

Abstract
Coronavirus (CoV) transcription requires a high-frequency recombination process that links newly synthesized minus-strand subgenomic RNA copies to the leader region, which is present only once, at the 5′ end of the genome. This discontinuous RNA synthesis step is based on the complementarity between the transcription-regulating sequences (TRSs) at the leader region and those preceding each gene in the nascent minus-strand RNA. Furthermore, the template switch requires the physical proximity of RNA genome domains located between 20,000 and 30,000 nucleotides apart. In this report, it is shown that the efficacy of this recombination step is promoted by novel additional long-distance RNA-RNA interactions between RNA motifs located close to the TRSs controlling the expression of each gene and their complementary sequences mapping close to the 5′ end of the genome. These interactions would bring together the motifs involved in the recombination process. This finding indicates that the formation of high-order RNA structures in the CoV genome is necessary to control the expression of at least the viral N gene. The requirement of these long-distance interactions for transcription was shown by the engineering of CoV replicons in which the complementarity between the newly identified sequences was disrupted. Furthermore, disruption of complementarity in mutant viruses led to mutations that restored complementarity, wild-type transcription levels, and viral titers by passage in cell cultures. The relevance of these high-order structures for virus transcription is reinforced by the phylogenetic conservation of the involved RNA motifs in CoVs.
INTRODUCTION
Coronavirus (CoV) genomes are plus-strand RNA molecules of around 30 kb (1, 2) and represent the largest known RNA viruses. The family Coronaviridae belongs to the Nidovirales order and shares a common genome organization with its members. The 5′ two-thirds of the genome codifies the replicase polyprotein, and the 3′ third includes the structural protein genes and several accessory genes (3). In contrast to the replicase gene, which is translated directly from the genome, the genes located at the 3′ end of the genome are translated from a collection of subgenomic mRNAs (sgmRNAs) of different sizes generated by a unique transcription mechanism. All of these sgmRNAs have a leader sequence, present only once, at the 5′ end of the genome, that is added in a discontinuous RNA synthesis step during the production of minus-strand sgRNAs (4).
Three major models have been described for sgmRNA synthesis in RNA viruses: (i) an internal initiation mechanism in which a minus-polarity copy of the viral genome is used as the template to synthesize sgmRNAs of plus polarity (5), (ii) a premature termination mechanism during minus-strand synthesis that generates sgRNAs that are then used to synthesize the plus-polarity sgmRNAs (6–8), and (iii) a discontinuous transcription mechanism with a premature termination mechanism that includes a template switch to add a copy of the leader sequence to the nascent minus-strand sgRNA. This discontinuous transcription mechanism is exclusive to the nidoviruses (4, 9, 10).
The discontinuous transcription mechanism resembles a similarity-assisted RNA recombination process (3, 9, 11), which requires homology between the acceptor and donor RNA sequences. In addition, the presence of hairpin structures in the acceptor RNA seems to be a requirement for recombination (12–14). Discontinuous transcription is essential for gene expression during the viral cycle, and it therefore takes place with a high frequency. It has been proposed that this high recombination frequency is facilitated by interactions that bring distant genomic sequences regulating transcription into close proximity (15).
Transmissible gastroenteritis virus (TGEV) is a member of the Coronaviridae family. Discontinuous transcription of sgmRNAs in TGEV is driven by the transcription-regulating sequences (TRSs), located at the 3′ end of the leader (TRS-L) and also preceding each gene (TRS-B) (Fig. 1A). TRSs include a conserved core sequence (CS) (5′-CUAAAC-3′) and variable 5′ and 3′ sequences flanking the CS (16). The TRS-L, located at the 5′ end of the genome, acts as the acceptor RNA during the template switch, while the TRS-B sequences act as the donor RNA (Fig. 1A). The complementarity between the TRS-L in the genomic RNA and the copy of the TRS-B (cTRS-B) in the nascent minus-strand RNA is a crucial factor for sgRNA production (9, 17–19). The relative amount of each sgmRNA during TGEV infection correlates with the free energy (ΔG) of the interaction between the TRS-L and the corresponding cTRS-B, with the exception of the N gene, which results in the most abundant sgmRNA despite having the lowest complementarity between the TRS-L and the cTRS-N (20). To maintain N gene sgmRNA levels, a long-distance RNA-RNA interaction between two complementary 9-nucleotide (nt) sequences (proximal and distal elements) (20) is necessary, probably by relocating an RNA motif, termed the active domain and 5′ flanking the distal element, immediately preceding the CS-N (21). In this report, we describe the identification of a novel long-distance RNA-RNA interaction between a 10-nt sequence within the active domain and a complementary RNA (cRNA) motif located at the 5′ end of the viral genome. This interaction could bring into physical proximity the leader sequence and the TRS-N, which are located more than 25,000 nt apart. This proximity would facilitate the template switch during the synthesis of the minus-strand sgRNAs by a high-frequency recombination mechanism. This is the first time that experimental support for physical proximity between the TRS-L and a TRS-B to facilitate discontinuous transcription has been provided.
Fig 1.
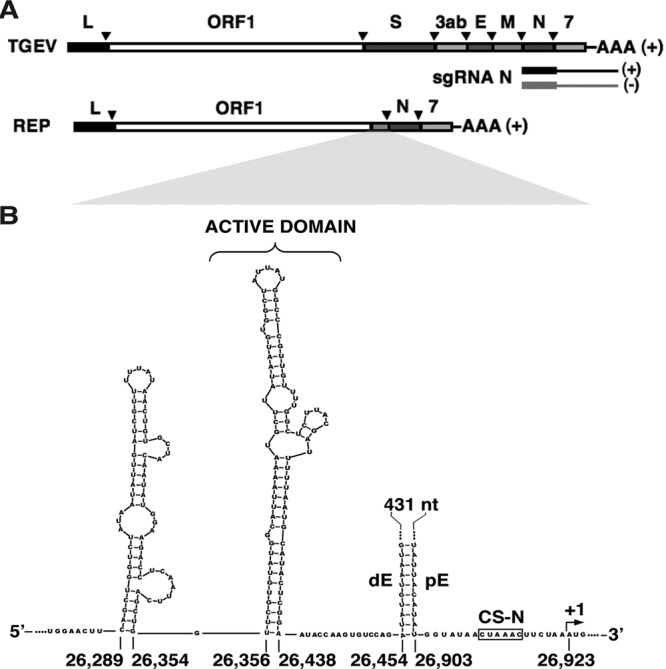
Genetic structure of TGEV infectious virus, replicon genomes, and subgenomic mRNAs. (A) (Top to bottom) Schemes representing the genetic structures of the TGEV genome (RNA of plus polarity), the N sgmRNA (plus polarity), the N sgRNA (minus polarity; first synthesized by the discontinuous mechanism), and a TGEV-derived replicon genome of plus polarity. L, leader sequence; ORF1ab, replicase genes; S, spike protein gene; 3ab, 3a and -b accessory genes; E, envelope protein gene; M, membrane protein gene; N, nucleocapsid protein gene; 7, accessory protein gene; AAA, poly(A). The inverted arrowheads indicate the positions of the TRSs. (B) Scheme showing the sequence and Mfold-predicted secondary structure of the sequences preceding the N gene in TGEV. CS-N, conserved core sequence of the TRS-N; pE, proximal element; dE, distal element; +1, first nucleotide of the N gene. The sequence between the dE and pE RNA motifs is represented by dots and a black line. The active domain hairpin is indicated. Numbers indicate the nucleotide positions in the TGEV genome.
