

Abstract
Herpes simplex virus (HSV) pathogenesis in mice differs based on availability of the principal entry receptors herpesvirus entry mediator (HVEM) and nectin-1 in a manner dependent upon route of inoculation. After intravaginal or intracranial inoculation of adult mice, nectin-1 is a major mediator of neurologic disease, while the absence of either receptor attenuates disease after ocular infection. We tested the importance of receptor availability and route of infection on disease in mouse models of neonatal HSV. We infected 7-day-old mice lacking neither or one principal HSV receptor or both principal HSV receptors with HSV-2 via a peripheral route (intranasal), via a systemic route (intraperitoneal), or by inoculation directly into the central nervous system (intracranial). Mortality, neurologic disease, and visceral dissemination of virus were significantly attenuated in nectin-1 knockout mice compared with HVEM knockout or wild-type mice after intranasal inoculation. Mice lacking both entry receptors (double-knockout mice) showed no evidence of disease after inoculation by any route. Nectin-1 knockout mice had delayed mortality after intraperitoneal inoculation relative to wild-type and HVEM knockout mice, but virus was able to spread to the brain and viscera in all genotypes except double-knockout mice. Unlike in adult mice, HVEM was sufficient to mediate disease in neonatal mice after direct intracranial inoculation, and the absence of HVEM delayed time to mortality relative to that of wild-type mice. Additionally, in wild-type neonatal mice inoculated intracranially, HSV antigen did not primarily colocalize with NeuN-positive neurons. Our results suggest that differences in receptor expression between adults and newborns may partially explain differences in susceptibility to HSV-2.
INTRODUCTION
Herpes simplex virus (HSV) causes neonatal infection in about 1 in 3,200 live births in the United States (1). More than half of infants with neonatal HSV disease have disseminated disease or encephalitis (2), which, despite effective antiviral treatment, results in the deaths of more than 25% of those with disseminated disease and in neurologic morbidity in more than two-thirds of survivors of encephalitis (3). Relative to other populations infected with HSV, newborns have the highest rates of dissemination and central nervous system (CNS) disease (4). Although differences in immune responses from those of adults have been implicated, precise reasons for the increased severity of disease in infants remain unknown (5).
Infection of susceptible human and mouse cells by HSV requires binding of the viral glycoprotein gD with one of its cell surface receptors (6, 7). HSV gD binds to three general classes of surface receptors, including herpesvirus entry mediator (HVEM), nectin-1 and −2, and specific sites in heparan sulfate (7). Of these, HVEM and nectin-1 appear to mediate viral entry most efficiently in both humans and mice (8, 9). The mouse receptors are orthologous to the human receptors, and HSV disease in mice resembles that in humans, allowing application of mouse models to the study of HSV pathogenesis in humans.
Recent studies with adult mice suggest that nectin-1 is important for development of neurologic disease after HSV type 2 (HSV-2) infection. After intravaginal inoculation, nectin-1 knockout (KO) mice had less severe neurologic disease, increased survival, and lower viral titers in dorsal root ganglia compared with wild-type (WT) or HVEM KO mice (10). Double-KO mice lacking both HVEM and nectin-1 were resistant to infection. Adult nectin-1 KO mice were resistant to encephalitis after direct intracerebral inoculation with HSV-2 (11). Interestingly, compared to that in WT mice, disease was attenuated in both HVEM KO and nectin-1 KO mice after corneal infection with HSV-1 (12).
In this study, we applied a murine model of neonatal HSV to compare disease in wild-type C57BL/6, HVEM KO, nectin-1 KO, and double-KO mice after inoculation by peripheral (intranasal [i.n.]), systemic (intraperitoneal [i.p.]), and direct intracranial (i.c.) routes. Our observations in this model support the importance of an interaction of HSV-2 with nectin-1 in the pathogenesis of neonatal HSV disease, which is most pronounced after intranasal inoculation. However, unlike in adult mice, HVEM is sufficient to mediate neurologic disease in neonatal mice, and nonneuronal cells appear to be the principal targets of infection in the CNS. Our data suggest that differences in HSV receptor availability may alter pathogenesis of disease in newborns relative to that in adults.
MATERIALS AND METHODS
Cells and viruses.
Vero cells were cultured in Dulbecco's modification of Eagle's medium (DMEM) plus 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin and were used for virus propagation and determination of virus titers. Plaque titrations were performed by standard methods.
HSV-2 strain 333 was originally isolated from a genital lesion and underwent limited passage in human cells (13). The virus was plaque purified and passaged no more than three times in Vero cells. The recombinant virus HSV-2/Δ7-15 was generated as described previously (14); it was purified and grown, and its titers were determined, on Vero cells in a manner identical to that for HSV-2 strain 333. This virus is unable to engage HVEM for entry (14).
Mouse HSV-2 infection and postinfection procedures.
