

Abstract
Stress granules (SGs) are cytoplasmic foci composed of stalled translation preinitiation complexes induced by environmental stress stimuli, including viral infection. Since viral propagation completely depends on the host translational machinery, many viruses have evolved to circumvent the induction of SGs or co-opt SG components. In this study, we found that expression of Japanese encephalitis virus (JEV) core protein inhibits SG formation. Caprin-1 was identified as a binding partner of the core protein by an affinity capture mass spectrometry analysis. Alanine scanning mutagenesis revealed that Lys97 and Arg98 in the α-helix of the JEV core protein play a crucial role in the interaction with Caprin-1. In cells infected with a mutant JEV in which Lys97 and Arg98 were replaced with alanines in the core protein, the inhibition of SG formation was abrogated, and viral propagation was impaired. Furthermore, the mutant JEV exhibited attenuated virulence in mice. These results suggest that the JEV core protein circumvents translational shutoff by inhibiting SG formation through an interaction with Caprin-1 and facilitates viral propagation in vitro and in vivo.
INTRODUCTION
In eukaryotic cells, environmental stresses such as heat shock, oxidative stress, UV irradiation, and viral infection trigger a sudden translational arrest, leading to stress granule (SG) formation (1). SGs are cytoplasmic foci composed of stalled translation preinitiation complexes and are postulated to play a critical role in regulating mRNA metabolism during stress via so-called “mRNA triage” (2). The initiation of SG formation results from phosphorylation of eukaryotic translation initiation factor 2α (eIF2α) at Ser51 by various kinases, including protein kinase R (PKR), PKR-like endoplasmic reticulum kinase (PERK), general control nonrepressed 2 (GCN2), and heme-regulated translation inhibitor (HRI), which are commonly activated by double-stranded RNA (dsRNA), endoplasmic reticulum (ER) stress, nutrient starvation, and oxidative stress, respectively. Phosphorylation of eIF2α reduces the amount of eIF2-GTP-tRNA complex and inhibits translation initiation, leading to runoff of elongating ribosomes from mRNA transcripts and the accumulation of stalled translation preinitiation complexes. Thus, SGs are defined by the presence of components of translation initiation machinery, including 40S ribosome subunits, poly(A)-binding protein (PABP), eIF2, eIF3, eIF4A, eIF4E, eIF4G, and eIF5. Then, primary aggregation occurs through several RNA-binding proteins (RBPs), including T-cell intracellular antigen-1 (TIA-1), TIA-1-related protein 1 (TIAR), and Ras-Gap-SH3 domain-binding protein (G3BP). These RBPs are independently self-oligomerized with the stalled initiation factors and with other RBPs, such as USP10, hnRNP Q, cytoplasmic activation/proliferation-associated protein-1 (Caprin-1), and Staufen and with nucleated mRNA-protein complex (mRNP) aggregations (3, 4). SG assembly begins with the simultaneous formation of numerous small mRNP granules which then progressively fuse into larger and fewer structures, a process known as secondary aggregation (5). The aggregation of TIA-1 or TIAR is regulated by molecular chaperones, such as heat shock protein 70 (Hsp70) (3), whereas that of G3BP is controlled by its phosphorylation at Ser149 (4). SG formation and disassembly in response to cellular stresses are strictly regulated by multiple factors.
Viral infection can certainly be viewed as a stressor for cells, and SGs have been reported in some virus-infected cells. Since the propagation of viruses is completely reliant on the host translational machinery, stress-induced translational arrest plays an important role in host antiviral defense. To antagonize this host defense, most viruses have evolved to circumvent SG formation during infection. For example, poliovirus (PV) proteinase 3C cleaves G3BP, leading to effective SG dispersion and virus propagation (6). Influenza A virus nonstructural protein 1 (NS1) has been shown to inactivate PKR and prevent SG formation (7). In the case of human immunodeficiency virus 1 (HIV-1) infection, Staufen1 is recruited in ribonucleoproteins for encapsidation through interaction with the Gag protein to prevent SG formation (8). In contrast, some viruses employ alternative mechanisms of translation initiation and promote SG formation to limit cap-dependent translation of host mRNA (9, 10). In addition, vaccinia virus induces cytoplasmic “factories” in which viral translation, replication, and assembly take place. These factories include G3BP and Caprin-1 to promote transcription of viral mRNA (11).
