

Abstract
Following entry of the HIV-1 core into target cells, productive infection depends on the proper disassembly of the viral capsid (uncoating). Although much is known regarding HIV-1 entry, the actions of host cell proteins that HIV-1 utilizes during early postentry steps are poorly understood. One such factor, transportin SR2 (TRN-SR2)/transportin 3 (TNPO3), promotes infection by HIV-1 and some other lentiviruses, and recent studies have genetically linked TNPO3 dependence of infection to the viral capsid protein (CA). Here we report that purified recombinant TNPO3 stimulates the uncoating of HIV-1 cores in vitro. The stimulatory effect was reduced by RanGTP, a known ligand for transportin family members. Depletion of TNPO3 in target cells rendered HIV-1 less susceptible to inhibition by PF74, a small-molecule HIV-1 inhibitor that induces premature uncoating. In contrast to the case for TNPO3, addition of the CA-binding host protein cyclophilin A (CypA) inhibited HIV-1 uncoating and reduced the stimulatory effect of TNPO3 on uncoating in vitro. In cells in which TNPO3 was depleted, HIV-1 infection was enhanced 4-fold by addition of cyclosporine, indicating that the requirement for TNPO3 in HIV-1 infection is modulated by CypA-CA interactions. Although TNPO3 was localized primarily to the cytoplasm, depletion of TNPO3 from target cells inhibited HIV-1 infection without reducing the accumulation of nuclear proviral DNA, suggesting that TNPO3 facilitates a stage of the virus life cycle subsequent to nuclear entry. Our results suggest that TNPO3 and cyclophilin A facilitate HIV-1 infection by coordinating proper uncoating of the core in target cells.
INTRODUCTION
Retroviruses such as HIV-1 contain viral RNA-protein (vRNP) complexes encased in a conical capsid shell. The capsid is composed of a polymer of CA subunits arranged in a hexagonal lattice (1–4). The viral core consists of the capsid and its contents. The core is packaged in an envelope, which is required for fusion of the virus with the cell. Upon fusion with the target cell, the core is released into the cytosol, where it is thought to undergo an uncoating process involving disassembly of the capsid (5), but the spatial and temporal aspects of uncoating are poorly understood. Recent genetic evidence suggests that uncoating occurs as early as 30 to 45 min postinfection (6, 7). Another study employed electron microscopy and detected intact cores at the nuclear membrane several hours postinfection, suggesting that uncoating occurs at a late stage (8). It is also presently not clear where uncoating occurs in the cell. Some evidence suggest that uncoating takes place in the cytoplasm (6, 9), while other evidence suggests that it takes place at nuclear pore complexes (NPCs) (8). Nonetheless, uncoating is tightly regulated by viral and, presumably, host cellular factors (reviewed in references 10 and 11). Viral proteins such as CA affect the process of uncoating. Mutations in CA that alter capsid stability generally impair HIV-1 infectivity (5) suggesting that optimal stability of the capsid is critical for productive infection. Host factors may also play a role in modulating HIV-1 uncoating (reviewed in references 12 and 13).
The host protein transportin 3 (TNPO3) was identified by genome-wide screens as an HIV-1 dependency factor (14, 15). TNPO3, a member of the importin β family of proteins, binds to serine/arginine-rich proteins (S/R proteins) and promotes their passage through the nuclear pore (16). Following translocation into the nucleus, the bound protein cargo is released upon binding of TNPO3 to RanGTP. Depletion of TNPO3 reduced HIV-1 infection in both dividing and nondividing cells (17), suggesting that an activity of TNPO3 is required for efficient infection even in cycling cells, where the virus might access the target cell DNA during mitosis without traversing nuclear pores. TNPO3 was also shown to bind to the viral integrase protein (IN) and to facilitate uptake of IN into the nucleus (17), though the relationship of IN binding to HIV-1 infection is unclear (18). Using a panel of retroviruses and HIV-1/murine leukemia virus (MLV) chimeric viruses, Krishnan et al. previously showed that TNPO3 dependency during infection was dictated by CA and was not quantitatively correlated with IN binding (19). This conclusion was also supported by the findings of other groups, who showed that point mutations in CA, including E45A, Q63A/Q67A, and N74D, among others, render HIV-1 insensitive to TNPO3 depletion (20, 21). Collectively, these results suggested that a function of the viral capsid is modulated by TNPO3 or vice versa.
The abundant host protein cyclophilin A (CypA) binds to the HIV-1 capsid and facilitates HIV-1 replication (22–24). Despite considerable investigation, the mechanism by which CypA stimulates replication is unknown. CypA appears to act at an early postentry stage of the virus infection before reverse transcription (25). It is a peptidyl-prolyl isomerase that binds to an exposed surface loop on CA (26), and it has been shown to catalyze cis-trans isomerization of the Gly89-Pro90 peptide bond on the CA protein (27, 28). Binding of CypA to CA causes nuclear magnetic resonance (NMR) chemical shift changes within and distal to the CypA-binding loop, suggesting that interaction with CA may result in conformational changes outside the CypA-binding loop and this might facilitate disassembly of the capsid (12, 27–30). Several other lines of evidence have also hinted at a role of CypA in HIV-1 uncoating (12, 27–35). In accordance with this evidence, Li et al. recently demonstrated that CypA modulates HIV-1 uncoating depending on the target cell type (36). There is also indirect evidence for a role of CypA in protection of the HIV-1 capsid from a putative cell-intrinsic host restriction factor (37–39). However, neither the identity of the putative restriction from which CypA protects the virus nor the mechanism by which it does this is known. Collectively, these findings are suggestive of a direct role of CypA in modulating capsid stability; however, there is no biochemical evidence for such an effect.
In this study, we employed biochemical and cell-based assays to understand the mechanism of action of TNPO3 and CypA in HIV-1 infection. We show that TNPO3 stimulates HIV-1 uncoating in vitro and stimulates the uncoating activity of a small-molecule capsid-targeting compound (PF74). PF74 was less effective at inhibiting HIV-1 infection of cells depleted of TNPO3. We also report that recombinant CypA directly inhibits HIV-1 uncoating in vitro. Furthermore, CypA binding to the capsid inhibited TNPO3-induced uncoating of HIV-1 in vitro and also made the virus more dependent on TNPO3 for infection. Our data indicate that TNPO3 and CypA can directly modulate the stability of the HIV-1 capsid.
MATERIALS AND METHODS
Plasmids and chemicals.
CA mutants were subcloned from HIV-1 proviral DNA construct R9 (40) by transfer of ApaI-SpeI or BssHII-SpeI DNA fragments into HIV-GFP, an envelope-defective pNL4-3-based HIV-1 reporter virus clone encoding green fluorescent protein (GFP) in place of Nef (41). The presence of these mutations in the final construct was confirmed by DNA sequencing. R9-ΔE-N74D was subcloned from proviral DNA construct R9-N74D by transfer of a BssHII-SpeI DNA fragment into the R9-ΔE vector (42). Plasmid pHCMV-G encodes a vesicular stomatitis virus G (VSV-G) protein (43) under the control of the human cytomegalovirus (CMV) promoter. The bacterial expression vector pGEX6P-3-TNPO3 was previously described (19). PF74 was synthesized and purified by the Chemical Synthesis Core, Vanderbilt Institute of Chemical Biology. Stocks of the compound were prepared by dissolution in dimethyl sulfoxide (DMSO) and stored at −80°C. Cyclosporine (CsA) was from Calbiochem. psPAX2, raltegravir (RAL), and efavirenz were obtained from the NIH AIDS Research and Reference Reagent Program.
Cells and viruses.
HeLa and 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Cellgro) supplemented with 10% fetal bovine serum (FBS), penicillin (50 IU/ml), and streptomycin (50 μg/ml) at 37°C with 5% CO2. Virus stocks were produced by calcium phosphate transfection of 293T cells (44). VSV-G-pseudotyped reporter virus particles were produced by cotransfection of 15 μg of wild-type (WT) or mutant HIV-GFP plasmid and 5 μg of pHCMV-G plasmid DNA. Two days after transfection, culture supernatants were harvested, clarified by filtration through 0.45-μm-pore-size filters, and frozen into aliquots at −80°C. The CA content of virus stocks was quantified by a p24-specific enzyme-linked immunosorbent assay (ELISA) as previously described (45).
Recombinant protein purification.
