

Abstract
Retroviruses integrate into cellular DNA nonrandomly. Lentiviruses such as human immunodeficiency virus type 1 (HIV-1) favor the bodies of active genes and gene-enriched transcriptionally active regions of chromosomes. The interaction between lentiviral integrase and the cellular protein lens epithelium-derived growth factor (LEDGF)/p75 underlies the targeting of gene bodies, whereas recent research has highlighted roles for the HIV-1 capsid (CA) protein and cellular factors implicated in viral nuclear import, including transportin 3 (TNPO3) and nucleoporin 358 (NUP358), in the targeting of gene-dense regions of chromosomes. Here, we show that CA mutations, which include the substitution of Asp for Asn74 (N74D), significantly reduce the dependency of HIV-1 on LEDGF/p75 during infection and that this difference correlates with the efficiency of viral DNA integration. The distribution of integration sites mapped by Illumina sequencing confirms that the N74D mutation reduces integration into gene-rich regions of chromosomes and gene bodies and reveals previously unrecognized roles for NUP153 (another HIV-1 cofactor implicated in viral nuclear import) and LEDGF/p75 in the targeting of the viral preintegration complex to gene-dense regions of chromatin. A role for the CA protein in determining the dependency of HIV-1 on LEDGF/p75 during infection highlights a connection between the viral capsid and chromosomal DNA integration.
INTRODUCTION
Reverse transcription, which converts the single-stranded RNA genome into linear double-stranded DNA, and the subsequent integration of this DNA molecule into a host cell chromosome are key steps in the early phase of the human immunodeficiency virus type 1 (HIV-1) replication cycle. The viral enzymes reverse transcriptase (RT) and integrase (IN), along with the genomic RNA, enter the cell enclosed in a cone-shaped shell or core comprised of the viral capsid (CA) protein, and reverse transcription is linked to the partial dissolution of the core through a process known as uncoating (1). 3′ processing of the viral DNA ends by IN converts the reverse transcription complex into the preintegration complex (PIC) (2, 3). The HIV-1 PIC is actively imported into the nucleus (4, 5) through nuclear pore complexes (NPCs). In the nucleus, the PICs locate to sites of chromosomal DNA for IN-mediated integration (see reference 6 for a recent overview of retroviral reverse transcription, nuclear import, and integration).
Retroviral integration is nonrandom, and different viruses have different integration specificities (reviewed in reference 7). Local DNA sequence preferences, which typically consist of ∼12-bp palindromes, have a modest effect on integration site selection (8–10). PICs also sense certain aspects of higher-order chromatin structure during integration. Lentiviruses, which include HIV-1, target the bodies of active genes and favor transcriptionally active gene-dense regions of chromosomes (11). The integration of Moloney murine leukemia virus (MLV), a gammaretrovirus, also favors active gene-dense regions, although to a lesser extent than does HIV-1 (12). In contrast to the lentiviruses, MLV preferentially integrates into promoter regions and affiliated CpG islands rather than the internal regions of genes (13). Viruses derived from other retroviral genera, including alpharetroviruses (14, 15), deltaretroviruses (8), and spumaviruses (16, 17), have less specificity for chromatin structural features than lentiviruses or MLV, showing weak preferences for gene promoter regions.
Although the mechanisms by which most retroviruses target chromatin during integration are unknown, the binding of lentiviral IN protein to the host binding protein lens epithelium-derived growth factor (LEDGF)/transcriptional coactivator p75 in large part defines the preference of this genus for the bodies of active genes. Cells depleted for LEDGF/p75 expression by RNA interference (RNAi) (18, 19) or gene knockout (20–22) support significantly lower overall HIV-1 integration than controls. The proviruses that do form under these conditions are found much less frequently in active genes (20–23), with an associated rise in integrations near promoters and affiliated CpG islands (20–22). LEDGF/p75 has a C-terminally located IN-binding domain (IBD) (24, 25) and N-terminal elements that bind chromatin (26, 27). Novel fusion proteins comprising the LEDGF/p75 IBD and foreign chromatin binding domains redirect HIV-1 to integrate at positions where the heterologous binding partner preferentially binds, demonstrating a direct role for the host factor in targeting lentiviral integration (28–30). Neither the preference for the local DNA sequence at the site of HIV-1 integration (20–22) nor the preference for gene-dense regions of chromosomes (12, 20) seems to be influenced by LEDGF/p75. In vitro experiments with prototype foamy virus showed that mutations in IN affected the preference for the local DNA sequence at the site of integration (31). This result suggests that the local target site sequence preferences of other retroviruses, including HIV-1, might also be dictated by IN. Recent work has suggested that viral and cellular factors implicated in PIC nuclear import, including CA (32, 33) and the host proteins transportin 3 (TNPO3) and nucleoporin 358 (NUP358) (12), play a role in determining the preference of HIV-1 to target gene-dense regions of chromosomes during integration.
Work with MLV–HIV-1 (MHIV) chimeric viruses that contain MLV proteins in place of their HIV-1 counterparts first showed that Gag structural determinants can play a role in HIV-1 integration targeting (12, 34). From the N terminus, HIV-1 Gag is composed of matrix (MA), CA, spacer peptide 1 (SP1), nucleocapsid (NC), SP2, and p6 proteins, whereas MLV is composed of MA, p12, CA, and NC (Fig. 1A). The MHIV-mMA12CA chimera, which has MLV MA, p12, and CA in place of HIV-1 MA and CA (Fig. 1A), integrated into genes at a frequency that was intermediate to those of HIV-1 and MLV (34) and, in contrast to the parental viruses, did not favor gene-dense regions of chromatin (12). The gene density targeting profile of MHIV-mMA12CA was similar to what was seen with HIV-1 when either TNPO3 or NUP358 was depleted from HeLa cells by RNAi (12). Work with additional MHIV chimeras, including MHIV-mMA12 (Fig. 1A), and with CA missense mutations, including N74D, revealed CA as the dominant genetic determinant of TNPO3 (35–38), NUP358 (32, 37), and NUP153 (32, 37, 39) dependency during HIV-1 infection. The N74D mutation, in particular, ablated integration targeting of the PIC to gene-dense regions of chromosomes (32), highlighting a link between PIC nuclear import, CA, and integration targeting. Here, we show that CA mutations alter the dependency of HIV-1 on LEDGF/p75 and that the IN-binding host factor concordantly influences the targeting of HIV-1 PICs to gene-dense regions of chromosomes. Furthermore, our results reveal a subsidiary role for NUP153 in the targeting of transcriptionally active regions of chromosomes during HIV-1 integration.
Fig 1.

Both CA and IN affect the dependence of HIV-1 infection on LEDGF/p75. (A) Schematic drawings of the parental HIV-1 (white boxes) and MLV (gray boxes) Gag and Pol proteins and the chimeric viruses. PR, protease (other abbreviations are defined in the text). Note that the MLV Gag p12 protein does not have a corresponding counterpart in HIV-1. (B) The absolute viral titers (left graph: black, E1f/+ cells; white, E2−/− cells) and relative viral titers of HIV-1, MLV, and the MHIV chimera in E2−/− knockout cells expressed as a percentage of the E1f/+ control cell infections (right graph) are shown. Data are means ± standard errors of the mean (SEM) for two independent experiments, each conducted in duplicate. (Right graph) Statistical comparison of relative titers versus WT HIV-1 (notation above bar) or between chimera viruses (brackets) was performed by paired two-tailed Student's t tests. n.s., not significant (P > 0.05); *, P < 0.05; **, P < 0.005. (C) Same as for panel B, except that the E6f/f WT and E6−/− knockout MEF cell pair was analyzed. In this case, the comparison of MHIV-mMA12CA to WT yielded a P value of 0.052.
MATERIALS AND METHODS
Plasmid constructs.
Plasmids that encode single-round luciferase reporter viruses included the MLV-HIVLAI chimera (33, 40), pNLX.Luc(R-) (41), and pHI-Luc (42). The backbone of pNLX.Luc(R-) was modified by BstEII-XmaI digestion, and the ends were repaired with the Klenow fragment of Escherichia coli DNA polymerase and religated to yield pNLX.Luc(R-)ΔAvrII. HIV-1 CA mutations were generated by site-directed mutagenesis of the HIV-1NL4-3-based packaging construct pHP-dI-N/A (39, 43) or pNLX.Luc(R-)ΔAvrII. The pN/N.Luc(R-) plasmid expressed the D64N/D116N IN active-site mutant virus, and the vesicular stomatitis virus G (VSV-G) glycoprotein was expressed from pCG-VSV-G (41). Sequencing was used to show that the PCR-mutated DNAs carried only the desired mutations.
