test (PRNT) in primary and secondary dengue virus infections: how alterations in assay conditions impact performance. Am. J. Trop. Med. Hyg. 81:825–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bachmann MF, Kalinke U, Althage A, Freer G, Burkhart C, Roost H, Aguet M, Hengartner H, Zinkernagel RM. 1997. The role of antibody concentration and avidity in antiviral protection. Science 276:2024–2027 [DOI] [PubMed] [Google Scholar]
- 77. Beckett CG, Tjaden J, Burgess T, Danko JR, Tamminga C, Simmons M, Wu SJ, Sun P, Kochel T, Raviprakash K, Hayes CG, Porter KR. 2011. Evaluation of a prototype dengue-1 DNA vaccine in a phase 1 clinical trial. Vaccine 29:960–968 [DOI] [PubMed] [Google Scholar]
- 78. Durbin AP, Whitehead SS. 2010. Dengue vaccine candidates in development. Curr. Topics Microbiol. Immunol. 338:129–143 [DOI] [PubMed] [Google Scholar]
- 79. Guirakhoo F, Kitchener S, Morrison D, Forrat R, McCarthy K, Nichols R, Yoksan S, Duan X, Ermak TH, Kanesa-Thasan N, Bedford P, Lang J, Quentin-Millet MJ, Monath TP. 2006. Live attenuated chimeric yellow fever dengue type 2 (ChimeriVax-DEN2) vaccine: phase I clinical trial for safety and immunogenicity: effect of yellow fever pre-immunity in induction of cross neutralizing antibody responses to all 4 dengue serotypes. Hum. Vaccin. 2:60–67 [DOI] [PubMed] [Google Scholar]
- 80. Guy B, Saville M, Lang J. 2010. Development of Sanofi Pasteur tetravalent dengue vaccine. Hum. Vaccin. 6:696–705 [DOI] [PubMed] [Google Scholar]
- 81. Kitchener S, Nissen M, Nasveld P, Forrat R, Yoksan S, Lang J, Saluzzo JF. 2006. Immunogenicity and safety of two live-attenuated tetravalent dengue vaccine formulations in healthy Australian adults. Vaccine 24:1238–1241 [DOI] [PubMed] [Google Scholar]
- 82. Morrison D, Legg TJ, Billings CW, Forrat R, Yoksan S, Lang J. 2010. A novel tetravalent dengue vaccine is well tolerated and immunogenic against all 4 serotypes in flavivirus-naive adults. J. Infect. Dis. 201:370–377 [DOI] [PubMed] [Google Scholar]
- 83. Poo J, Galan F, Forrat R, Zambrano B, Lang J, Dayan GH. 2011. Live-attenuated tetravalent dengue vaccine in dengue-naive children, adolescents, and adults in Mexico City: randomized controlled phase 1 trial of safety and immunogenicity. Pediatr. Infect. Dis. J. 30:e9–e17 doi:10.1097/INF.0b013e3181fe05af [DOI] [PubMed] [Google Scholar]
- 84. Raviprakash K, Wang D, Ewing D, Holman DH, Block K, Woraratanadharm J, Chen L, Hayes C, Dong JY, Porter K. 2008. A tetravalent dengue vaccine based on a complex adenovirus vector provides significant protection in rhesus monkeys against all four serotypes of dengue virus. J. Virol. 82:6927–6934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Simasathien S, Thomas SJ, Watanaveeradej V, Nisalak A, Barberousse C, Innis BL, Sun W, Putnak, Eckels KH, Hutagalung Y, Gibbons RV, Zhang C, De La Barrera R, Jarman RG, Chawachalasai W, Mammen MP., Jr 2008. Safety and immunogenicity of a tetravalent live-attenuated dengue vaccine in flavivirus naive children. Am. J. Trop. Med. Hyg. 78:426–433 [PubMed] [Google Scholar]
- 86. Sun W, Cunningham D, Wasserman SS, Perry J, Putnak JR, Eckels KH, Vaughn DW, Thomas SJ, Kanesa-Thasan N, Innis BL, Edelman R. 2009. Phase 2 clinical trial of three formulations of tetravalent live-attenuated dengue vaccine in flavivirus-naive adults. Hum. Vaccin. 5:33–40 [DOI] [PubMed] [Google Scholar]
- 87. Modis Y, Ogata S, Clements D, Harrison SC. 2005. Variable surface epitopes in the crystal structure of dengue virus type 3 envelope glycoprotein. J. Virol. 79:1223–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]