For purification of TNPO3, BL21 cells transformed with pGEX6P3-TNPO3 were grown in 3 liters of LB medium at 37°C to an A600 of 0.6 to 0.7. Protein was induced by culturing in the presence of 1 mM isopropyl-thio-β-d-galactopyranoside (IPTG) for 4 h. Bacteria were collected by centrifugation, resuspended in cold lysis buffer (150 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol [DTT], 50 mM Tris-HCl, pH 7.4), and lysed with a French press (1,200 lb/in2 pressure, 2 passes) followed by sonication (20% amplitude, 4 pulses of 15 s each) to shear bacterial DNA. The concentration of NaCl was adjusted to 500 mM, and the extract was clarified by centrifugation at 23,000 × g for 30 min at 4°C. The supernatant was incubated with 1.4 ml of a 50% slurry of glutathione-Sepharose beads (GE Healthcare) for 3 h at 4°C. After washing with an excess of lysis buffer, the beads were incubated with 60 U of PreScission protease (GE Healthcare) at 4°C for 16 to 24 h to remove the glutathione S-transferase (GST) tag. The beads were then pelleted, and the supernatant containing TNPO3 was collected. Beads were washed twice with lysis buffer containing 0.5 M NaCl, and the supernatants containing TNPO3 were collected. Fractions containing TNPO3 were pooled, and aliquots were flash-frozen in liquid N2 and stored at −80°C.
For purification of CypA, the CypA gene was amplified and ligated into pET21 vector DNA. Ros2(DE3) cells were transformed with pET21-CypA and grown in LB medium at 37°C to an A600 of 0.6 to 0.8. Proteins were induced with 1 mM IPTG at 18°C for 16 h. Cells were lysed by sonication in a buffer containing 25 mM sodium phosphate, pH 7.5. After centrifugation at 100,000 × g for 1 h, the supernatant was loaded onto a 10-ml HiTrap QP (GE Healthcare) column. Fractions containing CypA (flowthrough) were pooled, and the pH of the solution was adjusted to 5.5 with acetic acid. After centrifugation, the supernatant was loaded onto a 5-ml HiTrap SP (GE Healthcare) column and eluted using a 0 to 1 M NaCl gradient. Aggregates were removed with a Superdex 200 26/60 (GE Healthcare) gel filtration column equilibrated with a buffer containing 25 mM sodium phosphate (pH 6.5), 100 mM NaCl, 1 mM DTT, and 0.02% sodium azide.
For expression and purification of RanQ69L, BL21 transformed with pQE32-6×His-RanQ69L (46) was cultured at 28°C until the A600 was ∼0.5. Protein expression was induced by culturing in the presence of 1 mM IPTG for 4 h at 28°C. Cells that were pelleted at 6,300 × g for 10 to 15 min at 4°C were resuspended in 25 ml cold buffer A (50 mM HEPES [pH 7.0], 5 mM MgCl2, 100 mM NaCl, 5 mM DTT, 2 mM phenylmethylsulfonyl fluoride [PMSF]). Bacterial cells were lysed by sonication, and the lysate was clarified by ultracentrifugation at 50,000 × g for 3 h at 4°C. Clarified lysate was incubated with 1 ml of Ni-nitrilotriacetic acid (NTA) agarose beads equilibrated with buffer A containing 25 mM imidazole for 3 h at 4°C. The slurry was loaded onto a 2-ml column under gravity, washed 30 to 40 times with buffer A containing 25 mM imidazole, and eluted with 10× buffer A containing 200 mM imidazole. The eluate was applied to an SP Sepharose column (GE Healthcare) and eluted using a 0 to 1 M NaCl gradient. Peak fractions were pooled, concentrated, and further purified using an S200 Superdex gel filtration column (GE Healthcare) equilibrated with buffer D (20 mM HEPES [pH 7.0], 5 mM MgCl2, 100 mM NaCl, 5 mM DTT, 2 mM PMSF, 5% glycerol). Eluates were pooled, concentrated, aliquoted, and stored at −80°C. To charge H6-RanQ69L with GTP, 250 μg of H6-RanQ69L was incubated with 100 mM GTP at 4°C for 4 h. Unbound GTP was removed using P6 Biogel (Bio-Rad).
shRNA-mediated depletion of cellular TNPO3.
A Mission short hairpin RNA (shRNA) lentiviral vector (Sigma, no. TRCN0000038331; hairpin sequence, CCGGCGGCGCACAGAAATTA-TAGAACTCGAGTTCTATAATTTCTGTGCGCCGTTTTTG) was used to deplete TNPO3 in HeLa cells. The vector plasmid was cotransfected into 293T cells with pHCMV-G and psPAX2 (47) to produce lentiviral vector particles. The resulting viral particles were used to transduce HeLa cells, and stable populations of transduced cells were selected by culturing in puromycin (2 μg/ml) for 1 week prior to use in HIV-1 infection assays. TNPO3 depletion was confirmed by immunoblotting of whole-cell lysates. As an additional control, Mission pLKO.1-puro empty shRNA lentiviral vector (SHC001), used to transduce HeLa cells, was packaged as described above. During initial experiments, we observed that the selected cell populations gradually regained TNPO3 expression over several weeks. Therefore, the stable cells were cultured only briefly after selection, and multiple vials were cryopreserved. For each experiment, a new vial of cells was thawed and cultured for only a few days to help ensure that TNPO3 remained depleted.
Viral infectivity assays.
HeLa cells stably depleted of TNPO3 and control cells were plated at a density of 20,000 cells/well in 24-well plates. The next day, wild-type and mutant HIV-GFP viruses were titrated on the cells in the presence of Polybrene (8 μg/ml). In experiments with PF74, the compound was added to the cells at the same time as the virus. Twenty-four hours after transduction, the cells were supplemented with 1 ml of additional medium and cultured for another 24 h. Cells were then detached with trypsin, fixed in 2% paraformaldehyde, and analyzed for GFP expression with an Accuri C6 flow cytometer. The data presented as single values are within the linear range of the dose-titration curve.
Isolation of HIV-1 cores.
Cores were isolated from concentrated virions as described previously (48). Briefly, virus supernatants from transfected 293T cells were concentrated by centrifugation at 32,000 rpm at 4°C (175,000 × g at rmax) for 3 h. The pelleted viral particles were resuspended in 300 μl of 1× STE buffer (10 mM Tris-HCl [pH 7.4], 100 mM NaCl, and 1 mM EDTA) and incubated at 4°C for 2 to 4 h. The concentrated virions were then ultracentrifuged at 32,000 rpm (187,000 × g for 16 h at 4°C) through a 30 to 70% sucrose gradient overlaid with 1% Triton X-100. Fractions (1 ml) were collected from the top to the bottom of the sucrose gradient, and the fractions containing cores were identified by quantifying the CA content of the fractions by p24 ELISA. Core-associated CA was computed as quantity of CA in the core-containing fractions of the gradient as a percentage of the total CA in the gradient.
HIV-1 uncoating assay.
Samples of purified wild-type and mutant HIV-1 cores (50 μl) were diluted in 1× STE buffer (pH 7.4) (200 μl) containing bovine serum albumin (10 μg/ml) without or with recombinant proteins. The cores were then incubated at 37°C for indicated times, followed by ultracentrifugation at 125,000 × g at rmax (Beckman TLA-55 rotor at 45,000 rpm) for 20 min at 4°C. Supernatants were removed, and pellets were dissolved in sample diluent (10% donor calf serum and 0.5% Triton X-100 in phosphate-buffered saline [PBS]). The CA contents of supernatants and pellets were determined by p24 ELISA. The extent of uncoating was determined as the fraction of the total CA present in the supernatant. In some experiments PF74, dissolved in DMSO, was also added to the uncoating reaction mixture.
In experiments involving recombinant RanGTP protein, TNPO3 (0.75 μM) and RanQ69LGTP (1 μM) were preincubated either alone or together at 4°C for 15 min followed by 37°C for 15 min. The cores were then added, followed by further incubation at 37°C for 45 min. The extent of uncoating was determined as described above.
Cell fractionation and quantitative analysis of HIV-1 reverse transcription in target cells.
One day prior to infection, HeLa cells (150,000 per well) were plated in 12-well plates. Viruses were treated with 20 μg/ml DNase I and 10 mM MgCl2 at 37°C for 1 h to remove contaminating plasmid DNA. VSV-G-pseudotyped virus inocula equivalent to 15 ng of p24 were used to infect cells in the presence or absence of HIV-1 integrase inhibitor (raltegravir; 1 μM). Parallel infections were performed in the presence of efavirenz (1 μM) to define the residual plasmid DNA levels carried over from transfection. At 7 h or 24 h postinfection, cells were harvested with 0.25% trypsin. Trypsin was deactivated by addition of DMEM containing 10% FBS. Cells were pelleted by centrifugation at 300 × g for 5 min. Pellets were resuspended in 200 μl of PBS, and DNA was isolated using a DNeasy kit (Qiagen) according to the manufacturer's protocol. For quantification of viral DNA in the nucleus, cell pellets were lysed in buffer A (10 mM Tris [pH 8.0], 1.5 mM MgCl2, 10 mM KCl, 1 mM DTT, protease inhibitors) containing 0.1% Triton X-100 by incubation at 4°C for 15 min. Supernatants containing cytoplasmic fractions were discarded and the nuclear pellets were used to isolate DNA using a DNeasy kit. In parallel, whole-cell, cytoplasmic, and nuclear lysates were prepared from uninfected cells to check for cytoplasmic contamination of nuclear fractions and to study the subcellular localization of TNPO3. To prepare whole-cell lysates, cells were lysed in radioimmunoprecipitation assay buffer (50 mM Tris [pH 7.5], 1% Triton X-100, 250 mM NaCl, 5 mM EDTA, 0.1% SDS, 1% sodium deoxycholate, protease inhibitors); cytoplasmic lysates were prepared as described above, and nuclear lysates were prepared by isolating nuclei as described above and lysing them in SDS-PAGE sample buffer. Blots of whole-cell and nuclear lysates were probed with rabbit polyclonal anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) (Santa Cruz Biotechnology) and mouse monoclonal anti-Lamin B1 (Zymed) antibody, respectively. Blots were also probed for TNPO3 using mouse monoclonal anti-TNPO3 (Genway).