MATERIALS AND METHODS
Cells and viruses.
Baby hamster kidney (BHK) cells stably transformed with the porcine aminopeptidase N gene (pAPN) (22) and with the Sindbis virus replicon pSINrep1 (23) expressing TGEV N protein (BHK-N) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal calf serum (FCS) and with G418 (1.5 mg/ml) and puromycin (5 μg/ml) as selection agents for pAPN and pSINrep1, respectively. Recombinant TGEVs were grown in swine testis (ST) cells (24). ST cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS). TGEVs were obtained from an infective clone with the sequence of the isolated TGEV PUR46-MAD (25) and the spike protein of strain PTV (Purdue type strain) (26) by use of a reverse-genetic system. Virus titration was performed on ST cell monolayers as described previously (27).
Reverse-genetic system.
cDNAs encoding TGEV and replicons were constructed using pBAC-TGEV (25) (GenBank accession number AJ271965), which includes the whole TGEV genome sequence, and the replicon pBAC-REP1 (28). The pBAC vectors were transfected into BHK-N cells (described above). Cellular RNA polymerase II transcribes virus or replicon genomes from pBAC vectors, and viral RNA is then translated, initiating the viral cycle. To rescue infectious viruses from cDNA clones, transfected BHK-N cells were treated with trypsin at 6 h posttransfection (hpt) and plated over confluent ST cell monolayers.
Transfection.
BHK-N cells were grown to 95% confluence on 35-mm-diameter plates and then transfected with 4 μg of cDNA encoding a TGEV replicon or infectious virus, representing 100 molecules per cell, on average, by using 12 μg of Lipofectamine 2000 (Invitrogen) according to the manufacturer's specifications. The conditions in transfection experiments were strictly controlled, as follows: (i) the same number of cells per well was seeded (5 × 105 cells/well), (ii) the same amount of cDNA was always transfected (100 molecules per cell), and (iii) cDNA was purified using a large-construct kit (Qiagen) including an exonuclease treatment to remove bacterial DNA contamination and damaged plasmids, thus providing an ultrapure DNA plasmid for transfection.
DNase treatment of RNAs from transfection experiments.
To remove transfected cDNA-containing replicon genomes from samples before quantitative reverse transcription-PCR (qRT-PCR) analysis, 7 μg of each RNA in 100 μl was treated with 20 U of DNase I (Roche) for 30 min at 37°C. DNA-free RNAs were repurified using an RNeasy miniprep kit (Qiagen).
RNA analysis by quantitative RT-PCR.
Total intracellular RNA was extracted from transfected BHK-N cells at 24 hpt or from ST cells infected with recombinant TGEVs at 16 h postinfection. RNAs were purified with an RNeasy miniprep kit (Qiagen) according to the manufacturer's specifications. To remove the transfected replicon DNA prior to qRT-PCR analysis, each RNA sample was treated with DNase I (Roche). cDNAs were synthesized at 37°C for 2 h with MultiScribe reverse transcriptase (high-capacity cDNA reverse transcription kit; Applied Biosystems). Real-time qRT-PCR was used for quantitative analysis of genomic and subgenomic RNAs from infectious TGEV and TGEV-derived replicons. Oligonucleotides used for quantitative PCRs (Table 1) were designed with Primer Express software. SYBR green PCR master mix (Applied Biosystems) was used in the PCR step, according to the manufacturer's specifications. TaqMan assays were used to quantify the β-glucuronidase mRNA (GUS; Life Technologies) as an internal standard, as well as the viral gRNA (29). Detection was performed with an ABI Prism 7500 sequence detection system (Applied Biosystems). Data were analyzed with ABI Prism 7500 SDS, version 1.2.3, software. The relative quantifications were performed using the 2−ΔΔCT method (30). For each mutant sequence, two independent replicons were constructed. Each of these constructs was analyzed in two independent transfections, and RNA from each transfection experiment was analyzed twice by qRT-PCR. To minimize transfection variability, each pair of data used for comparison came from the same transfection and qRT-PCR experiment. The results shown in the graphs are averages for four independent transfection experiments analyzed twice by qRT-PCR.
Table 1.
Oligonucleotides used for quantitative RT-PCR analysis
| Amplicon | Forward primer namea | Forward primer sequence (5′ → 3′) | Reverse primer namea | Reverse primer sequence (5′ → 3′) |
|---|---|---|---|---|
| gRNA | RT-REP-VS | TTCTTTTGACAAAACATACGGTGAA | RT-REP-RS | CTAGGCAACTGGTTTGTAACATCTTT |
| N sgmRNA | Ldrt-VS | CGTGGCTATATCTCTTCTTTTACTTTAACTAG | N(82)-RS | TCTTCCGACCACGGGAATT |
| sgmRNA 7 | Ldrt-VS | CGTGGCTATATCTCTTCTTTTACTTTAACTAG | 7(38)-RS | AAAACTGTAATAAATACAGCATGGAGGAA |
The hybridization sites of the oligonucleotides within the TGEV genome are as follows: for RT-REP-VS, nt 4,829 to 4,853; for RT-REP-RS, nt 4,884 to 4,909; for Ldrt-VS, nt 25 to 56; for N(82)-RS, nt 26,986 to 27,004; and for 7(38)-RS, nt 28,086 to 28,114.
Plasmid constructs.
cDNAs of TGEV-derived replicons and infectious viruses (25, 28) were generated by PCR-directed mutagenesis. To generate the mutant replicons AD-ΔA, AD-ΔB, AD-ΔC, AD-ΔA-C′, AD-ΔA-B′12, AD-ΔA-B′9, AD-ΔA-B′4, and AD-ΔA-B′3, the plasmid pBAC-TGEV (25), containing the TGEV genome (GenBank accession no. AJ271965), was used as a template for the specific oligonucleotides shown in Table 2. All of the mutated fragments generated by PCR contained AvrII sites at both ends for introduction into the same site of TRS-N-ΔdE mutant cDNA (20). cDNAs of TGEV-derived replicons cB-218*/ΔB, cB-477*/ΔB, cB-218*/B, and cB-477*/B, as well as cDNA of rTGEV-cB*, were obtained by use of two overlapping fragments synthesized by PCR. The first one was generated with Oli 5′I and the specific reverse oligonucleotide (Table 2). The second fragment was generated with Oli 3′D and the specific forward oligonucleotide (Table 2). After overlapping PCR, the resulting fragments, with SfiI and ApaLI restriction sites at the 5′ and 3′ ends, respectively, were introduced into the same sites of a pBAC plasmid containing only the first 4,423 nt of the TGEV genome. From this intermediate plasmid, the SfiI-ClaI fragments were obtained and then introduced into the same sites of corresponding pBAC-TGEV-ΔCla plasmids lacking the ClaI-ClaI fragment (AD-ΔA and TRS-N-ΔdE for replicons and pBAC-TGEV for infectious clones). The ClaI-ClaI fragment was subsequently introduced into bacterial artificial chromosomes (BACs) to construct complete replicon or infectious cDNA clones including mutant sequences. The sequences including the active domain variations in replicons cB-218*/B*, cB-477*/B*, and cB*-477*/B*-14 were synthesized de novo (Geneart) with AvrII sites at both ends. Synthetic gene sequences including the corresponding mutations at the 5′ end of the genome (cB-218*/ΔB and cB-477*/ΔB) were introduced into the same sites of the plasmids. rTGEV-B* cDNA was constructed using an intermediate plasmid including the last 3 kb of the TGEV genome (AvrII-BamHI fragment from pBAC-TGEV [ 25]). A smaller fragment including AvrII-MfeI sites at the ends was generated by PCR using pBAC-TGEV as the template, with specific oligonucleotides (Table 2), and was reintroduced into the same sites of the intermediate plasmid and finally into the infectious cDNA clone.