Cells, viruses, and infections.
HEK293T, HeLa, and mouse embryo fibroblast (MEF) cells were propagated in Dulbecco's modified Eagle medium supplemented to contain 10% fetal bovine serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin. There were two matched pairs of Psip1 knockout MEFs: the E1f/+ control and E2−/− knockout and the E6f/f control and E6−/− knockout (22).
Single-round viruses were pseudotyped with the VSV-G envelope following transient transfection of HEK293T cells with two or three different plasmids as described previously (39). The levels of virus production were monitored using a 32P-based assay for exogenous RT activity, and MEFs (1.6 × 104/well) seeded 24 h before infection in 48-well plates were infected in duplicate with 2 × 105 RT counts per minute (cpm) for 24 h to determine single-round viral infectivities. At 48 h postinfection (hpi), cells were processed for duplicate luciferase assays, and results were expressed as relative light units (RLU) normalized to the concentration of the total protein in the cell extract (39). MHIV inocula were adjusted so that all infections were within approximately 2 log RLU/μg of control MEFs. Where applicable, cyclosporine (Sigma-Aldrich, St. Louis, MO) was added at the time of infection to a final concentration of 5 μM. Normalized infectivity data were log transformed, and statistical analyses were performed by paired two-tailed Student's t tests.
Quantitative PCR.
MEFs (1.6 × 105/well) seeded 24 h before infection in 6-well plates were incubated with 2 × 107 RT cpm of DNase-treated wild type (WT) or CA mutant virus for 2 h, after which the cells were washed with phosphate-buffered saline and replated into separate wells of a 12-well plate. DNA was extracted from the infected cells at various times using the QIAamp DNA minikit (Qiagen, Valencia, CA), and the DNA samples were analyzed in duplicate or triplicate by quantitative PCR (Q-PCR) to determine the total amount of HIV-1 DNA at 7 hpi. The amount of integrated proviruses was determined at 48 hpi using nested BBL-PCR (an assay that uses mouse B1, B2, and long interspersed element 1 [LINE-1] repeats) essentially as described previously (22, 39). DNA was prepared from parallel infections conducted in the presence of 25 μM azidothymidine (AZT) to determine the background residual transfected plasmid DNA in the Q-PCR assay. This background was subtracted from the experimental values (AZT-treated samples averaged 1.8% of the total and 7.7% of the BBL-PCR peak values). Plasmid DNA [pNLX.Luc(R-)] was used to generate the HIV-1 DNA standard curve, and a polyclonal population of E6f/f MEFs infected at a relatively low multiplicity of infection with an HIV-1-based pHI-puro single-round virus and selected for 3 weeks in 2 μg/ml puromycin was used to prepare the cellular DNA for the integration standard (39). Twofold dilutions of this DNA were mixed with DNA prepared from uninfected E6f/f cells to prepare the standard curve. The Q-PCR primers and probes for total HIV-1 detection have been described (39). The BBL-PCR primers and probes were also as described (22), except that AE3014 and AE3013 replaced the first- and second-round primers AE2257 and AE989, respectively, to increase the specificities of the amplifications (44).
Integration site libraries.
HeLa cells (2.5 × 104) were transfected in 48-well plates with 40 nM short-interfering (si) control or NUP153-targeting RNA 1 (39), pooled (2.5 × 106 cells), and infected 3 days later with 2.5 × 108 RT cpm of WT or N74D mutant virus derived from pNLX.Luc(R-)ΔAvrII. After 2 days, the cells were harvested, and DNA was purified as described above. The methods used to prepare the DNA fragments and to amplify and sequence the integration sites were similar to those described by Gillet et al. (45). Briefly, 5 μg of the DNA was sheared into ∼300- to 500-bp fragments by adaptive focused acoustics (Covaris Inc., Woburn, MA) and purified using AMPure XP magnetic beads (Beckman Coulter Genomics, Danvers, MA). The sheared DNA was end repaired, deoxyribosyladenine (dA) tailed, and ligated to a double-stranded DNA linker as described in Illumina's sequencing protocol. Both the first- and the second-round amplifications were done in multiple aliquoted reaction mixtures to increase the diversity of the integration site libraries. The resulting PCR products were sequenced using standard chemistry on an Illumina GA2x machine. Sequences that contained the 3′processed viral DNA through the canonical CA dinucleotide on one end and the ligated linker on the opposing end and that matched a genomic location at >95% identity for a minimal stretch of 20 bp were counted as HIV-1 integration sites. The genomic sequences were parsed for features such as genes, CpG islands, and transcriptional activity as described previously (28). Integration site libraries prepared from HIV-1-infected E1f/+ control and E2−/− knockout MEFs (22) were also analyzed by Illumina sequencing.
RESULTS AND DISCUSSION
MLV CA confers loss of LEDGF/p75 dependency during HIV-1 infection.
LEDGF/p75, which binds directly to lentiviral IN proteins, is an important cellular cofactor for HIV-1 integration (reviewed in references 46 and 47). Due to its central role in determining the distribution of integrated proviruses in chromosomes, we hypothesized that HIV-1-based constructs that carry changes in proteins other than IN, and that have altered integration site preferences, might also have altered infectivity profiles in cells depleted for LEDGF/p75. The MHIV-mMA12CA chimeric virus, which carries the section of the MLV gag gene that encodes MA, p12, and CA (Fig. 1A), is one such virus, as its integration site distribution differed significantly from each of its parents (12, 34). MEF cell lines in which the Psip1 gene (22), which encodes LEDGF/p75 (48), was inactivated were challenged with MHIV-mMA12CA and both parental viruses. Two different pairs of MEF cell lines, E1f/+, which expresses the LEDGF/p75 protein and the littermate-matched E2−/− knockout, as well as the LEDGF/p75-expressing cell line E6f/f and its matched E6−/− knockout, were used in the initial infectivity and integration experiments. Additional chimeric viruses that contained MLV IN in either the HIV-1 (MHIV-mIN) or MHIV-mMA12CA (MHIV-mMA12CAmIN) background or carried MLV MA and p12 in place of HIV-1 MA (MHIV-mMA12) (33, 40) were included to determine the contributions of these Gag determinants as well as IN under these experimental conditions (Fig. 1A). Because the infectivities of some of the chimeric viruses, especially those that express the MLV IN protein, are significantly lower than that of HIV-1 (33, 40), preliminary experiments were conducted to define the levels of the viral inocula that infected the LEDGF/p75-expressing control cells within approximately 2 orders of magnitude of the level needed for WT HIV-1 to infect them. Appropriate volumes of the viral stocks were then used to infect matched pairs of LEDGF/p75-expressing and knockout cells. The resulting absolute levels of viral infection, which are plotted with respect to the level of WT HIV-1 infection of LEDGF/p75-expressing cells (Fig. 1B and C, left panels), were regraphed as relative viral titers (percent infectivity in knockout cells compared to that in the matched LEDGF/p75-expressing cell line) (Fig. 1B and C, right panels) to simplify the interpretation of which mutation(s) alters the dependency on LEDGF/p75.
As shown previously (22), HIV-1 infectivity was significantly reduced by the Psip1 knockout, and MLV infectivity was either unaffected (in the E1f/+ and E2−/− cell pair) or affected to a much smaller extent than was HIV-1 infectivity (Fig. 1B and C, left panels, compare white and black bars). The enhanced HIV-1 infectivity defect in the E6−/− knockout cells (Fig. 1C) compared to E2−/− knockout cells correlates with a virus-specific transcriptional defect in the E6−/− cells (Y. Koh and A. Engelman, unpublished observations; also discussed below). The relative titers of the MHIV-mMA12 virus were very similar to those of HIV-1LAI in both sets of cells, suggesting that the HIV-1 MA protein plays no significant role in LEDGF/p75 dependency. LEDGF/p75 does not interact with or bind to MLV IN (49, 50), and as expected, the relative titers of MHIV-mIN and MLV were not significantly altered by the Psip1 knockout (Fig. 1B and C). The infectivity of MHIV-mMA12CA, which expresses HIV-1 IN and MLV MA and CA, was noticeably less dependent on LEDGF/p75 than either HIV-1LAI or MHIV-mMA12 (Fig. 1B and C). This reduced dependency of MHIV-mMA12CA on LEDGF/p75 was seen over a 100-fold range in the virus inoculum (data not shown). However, we noted that the MHIV-mMA12CA chimera was significantly more dependent on LEDGF/p75 than was the MHIV-mIN and that the addition of MLV IN to MHIV-mMA12CA caused significant further reductions in the dependency of the Gag chimera virus on the integration cofactor (Fig. 1B and C). These data confirm the primary role of IN in the dependency of HIV-1 infection on LEDGF/p75 and reveal a significant but secondary role for the CA protein.