Viral DNA was quantified by real-time PCR using an MX-3000p thermocycler (Stratagene) utilizing SYBR green chemistry (ABI). Late reverse transcription (LRT) products (U5-Gag) were detected using the forward primer MH531 (5′-TGTGTGCCCGTCTGTTGTGT-3′) and reverse primer MH532 (5′-GAGTCCTGCGTCGAGAGAGC-3′) (49). 2-LTR circles were detected using the forward primer MH535 (5′-AACTAGGGAACCCACTGCTTAAG-3′) and reverse primer MH536 (5′-TCCACAGATCAAGGATATCTTGTC-3′).
Immunoblotting analysis.
Cells were lysed with radioimmunoprecipitation assay buffer. Proteins were separated on 4 to 20% polyacrylamide gradient Tris-glycine gels (Bio-Rad) and were transferred onto nitrocellulose membranes (GE Healthcare), and membranes were blocked with 5% nonfat milk. The membranes were probed with mouse polyclonal anti-TNPO3 antibody (Genway) and rabbit polyclonal anti-GAPDH antibody (Santa Cruz Biotechnology). After washing four times with PBS containing 0.1% Tween 20 for 10 min, the membranes were probed with appropriate species-specific Alexa Fluor 680-conjugated secondary antibodies (Pierce). Protein bands were visualized using the Odyssey infrared imaging system (Li-Cor) and quantified using the instrument software.
Statistical analysis.
The effect of TNPO3 concentration on the uncoating of purified wild-type and mutant HIV-1 cores was evaluated using a dose-response analysis based on mixed-effects nonlinear regression. Regression coefficients were summarized using 95% confidence intervals (CI). Tests having P values of less than 0.05 and confidence intervals that failed to include the appropriate null value (e.g., zero) were considered statistically significant. The type of interaction between TNPO3 and PF74 as well as TNPO3 and CypA in their effect on viral uncoating was evaluated using mixed-effects nonlinear regression. An interaction factor was obtained, and regression coefficients were summarized using 95% CI. Tests having P values of less than 0.05 and confidence intervals that failed to include the appropriate null value (zero) were considered statistically significant. Other statistical analyses were performed with Student's two-sided t test.
RESULTS
Point mutations in CA modulate HIV-1 dependence on TNPO3 for infection.
To examine the role of CA in TNPO3 dependence of HIV-1 infection, HIV-1–eGFP reporter viruses encoding CA mutations were pseudotyped with VSV-G and titrated on HeLa cells stably depleted of TNPO3 (sh-TNPO3) (Fig. 1A), empty-vector-transduced HeLa (sh-empty), and untransduced HeLa cells. HIV-1 infection was measured by flow cytometric analysis of GFP expression. As previously observed (14, 17, 19, 21, 50, 51), infection by wild-type HIV-1 was markedly (10- to 20-fold) reduced in TNPO3-depleted cells (Fig. 1B). Consistent with previous reports (20, 21), infection by the N74D CA mutant virus was not affected by TNPO3 depletion (Fig. 1B). Consistent with other studies (20, 51, 52), but contrary to one report (53), we observed an equivalent reduction in TNPO3 dependence of the N74D mutant bearing the HIV-1 envelope glycoproteins (data not shown).
Fig 1.

Effects of CA substitutions on TNPO3 dependency of HIV-1 infection. (A) Quantitative immunoblot analysis of TNPO3-depleted HeLa cells. Cell extracts were immunoblotted with anti-TNPO3 and anti-GAPDH antibodies. Shown under each lane is the ratio of TNPO3 intensity to GAPDH intensity. (B) GFP reporter viruses were titrated on HeLa, sh-empty, and sh-TNPO3 cells. The percentage of GFP-positive cells is shown for each input viral dose (ng of p24). The data are representative of two independent experiments. (C) Control and TNPO3-depleted cells were challenged with CA mutant GFP reporter viruses. At 48 h, the percentage of GFP-positive cells was determined as a measure of infectivity. The ratio of infection of TNPO3-depleted relative to control cells is shown. Error bars represent the range of values from two independent experiments. Unfilled bars represent TNPO3-dependent viruses, black bars represent TNPO3-independent mutants, and gray bars represent partially TNPO3-dependent mutants. (D) Location of CA amino acid residues important for TNPO3 dependence of HIV-1 on the structural model of CA monomer extracted from the hexameric structure (Protein Data Bank [PDB] no. 3H4E). CA substitutions that do not alter TNPO3 dependence are shown in yellow, those that partially alter it are in orange, and those that completely alter it are in red.
We also analyzed the TNPO3 dependence of several CA mutants. The mutants selected for analysis have mutations distributed across the surface of CA. Several of these mutants exhibit deviations from wild-type capsid stability: P38A, Q63A/Q67A, and K203A particles contain unstable capsids (5), while E45A and E213A contain hyperstable capsids (5, 54, 55). The HIV-1 mutant “5mut” encodes five amino acid substitutions (Q67H, K70R, H87P, T107N, and L111I) in CA and was isolated by selection for resistance to the small-molecule HIV-1 inhibitor PF74 (56, 57). The infectivity of 5mut virus was not affected by TNPO3 depletion (Fig. 1C). Because 5mut contains 5 individual substitutions, we also tested HIV-1 mutants encoding each single substitution for TNPO3 dependence. Infection by the T107N mutant was not affected by depletion of TNPO3, while infection by the Q67H, K70R, and L111I mutants was partially reduced (Fig. 1C). In contrast, infection by the H87P mutant was reduced in TNPO3-depleted cells to an extent equivalent to that of the wild type.
Next, we tested a panel of mutants (Q4A, G116A, T119A, N139A, R143A, and P207A) which exhibit minimal impairment in infectivity and yield cores with intrinsic stability similar to that of the wild-type cores (58). Like the wild-type virus, the Q4A, G116A, T119A, R143A, and P207A mutants were dependent on TNPO3 for infection. N139A was partially dependent on TNPO3. Among other mutants, P38A and E213A were partially dependent on TNPO3 for infection, while K203A was not affected by TNPO3 depletion. (Fig. 1C). Consistent with previous reports (20, 21), infection by the E45A and Q63A/Q67A mutants, which exhibit altered kinetics of uncoating, was not affected by TNPO3 depletion. We recently identified a second-site suppressor mutation, R132T, that partially relieves the infectivity impairment exhibited by E45A (55). Infection by the E45A/R132T double mutant exhibited TNPO3 dependence between those of E45A and the wild type (Fig. 1C). This result suggests that the second-site suppressor mutant corrects an uncoating defect associated with E45A and concomitantly restores TNPO3 dependence of infection.
When visualized on the structural model of monomeric CA lattice, the CA substitutions that reduce the TNPO3 dependence of HIV-1 infection do not appear to localize to a specific region of CA (Fig. 1D). Thus, it would appear that the effects of the mutations are not readily explained by alteration of a specific binding surface on CA. Interestingly, many of the TNPO3-independent mutants have altered intrinsic capsid stability, suggesting a potential link between HIV-1 capsid stability and TNPO3 requirement for infection.
TNPO3 stimulates the uncoating of isolated HIV-1 cores in vitro.