Japanese encephalitis virus (JEV) belongs to the genus Flavivirus within the family Flaviviridae, which includes other mosquito-borne human pathogens, such as dengue virus (DENV), West Nile virus (WNV), and yellow fever virus, that frequently cause significant morbidity and mortality in mammals and birds (12). JEV has a single-stranded positive-sense RNA genome of approximately 11 kb. The genomic RNA carries a single large open reading frame, and a polyprotein translated from the genome is cleaved co- and posttranslationally by host and viral proteases to yield three structural proteins, the core, precursor membrane (PrM), and envelop (E) proteins, and seven nonstructural (NS) proteins, NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5 (13). PrM is further cleaved by the multibasic protease, furin, and matured to membrane (M) protein. The core, M, and E proteins are components of extracellular mature virus particles. NS proteins are not incorporated into particles and are thought to be involved in viral replication, which occurs in close association with ER-derived membranes (14). Previous reports have shown that WNV and DENV inhibit SG formation by sequestering TIA-1 and TIAR through specific interaction with viral RNA (15, 16). In addition, the membrane structure induced by WNV infection was suggested to prevent PKR activation and avoid induction of SG formation (17). In this study, we show that JEV core protein plays an important role in inhibition of SG formation. JEV core protein recruited several SG-associated proteins, including G3BP and USP10, through an interaction with Caprin-1 and suppressed SG formation. Furthermore, a mutant JEV carrying a core protein incapable of binding to Caprin-1 exhibited lower propagation in vitro and lower pathogenicity in mice than the wild-type (WT) JEV, suggesting that inhibition of SG formation by the core protein is crucial to antagonize host defense. These results reveal a novel strategy of JEV to inhibit SG formation through an interaction with Caprin-1 and facilitate viral propagation.
MATERIALS AND METHODS
Plasmids.
Plasmids encoding FLAG-tagged JEV core protein (pCAGPM-FLAG-Core) and hemagglutinin (HA)-tagged JEV proteins (pCAGPM-HA-JEV proteins) were generated as previously described (18, 19). The cDNA of the core protein of JEV AT31 (amino acid residues 2 to 105) was amplified from the pCAGPM-FLAG-Core plasmid by PCR and cloned into pET21b (Novagen-Merck, Darmstadt, Germany) for expression in bacteria as a His-tagged protein and in pCAG-MCS2-FOS for expression in mammalian cells as a FLAG-One-STrEP (FOS)-tagged protein. The resulting plasmids were designated pET21b-Core-His and pCAG-Core-FOS, respectively. The cDNA of the core protein of DENV2 (amino acid residues 2 to 100) was amplified from the pCAG/FLAG-DEN2C-HA plasmid (19) by PCR and cloned into pCAGPM-N-FLAG. The cDNA of human Caprin-1 was amplified from 293T cells by reverse transcription-PCR (RT-PCR) and cloned into pCAGPM-N-HA (20) and pGEX 6P-1 (GE Healthcare, Buckinghamshire, United Kingdom) for expression in bacteria as a glutathione S-transferase (GST) fusion protein and designated pCAGPM-HA-Caprin-1 and pGEX-GST-Caprin-1, respectively. The cDNAs of human G3BP1 and USP10 were also amplified from 293T cells by RT-PCR and cloned into pCAGPM-N-HA. The nucleotide residues of the adenine at 384, adenine at 385, cytosine at 387, and guanine at 388 of the JEV genome in pMWATG1 were replaced with guanine, cytosine, guanine, and cytosine, respectively, by PCR-based mutagenesis to change Lys97 and Arg98 of the core protein to Ala, yielding pMWAT/KR9798A. The cDNA of the mutant core protein was also cloned into pCAGPM-N-FLAG and pET21b. To generate stable cell lines expressing Aequorea coerulescens green fluorescent protein (AcGFP)-fused Caprin-1, the cDNA of human Caprin-1 was amplified by RT-PCR and cloned into pAcGFP N1 (Clontech, Mountain View, CA), and the Caprin-1-AcGFP gene was subcloned into the lentiviral vector pCSII-EF-RfA (21) and designated pCSII-EF-Caprin-1-AcGFP. All plasmids were confirmed by sequencing with an ABI Prism 3130 genetic analyzer (Applied Biosystems, Tokyo, Japan).