CA missense mutations or cyclosporine treatment reduces HIV-1 dependency on LEDGF/p75.
Our chimeric viral analysis revealed a role for the CA protein in determining the dependency of HIV-1 infection on LEDGF/p75. Because the HIV-based MHIV-mMA12CA chimera contained the heterologous MLV CA protein, we next analyzed missense mutant viruses to test whether minor changes in CA would alter HIV-1 dependency on LEDGF/p75. Eight CA mutations were selected based on their effects on the dependency of HIV-1 infection on host factors cyclophilin A (CypA), TNPO3, NUP358, and NUP153. The G89V and P90A mutations are within the CypA binding loop of the CA N-terminal domain and reduce CA binding to CypA (51, 52). These mutations also affect the dependency of the virus on TNPO3 (32, 35), NUP358 (32), and NUP153 (39). Cyclosporine (CsA) is a small molecule that binds the CypA active site (53), and the A92E and G94D mutations confer CsA dependence on HIV-1 replication in certain cell types (54, 55); the mutations marginally reduce TNPO3 dependency (35) and cause a modest increase in the dependency of infection on NUP153 (39). Three of the other mutant viruses have been shown to affect the stability of the assembled capsid core. The E45A mutation has a stabilizing effect, and T54A/N57A and Q63A/Q67A have destabilizing effects on the capsid core (56, 57). The infectivities of each of these mutants displayed less dependence on TNPO3 (35, 37, 38) or NUP153 (37, 39) than the WT virus. The final CA mutation, N74D, which was selected because it conferred resistance to the cytoplasmic restriction factor CPSF6-358 (37), reduced the dependency of HIV-1 infection on TNPO3 (32, 35, 37, 38), NUP358 (32, 37), and NUP153 (32, 37, 39). MEF cells were challenged with equal RT cpm of WT and mutant viral constructs that express luciferase. Although Thys et al. have indicated that pseudotyping with VSV-G significantly reduced the dependency of the N74D mutation on TNPO3 (58), subsequent reports showed that TNPO3 knockdown affected CA mutants carrying VSV-G or HIV-1 envelope glycoproteins similarly (32, 35, 38, 39). The IN active-site mutant virus D64N/D116N (N/N) was included as a control because it provides a measure of viral titer in the absence of functional IN activity due to weak expression from unintegrated viral DNA and/or low levels of host-mediated DNA integration (39, 42).
Previously published reports showed that the Psip1 knockout significantly decreased lentiviral DNA integration (20, 22); however, the relative titer of the N/N mutant virus in the E2−/−-E1f/+ MEF cell pair was unaffected (Fig. 2B, black bar) because the absolute titer of this mutant virus (∼0.4% of WT) (Fig. 2A, black bar) is independent of IN-mediated DNA recombination. The reduction in the relative titer of the N/N mutant virus in the E6−/−-E6f/f cell pair (Fig. 2D, black bar) is consistent with a transcriptional defect that we have identified for the HIV-1 long terminal repeat (LTR) in the E6−/− cell line and in a subset of other Psip1 knockout MEFs (Y. Koh and A. Engelman, unpublished data). Five of the tested CA mutants (Q63A/Q67A, N74D, G89V, P90A, and A92E) were less affected by the Psip1 knockout (∼2- to 2.5-fold) than was the WT virus (Fig. 2B and D, compare the black bars). Although the absolute titers of some of the CA mutants, including Q63A/Q67A and G89V, were reduced significantly compared to the WT (Fig. 2A and C, black bars), the dependence of the virus on LEDGF/p75 did not correlate with inherent infectivity; T54A/T57A, which was as dependent on LEDGF/p75 as the WT virus (Fig. 2B and D), had the lowest absolute titer of any of the CA mutants that we tested (∼1% of WT), whereas N74D, which was the least dependent on LEDGF/p75, had the greatest absolute titer (∼82% of WT) (Fig. 2A and C, black bars).
Fig 2.
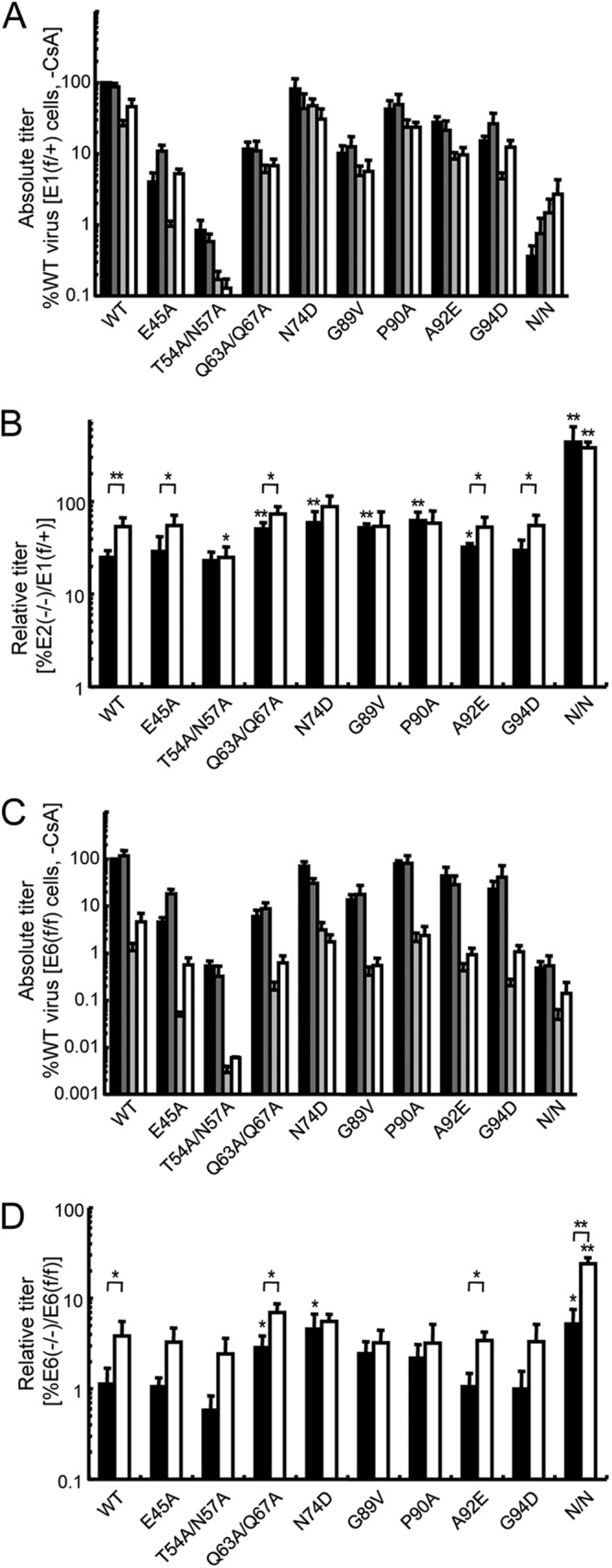
Infectivities of CA missense mutants in the presence and absence of LEDGF/p75. (A) Absolute titers of WT and indicated CA mutant viruses in E1f/+ WT and E2−/− cells. The black and dark-gray bars represent viral infectivities in E1f/+ cells minus versus plus CsA, respectively, expressed as percentages of the WT virus infection in the absence of CsA, which was set at 100%. Light-gray and white bars represent infectivities in E2−/− cells without versus with CsA, respectively. (B) Relative titers of WT and CA mutant viruses in knockout E2−/− cells are expressed as a percentage of their infectivity in control E1f/+ cells. White bars represent infections conducted in the presence of CsA. Results are averages of three to five experiments performed in duplicate, with error bars denoting 95% confidence intervals. Asterisks directly above the bars indicate statistical relevance in comparison to the WT virus (determined by paired two-tailed Student's t tests; *, P < 0.05; **, P < 0.005) without versus with CsA; asterisks above the brackets indicate relevant titer differences for the indicated viruses with versus without CsA. (C and D) Same as for panels A and B except the E6f/f and E6−/− MEF cell pair was analyzed in three independent experiments.