Animal care and use in this study were in accordance with institutional and NIH guidelines, and all studies were approved by the Northwestern University Animal Care and Use Committee. The mouse strains used have been previously described (10), including C57BL/6 (WT), HVEM KO (Tnfrsf14−/−), nectin-1 KO (Pvrl−/−), and double-KO (Tnfrsf14−/− × Pvrl1−/−) mice. Knockout strains were all on a C57BL/6 background. Mice were maintained under specific-pathogen-free conditions until transfer to a containment facility just prior to infection.
Breeding pairs were regularly monitored, with males separated from gravid females prior to delivery. Litters were infected at 7 days of age, which from an immunologic perspective corresponds most closely to humans at birth (15). HSV-2 was diluted in phosphate-buffered saline (PBS) containing 1% inactivated calf serum and 0.1% glucose (PBS-GCS) to deliver the target inoculum of 1 × 104 PFU/mouse for i.n. and i.p. inoculation and 1 × 103 PFU/mouse for i.c. inoculation of newborn mice. In experiments using i.c. inoculation of adult mice, 1 × 104 PFU/mouse was delivered by a method similar to that used for neonatal mice.
For i.n. inoculation, virus was delivered to pups via micropipette to both nares in a 5-μl total volume; each animal was held with the nose pointing upwards for at least 60 s after inoculation. Animals inoculated i.p. received virus in a 50-μl total volume, delivered with the needle tip pointing toward the spleen. For i.c. inoculation, a Hamilton syringe with a 26-gauge needle and a needle guard was used to inoculate a 5-μl total volume into one hemisphere of the brain in the approximate region of the hippocampus, using as a landmark a triangle formed by the nose, the ear, and the eye and injecting into the approximate center of that triangle. The left hemisphere was injected for pathogenesis and titer studies, while the right hemisphere was used for immunohistochemical studies. Each experiment also included at least one control mouse which was injected in an identical manner using the same volume of PBS-GCS.
Infected mice were monitored daily for signs of neurologic disease, including lethargy, motionlessness, and hunched posture. Mice displaying severe signs of illness were immediately sacrificed; other groups were sacrificed at various time points for measurements of viral spread.
Organs were harvested from infected and control pups at different times after infection. After i.n. and i.p. inoculation, brain, spleen, liver, and lung were harvested. Only brains were harvested after i.c. inoculation. Tissues were weighed, homogenized in DMEM with 5% inactivated calf serum and 1% ciprofloxacin, and sonicated. Tissue homogenates were stored at −70°C until titer determination.
Immunohistochemistry.
Selected mice inoculated by the i.c. route were sacrificed at 48 h to localize viral antigen within brain parenchyma, using methods similar to those previously described (11). Mice were deeply anesthetized with isoflurane and perfused transcardially first with PBS (pH 7.2), followed by 4% paraformaldehyde in PBS. The brain was removed and postfixed in 4% paraformaldehyde at 4°C. Coronal sections 50 μm thick were cut with a vibratome (Leica). Selected sections were washed with PBS and preincubated with 4% normal goat serum (NGS; Sigma-Aldrich) diluted in 0.1% Triton-X solution in PBS for 1.5 h at room temperature. Sections were then incubated overnight at 4°C with primary antibodies diluted 1:500 in working solution (2% NGS, 0.1% Triton-X in PBS). Antibodies used included rabbit polyclonal anti-HSV-2 (Dako) and mouse monoclonal anti-NeuN, a neuronal marker (Chemicon/Millipore). Sections were then washed six times for 10 min each time in PBS before incubation, for 1.5 h at room temperature, with secondary antibodies, including goat anti-rabbit IgG (H+L) Alexa 568 and goat anti-mouse IgG1 Alexa 488 diluted 1:1,000 (Invitrogen/Molecular Probes). Sections were washed six times for 10 min each time in PBS before being mounted on VWR frost-plus charged slides with Vectashield hard-set mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories). Images were taken with an Olympus Fluoview FV1000MPE confocal microscope.
Statistical tests.
Kaplan-Meier survival analysis was performed using the Gehan-Breslow test. Geometric means of values for viral infectious units in tissues were compared using the unpaired Student t test.
RESULTS
Mortality of newborn mice is dependent on route of inoculation and entry receptor availability.
Litters of mice were monitored daily after inoculation for signs of disease. In general, infected mice appeared similar to uninfected controls for the first several days after i.n. or i.p. inoculation or the first 24 h after i.c. inoculation. Infected and uninfected pups gained weight at similar rates until signs of illness developed (data not shown). Productively infected mice eventually demonstrated obvious neurologic distress, evidenced by difficulty in moving and lying on their sides, and were sacrificed as soon as these signs were apparent.
Initial experiments used i.n. inoculation to model a physiologically relevant route of exposure in human newborns (16). Mortality of wild-type and HVEM KO mice inoculated i.n. with HSV-2 occurred between 4 and 8 days after inoculation, leading to death in 70 to 85% of mice (Fig. 1A). In contrast, the overall survival rate of nectin-1 KO and double-KO mice was >80%, and at least one of the deaths observed in each of these cohorts was potentially attributable to poor postnatal weight gain and may not have been directly due to the effects of viral infection. These results show that mortality from i.n. infection with HSV-2 in newborn mice is significantly attenuated when HVEM is the only available receptor.