Cells and stress treatment.
Mammalian cell lines, Vero (African green monkey kidney), 293T (human kidney), Huh7 (human hepatocellular carcinoma), and HeLa (human cervical carcinoma), were maintained in Dulbecco's modified Eagle's minimal essential medium (DMEM) (Sigma, St. Louis, MO) supplemented with 100 U/ml penicillin, 100 mg/ml streptomycin, nonessential amino acids (Sigma), and 10% fetal bovine serum (FBS). The mosquito cell line C6/36 (Aedes albopictus) was grown in Leibovitz's L-15 medium with 10% FBS. Huh7 cells were transduced with a lentiviral vector expressing Caprin-1-AcGFP and AcGFP and designated Huh7/Caprin-1-AcGFP and Huh7/AcGFP, respectively. For induction of SGs, cells were treated with sodium arsenite at a final concentration of 1.0 mM in the culture medium for 30 min prior to fixation or lysis of the cells. SG formation was defined morphologically by immunostaining using anti-SG-related factor antibodies described below. Cell viability was determined by using CellTiter-Glo (Promega, Madison, WI) according to the manufacturer's instruction.
Viruses.
The wild-type and 9798A mutant of the JEV AT31 strain were generated by the transfection of pMWATG1 and pMWAT/KR9798A, respectively, as described previously (22). Viral infectivity was determined by an immunostaining focus assay as described previously (20), and the results are expressed in focus-forming units (FFU). JEV and DENV serotype 2 New Guinea C strain were amplified in C6/36 cells.
Antibodies.
Anti-JEV core rabbit polyclonal antibody (PAb) and anti-JEV NS3 mouse monoclonal antibody (MAb) were prepared as described previously (20, 23). Anti-JEV NS2B rabbit PAb was generated with synthetic peptides of JEV NS2B at Scrum, Inc. (Tokyo, Japan). Anti-DENV core protein rabbit PAb was prepared by using a GST-fused recombinant protein containing amino acid residues 2 to 100 of the DENV core protein. Anti-FLAG mouse MAb (M2) and rabbit PAb and anti-β-actin mouse MAb were purchased from Sigma. Anti-hnRNP Q mouse MAb (ab10687), anti-USP10 rabbit PAb (ab70895), and anti-eIF4B rabbit PAb (ab78916) were purchased from Abcam (Cambridge, United Kingdom). Anti-eIF2α, anti-phospho-eIF2α, and anti-eIF3A rabbit PAbs were purchased from Cell Signaling Technology (Danvers, MA). Anti-HA mouse MAb (HA11), anti-HA rat MAb (3F10), anti-His mouse MAb, anti-GFP mouse MAb (JL-8), anti-JEV envelope protein mouse MAb (6B4A-10), anti-G3BP mouse MAb, anti-TIA-1 goat PAb, anti-Caprin-1 rabbit PAb, and anti-dsRNA mouse MAb were purchased from Covance (Richmond, CA), Roche (Mannheim, Germany), R&D Systems (Minneapolis, MN), Clontech, Chemicon (Temecula, CA), BD Biosciences (Franklin Lakes, NJ), Santa Cruz (Santa Cruz, CA), Proteintech (Chicago, IL), and Biocenter (Szirak, Hungary), respectively. Alexa Fluor (AF)-conjugated secondary antibodies were purchased from Invitrogen (Carlsbad, CA).
Immunofluorescence microscopy.
Huh7 cells were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min at room temperature. After cells were quenched for 10 min with PBS containing 50 mM ammonium chloride (NH4Cl), they were permeabilized with 0.2% Triton X-100 in PBS for 10 min and blocked with PBS containing 2% bovine serum albumin (BSA) for 30 min at room temperature. The cells were then incubated with the antibodies indicated in the figure legends. Nuclei were stained with 4′,6′-diamidino-2-phenylindole (DAPI). The samples were examined by a Fluoview FV1000 laser scanning confocal microscope (Olympus, Tokyo, Japan).
Transfection, immunoprecipitation, and immunoblotting.