Either including CsA in the cell culture medium, which disrupts the CypA-CA interaction, or using viruses that have G89V or P90A mutations in CA increased the integration of HIV-1 DNA into gene-dense regions of chromosomes (32). The addition of CsA reduced the dependence of the WT virus on LEDGF/p75 by ∼2- to 3.5-fold in the knockout cells (Fig. 2A and C, compare white bar to light-gray bar). CsA also reduced the dependence of many of the CA mutant viruses on LEDGF/p75 (Fig. 2B and D, the statistically significant differences in relative titers are denoted by asterisks above the brackets). These data suggest that either pharmacological or genetic disruption of the CypA-CA interaction can significantly reduce the dependency of HIV-1 infection on LEDGF/p75. However, this relative increase in infectivity in the presence of CsA, or the corresponding increases in relative titers seen with the Q63A/Q67A and N74D mutants, failed to restore infectivity to the level seen with the WT virus in the presence of LEDGF/p75. In contrast, the N74D mutation could essentially completely overcome the requirement for TNPO3 (32, 35, 37, 38), NUP153 (37, 39), or NUP358 (32), host factors implicated in nuclear import that are predictably encountered before the PIC engages chromatin-associated LEDGF/p75.
CA mutations alter HIV-1 DNA integration.
The presence or absence of LEDGF/p75 influences the efficiency of HIV-1 DNA integration as well as the distribution of the residual proviruses with respect to bodies and promoter regions of active genes (46, 47). Prior studies of the effects of CA mutations have focused on the distribution of integration sites (32, 34). To determine if CA mutations might also influence the efficiency of integration, the reverse transcription and integration profiles of two relatively LEDGF/p75-independent CA mutant viruses, MHIV-mMA12CA and N74D (Fig. 1 and 2), were assessed by Q-PCR. Because the N74D mutation and the mMA12CA chimera were constructed in two different HIV-1 backgrounds (HIV-1NL4-3 for N74D and HIV-1LAI for mMA12CA), each WT strain was used as a positive control. Due to the transcriptional defect in the HIV-1 LTR identified in E6−/− knockout cells, the E2−/−-E1f/+ cell pair was used to simplify the interpretation of the results. PCR primers and probes were designed to detect the total amount of HIV-1 DNA (measured at 7 hpi, the approximate peak of reverse transcription [59]) and the number of integrated proviruses (at 48 h). The alu element, which is the chromosomal sequence routinely used in Q-PCR assays to measure HIV-1 integration in human cells, is primate specific (60). In the BBL-PCR assay, the mouse repeat elements B1, B2, and LINE-1 are used to measure the number of proviruses (22). Viral stocks were treated with DNase before the target cells were infected to reduce plasmid carryover from transfected cells. DNAs prepared from cells infected in the presence of 25 μM AZT were analyzed in parallel, and this background was subtracted from the experimental data sets. Absolute levels of DNA synthesis and integration (Fig. 3A and C) were regraphed as relative levels to assess the dependency of DNA formation on LEDGF/p75 (Fig. 3B and D).
Fig 3.

HIV-1 DNA synthesis and integration in the presence and absence of LEDGF/p75. (A) Total HIV-1 DNA levels at 7 hpi, expressed as the percentages of that of the indicated WT virus in E1f/+ cells. (B) The data shown in panel A were regraphed to highlight the DNA levels observed in the knockout cells relative to those in control LEDGF/75-expressing cells. The dotted line represents background values obtained from corresponding AZT-treated samples, which averaged 1.8% of the level seen in untreated cells. (C and D) Same as for panels A and B, except the BBL-PCR assay measured the number of integrated proviruses at 48 hpi; the AZT-plus background in panel D was 7.7% of the uninhibited control. (E) Absolute virus infectivities determined by processing fractions of the infected cells shown in panel A for luciferase activity and protein content at 48 hpi. (F) Relative titers of the viruses in E2−/− versus control E1f/+ cells. Panels A through D show averages and standard errors from duplicate Q-PCR assays that are representative of two independent experiments. Panels E and F average the results of two experiments, with the error bars denoting 95% confidence intervals. Although comparisons of relative amounts of mutant viral DNA (D) and titers (F) to the matched WT controls failed to achieve statistical significance in these experiments, we note that the fractional increase in N74D mutant viral DNA integration versus the WT virus, 2.1-fold (D), virtually matched the fractional difference in N74D-to-WT viral infectivity (1.9-fold) (F).
Consistent with our prior report (22), the total amount of HIV-1 DNA in infected cells at 7 hpi was not significantly affected by the LEDGF/p75 knockout (Fig. 3A). The amount of viral DNA synthesized by the N74D mutant was reduced about 2-fold from the WT viral vector independent of target cell type; however, the amount of DNA synthesized by the MHIV-mMA12CA chimera was reduced by about 2-fold in the knockout cells (Fig. 3A and B). The level of N74D mutant DNA integration in WT cells mirrored the level of reverse transcription (Fig. 3C). Because the level of N74D integration in the knockout cells was similar to the level of the WT virus in these cells, the CA mutant virus revealed about a 2-fold increase in the efficiency of viral DNA integration in the knockout cells relative to the WT virus (Fig. 3C and D). Comparing the absolute values of MHIV-mMA12CA DNA integration to those of its matched WT virus in the two cell types (Fig. 3C) revealed a similar 2-fold increase in the efficiency of mutant viral DNA integration in the knockout cells (Fig. 3D). Moreover, the fractional increase in integration frequency mirrored the increase in the relative titer of the N74D mutant virus (Fig. 3, compare D and F). However, the impact of the MHIV-mMA12CA mutation on the relative infectious titer was approximately 2.4-fold greater than the impact on provirus formation (Fig. 3D to F). From this we inferred that the impact of the N74D mutation on the overall integration frequency determined, in large part, the impact of the mutation on relative infectious titer. The difference seen between the effects on relative titer and integration frequency of MHIV-mMA12CA could be due to the low level of integration of the parental HIV-1LAI control in the knockout cells, which was less than 2-fold above the assay background, or to some other property of this mutant virus, for example its dependence on cell division for infection (12, 33).
Differential effects of the N74D CA mutation and NUP153 and LEDGF/p75 depletion on integration site distribution.
As discussed above, HIV-1 CA missense mutations, including N74D, reduce the dependency of HIV-1 replication and integration on cellular factors implicated in PIC nuclear import, including TNPO3, NUP358, and NUP153 (32, 35, 37–39). The distribution of HIV-1 integration sites was similarly affected by TNPO3 or NUP358 depletion (12) or the N74D mutation (32). We and others previously reported that knockdown of NUP153 reduced HIV-1 infection and integration (39, 61), but the effect of depleting this factor on the distribution of the integration sites has not been reported. Integration site libraries were prepared from NUP153-depleted and control cells, which were infected with WT HIV-1 or an isogenic N74D mutant virus (Fig. 4A). HeLa cells were used for this analysis to simplify a comparison of our results and those of other studies (12, 32, 34). As expected, siRNA-mediated knockdown of NUP153 reduced the titer of the WT virus approximately 10-fold without appreciably affecting the efficiency of infection by the mutant virus (39). Genomic DNA harvested 2 days postinfection was sheared by sonication, repaired, and amplified by ligation-mediated PCR for analysis using Illumina deep sequencing. Resulting sequences from each infection condition were parsed to remove duplicate insertion sites, yielding from approximately 1,300 (WT virus in NUP153 knockdown cells) to 23,000 (N74D virus in siRNA control-transfected cells) unique sites for the four infection conditions. Features of the sites, such as genes, gene density, start sites, and transcriptional activity, were compared among the four sets and to a computer-generated set of 10,000 random sites (Table 1; see Fig. 5 for statistical analyses).
Fig 4.