Fig 1.

Survival of neonatal mice of different genotypes infected with HSV-2 by the intranasal (A), intraperitoneal (B), and intracranial (C and D) routes. Panel A shows the symbol key for all plots. More than 15 mice per genotype were studied to generate the mortality data after i.n. inoculation, while other data are from 7 to 15 mice per genotype. (A) Kaplan-Meier analysis of mortality after i.n. inoculation demonstrates statistically significant differences in time to death for nectin-1 KO compared with WT (P = 0.0006) or HVEM KO (P = 0.007) mice and for double-KO versus WT (P = 0.0002) or HVEM KO (P = 0.002) mice. The difference in survival between WT and HVEM KO mice did not reach statistical significance (P = 0.08), and there was no difference between nectin-1 KO and double-KO mice (P = 0.78). (B) Kaplan-Meier analysis of mortality after i.p. inoculation identifies statistically significant differences in time to death for nectin-1 KO compared with WT (P < 0.0001) or HVEM KO (P = 0.0002) mice and for double-KO versus WT (P = 0.0002), HVEM KO (P = 0.0012), and nectin-1 KO mice (P = 0.015). The difference in survival between WT and HVEM KO mice did not reach statistical significance (P = 0.14). (C) Kaplan-Meier analysis of mortality after i.c. inoculation with HSV-2 (333) demonstrates statistically significant differences in time to death for HVEM KO, nectin-1 KO, and double-KO mice compared with WT mice (P < 0.0001). There was no difference between HVEM KO and nectin-1 KO mice (P = 0.66), while both groups were statistically different from double-KO mice (P < 0.0008). (D) Inoculation of WT or nectin-1 KO mice with HSV-2/Δ7–15, which is unable to engage HVEM, shows statistically significant difference in time to death for WT mice versus nectin-1 KO mice (P < 0.0001).
Decreased mortality after i.n. inoculation of mice lacking nectin-1 could be due to a relative lack of expression of HVEM in the nasal mucosa, leading to decreased viral replication in mucosal epithelia and diminished spread either directly into neurons or systemically throughout the animal. In order to test the importance of different entry receptors on HSV disease during systemic infection, we inoculated groups of newborn mice i.p. with virus. Inoculation i.p. led to mortality in WT and HVEM KO mice within 4 to 5 days (Fig. 1B). Although there was a delay in time to mortality in nectin-1 KO mice, none survived beyond 7 days after inoculation. In contrast, survival was 100% in double-KO mice for at least 10 days after i.p. inoculation. These results suggest that HVEM is sufficient to cause mortality in newborn mice during systemic infection, although mortality is delayed in the absence of nectin-1.
Severe neonatal disease due to HSV infection can be due to a combination of central nervous system (CNS) infection (encephalitis) and visceral dissemination (16). To directly test the entry receptor requirements of HSV-2 for replication in the CNS of newborn mice, we inoculated mice of different genotypes i.c. Mortality occurred in WT mice within 2 days of i.c. inoculation (Fig. 1C), while a significant delay in mortality was observed in HVEM KO and nectin-1 KO mice. However, nearly all WT and single-KO mice died within 5 days of i.c. inoculation, while all double-KO mice survived without symptoms of illness for the duration of the experiment.
To further confirm that disease in nectin-1 KO mice was due to engagement of HVEM by HSV-2, we inoculated separate groups of WT and nectin-1 KO mice with HSV-2/Δ7–15, a strain of HSV-2 (333) mutated to be unable to use HVEM for entry (14). Inoculation of WT mice with this virus i.c. led to mortality within 3 to 4 days (Fig. 1D), similar to our observations in HVEM KO mice inoculated with HSV-2 (333) (Fig. 1C). However, all nectin-1 KO mice inoculated i.c. with HSV-2/Δ7–15 survived without symptoms of illness for the duration of the experiment. Together, the results of the i.c. inoculation experiments show that HSV-2 is able to use either HVEM or nectin-1 to cause CNS disease in newborn mice but that disease is delayed in the absence of either receptor. Moreover, the absence of both receptors is protective against neurologic disease.
HSV replicates in brain tissue of newborn WT, HVEM KO, and nectin-1 KO mice.
The brains of some infected mice were homogenized and viral titers were determined by plaque assay to measure spread of virus to the CNS after different routes of inoculation. After i.n. inoculation, virus was detected in increasing numbers of WT and HVEM KO mice sacrificed over the first 5 days, with most mice having virus recovered from brains by days 4 to 5 (Fig. 2A). In contrast, virus was recovered from brain tissue in only a single nectin-1 KO mouse, on day 3 after inoculation. When virus was recovered, titers were similar regardless of the genotype studied.
Fig 2.