Several of the HIV-1 CA mutations (E45A, Q63A/Q67A, and 5mut) that rendered HIV-1 less sensitive to TNPO3 depletion also alter the intrinsic biochemical stability of the viral capsid (5, 57), suggesting a possible link between HIV-1 uncoating and TNPO3 function in HIV-1 infection. Therefore, we asked whether TNPO3 affects uncoating of purified HIV-1 cores in vitro. The extent of uncoating was determined by quantifying the fraction of the CA protein that dissociated from the cores during incubation. As previously reported (5), incubation of HIV-1 cores at 37°C led to shedding of the CA protein (Fig. 2A). Addition of recombinant TNPO3 at concentrations of up to 1 μM stimulated the uncoating reaction in a dose-dependent manner. To test the specificity of this effect, we purified cores from N74D virions, which are independent of TNPO3. Cores purified from N74D virions exhibited levels of core-associated CA similar to those of the cores from wild-type virions (Fig. 2B). To further examine the effects of the N74D substitution on capsid stability, the purified cores were incubated at 37°C for various periods, and the extent of uncoating was quantified. The rate of N74D uncoating in vitro was similar to that of the wild type (Fig. 2C). Collectively, these results indicate that the N74D mutation does not alter the intrinsic stability of the HIV-1 capsid. Relative to that of wild-type cores, uncoating of N74D cores appeared to be less responsive to stimulation by TNPO3 in vitro (Fig. 2D). Results from mixed-effects nonlinear regression indicated that the maximum percent uncoating (Emax) achievable through TNP03 was 61.7 (95% CI, 55.4 to 68.0) and 38.8 (95% CI, 31.9 to 45.6) for wild-type and N74D viruses, respectively. Hence, the maximum percent uncoating for N74D was 22.9 (95% CI, 13.6 to 32.3), less than that for the wild-type virus. The differences between the dose-response curves for WT and N74D were statistically significant (P < 0.001), indicating that the uncoating of wild-type cores by TNPO3 was greater than that of the N74D mutant cores. Thus, the N74D mutation was less responsive to TNPO3 in vitro. These results indicate that TNPO3 stimulates HIV-1 uncoating in vitro and that the effect is reduced by the N74D substitution.
Fig 2.

TNPO3 stimulates HIV-1 uncoating in vitro. (A) Purified HIV-1 cores were incubated at 37°C for 30 min in the absence or presence of the indicated concentrations of recombinant TNPO3. After incubation, the extent of uncoating was determined as the fraction of the total CA in the supernatant. Shown are the means ± standard errors of the means (SEM) from three independent experiments performed in duplicate. Asterisks above the bars indicate statistically significant differences in uncoating values in the absence or presence of the indicated TNPO3 concentrations. *, P < 0.05; **, P < 0.01. (B) The N74D substitution does not alter the intrinsic stability of the HIV-1 capsid. Cores from wild-type and N74D mutant HIV-1 particles were isolated by sucrose gradient centrifugation. The level of core-associated CA was determined from the CA contents of core-containing fractions as a percentage of total CA protein in the gradient. Shown are the means ± SEM from three separate experiments. (C) The N74D substitution does not affect HIV-1 uncoating in vitro. Wild-type and N74D cores were incubated at 37°C, and the extent of uncoating was quantified at each time point. Shown are the means ± SEM from two independent experiments performed in duplicate. (D) TNPO3 potently stimulates the uncoating of wild-type HIV-1 cores. Wild-type and N74D mutant cores were incubated at 37°C for 35 min in the absence or presence of the indicated concentrations of TNPO3, and the extent of uncoating was determined. Shown are the means ± SD from five independent experiments performed in duplicate. (E) RanQ69LGTP inhibits the ability of TNPO3 to stimulate uncoating of wild-type cores. Purified cores were incubated at 37°C for 45 min in the absence or presence of the indicated recombinant proteins, and the extent of uncoating was determined. Shown are means ± SEM from three independent experiments performed in duplicate. **, P < 0.01.
RanGTP is a cofactor for nuclear import mediated by importin β family members (reviewed in reference 59) and was recently shown to enhance binding of TNPO3 to HIV-1 CA (51). Therefore, we asked whether addition of RanGTP to TNPO3 modulates HIV-1 uncoating activity. The point mutation Q69L dramatically reduces the GTPase activity of Ran by several orders of magnitude, thereby effectively locking the protein into the GTP-bound state (60). Purified RanQ69L protein was charged with GTP, and in vitro uncoating assays were performed using purified HIV-1 cores with recombinant TNPO3 in the absence or presence of RanQ69LGTP. Preincubation of TNPO3 with RanQ69LGTP significantly decreased TNPO3-induced uncoating (Fig. 2E). Addition of RanQ69LGTP in the absence of TNPO3 had no significant effect on uncoating of HIV-1 cores. Thus, RanGTP specifically inhibited the ability of TNPO3 to stimulate the uncoating of HIV-1 cores.
TNPO3 potentiates the antiviral activity of a small-molecule antiviral compound that induces premature HIV-1 uncoating.
The small molecule PF74 inhibits HIV-1 infection by binding to the viral capsid, resulting in premature uncoating (57). PF74 potently stimulates the uncoating of HIV-1 cores in vitro and in target cells, thereby representing a potentially useful pharmacologic probe for studies of HIV-1 uncoating. To examine the potential effects of TNPO3 on HIV-1 uncoating in target cells, we asked whether depletion of TNPO3 would alter the sensitivity of HIV-1 infection to inhibition by PF74. We challenged control and TNPO3-depleted cells with HIV-1 in the presence of various concentrations of PF74. PF74 inhibited HIV-1 infection of control cells with a 50% inhibitory concentration (IC50) of approximately 0.15 μM, consistent with previous observations (56) (Fig. 3A). In TNPO3-depleted cells, the sensitivity of HIV-1 to PF74 was altered, albeit in an unusual manner: at lower concentrations, the curve was shifted slightly to the right, and at higher concentrations we reproducibly observed a reversal in the inhibition curve. Relative to wild-type HIV-1, the N74D mutant exhibited a 7-fold decrease in sensitivity to PF74 in HeLa cells (IC50 = 1.10 μM). Moreover, its sensitivity to PF74 was not affected by TNPO3 depletion (Fig. 3A). Similar to the case for N74D, other TNPO3-independent mutants, i.e., E45A, Q63A/Q67A, and 5mut, were equally sensitive to PF74 in control and TNPO3-depleted cells (Fig. 3B). Relative to wild-type HIV-1, the second-site suppressor mutation R132T, which partially restored TNPO3 dependence of E45A, rendered this mutant moderately less sensitive to PF74 in TNPO3-depleted cells than in control cells. Mutants P38A and E213A, which are partially dependent on TNPO3 for infection, had intermediate reductions in sensitivity to PF74 in TNPO3-depleted cells. Thus, TNPO3 sensitizes HIV-1 to inhibition by PF74, and this effect is correlated with the dependence of HIV-1 infection on TNPO3.
Fig 3.
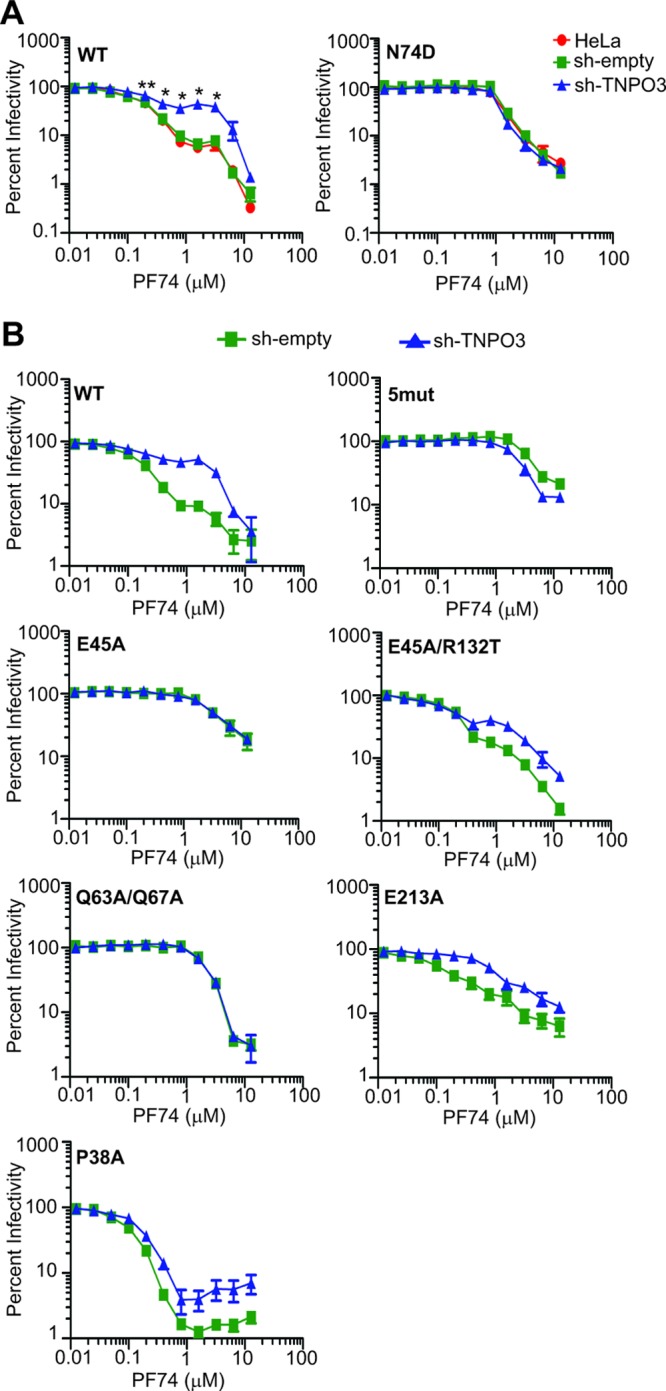
TNPO3 sensitizes HIV-1 to inhibition by PF74. (A) Cells were inoculated with wild-type or N74D mutant HIV-GFP reporter particles in the presence of the indicated concentrations of PF74. The extent of infection was determined 48 h later by quantifying GFP-positive cells. The results were normalized to the corresponding infections performed in the absence of PF74. Normalized values are represented as means ± SEM from four independent experiments performed in duplicate. Asterisks above each point represent the difference between sh-empty and sh-TNPO3 values at that particular concentration of PF74. *, P < 0.05; **, P < 0.01. (B) Analysis of additional HIV-1 CA mutants for the effects of TNPO3 depletion on sensitivity to inhibition by PF74. Data represent means ± SEM from three independent experiments performed in duplicate.