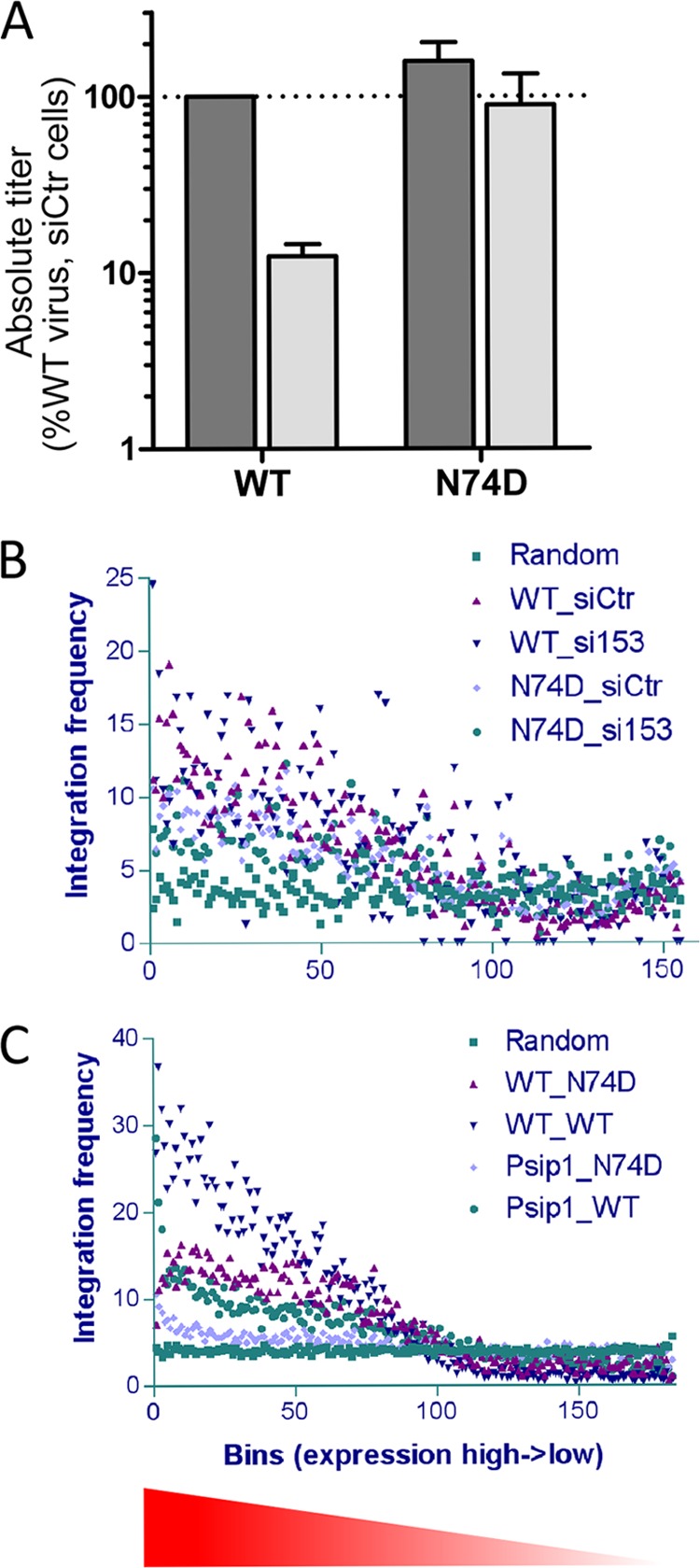
Infections and integration site preferences as functions of gene expression. (A) HeLa cells transfected with siNUP1531 (light-gray bars) or the siRNA control (siCtr) (dark-gray bars) (39) were infected with the indicated virus. The reported titers were calculated based on WT virus infection of siControl-transfected cells, which was set to 100%. Data are the means ± SEM from two independent experiments. NUP153 depletion yielded a significant reduction in WT viral titer (P = 0.026) as calculated by a one-sample one-tailed t test. (B) Integration frequency as a function of gene expression in HeLa cells. The x axis represents >16,000 well-characterized human genes placed into bins of 100 according to level of gene expression. The number of integrations in each bin is plotted for the four different infection conditions (A) (Table 1) versus the random calculated average. (C) As described for panel B, except integration as a function of gene expression was analyzed in mouse cells (Table 2).
Table 1.
Integration sites in control and knockdown HeLa cells
| Genomic feature | Value at each integration sitea |
||||
|---|---|---|---|---|---|
| WT_siCtr (n = 5,390) | WT_si153 (n = 1,318) | N74D_siCtr (n = 22,682) | N74D_si153 (n = 9,610) | Random (n = 10,000) | |
| Transcription unitsb | 3,604 (66.9) | 902 (68.4) | 13,582 (59.9) | 5,504 (57.3) | 3,960 (39.6) |
| No. of promoters (±1 kb) | 40 (0.74) | 15 (1.14) | 111 (0.49) | 49 (0.51) | 167 (1.67) |
| No. of CpG islands (±1 kb) | 17 (0.32) | 2 (0.15) | 19 (0.08) | 10 (0.10) | 81 (0.81) |
| Genes per 1 Mb | 12.2 | 10.9 | 5.7 | 5.5 | 7.7 |
| Transcriptional activityc | 400 | 316 | 120 | 119 | 173 |
Numbers in parentheses are percentage values. siCtr, siControl siRNA (39).
Refseq genes from human genome build hg19, UCSC Genome Bioinformatics website (http://genome.ucsc.edu/).
Total RNA tag counts per Mb surrounding each integration site.
Fig 5.
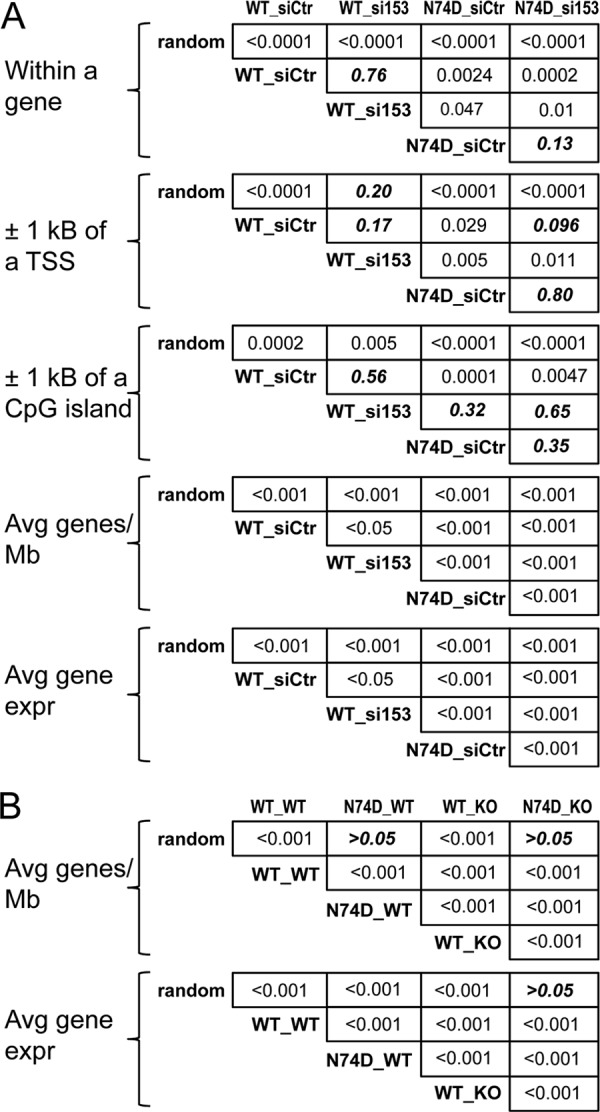
Statistical analyses of HIV-1 integration distributions. (A) Fisher's exact test was utilized to analyze the distributions of integration within genes, and near transcriptional start sites (TSSs) and CpG islands, in the indicated pairwise configurations for the data presented in Table 1. The Mann-Whitney U test (Wilcoxon rank sum test) was used to analyze the number of average genes/Mb and associated transcriptional activity. P values greater than 0.05 are in bold italic type. (B) Mann-Whitney U test results of pairwise analyses of integration into gene-dense regions of chromosomes and associated transcriptional activity in MEF cells (Table 2) are shown. Fisher's exact test of all pairwise combinations of integration values within genes as well as near promoters and CpG islands yielded P values of <0.0001 (matrices not shown).
Consistent with a recent report (32), the N74D mutation markedly reduced the tendency of HIV-1 DNA to integrate into gene-dense, transcriptionally active regions of chromosomes; the integration sites for this mutant had an average value of 5.7 genes/Mb, which is significantly less than the random calculated value of 7.7 (Table 1; Fig. 5A). The N74D mutation also significantly affected the distribution of integration sites with respect to the other evaluated markers, including within genes, near CpG islands or promoters, and transcriptional activity (Table 1; Fig. 4B and 5A). We note that Schaller et al. (32) previously reported similar significant responses for the N74D mutant virus, with the exception of integration near CpG islands. We conjecture that these effects of the N74D mutation on DNA integration site selection may be a contributing factor to the reduced replication capacity of this mutant, which can be observed under conditions of spreading viral replication (32, 62).