Plasmids were transfected into 293T or Huh7 cells by use of TransIT LT1 (Mirus, Madison, WI), and cells collected at 24 h posttransfection were subjected to immunostaining, immunoprecipitation, and/or immunoblotting as described previously (24). The immunoprecipitates were boiled in sodium dodecyl sulfate (SDS) sample buffer and subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA) and incubated with the appropriate antibodies. The immune complexes were visualized with SuperSignal West Femto substrate (Thermo Scientific, Rockford, IL) and detected by use of an LAS-3000 image analyzer system (Fujifilm, Tokyo, Japan).
FOS-tagged purification and mass spectrometry.
pCAG-Core-FOS or empty vector was transfected into 293T cells, harvested at 24 h posttransfection, washed with cold PBS, suspended in cell lysis buffer (20 mM Tris-HCl, pH 7.4, 135 mM NaCl, 1% Triton X-100, and protease inhibitor cocktail [Complete; Roche]), and centrifuged at 14,000 × g for 20 min at 4°C. The supernatant was pulled down using 50 μl of STrEP-Tactin Sepharose (IBA, Gottingen, Germany) equilibrated with cell lysis buffer for 2 h at 4°C. The affinity beads were washed three times with cell lysis buffer and suspended in 2× SDS-PAGE sample buffer. The proteins were subjected to SDS-PAGE, followed by Coomassie brilliant blue (CBB) staining using CBB Stain One (Nakalai Tesque, Kyoto, Japan). The gels were divided into 10 pieces, and each fraction was trypsinized and subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis to identify coimmunoprecipitated proteins. All of the proteins in gels were identified comprehensively, and the proteins detected in cells transfected with pCAG-Core-FOS but not in those with empty vector were regarded as candidates for binding partners of JEV core.
Gene silencing.
A commercially available small interfering RNA (siRNA) pool targeting Caprin-1 (siGENOME SMARTpool, human Caprin1) and control nontargeting siRNA were purchased from Dharmacon (Buckinghamshire, United Kingdom) and transfected into 293T cells using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's protocol.
Preparation of recombinant proteins and GST pulldown assay.
His-tagged JEV core protein (core-His) was purified as described in a previous report (25). Briefly, core-His was expressed in Escherichia coli (E. coli) Rosetta-gami 2(DE3) strain cells (Novagen-Merck) transformed with pET21b-Core-His (WT or 9798A). Bacteria grown to an optical density at 600 nm of 0.6 were induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG), incubated for 5 h at 37°C with shaking, collected by centrifugation at 6,000 × g for 10 min, lysed in 10 ml of bacteria lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, and protease inhibitor cocktail [Complete; Roche]) by sonication on ice, and centrifuged at 10,000 × g for 15 min. The supernatant containing core-His was subjected to ammonium sulfate fractionation, followed by cation exchange chromatography with a HiTrap SP column (GE Healthcare). The eluted core-His recombinant protein was dialyzed with 50 mM Tris-HCl buffer containing 150 mM NaCl at 4°C overnight. GST-fused Caprin-1 (GST-Caprin-1) was expressed in E. coli BL21(DE3) cells transformed with pGEX-GST-Caprin-1. Bacteria grown to an optical density at 600 nm of 1.0 were induced with 0.1 mM IPTG, incubated for 5 h at 25°C with shaking, collected by centrifugation at 6,000 × g for 10 min, lysed in 10 ml of bacteria lysis buffer by sonication on ice, and centrifuged at 10,000 × g for 15 min. The supernatant was mixed with 200 μl of glutathione-Sepharose 4B beads (GE Healthcare) equilibrated with bacteria lysis buffer for 1 h at room temperature, and then the beads were washed five times with lysis buffer. Twenty micrograms of GST-Caprin-1 or GST was mixed with equal volumes of the purified core-His for 2 h at 4°C with gentle agitation. The beads were washed five times with bacteria lysis buffer and then suspended in SDS-PAGE sample buffer.
Mouse experiments.
Experimental infections were approved by the Committee for Animal Experiment of RIMD, Osaka University (H19-2-0). Female ICR mice (3 weeks old) were purchased from CLEA Japan (Tokyo, Japan) and kept in specific pathogen-free environments. Groups of mice (n = 10) were intraperitoneally inoculated with 5 × 104 FFU (100 μl) of the viruses. The mice were observed for 3 weeks after inoculation to determine survival rates. To examine viral growth in the brain, 5 × 104 FFU of the viruses were intraperitoneally administered to the groups of mice (n = 3). At 7 days postinfection, mice were euthanized, and the cerebrums were collected. The infectious titers in the homogenates of the cerebrums were determined in Vero cells as described above.