HSV-2 titers in brain tissue after various routes of inoculation in newborn mice of different genotypes. Each symbol represents PFU per g tissue from an individual mouse, with horizontal lines indicating geometric means for each group. Symbols plotted in the gray box represent values below the detection limit of the assay. Virus was not detected in any control mice inoculated with vehicle by the same route or in any double-KO mice inoculated with virus. (A) Time course of HSV-2 spread into brain tissue in mice inoculated i.n. (B) Titers of HSV-2 in brain tissue at time of sacrifice for different genotypes of mice inoculated i.p. Mice were sacrificed after demonstrating severe neurologic symptoms: days 4 or 5 for WT and HVEM KO mice and days 4 to 8 for nectin-1 KO mice. (C) Titers of HSV-2 in brain tissue at time of sacrifice for different genotypes of mice inoculated i.c. Mice were sacrificed after demonstrating severe neurologic symptoms: day 2 for WT mice and days 3 to 5 for HVEM KO and nectin-1 KO mice. (D) Titers of HSV-2 in brain tissue at time of sacrifice for WT or nectin-1 KO mice inoculated i.c. with HSV-2/Δ7–15. WT mice were sacrificed on days 3 to 4 after demonstrating severe neurologic symptoms, while nectin-1 KO mice were sacrificed on day 5 after remaining asymptomatic.
Spread of virus into the CNS after systemic (i.p.) inoculation was measured in mice at the time of sacrifice due to neurologic symptoms. One cohort of asymptomatic nectin-1 KO mice was also sacrificed 5 days after inoculation, and asymptomatic double-KO mice were sacrificed either 5 or 10 days after inoculation. Nearly all WT, HVEM KO, or nectin-1 KO mice had virus recovered at similar levels regardless of genotype (Fig. 2B), despite the increased length of time observed for nectin-1 KO mice to demonstrate neurologic symptoms (Fig. 1B). Of the individual mice in each genotype which did not have measurable virus recovered from brain after i.p. inoculation, the WT mouse had severe symptoms of illness, while the HVEM and nectin-1 KO mice were asymptomatic at sacrifice 5 days after inoculation.
Recovery of virus from brains of mice infected i.c. was similar to that observed for i.p. inoculation. Nearly all WT, HVEM KO, and nectin-1 KO mice infected i.c. had virus recovered from the brain at similar levels at the time of sacrifice (Fig. 2C). It is not clear why a single HVEM KO mouse did not have recoverable virus, as this mouse had neurologic symptoms. Mutant virus able to engage HVEM only was recovered at levels in WT mice similar to those observed for WT virus but was not generally recovered from brains of nectin-1 KO mice (Fig. 2D). Interestingly, 2 of 14 nectin-1 KO mice inoculated i.c. with HSV-2/Δ7–15 also had virus recovered from the brain 5 days after inoculation, despite none of these mice demonstrating neurologic symptoms. The titers in these mice were lower than in mice demonstrating neurologic symptoms but suggest either that low levels of replication may occur in some circumstances via the use of a receptor other than HVEM or nectin-1 for entry or, alternatively, that HSV-2/Δ7–15 may be able to use HVEM at low levels for entry. However, the latter explanation is not supported by our prior studies with this virus (14).
Virus was not detected from brains of any double-KO mice sacrificed 5 days or more after inoculation by any route (data not shown), suggesting that either HVEM or nectin-1 is generally necessary for productive infection to be sustained.
HSV disseminates to visceral organs in newborn mice after i.n. or i.p. inoculation.
Neonatal HSV infection encompasses overlapping clinical presentations which can include dissemination of virus to visceral organs (2). We measured viral spread to spleen, liver, and lung after either i.n. or i.p. inoculation (Fig. 3). After i.n. inoculation, about half of WT and HVEM KO mice had virus detected in spleen and liver, while most had virus detected in lung tissue. Virus was not detected in visceral organs of any nectin-1 KO newborn mice sacrificed 5 days after inoculation (Fig. 3A to C), though we sporadically detected virus in these organs from individual nectin-1 KO mice sacrificed at earlier time points after inoculation (data not shown). This observation supports the concept that nectin-1 is needed for efficient infection and replication at the nasal mucosa. In contrast, virus was frequently detected in visceral organs of pups inoculated i.p. (Fig. 3D to F) as long as one of the principal entry receptors was present, suggesting that neither HVEM nor nectin-1 is required for virus to spread throughout the newborn mouse after inoculation by a systemic route.
Fig 3.

HSV-2 titers in spleen, liver, and lung after i.n. or i.p. inoculation of mice of different genotypes. Each symbol represents PFU per g tissue from an individual mouse, with horizontal lines indicating geometric means for each group. Symbols plotted in the gray box represent values below the detection limit of the assay. Virus was not detected in any control mice inoculated with vehicle by the same route or in any double-KO mice inoculated with virus. (A to C) Titers of HSV-2 in spleen, liver, and lung tissue 4 to 5 days after infection for different genotypes of mice inoculated i.n. (D to F) Titers of HSV-2 in spleen, liver, and lung tissue at time of sacrifice for different genotypes of mice inoculated i.p. Mice were sacrificed after demonstrating severe neurologic symptoms: days 4 or 5 for WT and HVEM KO mice and days 4 to 8 for nectin-1 KO mice.
HSV antigen does not colocalize with neurons in brains of newborn mice after i.c. inoculation.