Depletion of NUP153 from target cells also caused statistically significant reductions in the tendencies of viral DNA to integrate into gene-dense transcriptionally active regions of chromosomes, although these effects were less dramatic than the effects caused by the N74D mutation (Table 1; Fig. 4B and 5A). Although it is somewhat difficult to compare results across studies, the reduction in the tendency of HIV-1 to target gene-dense regions upon NUP153 depletion would appear to be more modest than the magnitude of the reductions caused by either TNPO3 or NUP358 depletion (12). NUP153 depletion did not significantly influence the distribution of WT HIV-1 integration sites with respect to any of the other evaluated markers (Table 1; Fig. 5A). Because the N74D mutant virus infects control and NUP153 knockdown cells with similar efficiencies (Fig. 4A), the distribution of the integration sites of the mutant virus in cells depleted for NUP153 predictably mimicked the proviral distributions observed with siControl-treated cells (Table 1; Fig. 4B and 5A). We noted that neither NUP153 depletion nor the N74D mutation affected the local DNA sequence specificity at the site of HIV-1 integration (data not shown).
Although prior reports failed to ascribe a role for LEDGF/p75 in the targeting of HIV-1 integration to gene-dense regions of chromosomes (12, 20), we found that mutations in CA can significantly alter the dependency of virus infection on the integration cofactor (Fig. 1 and 2), prompting us to reevaluate the role of LEDGF/p75 in this process. Integration site libraries were prepared from matched LEDGF/p75-expressing E1f/+ and E2−/− knockout cells infected with either the WT or N74D CA mutant virus, which yielded approximately 92,500 (WT infection of Psip1 knockout cells) to 524,000 (N74D virus in WT cells) unique sequences among the four infection conditions (Table 2). As expected (20–22), the integration of the WT virus within genes was reduced significantly, from 73.5% to 49.4%, by the knockout, with concomitant increases in targeting to CpG islands and gene promoter regions (Table 2). The integration distribution of the N74D mutant virus was also significantly affected by the knockout, with integration into genes dropping from 59.0% in the presence of LEDGF/p75 to 39.0% in its absence (Table 2). Thus, although the efficiency of N74D mutant virus integration is less affected by the Psip1 knockout than is the efficiency of WT virus integration, the distribution of mutant viral DNA integration, in large part, remains LEDGF/p75 dependent (Fig. 3C and D). Accordingly, N74D, like the WT virus, favors gene promoter regions and CpG islands in the absence of LEDGF/p75, even though the mutant virus tends to avoid these elements in the presence of LEDGF/p75 (Tables 1 and 2; Fig. 5). As observed in HeLa cells, the N74D mutation significantly reduced the tendency of the PIC to target active genes (Fig. 4B and C) and greatly reduced integration into transcriptionally active, gene-dense regions of mouse chromosomes (Tables 1 and 2; Fig. 5). The Psip1 knockout, moreover, reduced the average gene density for integration of the WT virus from 18.2 to 14.8 genes/Mb, a statistically significant difference (Table 2; Fig. 5B). We suggest that the difference in the significance of targeting of transcriptionally active gene-enriched regions of chromosomes observed here compared to previous reports (12, 20) may be due to different genomic annotations (Marshall et al. [20] utilized mouse genome build mm8, whereas mm9 was utilized here) and/or the deeper coverage in our integration site libraries.
Table 2.
Integration sites in control and LEDGF/p75 knockout MEFs
| Genomic feature | Value at each integration sitea |
||||
|---|---|---|---|---|---|
| WT_WT (n = 273,049) | N74D_WT (n = 524,372) | WT_Psip1−/− (n = 92,475) | N74D_Psip1−/− (n = 440,756) | Random (n = 100,000) | |
| Transcription unitsb,c | 200,760 (73.5) | 309,381 (59.0) | 45,695 (49.4) | 171,574 (39.0) | 33,031 (33.0) |
| No. of promoters (±1 kb)c | 1,866 (0.68) | 2,636 (0.50) | 3,540 (3.83) | 10,065 (2.28) | 1,595 (1.60) |
| No. of CpG islands (±1 kb)c | 1,428 (0.52) | 882 (0.17) | 2,422 (2.62) | 4,884 (1.11) | 728 (0.73) |
| Genes per 1 Mb | 18.2 | 7.7 | 14.8 | 8.0 | 8.7 |
| Transcriptional activityd | 587 | 222 | 464 | 231 | 249 |
Percentage values are indicated in parentheses.
Refseq genes from mouse genome build mm9, UCSC Genome Bioinformatics website (http://genome.ucsc.edu/).
Pairwise comparisons by Fisher's exact test in all cases yielded P values of <0.0001.
Total RNA tag counts per Mb surrounding each integration site.
Conclusions.
This study extends our knowledge of the effects that CA mutations and host cell factors have on the efficiency of HIV-1 integration and distribution of the resulting proviruses. In addition to affecting the targeting of gene-dense regions of chromosomes (32), we have shown that CsA treatment and CA mutations reduced the dependency of the virus on LEDGF/p75 (Fig. 1 and 2), which correlates with their effects on integration efficiency (Fig. 3). The N74D mutation reduced integration into the bodies of active genes and eliminated the targeting of transcriptionally active gene-dense regions of chromosomes, whereas NUP153 played a subsidiary role in the targeting of gene-dense regions (Tables 1 and 2; Fig. 4 and 5). By analyzing relatively large data sets, we have shown that chromatin-associated LEDGF/p75 also plays a role in the targeting of HIV-1 integration into gene-dense regions of chromosomes (Table 2; Fig. 5B). It would appear that LEDGF/p75 plays the dominant role in targeting integration into the bodies of active genes, whereas host factors associated with PIC nuclear import play the predominant role in the targeting of gene-dense regions of chromosomes (12). An analysis of cells depleted for nuclear transport factors by gene knockout, which have been described for NUP358 (63), would help to clarify potential contributions of low levels of nucleocytoplasmic transport proteins to HIV-1 infection and viral DNA integration.
The relative insensitivity of HIV-1 CA mutant viruses to LEDGF/p75 ablation supports at least two potential models. Based on the relatively large gain in infectious titer by the mMA12CA chimeric virus, we suggest that the HIV-1 CA protein, either indirectly or through a direct interaction with IN, impacts the functional interaction between LEDGF/p75 and the viral recombinase. Concordantly, relatively minor CA changes, like the N74D missense mutation, in large part retain the IN-LEDGF/p75 interaction. Our data also raise the alternative possibility that HIV-1 CA might interact with a cellular factor that bridges it to LEDGF/p75. Given the similarity between the effects of a TNPO3 knockdown and the N74D mutation on HIV-1 integration site selectivity (12, 32), a potential role for TNPO3 or TNPO3-interacting factors should be considered. Notably, it has also been reported that HIV-1 CA can enter the nucleus during infection but that the transport of the N74D mutant CA into the nucleus is diminished relative to the WT virus (38). Whether the mode of trafficking through the NPC or the presence of CA in the PIC, or both, influences the subsequent interactions of IN with nuclear factors, including LEDGF/p75, will be important to determine.
An obvious limitation of our approach is the possibility that knockdown or knockout of the cell factors could have an indirect effect on the distribution of the integrated proviruses. NUP-associated regions of chromatin, which encompass approximately 25% of the Drosophila genome (64), are enriched for markers of active transcription, including RNA polymerase II occupancy and histone H4K16 acetylation. It therefore seems likely that depletion of NPC components could significantly affect chromatin organization and/or the distribution of the chromosomes within the nucleus, which could, in turn, affect the efficiency and/or distribution of viral DNA integration. Although LEDGF/p75 is a constitutive chromatin component and its depletion influences the expression of large numbers of cellular genes (23), the fact that novel LEDGF/p75 fusion proteins that contain foreign chromatin binding domains quantitatively redirect HIV-1 integration (28–30) shows that LEDGF/p75 has a direct role in HIV-1 DNA integration targeting.
Fortunately, the question of whether a host gene knockdown can indirectly influence integration site selection does not apply to the data obtained with the N74D mutant virus. Biochemical studies that characterize the details of virus-host interactions can also help to delineate direct versus indirect effects. Two different NUPs, NUP153 (65) and NUP62 (66), have been shown to bind to HIV-1 IN, suggesting that it may be possible to configure novel targeting constructs based on these factors as well. Our finding that mutations in CA, which is the key HIV-1 determinant of PIC nuclear import (33, 57), alter the dependency of the virus on cellular LEDGF/p75 supports a model whereby the mechanism of PIC trafficking is linked to the efficiency and specificity of viral DNA integration.
ACKNOWLEDGMENTS
We thank Charles Bangham for sharing the method-of-integration-site library construction prior to publication, Greg Towers for stimulating discussion in the early phase of this project, and Masahiro Yamashita and Michael Emerman for their generous gift of parental and MHIV chimera reporter constructs.