To further examine the interplay between PF74 and TNPO3, we quantified the effect of TNPO3 on PF74-induced uncoating in vitro. TNPO3 stimulated the uncoating of cores by ∼2.5-fold (Fig. 4). A subinhibitory concentration of PF74 (0.02 μM) alone did not stimulate uncoating, but addition of TNPO3 increased uncoating by ∼3.5-fold (Fig. 4). A similar effect was observed at a higher concentration of PF74, with 1.5-fold and 5-fold increases over basal uncoating with PF74 alone and PF74 plus TNPO3, respectively. In sharp contrast, PF74 and TNPO3 effects on uncoating of N74D mutant cores were additive (Fig. 4). Results from mixed-effects nonlinear regression indicated a synergistic interaction between TNPO3 and PF74 for the wild-type virus. For each micromolar increase in the concentration of PF74, the percent HIV-1 uncoating increased by 70.44 (95% CI, 40.06 to 100.82) per micromolar of TNPO3. The interaction between TNPO3 and PF74 (interaction effect) was not significant for N74D: 12.88 (95% CI, −25.11 to 50.86). The difference in the interaction effect for the wild type versus N74D was significant: 57.57 (95% CI, 6.22 to 108.91). Taken together, these data demonstrate that TNPO3 sensitizes HIV-1 to the antiviral activity of PF74 during infection and that TNPO3 potentiates HIV-1 uncoating induced by PF74 in vitro.
Fig 4.

TNPO3 potentiates HIV-1 uncoating induced by PF74 in vitro. HIV-1 cores were incubated at 37°C for 45 min in the presence or absence of TNPO3 and PF74, and the extent of uncoating was quantified. Shown are the means ± SEM from three independent experiments performed in duplicate. The dashed lines represent basal uncoating in the absence of PF74 and TNPO3.
TNPO3 is required at a post-nuclear entry stage of HIV-1.
Initial studies of TNPO3-depleted cells revealed a block to HIV-1 infection at nuclear entry (15, 17, 50), yet contradictory findings have been reported (20, 51). To confirm which step of the HIV-1 life cycle is affected by depletion of TNPO3, we used quantitative PCR (qPCR) to determine the extent of reverse transcription following infection of cells with either wild-type or N74D mutant virus. In agreement with previous studies (14, 17, 20, 50, 51), we detected no difference in the amount of the late reverse transcription (LRT) products synthesized in sh-empty and sh-TNPO3 cells at both 7 and 24 h postinfection (Fig. 5A), confirming that HIV-1 reverse transcription is not dependent on TNPO3. As an initial approach to test the efficiency of HIV-1 nuclear entry in TNPO3-depleted cells, we quantified the levels of 2-LTR circles that accumulated in the presence or absence of an integrase inhibitor, raltegravir (RAL). Comparable levels of 2-LTR circles accumulated in sh-empty and sh-TNPO3 cells inoculated with wild-type and N74D HIV-1 (Fig. 5B), suggesting that HIV-1 nuclear entry is not affected by TNPO3 depletion. Inclusion of RAL during infection resulted in a 4-fold increase in accumulation of 2-LTR circles for both wild-type and N74D HIV-1 in control cells at 24 h, as expected. However, RAL did not produce an increase in wild-type 2-LTR circles in sh-TNPO3 cells, nor did they accumulate to the level observed in RAL-treated control cells. The lack of an effect of RAL on the wild-type virus in sh-TNPO3 cells suggested that HIV-1 is impaired for integration in these cells; however, the decreased accumulation relative to that in control cells suggested the possibility of an additional impairment in nuclear entry or in the formation or stability of 2-LTR circles.
Fig 5.

TNPO3 depletion affects a step in HIV-1 infection after nuclear entry. (A and B) Cells were challenged with wild-type or N74D virus in the presence or absence of raltegravir. DNA was isolated from these cells at 7 and 24 h postinfection, followed by quantification of total viral DNA (A) and 2-LTR circles (B) by qPCR. The data represent means ± SEM from three independent experiments. (C) Immunoblot analysis was performed to ensure the integrity of subcellular fractionation by using a cytoplasmic protein, GAPDH, and a nuclear protein, lamin B1. (D) Infection was performed as for panels A and B, and cells were lysed and fractionated into cytoplasmic and nuclear fractions at 7 h and 24 h postinfection. Total viral DNA in the nuclear fractions was quantified by qPCR. Data represents means ± SEM from two separate experiments. (E) Whole-cell (Total), cytoplasmic (cyto), and nuclear (Nuc) lysates were resolved by SDS-PAGE and immunoblotted for TNPO3. GAPDH and lamin B1 were used as markers to ensure the integrity of subcellular fractionation. The right panel shows the quantitative distribution of TNPO3 in cytoplasmic and nuclear fractions of sh-empty and sh-TNPO3 cells. Data represents means ± standard deviations (SD) from three independent experiments.
While 2-LTR circles are often relied upon as a measure of HIV-1 entry into the nucleus, it is conceivable that perturbation of nuclear host factors such as TNPO3 might also directly alter 2-LTR circle formation or stability once the viral DNA has entered the nucleus. To more directly assess the effect of TNPO3 on HIV-1 nuclear entry, we purified nuclei from control and TNPO3-depleted cells inoculated with wild-type virus. Nuclear DNA was extracted and assayed for LRT products by qPCR. The quality of fractionation was confirmed by preparing whole-cell or nuclear lysates from same number of cells and determining the relative quantities of GAPDH (a cytoplasmic protein) in relation to lamin B1 (a nuclear protein) in these lysates (Fig. 5C). We observed that similar levels of LRT products were present in nuclear fractions from sh-empty and sh-TNPO3 cells, indicating that TNPO3 depletion did not affect the levels of total nuclear HIV-1 DNA. Because TNPO3 did not affect the levels of LRT or nuclear HIV-1 DNA (Fig. 5D), we conclude that HIV-1 efficiently enters the nucleus in TNPO3-depleted cells but is impaired for integration (14, 20, 51). Furthermore, the impairment in accumulation of 2-LTR circles in TNPO3-depleted cells in the presence of RAL suggests that TNPO3 depletion results in decreased efficiency of 2-LTR circle formation and/or stability. This effect of RAL was not observed with the N74D mutant. Our observations are consistent with the recent findings of Valle-Casuso et al. (61) as well as De Iaco and Luban (20), who also concluded that TNPO3 is not required for HIV-1 entry into the nucleus.
To study the subcellular distribution of TNPO3 in the cells, we analyzed cytoplasmic, nuclear, and whole-cell lysates for TNPO3 by Western blotting. Analysis of the band intensities demonstrated that approximately 95% of TNPO3 was present in the cytoplasmic fraction (Fig. 5E). These results are in accord with the recent report of Valle-Casuso et al. (61). We conclude that TNPO3 is localized primarily to the cytoplasm of HeLa cells. TNPO3 continuously shuttles between the cytoplasm and nucleus, and a predominant localization in the cytoplasm might simply be due to a higher retention time in the cytoplasm upon binding to its cargoes.
Cyclophilin A stabilizes the HIV-1 capsid and antagonizes TNPO3 acceleration of uncoating in vitro.
The host protein cyclophilin A (CypA) enhances HIV-1 infection by a poorly understood mechanism. CypA binds to the viral capsid following cell entry. Mutations in CA, including the G89V substitution, lower CypA binding affinity and reduce viral infectivity. A recent study demonstrated that depending on the target cell type, the binding of CypA either stabilizes or destabilizes the HIV-1 capsid in the cells (36). However, biochemical evidence for a direct action of CypA on HIV-1 capsid stability is lacking. Therefore, we tested the effects of recombinant CypA on uncoating of purified HIV-1 cores in vitro. CypA inhibited uncoating of wild-type cores in a dose-dependent manner, with an ∼50% reduction at a concentration of 40 μM (Fig. 6A). This potency is within the range to be physiologically relevant, as the reported affinity of CypA for CA is 18 μM (62), and intracellular concentrations of CypA are typically around 20 μM (63, 64). In contrast, CypA did not alter the uncoating of G89V mutant cores (Fig. 6A), demonstrating that the effect of CypA on uncoating requires binding to CA.