Table 2.
Oligonucleotides used for site-directed mutagenesis
| Mutant(s) | Oligonucleotide | Sequence (5′ → 3′)a |
|---|---|---|
| AD-ΔA-B′12, AD-ΔA-B′9, AD-ΔA-B′4, AD-ΔA-B′3, AD-ΔA-C′, AD-ΔA, AD-ΔB, AD-ΔC | Rep Mut 3 VS | TTCCTAGGTGGAACTTCAGCTGGTCTATAATATTGATC |
| AD-ΔA | ΔA-RS | CCTAGGCTGCAATACTAAAGCCGAACATTACATATCTGGACACTTGGTATTCCGAGTATGCATTAAAAATCGTAAGAGCCAAAATAAGCATTTTAATGCCATACACGAAC |
| AD-ΔB | ΔB-RS | CCTAGGCTGCAATACTAAAGCCGAACATTACATATCTGGACACTTGGTATTCCGAGTATGCATTAAACAACGGGCCATAATAGCCACATTATTTAATGCCATACACGAAC |
| AD-ΔC | ΔC-RS | CGAACATTACATATCTGGACACTTGGTATTAATCGTAAGAGCCAAAACAACGGGCCATAATAGCCACATTATAAGCATACCAGCTGAATTGAGGTCTTCCATATTGTAGC |
| AD-ΔA-C′ | 3′AD-ΔA-C′-RS | CATTACATATCTGGACACTTGGTATTAATACAAGCCCACCTAAATCGTAAGAGCCAAAATAAGCATTAGGTGGCGCTTGAATTACCAGCTGAATTGAGGTCTTCC |
| AD-ΔC, AD-ΔA-C′, AD-ΔA-B′12, AD-ΔA-B′9 | ΔC-3′-RS | CCTAGGCTGCAATACTAAAGCCGAACATTACATATCTGGACACTTGGTATT |
| AD-ΔA-B′12 | 3′AD-ΔA-B′12-RS | CATTACATATCTGGACACTTGGTATTCCGAGTATGCATTAAAGACCACAAAGATGCCAATAAGCAATTTAATGCC |
| AD-ΔA-B′9 | 3′AD-ΔA-B′9-RS | CATTACATATCTGGACACTTGGTATTCCGAGTATGCATTAAAAAAGAAGTCTGCCAAAATAAGCATTTTAATGCC |
| AD-ΔA-B′4 | 3′AD-ΔA-B′4-RS | AACCTAGGCTGCAATACTAAAGCCGAACATTACATATCTGGACACTTGGTATTCCGAGTATGCATTAAAAATCAATTGAGCCAAAATAAGC |
| AD-ΔA-B′3 | 3′AD-ΔA-B′3-RS | AACCTAGGCTGCAATACTAAAGCCGAACATTACATATCTGGACACTTGGTATTCCGAGTATGCATTAAAAATTGTTAAAGCCAAAATAAGC |
| cB-218*/ΔB, cB-218*/B | 218-VS | CGGTGCAGTAGGGTTCCGTCCCTATTAACTATCTCTGTTAGTAGTAGCGAGTGCGGTTCCG |
| cB-218*/ΔB, cB-218*/B | 218-RS | AATAGGGACGGAACCCTACTGCACCG |
| cB-477*/ΔB, cB-477*/B | 477-VS | CTGTCGTGACCTAGTTGATTGCGACAGGAAAGATCACTACGTCATTGGTG |
| cB-477*/ΔB, cB-477*/B, rTGEV-cB* | 477-RS | GCAATCAACTAGGTCACGACAG |
| rTGEV-cB* | 5′-482 1mut-VS | CTGTCGTGACCTAGTTGATTGCGATCGGAAGGATCACTACGTCATTGGTG |
| rTGEV-B* | 3′-AvrII-VS | TGACGTTTCCTAGGGCATTGACTGTCATAG |
| AD-26418-1mut-RS | AACAATTGCACCTGCAATACTAAAGCCGAACATTACATATCTGGACACTTGGTATTCCGAGTATGCATTAAAAATTGTAAGAGCCAAAACAACGGGCC |
The mutated nucleotides are shown in bold. Restriction sites are underlined (AvrII, CCTAGG; EcoRI, GAATTC; and MfeI, CAATTG).
RNA affinity chromatography.
The capture of proteins binding specific RNAs was performed using 5′-biotinylated RNAs linked to a streptavidin-conjugated solid matrix (Dynabeads; Invitrogen). Proteins were extracted from TGEV-infected human Huh7 cells by use of the following extraction buffer: 2.5 mM HEPES, pH 7.9, 2.5 mM KCl, 25 μM EDTA, 0.25 mM dithiothreitol (DTT), and protease inhibitor (Roche). Sixty microliters of solid matrix (10 μg/μl) was used per RNA binding assay. The solid matrix was washed twice with H-BW solution (50 mM HEPES, pH 7.9, 150 mM KCl, 5% glycerol, 0.01% NP-40). Protein extracts diluted in H-BW solution (500 μg of total protein per RNA binding assay) were precleared three times by incubation with the solid matrix in an orbital shaker for at least 5 h each time at 4°C. For RNA binding, 60 μl of solid matrix per RNA binding assay was first washed twice with solution BW (5 mM Tris-HCl, pH 7.5, 1 mM EDTA, 1 M NaCl) and then incubated with the biotinylated RNAs (8 μg) in BW solution for 30 min at room temperature. Immobilized RNAs were washed twice with H-BW. Five hundred micrograms of protein extract and different amounts of nonspecific competitor (0.5 or 1.25 μg tRNAs/μg protein) tRNA (baker's yeast; Sigma) were incubated overnight in an orbital shaker at 4°C. After three washes with H-BW solution, the proteins were eluted with NuPAGE LDS sample buffer (Invitrogen) supplemented with 100 mM DTT for 10 min at room temperature. Proteins were resolved in NuPAGE 4 to 12% Bis-Tris gels (Invitrogen) with NuPAGE MOPS-SDS running buffer (Invitrogen) (2.5 mM morpholinepropanesulfonic acid [MOPS], 2.5 mM Tris base, 0.005% SDS, 0.05 mM EDTA, pH 7.7). Finally, gels were stained with Coomassie Simply Blue Safe stain (Invitrogen). Images were taken with Image Lab V3.0 (Bio-Rad).
In silico analysis.
Potential base pairing score calculations were performed as previously described (19). ΔG calculations were performed using the two-state hybridization server (http://mfold.rna.albany.edu//?q=DINAMelt/Two-state-melting) (31). Secondary structure predictions were performed using the Mfold server for nucleic acid folding and hybridization prediction (http://mfold.rna.albany.edu/?q=mfold/RNA-Folding-Form) (32). The analysis of sequences was performed using DNASTAR Lasergene software 7.0. Comparison of RNA sequences was performed using ClustalW2/EBI (http://www.ebi.ac.uk/Tools/clustalw2/index.html). To identify sequences complementary to the B RNA motif within the TGEV genome, DOT-PLOT MAKER v1.0 software was used (http://bioinfogp.cnb.csic.es/tools/dotplot/index).