This work was supported by the National Cancer Institute's Intramural Center for Cancer Research, which supports the HIV Drug Resistance Program (V.N.K. and S.H.H.), and National Institutes of Health (NIH) grants AI039394 and AI052014 (to A.E.).
The contents of this article or mention of commercial reagents is in no way meant to reflect the options of, or endorsement by, the NIH or U.S. Government.
Footnotes
Published ahead of print 24 October 2012
REFERENCES
- 1. Hulme AE, Perez O, Hope TJ. 2011. Complementary assays reveal a relationship between HIV-1 uncoating and reverse transcription. Proc. Natl. Acad. Sci. U. S. A. 108:9975–9980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen H, Engelman A. 2001. Asymmetric processing of human immunodeficiency virus type 1 cDNA in vivo: implications for functional end coupling during the chemical steps of DNA transposition. Mol. Cell. Biol. 21:6758–6767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miller MD, Farnet CM, Bushman FD. 1997. Human immunodeficiency virus type 1 preintegration complexes: studies of organization and composition. J. Virol. 71:5382–5390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bukrinsky M, Sharova N, Dempsey M, Stanwick T, Bukrinskaya A, Haggerty S, Stevenson M. 1992. Active nuclear import of human immunodeficiency virus type 1 preintegration complexes. Proc. Natl. Acad. Sci. U. S. A. 89:6580–6584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Katz RA, Greger JG, Boimel P, Skalka AM. 2003. Human immunodeficiency virus type 1 DNA nuclear import and integration are mitosis independent in cycling cells. J. Virol. 77:13412–13417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Engelman A. 2010. Reverse transcription and integration, p 129–159 In Kurth R, Bannert N. (ed), Retroviruses: molecular biology, genomics and pathogenesis. Caister Academic Press, Norfolk, United Kingdom [Google Scholar]
- 7. Bushman F, Lewinski M, Ciuffi A, Barr S, Leipzig J, Hannenhalli S, Hoffmann C. 2005. Genome-wide analysis of retroviral DNA integration. Nat. Rev. Microbiol. 3:848–858 [DOI] [PubMed] [Google Scholar]
- 8. Derse D, Crise B, Li Y, Princler G, Lum N, Stewart C, McGrath CF, Hughes SH, Munroe DJ, Wu X. 2007. HTLV-1 integration target sites in the human genome: comparison with other retroviruses. J. Virol. 81:6731–6741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Holman AG, Coffin JM. 2005. Symmetrical base preferences surrounding HIV-1, avian sarcoma/leukosis virus, and murine leukemia virus integration sites. Proc. Natl. Acad. Sci. U. S. A. 102:6103–6107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu X, Li Y, Crise B, Burgess SM, Munroe DJ. 2005. Weak palindromic consensus sequences are a common feature found at the integration target sites of many retroviruses. J. Virol. 79:5211–5214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schroder ARW, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. 2002. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 110:521–529 [DOI] [PubMed] [Google Scholar]
- 12. Ocwieja KE, Brady TL, Ronen K, Huegel A, Roth SL, Schaller T, James LC, Towers GJ, Young JA, Chanda SK, König R, Malani N, Berry CC, Bushman FD. 2011. HIV integration targeting: a pathway involving Transportin-3 and the nuclear pore protein RanBP2. PLoS Pathog. 7:e1001313 doi:10.1371/journal.ppat.1001313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu X, Li Y, Crise B, Burgess SM. 2003. Transcription start regions in the human genome are favored targets for MLV integration. Science 300:1749–1751 [DOI] [PubMed] [Google Scholar]
- 14. Mitchell RS, Beitzel BF, Schroder ARW, Shinn P, Chen H, Berry CC, Ecker JR, Bushman FD. 2004. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2:e234 doi:10.1371/journal.pbio.0020234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Narezkina A, Taganov KD, Litwin S, Stoyanova R, Hayashi J, Seeger C, Skalka AM, Katz RA. 2004. Genome-wide analyses of avian sarcoma virus integration sites. J. Virol. 78:11656–11663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nowrouzi A, Dittrich M, Klanke C, Heinkelein M, Rammling M, Dandekar T, von Kalle C, Rethwilm A. 2006. Genome-wide mapping of foamy virus vector integrations into a human cell line. J. Gen. Virol. 87:1339–1347 [DOI] [PubMed] [Google Scholar]
- 17. Trobridge GD, Miller DG, Jacobs MA, Allen JM, Kiem H-P, Kaul R, Russell DW. 2006. Foamy virus vector integration sites in normal human cells. Proc. Natl. Acad. Sci. U. S. A. 103:1498–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Llano M, Saenz DT, Meehan A, Wongthida P, Peretz M, Walker WH, Teo W, Poeschla EM. 2006. An essential role for LEDGF/p75 in HIV integration. Science 314:461–464 [DOI] [PubMed] [Google Scholar]
- 19. Vandekerckhove L, Christ F, Van Maele B, De Rijck J, Gijsbers R, Van den Haute C, Witvrouw M, Debyser Z. 2006. Transient and stable knockdown of the integrase cofactor LEDGF/p75 reveals its role in the replication cycle of human immunodeficiency virus. J. Virol. 80:1886–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marshall HM, Ronen K, Berry C, Llano M, Sutherland H, Saenz D, Bickmore W, Poeschla E, Bushman FD. 2007. Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS One 2:e1340 doi:10.1371/journal.pone.0001340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schrijvers R, De Rijck J, Demeulemeester J, Adachi N, Vets S, Ronen K, Christ F, Bushman FD, Debyser Z, Gijsbers R. 2012. LEDGF/p75-independent HIV-1 replication demonstrates a role for HRP-2 and remains sensitive to inhibition by LEDGINs. PLoS Pathog. 8:e1002558 doi:10.1371/journal.ppat.1002558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shun M-C, Raghavendra NK, Vandegraaff N, Daigle JE, Hughes S, Kellam P, Cherepanov P, Engelman A. 2007. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 21:1767–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ciuffi A, Llano M, Poeschla E, Hoffmann C, Leipzig J, Shinn P, Ecker JR, Bushman F. 2005. A role for LEDGF/p75 in targeting HIV-DNA integration. Nat. Med. 11:1287–1289 [DOI] [PubMed] [Google Scholar]
- 24. Cherepanov P, Devroe E, Silver PA, Engelman A. 2004. Identification of an evolutionarily-conserved domain in LEDGF/p75 that binds HIV-1 integrase. J. Biol. Chem. 279:48883–48892 [DOI] [PubMed] [Google Scholar]
- 25. Vanegas M, Llano M, Delgado S, Thompson D, Peretz M, Poeschla E. 2005. Identification of the LEDGF/p75 HIV-1 integrase-interaction domain and NLS reveals NLS-independent chromatin tethering. J. Cell Sci. 118:1733–1743 [DOI] [PubMed] [Google Scholar]
- 26. Llano M, Vanegas M, Hutchins N, Thompson D, Delgado S, Poeschla EM. 2006. Identification and characterization of the chromatin-binding domains of the HIV-1 integrase interactor LEDGF/p75. J. Mol. Biol. 360:760–773 [DOI] [PubMed] [Google Scholar]
- 27. Turlure F, Maertens G, Rahman S, Cherepanov P, Engelman A. 2006. A tripartite DNA-binding element, comprised of the nuclear localization signal and two AT-hook motifs, mediates the association of LEDGF/p75 with chromatin in vivo. Nucleic Acids Res. 34:1653–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ferris AL, Wu X, Hughes CM, Stewart C, Smith SJ, Milne TA, Wang GG, Shun M-C, Allis CD, Engelman A, Hughes SH. 2010. Lens epithelium-derived growth factor fusion proteins redirect HIV-1 DNA integration. Proc. Natl. Acad. Sci. U. S. A. 107:3135–3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gijsbers R, Ronen K, Vets S, Malani N, De Rijck J, McNeely M, Bushman FD, Debyser Z. 2010. LEDGF hybrids efficiently retarget lentiviral integration into heterochromatin. Mol. Ther. 18:552–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Silvers RM, Smith JA, Schowalter M, Litwin S, Liang Z, Geary K, Daniel R. 2010. Modification of integration site preferences of an HIV-1-based vector by expression of a novel synthetic protein. Hum. Gene Ther. 21:337–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maertens GN, Hare S, Cherepanov P. 2010. The mechanism of retroviral integration from X-ray structures of its key intermediates. Nature 468:326–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schaller T, Ocwieja KE, Rasaiyaah J, Price AJ, Brady TL, Roth SL, Hué S, Fletcher AJ, Lee K, KewalRamani VN, Noursadeghi M, Jenner RG, James LC, Bushman FD, Towers GJ. 2011. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 7:e1002439 doi:10.1371/journal.ppat.1002439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yamashita M, Emerman M. 2004. Capsid is a dominant determinant of retrovirus infectivity in nondividing cells. J. Virol. 