Fig 6.

Cyclophilin A stabilizes HIV-1 cores and inhibits TNPO3-enhanced uncoating. (A) Wild-type and G89V cores were incubated at 37°C for 60 min in the presence or absence of the indicated concentrations of cyclophilin A. Following incubation, the extent of uncoating was determined. Shown are the means ± SEM from two independent experiments performed in duplicate. Asterisks above the bars indicate differences in uncoating in the absence or presence of the indicated concentration of cyclophilin A. *, P < 0.05; **, P < 0.01. (B) Uncoating of HIV-1 cores was assayed in the presence or absence of the indicated recombinant proteins (TNPO3 [1 μM] or CypA [20 μM]). Shown are the means ± SEM from three independent experiments performed in duplicate. (C and D) Cells were infected with wild-type (C and D), N74D mutant (C), or G89V mutant (D) particles, and the extent of infection was quantified by flow cytometry for GFP expression. Results are expressed as means ± SEM from three independent experiments. Asterisks indicate differences between the fold changes in infectivity in control versus sh-TNPO3 cells. *, P < 0.05.
These data indicate that CypA and TNPO3 exhibit opposing effects on HIV-1 uncoating in vitro. Therefore, we examined the effects of both CypA and TNPO3 on the uncoating of HIV-1 cores. Under the specific conditions used, TNPO3 stimulated uncoating of wild-type cores by ∼100%, while CypA reduced the extent of HIV-1 uncoating by 20% (Fig. 6B). Addition of CypA decreased TNPO3-induced uncoating by ∼80%. As expected, TNPO3-induced uncoating of G89V cores was not much affected by CypA addition (Fig. 6B). Results from mixed-effects nonlinear regression presented strong evidence for an antagonistic interaction between TNPO3 and CypA for the wild-type virus. The evidence was weaker for the G89V virus. For each micromolar increase in the concentration of CypA, the percent uncoating of HIV-1 cores decreased by −0.34 (95% CI, −0.57 to −0.12) per micromolar of TNPO3. The antagonistic interaction between TNPO3 and CypA was borderline significant for G89V cores: −0.26 (95% CI, −0.53 to 0.011). To determine the effects of interplay between TNPO3 and CypA on HIV-1 infection, we inoculated control and TNPO3-depleted cells with wild-type or N74D virus in the presence or absence of CsA (5 μM), an inhibitor that prevents binding of CypA to CA. Infection by wild-type HIV-1 was 13-fold lower in TNPO3-depleted cells than in control cells (Fig. 6C). However, in the presence of CsA, the decrease in infectivity was only 7.5-fold. Furthermore, infectivity of G89V was reduced by 3-fold in TNPO3-depleted cells, in contrast to the 13-fold reduction observed for the wild type (Fig. 6D). Viewed another way, addition of CsA markedly stimulated HIV-1 infection of TNPO3-depleted cells. Collectively, our results demonstrate that binding of CypA to CA stabilizes HIV-1 cores in vitro and increases the requirement for TNPO3 in HIV-1 infection.
DISCUSSION
In the present study, we demonstrate that TNPO3 stimulates uncoating of purified HIV-1 cores in vitro. The point mutation N74D in CA, which renders the virus independent of TNPO3, reduced the effect of TNPO3 on HIV-1 uncoating in vitro. RanGTP, a regulator of nucleocytoplasmic transport, inhibited the ability of TNPO3 to stimulate uncoating of HIV-1 cores. RanGTP acts as a molecular switch during nucleocytoplasmic transport. After nuclear import of the importin-cargo complex, RanGTP associates with the Ran-binding domain of the importin. This process triggers a conformational change in importin and subsequently relieves the interaction between importin and cargo by reducing the affinity of the latter to the cargo-binding domain of importin (reviewed in reference 59). In the case of TNPO3, the C-terminal cargo-binding domain was recently reported to be important for CA binding (51). Our observation that RanGTP inhibits TNPO3-stmulated uncoating is consistent with this finding and also demonstrates specificity of the effect of TNPO3 on HIV-1 uncoating in vitro.
We also observed that the antiviral activity of a small-molecule capsid-targeting compound, PF74, was decreased in TNPO3-depleted cells. Reduction in PF74 sensitivity in TNPO3-depleted cells was associated with TNPO3 dependence of the virus. PF74 inhibits HIV-1 infection at an early stage of HIV-1 infection by inducing premature uncoating of the viral capsid in vitro and in target cells (56, 57). We observed that the uncoating-stimulating action of PF74 on HIV-1 cores in vitro was markedly stimulated by TNPO3. The extent of uncoating by PF74 plus TNPO3 was greater than the sum of the individual effects of each. These results suggest that TNPO3 sensitizes HIV-1 to PF74 by synergizing with PF74 in stimulating uncoating of HIV-1 capsid. Thus, in normal cells, PF74 induces premature uncoating and markedly reduces infectivity, while in TNPO3-depleted cells, the compound may be less effective in stimulating uncoating. We have previously concluded that premature uncoating is detrimental to infection, as mutations in CA that reduce the stability of the capsid are associated with impaired infectivity (5). Therefore, we propose that TNPO3 stimulates uncoating in the cells and thereby enhances the antiviral activity of PF74, which induces premature uncoating of the viral capsid, and this leads to a marked reduction in infectivity.
In accord with recent reports (20, 61), we observed that TNPO3 is apparently not required for HIV-1 entry into the nucleus. Depletion of TNPO3 had no apparent effect on the efficiency of reverse transcription, nor did it appear to reduce the levels of 2-LTR circles. Analysis of viral DNA in nuclear and cytoplasmic fractions of cells revealed accumulation of HIV-1 DNA in the nucleus, suggesting that impaired nuclear entry does not account for the marked impairment in HIV-1 infection observed in TNPO3-depleted cells. These observations suggest that a major block to infection of TNPO3-depleted cells lies between nuclear entry and integration. Depletion of TNPO3 has also been reported to alter integration site selection (65), indicating that the host protein can have both qualitative and quantitative effects on integration.
A nuclear function of TNPO3 in HIV-1 infectivity was also proposed by Zhou et al. (51). In their study, the authors observed that HIV-1 CA accumulates in the nuclei of cells depleted of TNPO3; this effect was specific to wild-type HIV-1, since the N74D mutant CA did not exhibit this phenotype. TNPO3 was found to associate with CA and tRNA in pulldown assays using viral lysates, although CA mutants were not examined, nor was a direct interaction between CA and TNPO3 demonstrated. The authors proposed that TNPO3 performs a nuclear maturation step by removing CA bound to preintegration complexes (PICs) and exporting CA and tRNA out of the nucleus. More recently, Valle-Casuso et al. reported that TNPO3 binds to HIV-1 CA-NC complexes in vitro (61). These findings together with our observation that TNPO3 can directly stimulate HIV-1 uncoating in vitro suggest that the host protein may promote HIV-1 uncoating in the nucleus, thus allowing export of the dissociated CA protein.
Our study also provides new insight into CypA action in HIV-1 infection. The mechanism of action of the CA-binding host protein CypA in the early stages of HIV-1 infection has been unclear despite intensive study. Towers and coworkers reported that CypA modulates the sensitivity of HIV-1 to host restriction factors, which is suggestive of a protective role in the cytoplasm (39). Furthermore, Li et al. showed that CypA regulates the extent of uncoating in a cell type-dependent manner (36). In the present study, we observed that CypA directly stabilizes the HIV-1 capsid in vitro and that binding of CypA to the incoming capsid modulates the dependence of HIV-1 infection on TNPO3. Thus, inhibiting the CA-CypA interaction either by CsA or by the point mutation G89V reduced the dependence of HIV-1 on TNPO3 in target cells. Additionally, CypA also counteracted TNPO3-stimulated uncoating of the capsid in vitro. These observations suggest that CypA and TNPO3 exhibit opposite effects on HIV-1 infectivity in HeLa cells, likely by modulating uncoating. Since CypA exhibits a cell type-specific effect on HIV-1 uncoating (36), it will be interesting to study its effect on the TNPO3 dependence of HIV-1 in cell types in which the virus is more dependent on CypA for infection.
Taken together, our results are consistent with a model in which the binding of CypA to the incoming HIV-1 capsid prevents premature uncoating, allowing for efficient reverse transcription in the cytoplasm (25, 66) and/or intracytoplasmic transport to the nucleus. The CypA-bound capsid may subsequently engage TNPO3, localized to the cytoplasm, and enter the nucleus, where CypA may then dissociate, thereby allowing TNPO3 to induce uncoating required for integration. Following uncoating in the nucleus, RanGTP may then bind to the CA-TNPO3 complex and dissociate it, thus allowing export of CA to the cytoplasm (51). This model suggests that HIV-1 uncoating is not completed in the cytoplasm of the infected cells, and it is consistent with reports that intact cores can be observed docking to the nuclear pore (8) and that CA can be observed in the nuclei of infected cells (51).