RESULTS
Structural requirements of the active domain for N gene transcription.
We previously described that the active domain, including two hairpins, is essential for N gene transcription in TGEV. Since only the most 3′ hairpin is necessary for N gene transcription (21), the term “active domain” refers to this hairpin, comprised of nt 26,356 to 26,438 of the TGEV genome (33) (Fig. 1B). To study the sequence and structural requirements of the active domain, its Mfold-predicted structure was divided into the apical (A), intermediate (B), and basal (C) regions (Fig. 2). Three mutant TGEV genome-derived replicons were constructed in order to analyze the contribution of each region to the active domain function. Regions A, B, and C were deleted in mutants AD-ΔA, AD-ΔB, and AD-ΔC, respectively (Fig. 2). BHK cells constitutively expressing the TGEV N protein (BHK-N cells) were transfected with the corresponding cDNAs, and N sgmRNA levels were evaluated by qRT-PCR. The AD-ΔA mutant showed N gene transcription levels similar to those of the replicon including the complete active domain, previously named dE-173-20 (21), indicating that region A is not necessary for active domain function (Fig. 2). In contrast, N sgmRNA levels in the AD-ΔB and -ΔC mutants were similar to those in the replicon TRS-N-ΔdE, lacking the whole active domain (Fig. 2), indicating that regions B and C are required to maintain N gene transcription levels.
Fig 2.
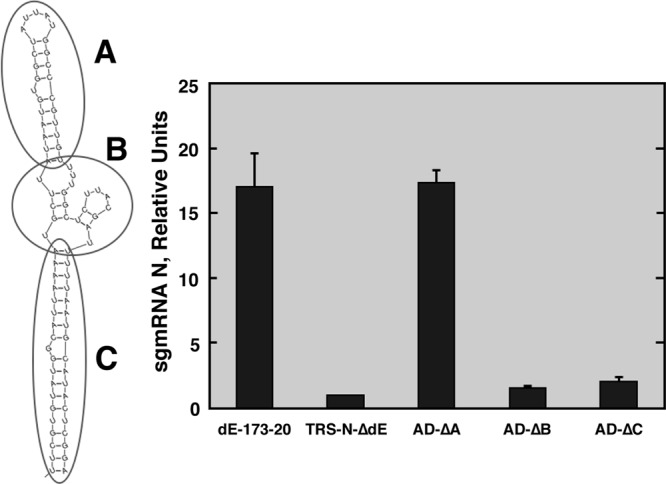
Relevance of active domain regions. (Left) Mfold secondary structure prediction of the active domain. Regions A, B, and C are indicated by circles. (Right) qRT-PCR analysis of N sgmRNA levels normalized to gRNA (N sgmRNA/gRNA) and referred to the level in the reference replicon TRS-N-ΔdE, which was set at 1. dE-173-20, positive control. The data are averages for four independent transfection experiments analyzed twice by qRT-PCR. Error bars represent standard deviations.
Requirement of active domain region C secondary structure for N gene transcription.
According to Mfold predictions, the active domain region C adopted a secondary structure consisting of a long and stable stem. In order to determine whether this secondary structure was required for active domain function, the sequence of region C was replaced by a nonrelated primary sequence adopting a similar secondary structure according to Mfold prediction (AD-ΔA-C′ replicon) (Fig. 3). The AD-ΔA-C′ mutant showed N sgmRNA levels similar to those of the positive control, AD-ΔA (Fig. 3). Additionally, the AD-ΔC mutant (Fig. 2), in which region C was replaced by adjacent sequences with a nonrelated primary sequence or secondary structure, failed to maintain N gene transcription levels. These results indicated that the active domain function required a region C with stable secondary structure but not a specific primary sequence.
Fig 3.
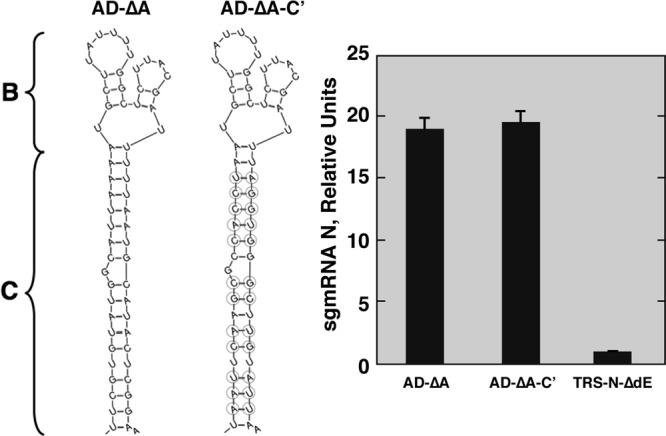
Relevance of region C secondary structure for active domain activity. (Left) Mfold secondary structure predictions of active domains in mutants AD-ΔA and AD-ΔA-C′. Regions B and C are indicated. The mutated nucleotides in AD-ΔA-C′ region C are shown inside circles. (Right) qRT-PCR analysis of N sgmRNA levels (N sgmRNA/gRNA) in relation to the level in the reference replicon TRS-N-ΔdE, which was set at 1. 3′AD-ΔA, positive control. Data are averages for four independent transfection experiments analyzed twice by qRT-PCR. Error bars represent standard deviations.
Relevance of active domain region B primary sequence for optimal N gene transcription.
The requirement for region B to maintain N gene transcription levels was shown above (Fig. 2). To analyze whether the region B requirement was dependent on its secondary structure or primary sequence, four new mutant replicons, containing 12-, 9-, 4-, and 3-nucleotide substitutions (AD-ΔA-B′12, -B′9, -B′4, and -B′3, respectively), were constructed (Fig. 4). All replicons showed N sgmRNA levels similar to those of the reference replicon TRS-N-ΔdE, with no active domain, and significantly lower than those of the positive control, AD-ΔA, which included the wild-type region B sequence (Fig. 4). The significant reductions in N sgmRNA levels of the AD-ΔA-B′4 and AD-ΔA-B′3 mutants, which included mutations in the primary sequence of region B without affecting its secondary structure prediction, suggested that the primary sequence of the active domain region B, named the B motif (nt 26,408 to 26,421), is essential for maintaining N gene transcription levels. This requirement could be due either to the interaction of this RNA motif with viral and host proteins or to RNA-RNA interactions with other RNA sequences in the viral genome.
Fig 4.

Relevance of active domain region B primary sequence for N gene transcription. (Top) Schemes with Mfold secondary structure predictions for the active domain region B sequences included in different mutants. The changed nucleotides in region B are shown inside circles. (Bottom) qRT-PCR analysis of N sgmRNA levels (N sgmRNA/gRNA) in relation to the level in the reference replicon TRS-N-ΔdE, which was set at 1. 3′AD-ΔA, positive control. Data are averages for four independent transfection experiments analyzed twice by qRT-PCR. Error bars represent standard deviations.
Involvement of the B motif included in the active domain in RNA-RNA interactions.