HSV-2 infection in different regions of the brain was assessed by immunohistochemical evaluation of brain sections from WT mice sacrificed 48 h after inoculation. Unlike in adult mice, in which viral antigen primarily colocalized with NeuN-positive neuronal cells (Fig. 4F) (11), we were unable to identify NeuN-positive cells which were also positive for HSV antigen in brains of neonatal mice (Fig. 4A to E). This observation suggests that CNS disease in newborns caused by HSV-2 may be mediated by infection of nonneuronal cells.
Fig 4.
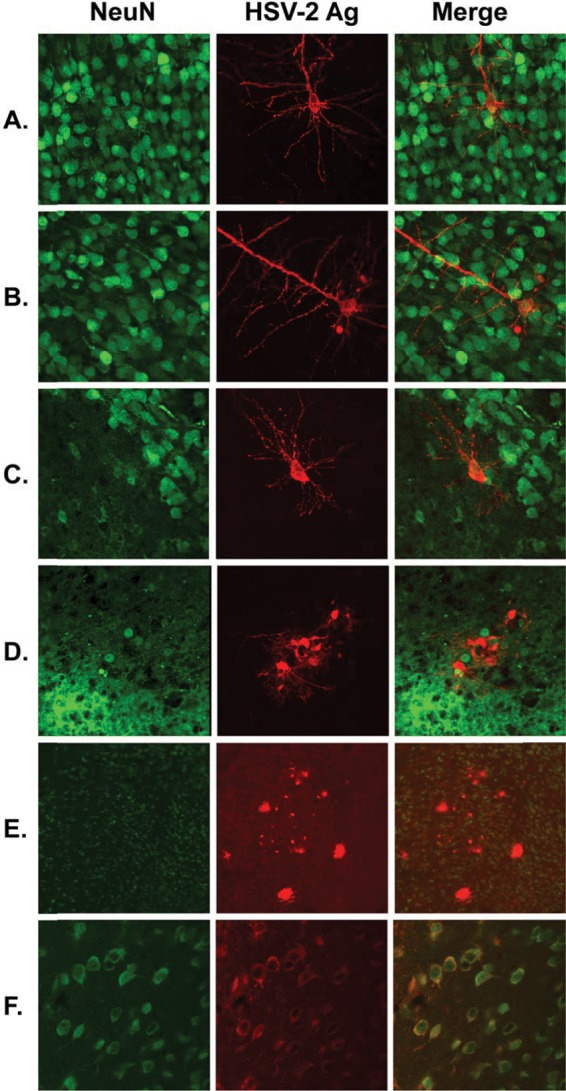
Localization of infected cells in brains of mice after i.c. inoculation. Brains were harvested from newborn or adult WT mice 48 h after inoculation and stained for the neuronal marker NeuN (green) and HSV antigen (Ag) (red) according to the methods described in the text. Representative images are from areas of newborn cortex (A to B), newborn hypothalamus (C), newborn thalamus (D to E), and adult hippocampus (F).
DISCUSSION
We studied pathogenesis of neonatal HSV-2 disease in mouse models of infection. Our main finding is that receptor requirements for HSV-2 infection of newborns differ from those previously found in experiments using adult mice (11). In the prior study (11), adult nectin-1 KO mice inoculated i.c. with HSV-2 demonstrated no clinical symptoms for as long as 14 days. Those prior experiments (11) also included immunohistochemical analyses of brains from i.c.-inoculated adult nectin-1 KO mice which identified HSV antigen only in nonparenchymal cells lining the walls of the ventricles, and only in mice studied 24 h after inoculation, after which virus was apparently cleared. In contrast, we found in the current study that newborn nectin-1 KO mice were susceptible to neurologic disease after direct i.c. inoculation with HSV-2, albeit in a delayed fashion (Fig. 1C), with virus able to replicate in the brain to levels similar to those in newborn WT or HVEM KO mice (Fig. 2C). We were also able to demonstrate in this study that disease in nectin-1 KO mice required interaction of the virus with HVEM, as mice inoculated with a mutant virus unable to engage HVEM exhibited no evidence of neurologic disease (Fig. 1D), and mutant virus was not recovered from most i.c.-inoculated nectin-1 KO mice (Fig. 2D).
There are several possible explanations for the observation that HVEM can mediate disease in newborn but not adult mice. HVEM expression in the brain may be developmentally regulated, such that newborn but not adult cells are susceptible to HSV infection mediated by HVEM. Precise temporal and spatial expression of cell surface molecules is important in the developing nervous system (17–19) and may include receptors which have functions in immune signaling (19), such as HVEM. Alternatively, there may be HVEM-expressing cells in the newborn brain that are simply not present in the adult brain; for example, certain neurons in the developing brain undergo autophagy or apoptosis for proper brain development (20, 21). It could be speculated that cells which may express HVEM in the newborn CNS are among those which are removed during development. Lastly, it is possible that both newborn and adult brains may contain HVEM-expressing cells but that adult cells may be resistant to HSV infection for other reasons, such as rapid apoptosis after viral infection or other mechanisms which may confer resistance to viral replication. Although HSV infection inhibits apoptosis in many cell types, including neurons (22, 23), it is known, for example, that immature murine and human dendritic cells rapidly undergo apoptosis after HSV infection (24, 25). One might hypothesize that adult HVEM-expressing cells in the CNS might undergo apoptosis more quickly than similar neonatal cells, leading to a relative limitation of viral replication. It is also plausible that other mechanisms of intrinsic cellular antiviral resistance may also differ between HVEM-expressing cells in the adult and newborn CNS, such as ND10-mediated restriction of viral transcription (26).