While our biochemical data suggest that TNPO3 directly promotes uncoating of HIV-1, it remains possible that TNPO3 acts indirectly by modulating the levels or intracellular localization of another host factor. Of particular interest is CPSF6: expression of a truncated form of this protein (CPSF6-358) blocks HIV-1 infection by targeting the capsid, and mutations in CA that confer resistance to CPSF6-358 also render HIV-1 less dependent on TNPO3 expression (21). Thus, sensitivity to CPSF6-358 inhibition appears to be tightly correlated with a requirement for TNPO3 in HIV-1 infection. CPSF6 is an SR protein (67) and may be a substrate for TNPO3-dependent nuclear transport, thereby potentially sequestering CPSF6 from the incoming HIV-1 core. Interestingly, PF74 was recently demonstrated to completely inhibit binding of CPSF6313-327 peptide to CA-NTD (68).
The viability of TNPO3 as a potential therapeutic target is currently unknown. Karyopherins such as TNPO3 play critical roles in regulating nuclear trafficking, making it likely that their inhibition will result in cellular toxicity. In the course of our studies, we observed that depletion of TNPO3 via shRNA transduction is of limited durability; the stably selected populations regain TNPO3 expression during 2 to 3 weeks of culture, indicative of a growth disadvantage. We were able to observe a strong effect of TNPO3 depletion on HIV-1 infection only during a limited temporal window of culturing the cells after shRNA transduction and selection. Additional studies of the molecular mechanism of TNPO3 action in retrovirus infection may reveal novel approaches to selectively interfere with viral or cellular targets that are specific for HIV-1.
ACKNOWLEDGMENTS
We thank Ernest Yufenyuy and Andrew Flyak for assistance with the structural model for CA and cell fractionation experiments, respectively. The following reagents were obtained from the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: HIV-1 p24 hybridoma (183-H12-5C) from Bruce Chesebro, HIV-1-IG from NABI and NHLBI, psPAX2 (catalog number 11348) from Didier Trono, and Efavirenz and Raltegravir (catalog number 11680) from Merck & Company, Inc.
This work was supported by NIH grants AI076121 (C.A.), GM082251 (J.A.), and AI052014 (A.E.).
Footnotes
Published ahead of print 24 October 2012
REFERENCES
- 1. Ganser-Pornillos BK, Cheng A, Yeager M. 2007. Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell 131:70–79 [DOI] [PubMed] [Google Scholar]
- 2. Li S, Hill CP, Sundquist WI, Finch JT. 2000. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 407:409–413 [DOI] [PubMed] [Google Scholar]
- 3. Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M. 2009. X-ray structures of the hexameric building block of the HIV capsid. Cell 137:1282–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pornillos O, Ganser-Pornillos BK, Yeager M. 2011. Atomic-level modelling of the HIV capsid. Nature 469:424–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Forshey BM, von Schwedler U, Sundquist WI, Aiken C. 2002. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 76:5667–5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hulme AE, Perez O, Hope TJ. 2011. Complementary assays reveal a relationship between HIV-1 uncoating and reverse transcription. Proc. Natl. Acad. Sci. U. S. A. 108:9975–9980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perez-Caballero D, Hatziioannou T, Zhang F, Cowan S, Bieniasz PD. 2005. Restriction of human immunodeficiency virus type 1 by TRIM-CypA occurs with rapid kinetics and independently of cytoplasmic bodies, ubiquitin, and proteasome activity. J. Virol. 79:15567–15572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arhel NJ, Souquere-Besse S, Munier S, Souque P, Guadagnini S, Rutherford S, Prevost MC, Allen TD, Charneau P. 2007. HIV-1 DNA Flap formation promotes uncoating of the pre-integration complex at the nuclear pore. EMBO J. 26:3025–3037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McDonald D, Vodicka MA, Lucero G, Svitkina TM, Borisy GG, Emerman M, Hope TJ. 2002. Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 159:441–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aiken C. 2006. Viral and cellular factors that regulate HIV-1 uncoating. Curr. Opin. HIV AIDS 1:194–199 [DOI] [PubMed] [Google Scholar]
- 11. Arhel N. 2010. Revisiting HIV-1 uncoating. Retrovirology 7:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Luban J. 2007. Cyclophilin A, TRIM5, and resistance to human immunodeficiency virus type 1 infection. J. Virol. 81:1054–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Strebel K, Luban J, Jeang KT. 2009. Human cellular restriction factors that target HIV-1 replication. BMC Med. 7:48 doi:10.1186/1741-7015-7-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ. 2008. Identification of host proteins required for HIV infection through a functional genomic screen. Science 319:921–926 [DOI] [PubMed] [Google Scholar]
- 15. Konig R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, Chiang CY, Tu BP, De Jesus PD, Lilley CE, Seidel S, Opaluch AM, Caldwell JS, Weitzman MD, Kuhen KL, Bandyopadhyay S, Ideker T, Orth AP, Miraglia LJ, Bushman FD, Young JA, Chanda SK. 2008. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 135:49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kataoka N, Bachorik JL, Dreyfuss G. 1999. Transportin-SR, a nuclear import receptor for SR proteins. J. Cell Biol. 145:1145–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Christ F, Thys W, De Rijck J, Gijsbers R, Albanese A, Arosio D, Emiliani S, Rain JC, Benarous R, Cereseto A, Debyser Z. 2008. Transportin-SR2 imports HIV into the nucleus. Curr. Biol. 18:1192–1202 [DOI] [PubMed] [Google Scholar]
- 18. Cribier A, Segeral E, Delelis O, Parissi V, Simon A, Ruff M, Benarous R, Emiliani S. 2011. Mutations affecting interaction of integrase with TNPO3 do not prevent HIV-1 cDNA nuclear import. Retrovirology 8:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krishnan L, Matreyek KA, Oztop I, Lee K, Tipper CH, Li X, Dar MJ, Kewalramani VN, Engelman A. 2010. The requirement for cellular transportin 3 (TNPO3 or TRN-SR2) during infection maps to human immunodeficiency virus type 1 capsid and not integrase. J. Virol. 84:397–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. De Iaco A, Luban J. 2011. Inhibition of HIV-1 infection by TNPO3 depletion is determined by capsid and detectable after viral cDNA enters the nucleus. Retrovirology 8:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, Takemura T, Shelton K, Taniuchi I, Li Y, Sodroski J, Littman DR, Coffin JM, Hughes SH, Unutmaz D, Engelman A, KewalRamani VN. 2010. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7:221–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Franke EK, Yuan HEH, Luban J. 1994. Specific incorporation of cyclophilin A into HIV-1 virions. Nature 372:359–362 [DOI] [PubMed] [Google Scholar]
- 23. Luban J, Bossolt KL, Franke EK, Kalpana GV, Goff SP. 1993. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 73:1067–1078 [DOI] [PubMed] [Google Scholar]
- 24. Thali M, Bukovsky A, Kondo E, Rosenwirth B, Walsh CT, Sodroski J, Gottlinger HG. 1994. Functional association of cyclophilin A with HIV-1 virions. Nature 372:363–365 [DOI] [PubMed] [Google Scholar]
- 25. Braaten D, Franke EK, Luban J. 1996. Cyclophilin A is required for an early step in the life cycle of human immunodeficiency virus type 1 before the initiation of reverse transcription. J. Virol. 70:3551–3560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gamble TR, Vajdos FF, Yoo S, Worthylake DK, Houseweart M, Sundquist WI, Hill CP. 1996. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87:1285–1294 [DOI] [PubMed] [Google Scholar]
- 27. Bosco DA, Eisenmesser EZ, Pochapsky S, Sundquist WI, Kern D. 2002. Catalysis of cis/trans isomerization in native HIV-1 capsid by human cyclophilin A. Proc. Natl. Acad. Sci. U. S. A. 99:5247–5252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bosco DA, Kern D. 2004. Catalysis and binding of cyclophilin A with different HIV-1 capsid constructs. Biochemistry 43:6110–6119 [DOI] [PubMed] [Google Scholar]
- 29. Howard BR, Vajdos FF, Li S, Sundquist WI, Hill CP. 2003. Structural insights into the catalytic mechanism of cyclophilin A. Nat. Struct. Biol. 10:475–481 [DOI] [PubMed] [Google Scholar]