The possibility of differential protein binding to the B motif with a native or mutated sequence was analyzed by RNA affinity chromatography. No proteins specifically interacting with the B motif with a native or mutated sequence were identified by mass spectrometry (Fig. 5). The alternative possibility of an interaction of the B motif with a cRNA sequence (cB) present within the viral genome was then analyzed using the program DOT-PLOT MAKER v1.0. The criterion to select potential RNA-RNA interactions was an associated stability (ΔG) similar to or higher than that of the described interaction between proximal and distal elements (−8.6 kcal/mol) required to maintain N gene transcription (20). Two potential B motif interaction sites, located at the genome's 5′ end (nt 218 to 229 and 477 to 486), were identified in the TGEV genome. To analyze the functional relevance of the postulated RNA-RNA interactions between the B and cB motifs, two mutant replicons were constructed (Fig. 6A and B). In the cB-218*/B mutant, the sequence of interest, located at nt 218 to 229 (5′-UCGUAAGUCGCC-3′), was modified at 8 positions (underlined) to disrupt the complementarity with the active domain B motif: 5′-AACUAUCUCUGU-3′. Similarly, in replicon cB-477*/B, the original sequence, located at nt 477 to 486 (5′-GAUCGUAAGG-3′), was mutated to 5′-GACAGGAAAG-3′ (Fig. 6A and B). Since the cB-477 RNA motif was included within the viral ORF1a coding sequence, the mutations were designed to preserve the amino acid sequence (Fig. 6B). Furthermore, since mutations at the 5′ end of the genome could have a general effect on RNA synthesis, two reference replicons (218*/ΔB and cB-477*/ΔB) were constructed to lack the active domain and include the same mutations as those described above, at positions 218 to 229 and 477 to 486, respectively (Fig. 6A and B). After transfection into BHK-N cells, N sgmRNA levels transcribed by replicon cB-218*/B, with a mutated sequence from nt 218 to 229, were similar to those of the positive control (Fig. 6C) and significantly higher than those of the reference replicons cB-218*/ΔB and wt/ΔB (Fig. 6C). Accordingly, mutations in the cB-218*/B* replicon to restore the complementarity between the mutated cB-218* sequence and the B motif (the original sequence of the B motif [5′-GGCUCUUACGA-3′] was changed to 5′-GCAGAGAUAGUU-3′) did not maintain the original N sgmRNA levels, indicating that the cB sequence motif in nt 218 to 229 is not required for N gene transcription.
Fig 5.

Analysis of proteins interacting with region B of the active domain. (Left) Schemes showing Mfold secondary structure predictions for the biotinylated RNAs used in RNA affinity chromatography assays. (Right) The RNA-captured proteins were separated in a polyacrylamide gradient gel (4 to 12%) and stained with Coomassie blue. The name of the RNA sequence used in each case is indicated, along with the amount (μg) of tRNA per μg of total protein used as a nonspecific competitor. Proteins interacting with the wild-type sequence (AD-B) but not the mutant sequence (AD-B′9) were not found. This result did not rule out that specific RNA-protein interactions were taking place, but these would be under the detection limit of this technique.
Fig 6.

Involvement of the active domain in an RNA-RNA interaction with the 5′ end of the TGEV genome. (A) Schemes of the genetic structures of the mutant replicons. The sequence positions in the genome are indicated on the top line. Letters above the boxes indicate the proximal element (pE), distal element (dE), N gene (N), and B motif in the active domain (nt 26,412 to 26,421). The asterisks indicate that the sequence was mutated as described in the text (nt 218 to 229 and 477 to 486). (B) Depiction of the possible RNA-RNA interactions between the B motif in the active domain (nt 26,412 to 26,421) and the sequences at nt 218 to 229 or nt 477 to 486 in the different mutants. (C) Analysis by qRT-PCR of N sgmRNA levels normalized to gRNA in relation to reference levels in replicons wt/ΔB, cB-218*/ΔB, and cB-477*/ΔB, which were set at 1. wt/B, positive control. Data are averages for four independent transfection experiments analyzed twice by qRT-PCR. Error bars represent standard deviations. (D) Analysis by qRT-PCR of sgmRNA 7 levels normalized to gRNA in relation to reference levels in replicons wt/ΔB, cB-218*/ΔB, and cB-477*/ΔB, which were set at 1. wt/B, positive control. Data are averages for four independent transfection experiments analyzed twice by qRT-PCR. Error bars represent standard deviations.
In contrast, the mutations introduced into replicon cB-477*/B (nt 477 to 486) reduced N sgmRNA levels (Fig. 6C). Therefore, the cB-477 RNA motif at the 5′ end of the viral genome is involved in N gene transcriptional regulation. To confirm the RNA-RNA interaction between the cB-477 and B motifs, a new replicon (cB-477*/B*) was constructed by introducing point mutations in the B motif (5′-UCUUACGAUU-3′ was changed to 5′-UUUUCCUGUU-3′) in order to restore the complementarity with the mutated cB-477* motif. In addition, the mutant replicon cB-477*/B*-14 was constructed, with the double objective of restoring and extending the complementarity between the cB-477 and B RNA motifs from nt 10 to 14 (the B motif sequence [5′-UGGCUCUUACGAUU-3′] was changed to 5′-UGAUCUUUCCUGUC-3′). N gene transcription levels in both the cB-477*/B* and cB-477*/B*-14 mutants were even higher than those in the positive control (Fig. 6C), indicating that the restoration of the complementarity between the cB-477 (nt 477 to 486) and B (nt 26,412 to 26,421) RNA motifs recovered N gene transcription levels. Furthermore, these results indicated that the complementarity, not the primary sequences, of the cB-477 and B motifs was required for transcriptional regulation of N sgmRNA.
To evaluate the effect of the RNA-RNA interaction between the cB-477 and B motifs on the transcription of a gene located downstream of the N gene, the levels of sgmRNA 7 were analyzed by qRT-PCR with replicons including mutations in the B and cB-477 motifs. Deletion of the B motif (replicons wt/ΔB, cB-218*/ΔB, and cB-477*/ΔB) or disruption of the cB-477–B interaction (replicons cB-218*/B* and cB-477*/B) did not significantly affect sgmRNA 7 levels compared to those with the wild-type sequence (wt/B) (Fig. 6D). Therefore, according to the data, the new RNA-RNA interaction described here essentially affects the transcription of the N gene.
Relevance of RNA-RNA interaction between the cB motif at the 5′ end of the genome and the B motif in the active domain in the viral infective cycle.
To confirm that the RNA-RNA interaction between the cB-477 and B RNA motifs was relevant for the virus life cycle, two recombinant viruses, rTGEV-cB* and rTGEV-B*, were generated (Fig. 7A) and included one point mutation each, at positions 482 and 26,418, respectively, that decreased the complementarity between the cB and B RNA motifs without introducing amino acid changes. Viruses with the wild-type and mutated sequences were obtained after full-length cDNA transfection. A region extending 1,000 nt over the mutated positions of the recombinant viruses was sequenced after one virus passage in ST cells. Only the originally introduced mutations were observed in mutant viruses (Fig. 7B). The N sgmRNA levels and viral titers for both rTGEV-cB* and -B* were significantly reduced (4-fold for the N sgmRNA level and 100-fold for the viral titer) in comparison to those of the wild type (Fig. 7C), whereas viral replication was not reduced (Fig. 7D). After 11 serial passages in cell culture, the sequences of interest were sequenced. The sequence at position 482 in rTGEV-cB* had reverted to the wild-type sequence (Fig. 7B), and more importantly, viral titers and N sgmRNA amounts were also restored to the wild-type levels (Fig. 7C). In the mutant virus rTGEV-B*, a double sequence (C/U) at position 480 was observed, corresponding to the wild-type nt (C) and to the new sequence (U), restoring the complementarity with a G → A mutation at position 26,418 of the rTGEV-B* genome. Although at passage 11 both sequences coexisted in the virus population, the C → U substitution was dominant by passage 18 (Fig. 7B). Interestingly, this new mutation at position 480 in rTGEV-B* restored the wild-type viral titers and N sgmRNA levels (Fig. 7C). These results showed the functional relevance of the RNA-RNA interaction between the B and cB RNA motifs in the TGEV infectious cycle.