A second important observation from this work is that unlike findings in adult mice (Fig. 4F) (11), NeuN-positive cells in the newborn brain were not the primary targets of HSV infection after i.c. inoculation (Fig. 4A to E). Although morphologically some of the infected cells resembled neurons, detection of NeuN generally indicates postmitotic (“mature”) neurons, at least in the adult brain (27). It is possible that we detected HSV antigen in developing neurons which were NeuN negative, though this would not explain why we were unable to detect HSV antigen in any NeuN-positive cells. HSV infection could downregulate NeuN expression, but in this case we would expect to see at least some NeuN expression in at least some infected cells, particularly at the relatively early time point studied (48 h after inoculation). More intriguing is the hypothesis that nonneuronal cells are the primary target of HSV infection in the newborn CNS. Studies of mouse brain development suggest that in the CA1 region of the hippocampus, total numbers of neurons are relatively constant between 1 and 8 weeks of age, while 1-week-old mice have significantly higher numbers of astrocytes and microglia and significantly fewer oligodendrocytes than 8-week-old mice (28). Such glial cells could be a primary target of HSV infection in the newborn CNS, which might suggest a difference in pathophysiology of neurologic disease in newborns relative to adults. We are actively pursuing further experiments intended to identify the cell type infected in the newborn CNS and the responses of these cells after infection.
Our observation that neurologic disease is delayed in newborn HVEM KO mice relative to WT mice inoculated i.c. (Fig. 1C) represents the first infectious model that has shown a difference in pathogenesis of HSV-2 infection on the basis of HVEM expression. As noted above, prior studies using intravaginal or intracranial inoculation did not show any effect of knocking out HVEM on HSV-2 disease in mice (10, 11). In an ocular scarification infection model using adult mice, Karaba et al. demonstrated that disease due to HSV-1 was attenuated in the absence of HVEM expression (12). However, studies using HSV-2 in this model led to results which differ from those with HSV-1, showing no dependence of disease characteristics on HVEM expression (A. H. Karaba, S. J. Kopp, and R. Longnecker, submitted for publication). The role of HVEM in infections with HSV is complex, as HVEM has a natural function in immune signaling in addition to its role in HSV entry (29). Previous experiments comparing vaginal mucosal chemokine responses after infection with HSV-2/Δ7-15 compared to wild-type virus suggest that HSV may alter signals mediated by HVEM (14) and may subsequently affect HSV-specific T-cell recall responses at the mucosa (30). Functions for HVEM in either neurodevelopment or neuroinflammatory responses have not been described, and efforts to identify the cell types expressing HVEM in the developing brain and the responses of those cells to HSV infection may inform future experiments in those areas.
Along with direct i.c. infection of brain, we studied physiologically relevant (i.n.) and systemic (i.p.) routes of inoculation. In both situations, disease phenotype was influenced by nectin-1 expression, with significantly less mortality in nectin-1 KO mice after i.n. inoculation than in WT and HVEM KO mice (Fig. 1A) and a delay in mortality after i.p. inoculation (Fig. 1B). After i.n. inoculation, spread of virus to brain (Fig. 2A) and viscera (Fig. 3A to C) was significantly lower in the absence of nectin-1. After i.p. inoculation, however, virus was able to spread to the brain (Fig. 2B) and viscera (Fig. 3D to F) equally well in nectin-1 KO, HVEM KO, and WT mice.
Our results after i.n. inoculation suggest that HVEM is not expressed to a significant degree in nasal mucosal tissue of newborn mice. An alternative explanation is that cells expressing HVEM are present but resistant to HSV replication, and the virus must infect nectin-1 expressing cells to efficiently replicate and spread in the newborn nasal mucosa. This result is reminiscent of observations made after intravaginal infection of adult mice, after which mucosal replication was diminished (but not absent) in the absence of nectin-1, leading to a delay in time to mortality (10). Similarly, knockdown of nectin-1 with a cholesterol-conjugated small interfering RNA (siRNA) has been shown to provide protection against vaginal HSV-2 challenge in mice (31). A clinically relevant implication of these collective results is that inhibitors of the interaction of HSV gD with nectin-1 could be of therapeutic benefit for patients at risk of mucosal exposure to HSV, such as infants potentially exposed to HSV at the time of delivery.