- 30. Ylinen LM, Schaller T, Price A, Fletcher AJ, Noursadeghi M, James LC, Towers GJ. 2009. Cyclophilin A levels dictate infection efficiency of human immunodeficiency virus type 1 capsid escape mutants A92E and G94D. J. Virol. 83:2044–2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Berthoux L, Sebastian S, Sokolskaja E, Luban J. 2005. Cyclophilin A is required for TRIM5α-mediated resistance to HIV-1 in Old World monkey cells. Proc. Natl. Acad. Sci. U. S. A. 102:14849–14853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Javanbakht H, Diaz-Griffero F, Yuan W, Yeung DF, Li X, Song B, Sodroski J. 2007. The ability of multimerized cyclophilin A to restrict retrovirus infection. Virology 367:19–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Keckesova Z, Ylinen LM, Towers GJ. 2006. Cyclophilin A renders human immunodeficiency virus type 1 sensitive to Old World monkey but not human TRIM5 alpha antiviral activity. J. Virol. 80:4683–4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stremlau M, Perron M, Lee M, Li Y, Song B, Javanbakht H, Diaz-Griffero F, Anderson DJ, Sundquist WI, Sodroski J. 2006. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc. Natl. Acad. Sci. U. S. A. 103:5514–5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yap MW, Mortuza GB, Taylor IA, Stoye JP. 2007. The design of artificial retroviral restriction factors. Virology 365:302–314 [DOI] [PubMed] [Google Scholar]
- 36. Li Y, Kar AK, Sodroski J. 2009. Target cell type-dependent modulation of human immunodeficiency virus type 1 capsid disassembly by cyclophilin A. J. Virol. 83:10951–10962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hatziioannou T, Perez-Caballero D, Cowan S, Bieniasz PD. 2005. Cyclophilin interactions with incoming human immunodeficiency virus type 1 capsids with opposing effects on infectivity in human cells. J. Virol. 79:176–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sokolskaja E, Sayah DM, Luban J. 2004. Target cell cyclophilin A modulates human immunodeficiency virus type 1 infectivity. J. Virol. 78:12800–12808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Towers GJ, Hatziioannou T, Cowan S, Goff SP, Luban J, Bieniasz PD. 2003. Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat. Med. 9:1138–1143 [DOI] [PubMed] [Google Scholar]
- 40. Gallay P, Hope T, Chin D, Trono D. 1997. HIV-1 infection of nondividing cells through the recognition of integrase by the importin/karyopherin pathway. Proc. Natl. Acad. Sci. U. S. A. 94:9825–9830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. He J, Chen Y, Farzan M, Choe H, Ohagen A, Gartner S, Busciglio J, Yang X, Hofmann W, Newman W, Mackay CR, Sodroski J, Gabuzda D. 1997. CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature 385:645–649 [DOI] [PubMed] [Google Scholar]
- 42. Zhou J, Aiken C. 2001. Nef enhances human immunodeficiency virus type 1 infectivity resulting from intervirion fusion: evidence supporting a role for Nef at the virion envelope. J. Virol. 75:5851–5859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yee JK, Friedmann T, Burns JC. 1994. Generation of high-titer pseudotyped retroviral with very broad host range. Methods Cell Biol. 43:99–112 [DOI] [PubMed] [Google Scholar]
- 44. Chen C, Okayama H. 1987. High-efficiency tranformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 7:2745–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wehrly K, Chesebro B. 1997. p24 antigen capture assay for quantification of human immunodeficiency virus using readily available inexpensive reagents. Methods 12:288–293 [DOI] [PubMed] [Google Scholar]
- 46. Izaurralde E, Kutay U, von Kobbe C, Mattaj IW, Gorlich D. 1997. The asymmetric distribution of the constituents of the Ran system is essential for transport into and out of the nucleus. EMBO J. 16:6535–6547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. 1997. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 15:871–875 [DOI] [PubMed] [Google Scholar]
- 48. Shah VB, Aiken C. 2011. Uncoating of HIV-1 cores. J. Vis. Exp. 57:e3384 doi:10.3791/3384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Butler SL, Hansen MS, Bushman FD. 2001. A quantitative assay for HIV DNA integration in vivo. Nat. Med. 7:631–634 [DOI] [PubMed] [Google Scholar]
- 50. Logue EC, Taylor KT, Goff PH, Landau NR. 2011. The cargo-binding domain of transportin 3 is required for lentivirus nuclear import. J. Virol. 85:12950–12961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou L, Sokolskaja E, Jolly C, James W, Cowley SA, Fassati A. 2011. Transportin 3 promotes a nuclear maturation step required for efficient HIV-1 integration. PLoS Pathog. 7:e1002194 doi:10.1371/journal.ppat.1002194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Matreyek KA, Engelman A. 2011. The requirement for nucleoporin NUP153 during human immunodeficiency virus type 1 infection is determined by the viral capsid. J. Virol. 85:7818–7827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Thys W, De Houwer S, Demeulemeester J, Taltynov O, Vancraenenbroeck R, Gerard M, De Rijck J, Gijsbers R, Christ F, Debyser Z. 2011. Interplay between HIV entry and transportin-SR2 dependency. Retrovirology 8:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Byeon IJ, Meng X, Jung J, Zhao G, Yang R, Ahn J, Shi J, Concel J, Aiken C, Zhang P, Gronenborn AM. 2009. Structural convergence between Cryo-EM and NMR reveals intersubunit interactions critical for HIV-1 capsid function. Cell 139:780–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang R, Shi J, Byeon IJ, Ahn J, Sheehan JH, Meiler J, Gronenborn AM, Aiken C. 2012. Second-site suppressors of HIV-1 capsid mutations: restoration of intracellular activities without correction of intrinsic capsid stability defects. Retrovirology 9:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Blair WS, Pickford C, Irving SL, Brown DG, Anderson M, Bazin R, Cao J, Ciaramella G, Isaacson J, Jackson L, Hunt R, Kjerrstrom A, Nieman JA, Patick AK, Perros M, Scott AD, Whitby K, Wu H, Butler SL. 2010. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog. 6:e1001220 doi:10.1371/journal.ppat.1001220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shi J, Zhou J, Shah VB, Aiken C, Whitby K. 2011. Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J. Virol. 85:542–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. von Schwedler UK, Stray KM, Garrus JE, Sundquist WI. 2003. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J. Virol. 77:5439–5450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lui K, Huang Y. 2009. RanGTPase: a key regulator of nucleocytoplasmic trafficking. Mol. Cell. Pharmacol. 1:148–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bischoff FR, Klebe C, Kretschmer J, Wittinghofer A, Ponstingl H. 1994. RanGAP1 induces GTPase activity of nuclear Ras-related Ran. Proc. Natl. Acad. Sci. U. S. A. 91:2587–2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Valle-Casuso JC, Di Nunzio F, Yang Y, Reszka N, Lienlaf M, Arhel N, Perez P, Brass AL, Diaz-Griffero F. 2012. TNPO3 is required for HIV-1 replication after nuclear import but prior to integration and binds the HIV-1 core. J. Virol. 86:5931–5936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yoo S, Myszka DG, Yeh C, McMurray M, Hill CP, Sundquist WI. 1997. Molecular recognition in the HIV-1 capsid/cyclophilin A complex. J. Mol. Biol. 269:780–795 [DOI] [PubMed] [Google Scholar]
- 63. Harding MW, Handschumacher RE, Speicher DW. 1986. Isolation and amino acid sequence of cyclophilin. J. Biol. Chem. 261:8547–8555 [PubMed] [Google Scholar]
- 64. Schonbrunner ER, Mayer S, Tropschug M, Fischer G, Takahashi N, Schmid FX. 1991. Catalysis of protein folding by cyclophilins from different species. J. Biol. Chem. 266:3630–3635 [PubMed] [Google Scholar]
- 65. Ocwieja KE, Brady TL, Ronen K, Huegel A, Roth SL, Schaller T, James LC, Towers GJ, Young JA, Chanda SK, Konig R, Malani N, Berry CC, Bushman FD. 2011. HIV integration targeting: a pathway involving transportin-3 and the nuclear pore protein RanBP2. PLoS Pathog. 7:e1001313 doi:10.1371/journal.ppat.1001313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ptak RG, Gallay PA, Jochmans D, Halestrap AP, Ruegg UT, Pallansch LA, Bobardt MD, de Bethune MP, Neyts J, De Clercq E, Dumont JM, Scalfaro P, Besseghir K, Wenger RM, Rosenwirth B. 2008. Inhibition of human immunodeficiency virus type 1 replication in human cells by Debio-025, a novel cyclophilin binding agent. Antimicrob. Agents Chemother. 52:1302–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ruegsegger U, Blank D, Keller W. 1998. Human pre-mRNA cleavage factor Im is related to spliceosomal SR proteins and can be reconstituted in vitro from recombinant subunits. Mol. Cell 1:243–253 [DOI] [PubMed] [Google Scholar]
- 68. Price AJ, Fletcher AJ, Schaller T, Elliott T, Lee K, Kewalramani VN, Chin JW, Towers GJ, James LC. 2012. CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog. 8:e1002896 doi:10.1371/journal.ppat.1002896 [DOI] [PMC free article] [PubMed] [Google Scholar]