Fig 7.
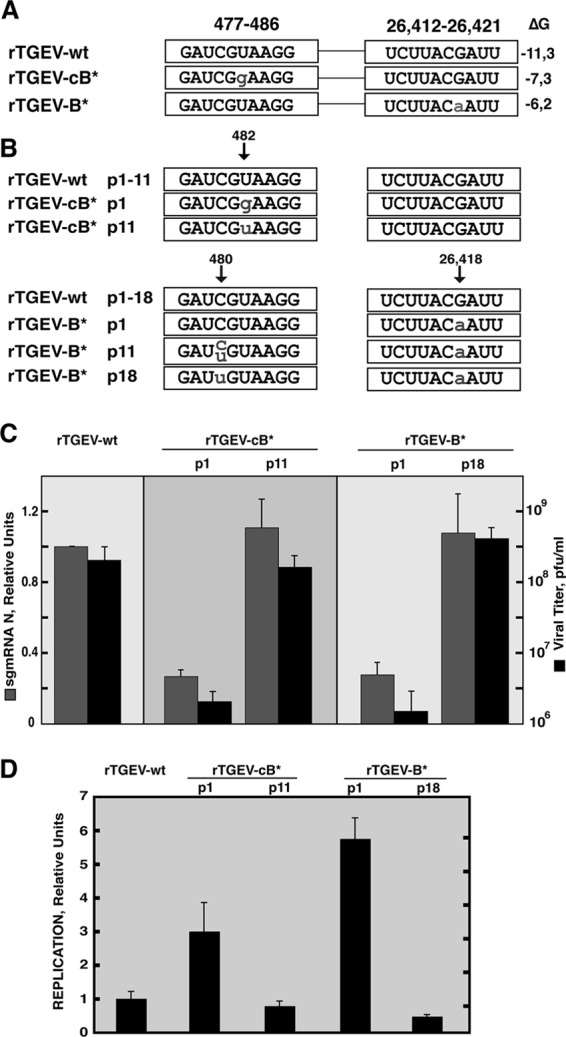
Relevance of the RNA-RNA interaction between the active domain and the 5′ end of the genome in the TGEV infective cycle. (A) To the left are the names of the recombinant viruses. Viral sequences at nt 477 to 486 and 26,412 to 26,421 are shown inside boxes. Lowercase letters indicate mutated positions. To the right is the stability of the RNA-RNA interaction, measured as ΔG (kcal/mol). (B) Viral sequences at nt 477 to 486 and 26,412 to 26,421 along passages in cell culture. Reversion to the wild-type sequence and substitutions are indicated by lowercase letters. The “p” and number(s) in the names of the recombinant viruses indicate the passage number(s). (C) Analysis of N sgmRNA levels normalized to gRNA (gray columns), with reference to the level in rTGEV-wt, which was set at 1. Data are averages for four independent infection experiments analyzed twice by qRT-PCR. Viral titers are shown by black columns. Error bars represent standard deviations. (D) Analysis by qRT-PCR of replication levels in mutant viruses rTGEV-cB* and rTGEV-B*. Viral replication was quantified as gRNA levels normalized to the amount of cellular GUS mRNA (gRNA/GUS B) in relation to the level in rTGEV-wt, which was set at 1. ST cells were infected at a multiplicity of infection of 1, and RNA was extracted at 8 h postinfection in order to avoid reinfections. Data are averages for four independent infection experiments analyzed twice by qRT-PCR. Error bars represent standard deviations.
Conservation of genomic RNA motifs involved in TGEV N gene transcription.
The sequences of RNA motifs B and cB involved in the novel RNA-RNA interaction described above are conserved among the Alphacoronavirus I species, comprising TGEV, canine coronavirus (CCoV), feline coronavirus (FCoV) type I, and FCoV type II or feline infectious peritonitis virus (FIPV) (Fig. 8A). Additionally, the secondary structure of the 5′-end genomic region, including the cB motif at the apical loop, is conserved according to Mfold predictions (Fig. 8B), suggesting a functional conservation in these CoV species. Interestingly, in the cB RNA motifs of CCoV (nt 477 to 486) and FCoV types I and II (nt 474 to 483), a single nucleotide change was found in comparison with the TGEV sequence, replacing a G-U pair with an A-U pair (Fig. 8A). In FCoV, another nucleotide change was observed at position 26,271, replacing U with G and thus disrupting the G-U pair present in TGEV and CCoV (Fig. 8A). However, none of these nt changes significantly affected the stability of the RNA-RNA interaction (Fig. 8A). The sequence of the nsp1 coding region containing the cB motif was mainly conserved at the nucleotide and amino acid levels in Alphacoronavirus I species. A substitution (G → A) in the first nucleotide of the cB motif in the canine and feline CoVs with respect to TGEV led to an amino acid change (Asp → Asn) in the nsp1 protein. In contrast, the coding sequence of the M protein containing the B motif was conserved at both the nucleotide and amino acid levels in Alphacoronavirus I species (Fig. 8A). Similar structures at similar genome positions have not been found either in other species of the Alphacoronavirus genus (human coronaviruses [HCoVs] 229E and NL63) or in Beta- and Gammacoronavirus species.
Fig 8.
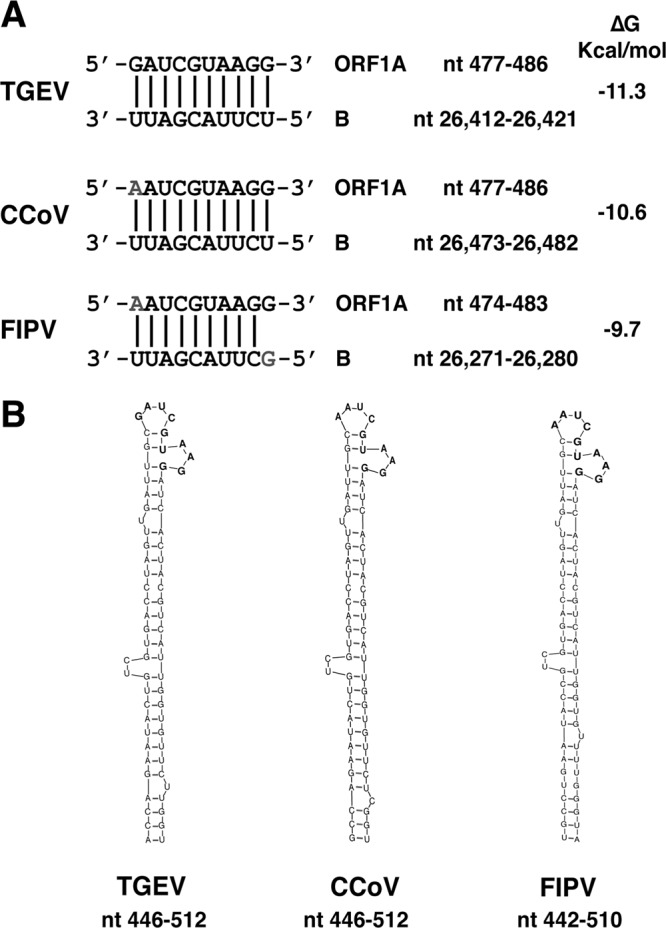
Conservation in Alphacoronavirus I species of the long-distance RNA-RNA interaction between the active domain and the 5′ end of the genome. (A) Names of viruses and RNA sequences in the 5′ end of the genome (cB motif into ORF1a) and the active domain (B motif) that are involved in the RNA-RNA interaction (nucleotide positions are indicated by numbers). Vertical lines indicate base pairs. The letters in gray represent nucleotide variations in relation to the TGEV sequence. The predicted stability for each interaction is indicated as ΔG (kcal/mol). (B) Schemes showing Mfold secondary structure prediction of the region in the 5′ end of the genome that contains nt 477 to 486 in TGEV and CCoV and nt 474 to 483 in FIPV.