78:5670–5678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lewinski MK, Yamashita M, Emerman M, Ciuffi A, Marshall H, Crawford G, Collins F, Shinn P, Leipzig J, Hannenhalli S, Berry CC, Ecker JR, Bushman FD. 2006. Retroviral DNA integration: viral and cellular determinants of target-site selection. PLoS Pathog. 2:e60 doi:10.1371/journal.ppat.0020060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. De Iaco A, Luban J. 2011. Inhibition of HIV-1 infection by TNPO3 depletion is determined by capsid and detectable after viral cDNA enters the nucleus. Retrovirology 8:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krishnan L, Matreyek KA, Oztop I, Lee K, Tipper CH, Li X, Dar MJ, Kewalramani VN, Engelman A. 2010. The requirement for cellular transportin 3 (TNPO3 or TRN-SR2) during infection maps to human immunodeficiency virus type 1 capsid and not integrase. J. Virol. 84:397–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, Takemura T, Shelton K, Taniuchi I, Li Y, Sodroski J, Littman DR, Coffin JM, Hughes SH, Unutmaz D, Engelman A, KewalRamani VN. 2010. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7:221–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou L, Sokolskaja E, Jolly C, James W, Cowley SA, Fassati A. 2011. Transportin 3 promotes a nuclear maturation step required for efficient HIV-1 integration. PLoS Pathog. 7:e1002194 doi:10.1371/journal.ppat.1002194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Matreyek KA, Engelman A. 2011. The requirement for nucleoporin NUP153 during human immunodeficiency virus type 1 infection is determined by the viral capsid. J. Virol. 85:7818–7827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yamashita M, Emerman M. 2005. The cell cycle independence of HIV infections is not determined by known karyophilic viral elements. PLoS Pathog. 1:e18 doi:10.1371/journal.ppat.0010018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lu R, Limon A, Devroe E, Silver PA, Cherepanov P, Engelman A. 2004. Class II integrase mutants with changes in putative nuclear localization signals are primarily blocked at a postnuclear entry step of human immunodeficiency virus type 1 replication. J. Virol. 78:12735–12746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nakajima N, Lu R, Engelman A. 2001. Human immunodeficiency virus type 1 replication in the absence of integrase-mediated DNA recombination: definition of permissive and nonpermissive T-cell lines. J. Virol. 75:7944–7955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chang LJ, Urlacher V, Iwakuma T, Cui Y, Zucali J. 1999. Efficacy and safety analyses of a recombinant human immunodeficiency virus type 1 derived vector system. Gene Ther. 6:715–728 [DOI] [PubMed] [Google Scholar]
- 44. Engelman A, Oztop I, Vandegraaff N, Raghavendra NK. 2009. Quantitative analysis of HIV-1 preintegration complexes. Methods 47:283–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gillet NA, Malani N, Melamed A, Gormley N, Carter R, Bentley D, Berry C, Bushman FD, Taylor GP, Bangham CRM. 2011. The host genomic environment of the provirus determines the abundance of HTLV-1–infected T-cell clones. Blood 117:3113–3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Engelman A, Cherepanov P. 2008. The lentiviral integrase binding protein LEDGF/p75 and HIV-1 replication. PLoS Pathog. 4:e1000046 doi:10.1371/journal.ppat.1000046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Poeschla EM. 2008. Integrase, LEDGF/p75 and HIV replication. Cell. Mol. Life Sci. 65:1403–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ge H, Si Y, Roeder RG. 1998. Isolation of cDNAs encoding novel transcription coactivators p52 and p75 reveals an alternate regulatory mechanism of transcriptional activation. EMBO J. 17:6723–6729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Busschots K, Vercammen J, Emiliani S, Benarous R, Engelborghs Y, Christ F, Debyser Z. 2005. The interaction of LEDGF/p75 with integrase is lentivirus-specific and promotes DNA binding. J. Biol. Chem. 280:17841–17847 [DOI] [PubMed] [Google Scholar]
- 50. Llano M, Vanegas M, Fregoso O, Saenz D, Chung S, Peretz M, Poeschla EM. 2004. LEDGF/p75 determines cellular trafficking of diverse lentiviral but not murine oncoretroviral integrase proteins and is a component of functional lentiviral preintegration complexes. J. Virol. 78:9524–9537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Braaten D, Franke EK, Luban J. 1996. Cyclophilin A is required for an early step in the life cycle of human immunodeficiency virus type 1 before the initiation of reverse transcription. J. Virol. 70:3551–3560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yoo S, Myszka DG, Yeh C, McMurray M, Hill CP, Sundquist WI. 1997. Molecular recognition in the HIV-1 capsid/cyclophilin A complex. J. Mol. Biol. 269:780–795 [DOI] [PubMed] [Google Scholar]
- 53. Harding MW, Handschumacher RE. 1988. Cyclophilin, a primary molecular target for cyclosporine. Structural and functional implications. Transplantation 46(2 Suppl):29S–35S [DOI] [PubMed] [Google Scholar]
- 54. Aberham C, Weber S, Phares W. 1996. Spontaneous mutations in the human immunodeficiency virus type 1 gag gene that affect viral replication in the presence of cyclosporins. J. Virol. 70:3536–3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Braaten D, Aberham C, Franke EK, Yin L, Phares W, Luban J. 1996. Cyclosporine A-resistant human immunodeficiency virus type 1 mutants demonstrate that Gag encodes the functional target of cyclophilin A. J. Virol. 70:5170–5176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Forshey BM, von Schwedler U, Sundquist WI, Aiken C. 2002. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 76:5667–5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yamashita M, Perez O, Hope TJ, Emerman M. 2007. Evidence for direct involvement of the capsid protein in HIV infection of nondividing cells. PLoS Pathog. 3:1502–1510 doi:10.1371/journal.ppat.0030156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Thys W, De Houwer S, Demeulemeester J, Taltynov O, Vancraenenbroeck R, Gerard M, De Rijck J, Gijsbers R, Christ F, Debyser Z. 2011. Interplay between HIV entry and transportin-SR2 dependency. Retrovirology 8:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kim SY, Byrn R, Groopman J, Baltimore D. 1989. Temporal aspects of DNA and RNA synthesis during human immunodeficiency virus infection: evidence for differential gene expression. J. Virol. 63:3708–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Deininger PL, Batzer MA. 1999. Alu repeats and human disease. Mol. Genet. Metab. 67:183–193 [DOI] [PubMed] [Google Scholar]
- 61. Konig R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, Chiang CY, Tu BP, De Jesus PD, Lilley CE, Seidel S, Opaluch AM, Caldwell JS, Weitzman MD, Kuhen KL, Bandyopadhyay S, Ideker T, Orth AP, Miraglia LJ, Bushman FD, Young JA, Chanda SK. 2008. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 135:49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ambrose Z, Lee K, Ndjomou J, Xu H, Oztop I, Matous J, Takemura T, Unutmaz D, Engelman A, Hughes SH, KewalRamani VN. 2012. Human immunodeficiency virus type 1 capsid mutation N74D alters cyclophilin A dependence and impairs macrophage infection. J. Virol. 86:4708–4714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dawlaty MM, Malureanu L, Jeganathan KB, Kao E, Sustmann C, Tahk S, Shuai K, Grosschedl R, van Deursen JM. 2008. Resolution of sister centromeres requires RanBP2-mediated SUMOylation of topoisomerase IIalpha. Cell 133:103–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vaquerizas JM, Suyama R, Kind J, Miura K, Luscombe NM, Akhtar A. 2010. Nuclear pore proteins nup153 and megator define transcriptionally active regions in the Drosophila genome. PLoS Genet. 6:e1000846 doi:10.1371/journal.pgen.1000846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Woodward CL, Prakobwanakit S, Mosessian S, Chow SA. 2009. Integrase interacts with nucleoporin NUP153 to mediate the nuclear import of human immunodeficiency virus type 1. J. Virol. 83:6522–6533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ao Z, Jayappa KD, Wang B, Zheng Y, Wang X, Peng J, Yao X. 2012. Contribution of host nucleoporin 62 in HIV-1 integrase chromatin association and viral DNA integration. J. Biol. Chem. 287:10544–10555 [DOI] [PMC free article] [PubMed] [Google Scholar]