HSV DNA is commonly detected in blood samples from infected newborns and may persist for weeks after infection (32–34). Systemic dissemination, which we and others have observed in mouse models after i.n. inoculation (35, 36), is likely secondary to hematogenous spread of virus. Although our model of systemic infection demonstrated that nectin-1 is not required for disease and viral spread, the delay in mortality in newborn nectin-1 KO mice (Fig. 1B) might still support efforts to inhibit the gD-nectin-1 interaction as a therapeutic approach, likely in combination with drugs that inhibit viral replication (e.g., acyclovir).
In summary, we have identified differences in HSV entry receptor requirements for pathogenesis of HSV-2 disease in newborn mice compared with previously reported data for adults (10–12), and we found that different cells are the principal targets of infection in newborn mice and adult mice after direct i.c. inoculation. These observations may in part account for differences in susceptibility to infection of newborns and adults, and they continue to support a possible therapeutic role for compounds which inhibit the gD-nectin-1 interaction, as has been previously suggested (10).
ACKNOWLEDGMENTS
We thank Nanette Susmarski for providing cell culture expertise and assistance in viral titering. We also appreciate helpful comments and guidance from Patricia Spear, Richard Longnecker, Gregory Smith, Anne Rowley, and members of the Longnecker and Smith Labs.
Footnotes
Published ahead of print 24 October 2012
REFERENCES
- 1. Thompson C, Whitley R. 2011. Neonatal herpes simplex virus infections: where are we now? Adv. Exp. Med. Biol. 697:221–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kimberlin DW. 2004. Neonatal herpes simplex infection. Clin. Microbiol. Rev. 17:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kimberlin DW, Lin CY, Jacobs RF, Powell DA, Frenkel LM, Gruber WC, Rathore M, Bradley JS, Diaz PS, Kumar M, Arvin AM, Gutierrez K, Shelton M, Weiner LB, Sleasman JW, de Sierra TM, Soong SJ, Kiell J, Lakeman FD, Whitley RJ. 2001. Natural history of neonatal herpes simplex virus infections in the acyclovir era. Pediatrics 108:223–229 [DOI] [PubMed] [Google Scholar]
- 4. Schiffer JT, Corey L. 2009. Herpes simplex virus. In Mandell GL, Bennett JE, Dolin R. (ed), Mandell, Douglas, and Bennett's principles and practice of infectious diseases, 7th ed, p 1943–1962 Churchill Livingstone, Philadelphia, PA [Google Scholar]
- 5. Muller WJ, Jones CA, Koelle DM. 2010. Immunobiology of herpes simplex virus and cytomegalovirus infections of the fetus and newborn. Curr. Immunol. Rev. 6:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Spear PG. 2004. Herpes simplex virus: receptors and ligands for cell entry. Cell. Microbiol. 6:401–410 [DOI] [PubMed] [Google Scholar]
- 7. Spear PG, Eisenberg RJ, Cohen GH. 2000. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 275:1–8 [DOI] [PubMed] [Google Scholar]
- 8. Lopez M, Cocchi F, Menotti L, Avitabile E, Dubreuil P, Campadelli-Fiume G. 2000. Nectin2alpha (PRR2alpha or HveB) and nectin2delta are low-efficiency mediators for entry of herpes simplex virus mutants carrying the Leu25Pro substitution in glycoprotein D. J. Virol. 74:1267–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shukla D, Liu J, Blaiklock P, Shworak N, Bai X, Esko J, Cohen G, Eisenberg R, Rosenberg R, Spear P. 1999. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99:13–22 [DOI] [PubMed] [Google Scholar]
- 10. Taylor JM, Lin E, Susmarski N, Yoon M, Zago A, Ware CF, Pfeffer K, Miyoshi J, Takai Y, Spear PG. 2007. Alternative entry receptors for herpes simplex virus and their roles in disease. Cell Host Microbe 2:19–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kopp SJ, Banisadr G, Glajch K, Maurer UE, Grunewald K, Miller RJ, Osten P, Spear PG. 2009. Infection of neurons and encephalitis after intracranial inoculation of herpes simplex virus requires the entry receptor nectin-1. Proc. Natl. Acad. Sci. U. S. A. 106:17916–17920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karaba AH, Kopp SJ, Longnecker R. 2011. Herpesvirus entry mediator and nectin-1 mediate herpes simplex virus 1 infection of the murine cornea. J. Virol. 85:10041–10047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Westmoreland D, Rapp F. 1976. Host range temperature-sensitive mutants of herpes simplex virus type 2. J. Virol. 18:92–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yoon M, Kopp SJ, Taylor JM, Storti CS, Spear PG, Muller WJ. 2011. Functional interaction between herpes simplex virus type 2 gD and HVEM transiently dampens local chemokine production after murine mucosal infection. PLoS One 6:e16122 doi:10.1371/journal.pone.0016122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Adkins B, Leclerc C, Marshall-Clarke S. 2004. Neonatal adaptive immunity comes of age. Nat. Rev. Immunol. 4:553–564 [DOI] [PubMed] [Google Scholar]