DISCUSSION
The sequence and secondary structure requirements of the active domain, a TGEV genome region essential for N gene transcription regulation, have been shown in this report. The mechanism of action of this motif implies a novel long-distance interaction between RNA motifs mapping almost 26,000 nt apart in the coronavirus genome. This interaction between the B motif in the active domain and the complementary cB motif next to the 5′ end of the genome was essential to maintain N gene transcription levels in both TGEV-derived replicons and infectious viruses (Fig. 6 and 7). In contrast, transcription of sgmRNA 7 in mutant replicons (Fig. 6) and viruses (data not shown) was not significantly affected by the novel RNA-RNA interaction, suggesting that the interaction between B and cB RNA motifs is required mainly for N gene transcription. These results led us to propose a new model for N gene transcription regulation. According to this model, the RNA-RNA interaction between the proximal and distal elements, located within the transcription-regulating sequences of the N gene, relocates the active domain with a proper conformation to a site immediately preceding the N gene CS (Fig. 9). The B motif within the active domain interacts with the complementary cB motif sequence at the 5′ end of the genome (nt 477 to 486), bringing in close proximity the TRS-L and TRS-N sequences, which guide transcription by a recombination process (Fig. 9). The physical proximity of CS-L and CS-N sequences favors the template switch necessary for discontinuous transcription, increasing the frequency of this long-distance recombination. Our data represent the first evidence of a mechanism that brings into physical proximity the leader sequence at the 5′ end of the genome and the TRS preceding a gene close to the 3′ end, which map 26,000 nt apart in the coronavirus genome. We postulate that this network of RNA-RNA interactions drives the genome to adopt a high-order structure that facilitates discontinuous transcription in nidoviruses. The previously proposed working model of coronavirus transcription (17) includes three steps. The first step is the formation of precomplexes in the genomic RNA that locate the TRS-L in close proximity to the TRS-Bs at the 3′ end of the genome, promoting a stop of synthesis of the nascent minus-strand RNA and a high-frequency recombination. The novel finding reported in this work fits very well into the first step of the model, since the interaction described for the B and cB motifs locates the TRS-L in close proximity to the TRS-N. The second step is the formation of a complex among the TRS-L, the TRS-B, and the nascent cTRS-B. The complementarity between the TRS-L and the cTRS-B is evaluated, and if this complementarity is above a minimum threshold, a certain number of newly synthesized negative RNAs undergo a template switch to the TRS-L. The third step resumes the synthesis of RNA to obtain a complete negative-strand sgRNA.
Fig 9.

Model for N gene transcription regulation mediated by the formation of genome high-order structures. The black line represents the TGEV genome. The thicker line indicates the leader sequence in the 5′ end of the genome. The gray line with arrowheads represents the minus-polarity nascent RNA. The dotted gray line represents the copy of the leader that is added to the nascent RNA after the template switch. AD, active domain secondary structure prediction. The RNA-RNA interactions between the proximal and distal elements (pE, proximal element; dE, distal element) and between the B motif in the active domain (nt 26,412 to 26,421; B-M) and the cB motif in the 5′ end of the genome (nt 477 to 486; cB-M) are represented in the plus-strand genomic RNA. CS-L, conserved core sequence of the leader; CS-N, conserved core sequence of the N gene; cCS-N, copy of the CS-N. The arrow indicates the template switch to the leader sequence during discontinuous transcription.
It is probable that similar interactions are involved in the transcription of the other coronavirus genes. The conservation of both the primary sequence and secondary structure of the new RNA motifs involved in this network of RNA-RNA interactions within the Alphacoronavirus I species supports the existence and relevance of these interactions in coronavirus transcription.
Interestingly, in the nonrelated tombusvirus tomato bushy stunt virus (TBSV), RNA-RNA interactions relocate, just before each gene, a secondary structure that acts as a physical barrier mediating premature termination during the synthesis of the minus-strand subgenomic RNAs (7, 34). In TGEV, the secondary structure of the active domain and the high-order structure formed by the RNA-RNA interactions could also promote the slowdown and stop of the transcription complex at the CS-N. In addition to premature termination, CoV discontinuous transcription also requires a template switch, a recombination process guided by the homology between the TRS-L and the TRS-Bs. To achieve a high-frequency recombination during transcription, physical proximity between the leader sequence and the TRS-Bs would be extremely helpful. This proximity is probably promoted by the described long-distance RNA-RNA interaction between the active domain and the genome's 5′ end. This paper shows evidence for the first time of a molecular mechanism that could bring into physical proximity sequences present in the exceptionally long coronavirus RNA genomes that are fused during discontinuous transcription. Although RNA-RNA interactions leading to genomic high-order structures are relevant for essential viral processes in other RNA virus families (35–37), this work provides the first evidence for a long-distance (26,000 nt) RNA-RNA interaction regulating discontinuous transcription in nidoviruses.
ACKNOWLEDGMENTS
This work was supported by grants from the Ministry of Science and Innovation of Spain (BIO2010-16707 and PET2008-0310) and the European Community's Seventh Framework Programme (FP7/2007-2013), under the projects EMPERIE (EC grant agreement 223498) and PoRRSCon (EC grant agreement 245141). P.A.M.-G. received a fellowship from the Ministry of Science and Innovation of Spain (BES-2008-001932).
We gratefully acknowledge C. M. Sanchez, M. Gonzalez, and S. Ros for technical assistance.