- 16. Corey L, Wald A. 2009. Maternal and neonatal herpes simplex virus infections. N. Engl. J. Med. 361:1376–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maness PF, Schachner M. 2007. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat. Neurosci. 10:19–26 [DOI] [PubMed] [Google Scholar]
- 18. Moore R, Larue L. 2004. Cell surface molecules and truncal neural crest ontogeny: a perspective. Birth Defects Res. C Embryo Today 72:140–150 [DOI] [PubMed] [Google Scholar]
- 19. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. 2007. The classical complement cascade mediates CNS synapse elimination. Cell 131:1164–1178 [DOI] [PubMed] [Google Scholar]
- 20. Cecconi F, Di Bartolomeo S, Nardacci R, Fuoco C, Corazzari M, Giunta L, Romagnoli A, Stoykova A, Chowdhury K, Fimia GM, Piacentini M. 2007. A novel role for autophagy in neurodevelopment. Autophagy 3:506–508 [DOI] [PubMed] [Google Scholar]
- 21. Yuan J, Yankner BA. 2000. Apoptosis in the nervous system. Nature 407:802–809 [DOI] [PubMed] [Google Scholar]
- 22. Ahmed M, Lock M, Miller CG, Fraser NW. 2002. Regions of the herpes simplex virus type 1 latency-associated transcript that protect cells from apoptosis in vitro and protect neuronal cells in vivo. J. Virol. 76:717–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Perkins D, Pereira EF, Gober M, Yarowsky PJ, Aurelian L. 2002. The herpes simplex virus type 2 R1 protein kinase (ICP10 PK) blocks apoptosis in hippocampal neurons, involving activation of the MEK/MAPK survival pathway. J. Virol. 76:1435–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bosnjak L, Miranda-Saksena M, Koelle DM, Boadle RA, Jones CA, Cunningham AL. 2005. Herpes simplex virus infection of human dendritic cells induces apoptosis and allows cross-presentation via uninfected dendritic cells. J. Immunol. 174:2220–2227 [DOI] [PubMed] [Google Scholar]
- 25. Jones CA, Fernandez M, Herc K, Bosnjak L, Miranda-Saksena M, Boadle RA, Cunningham A. 2003. Herpes simplex virus type 2 induces rapid cell death and functional impairment of murine dendritic cells in vitro. J. Virol. 77:11139–11149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cuchet-Lourenço D, Boutell C, Lukashchuk V, Grant K, Sykes A, Murray J, Orr A, Everett RD. 2011. SUMO pathway dependent recruitment of cellular repressors to herpes simplex virus type 1 genomes. PLoS Pathog. 7:e1002123 doi:10.1371/journal.ppat.1002123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kempermann G, Jessberger S, Steiner B, Kronenberg G. 2004. Milestones of neuronal development in the adult hippocampus. Trends Neurosci. 27:447–452 [DOI] [PubMed] [Google Scholar]
- 28. Kimoto H, Eto R, Abe M, Kato H, Araki T. 2009. Alterations of glial cells in the mouse hippocampus during postnatal development. Cell. Mol. Neurobiol. 29:1181–1189 [DOI] [PubMed] [Google Scholar]
- 29. Sedý J, Spear P, Ware C. 2008. Cross-regulation between herpesviruses and the TNF superfamily members. Nat. Rev. Immunol. 8:861–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kopp SJ, Storti CS, Muller WJ. 2012. Herpes simplex virus-2 glycoprotein interaction with HVEM influences virus-specific recall cellular responses at the mucosa. Clin. Dev. Immunol. doi:10.1155/2012/284104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu Y, Navarro F, Lal A, Basar E, Pandey RK, Manoharan M, Feng Y, Lee SJ, Lieberman J, Palliser D. 2009. Durable protection from herpes simplex virus-2 transmission following intravaginal application of siRNAs targeting both a viral and host gene. Cell Host Microbe 5:84–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Diamond C, Mohan K, Hobson A, Frenkel L, Corey L. 1999. Viremia in neonatal herpes simplex virus infections. Pediatr. Infect. Dis. J. 18:487–489 [DOI] [PubMed] [Google Scholar]
- 33. Kimura H, Futamura M, Kito H, Ando T, Goto M, Kuzushima K, Shibata M, Morishima T. 1991. Detection of viral DNA in neonatal herpes simplex virus infections: frequent and prolonged presence in serum and cerebrospinal fluid. J. Infect. Dis. 164:289–293 [DOI] [PubMed] [Google Scholar]
- 34. Malm G, Forsgren M. 1999. Neonatal herpes simplex virus infections: HSV DNA in cerebrospinal fluid and serum. Arch. Dis. Child. Fetal Neonatal Ed. 81:F24–F29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kern ER, Overall JC, Jr, Glasgow LA. 1973. Herpesvirus hominis infection in newborn mice. I. An experimental model and therapy with iododeoxyuridine. J. Infect. Dis. 128:290–299 [DOI] [PubMed] [Google Scholar]
- 36. Kern ER, Richards JT, Overall JC., Jr 1986. Acyclovir treatment of disseminated herpes simplex virus type 2 infection in weanling mice: alteration of mortality and pathogenesis. Antiviral Res. 6:189–195 [DOI] [PubMed] [Google Scholar]