Footnotes
Published ahead of print 10 October 2012
REFERENCES
- 1. de Groot RJ, Baker SC, Baric R, Enjuanes L, Gorbalenya AE, Holmes KV, Perlman S, Poon L, Rottier PJM, Talbot PJ, Woo PCY, Ziebuhr J. 2011. Coronaviridae, p 774–796 In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ. (ed), Virus taxonomy: ninth report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, San Diego, CA [Google Scholar]
- 2. Gorbalenya AE, Enjuanes L, Ziebuhr J, Snijder EJ. 2006. Nidovirales: evolving the largest RNA virus genome. Virus Res. 117:17–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Enjuanes L, Gorbalenya AE, de Groot RJ, Cowley JA, Ziebuhr J, Snijder EJ. 2008. The Nidovirales, p 419–430 In Mahy BWJ, Van Regenmortel M, Walker P, Majumder-Russell D. (ed), Encyclopedia of virology, 3rd ed Elsevier Ltd, Oxford, United Kingdom [Google Scholar]
- 4. Enjuanes L, Almazan F, Sola I, Zuniga S. 2006. Biochemical aspects of coronavirus replication and virus-host interaction. Annu. Rev. Microbiol. 60:211–230 [DOI] [PubMed] [Google Scholar]
- 5. Miller WA, Dreher TW, Hall TC. 1985. Synthesis of brome mosaic virus subgenomic RNA in vitro by internal initiation on (−)-sense genomic RNA. Nature 313:68–70 [DOI] [PubMed] [Google Scholar]
- 6. Lindenbach BD, Sgro JY, Ahlquist P. 2002. Long-distance base pairing in flock house virus RNA1 regulates subgenomic RNA3 synthesis and RNA2 replication. J. Virol. 76:3905–3919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang S, Mortazavi L, White KA. 2008. Higher-order RNA structural requirements and small-molecule induction of tombusvirus subgenomic mRNA transcription. J. Virol. 82:3864–3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. White KA. 2002. The premature termination model: a possible third mechanism for subgenomic mRNA transcription in (+)-strand RNA viruses. Virology 304:147–154 [DOI] [PubMed] [Google Scholar]
- 9. Pasternak AO, van den Born E, Spaan WJ, Snijder EJ. 2001. Sequence requirements for RNA strand transfer during nidovirus discontinuous subgenomic RNA synthesis. EMBO J. 20:7220–7228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sawicki SG, Sawicki DL. 1995. Coronaviruses use discontinuous extension for synthesis of subgenome-length negative strands. Adv. Exp. Med. Biol. 380:499–506 [DOI] [PubMed] [Google Scholar]
- 11. Brian DA, Spaan WJM. 1997. Recombination and coronavirus defective interfering RNAs. Semin. Virol. 8:101–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Draghici HK, Varrelmann M. 2010. Evidence for similarity-assisted recombination and predicted stem-loop structure determinant in potato virus X RNA recombination. J. Gen. Virol. 91:552–562 [DOI] [PubMed] [Google Scholar]
- 13. Dufour D, Mateos-Gomez PA, Enjuanes L, Gallego J, Sola I. 2011. Structure and functional relevance of a transcription-regulating sequence involved in coronavirus discontinuous RNA synthesis. J. Virol. 85:4963–4973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nagy PD, Simon AE. 1997. New insights into the mechanisms of RNA recombination. Virology 235:1–9 [DOI] [PubMed] [Google Scholar]
- 15. Sola I, Mateos-Gomez PA, Almazan F, Zuniga S, Enjuanes L. 2011. RNA-RNA and RNA-protein interactions in coronavirus replication and transcription. RNA Biol. 8:237–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alonso S, Izeta A, Sola I, Enjuanes L. 2002. Transcription regulatory sequences and mRNA expression levels in the coronavirus transmissible gastroenteritis virus. J. Virol. 76:1293–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sola I, Moreno JL, Zuñiga S, Alonso S, Enjuanes L. 2005. Role of nucleotides immediately flanking the transcription-regulating sequence core in coronavirus subgenomic mRNA synthesis. J. Virol. 79:2506–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van Marle G, Dobbe JC, Gultyaev AP, Luytjes W, Spaan WJM, Snijder EJ. 1999. Arterivirus discontinuous mRNA transcription is guided by base pairing between sense and antisense transcription-regulating sequences. Proc. Natl. Acad. Sci. U. S. A. 96:12056–12061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zuñiga S, Sola I, Alonso S, Enjuanes L. 2004. Sequence motifs involved in the regulation of discontinuous coronavirus subgenomic RNA synthesis. J. Virol. 78:980–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moreno JL, Zuniga S, Enjuanes L, Sola I. 2008. Identification of a coronavirus transcription enhancer. J. Virol. 82:3882–3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mateos-Gomez PA, Zuniga S, Palacio L, Enjuanes L, Sola I. 2011. Gene N proximal and distal RNA motifs regulate coronavirus nucleocapsid mRNA transcription. J. Virol. 85:8968–8980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Delmas B, Gelfi J, Sjöström H, Noren O, Laude H. 1993. Further characterization of aminopeptidase-N as a receptor for coronaviruses. Adv. Exp. Med. Biol. 342:293–298 [DOI] [PubMed] [Google Scholar]
- 23. Frolov I, Hoffman TA, Prágai BM, Dryga SA, Huang HV, Schlesinger S, Rice CM. 1996. Alphavirus-based expression vectors: strategies and applications. Proc. Natl. Acad. Sci. U. S. A. 93:11371–11377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McClurkin AW, Norman JO. 1966. Studies on transmissible gastroenteritis of swine. II. Selected characteristics of a cytopathogenic virus common to five isolates from transmissible gastroenteritis. Can. J. Comp. Med. Vet. Sci. 30:190–198 [PMC free article] [PubMed] [Google Scholar]
- 25. Almazan F, González JM, Pénzes Z, Izeta A, Calvo E, Plana-Durán J, Enjuanes L. 2000. Engineering the largest RNA virus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. U. S. A. 97:5516–5521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sanchez CM, Izeta A, Sánchez-Morgado JM, Alonso S, Sola I, Balasch M, Plana-Durán J, Enjuanes L. 1999. Targeted recombination demonstrates that the spike gene of transmissible gastroenteritis coronavirus is a determinant of its enteric tropism and virulence. J. Virol. 73:7607–7618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Correa I, Jiménez G, Suñé C, Bullido MJ, Enjuanes L. 1988. Antigenic structure of the E2 glycoprotein from transmissible gastroenteritis coronavirus. Virus Res. 10:77–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Almazan F, Galan C, Enjuanes L. 2004. The nucleoprotein is required for efficient coronavirus genome replication. J. Virol. 78:12683–12688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sola I, Galán C, Mateos-Gómez PA, Palacio L, Zúñiga S, Cruz JL, Almazán F, Enjuanes L. 2011. The polypyrimidine tract-binding protein affects coronavirus RNA accumulation levels and relocalizes viral RNAs to novel cytoplasmic domains different from replication-transcription sites. J. Virol. 85:5136–5149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 31. Mathews DH, Sabina J, Zuker M, Turner DH. 1999. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 288:911–940 [DOI] [PubMed] [Google Scholar]
- 32. Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31:3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Penzes Z, Gonzalez JM, Calvo E, Izeta A, Smerdou C, Méndez A, Sanchez CM, Sola I, Almazan F, Enjuanes L. 2001. Complete genome sequence of transmissible gastroenteritis coronavirus PUR46-MAD clone and evolution of the Purdue virus cluster. Virus Genes 23:105–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lin HX, White KA. 2004. A complex network of RNA-RNA interactions controls subgenomic mRNA transcription in a tombusvirus. EMBO J. 23:3365–3374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu Y, Wimmer E, Paul AV. 2009. Cis-acting RNA elements in human and animal plus-strand RNA viruses. Biochim. Biophys. Acta 1789:495–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miller WA, White KA. 2006. Long-distance RNA-RNA interactions in plant virus gene expression and replication. Annu. Rev. Phytopathol. 44:447–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu B, Pogany J, Na H, Nicholson BL, Nagy PD, White KA. 2009. A discontinuous RNA platform mediates RNA virus replication: building an integrated model for RNA-based regulation of viral processes. PLoS Pathog. 5:e1000323 doi:10.1371/journal.ppat.1000323 [DOI] [PMC free article] [PubMed] [Google Scholar]