RESULTS
JEV infection confers resistance to SG induction.
To examine the formation of SGs in cells infected with JEV, Huh7 cells were infected with JEV at a multiplicity of infection (MOI) of 0.5, and the expression of JEV proteins and an accepted marker for SGs, G3BP, was determined by immunofluorescence analysis at 24 h postinfection. G3BP was mainly accumulated in the perinuclear region and partially colocalized with the JEV core protein, while only partial colocalization with the NS2B protein was also observed (Fig. 1A, left). In addition, a few small G3BP-positive foci were scattered in the cytoplasm. This accumulation of G3BP was observed in not only Huh7 cells but also other cell lines, i.e., 293T and HeLa cells, infected with JEV (Fig. 1B). However, the expression level of G3BP in cells infected with JEV was comparable to that in mock-infected cells (Fig. 1C). To further investigate SG induction by JEV infection, expression of TIA-1, another SG marker, was examined. Although accumulation of TIA-1 in the perinuclear region was not observed, a few TIA-1-positive foci were observed in the JEV-infected cells and were colocalized with G3BP and JEV NS2B, indicating that SG foci were induced in cells infected with JEV (Fig. 1D). The accumulation of G3BP and the aggregation of TIA-1, indicating SG formation, appeared at 24 h postinfection in accord with the expression of viral proteins (Fig. 1E). We further examined the dynamics of other SG-associated factors in cells infected with JEV. Each factor formed clear SGs in cells treated with sodium arsenite, a potent SG inducer eliciting oxidative stress (Fig. 2). As shown in Fig. 1F, three distinct patterns of the subcellular localization of SG components were observed. USP10 and Caprin-1 were accumulated in the perinuclear region and also formed a few small foci scattered throughout the cytoplasm, as seen for G3BP; TIA-1 and hnRNP Q formed cytoplasmic foci but were not accumulated in the perinuclear region; and subcellular localization of eIF3A and eIF4B was not changed. The cytoplasmic foci were confirmed as SGs by immunofluorescence analyses using specific antibodies to SG-associated factors (data not shown). Taken together, these results indicate that JEV infection induces accumulation of several RBPs and formation of a few SGs.
Fig 1.

Dynamics of SG-associated factors during JEV infection. (A) Huh7 cells infected with JEV at an MOI of 0.5 were treated with or without 1.0 mM sodium arsenite for 30 min at 37°C, and the levels of expression of G3BP and JEV core protein/NS2B were determined at 24 h postinfection by immunofluorescence analysis with mouse anti-G3BP MAb and rabbit anti-core protein or anti-NS2B PAb, followed by AF488-conjugated anti-mouse IgG (Invitrogen) and AF594-conjugated anti-rabbit IgG, respectively. Cell nuclei were stained with DAPI (blue). (B) Cellular localizations of G3BP and JEV NS2B in 293T and HeLa cells infected with JEV were determined at 24 h postinfection by immunofluorescence analysis with mouse anti-G3BP MAb and rabbit anti-NS2B PAb, followed by AF488-conjugated anti-mouse IgG and AF594-conjugated anti-rabbit IgG, respectively. Cell nuclei were stained with DAPI (blue). (C) Phosphorylation of eIF2α in cells prepared as described in panel A was determined by immunoblotting using the indicated antibodies. The band intensities were quantified by ImageJ software (NIH, Bethesda, MD), and the relative levels for the indicated proteins are shown based on the level of the mock-infected cells. (D) Cellular localizations of G3BP, NS2B, and TIA-1 in Huh7 cells infected with JEV were determined at 24 h postinfection by immunofluorescence analysis with mouse anti-G3BP MAb, rabbit anti-NS2B PAb, and goat anti-TIA-1 PAb, followed by AF488-conjugated anti-mouse IgG, AF594-conjugated anti-rabbit IgG, and AF633-conjugated anti-goat IgG, respectively. Cell nuclei were stained with DAPI (gray). (E) Dynamics of G3BP and TIA-1 during JEV infection. Huh7 cells infected with JEV were immunostained at 0, 12, and 24 h postinfection (hpi) with mouse anti-G3BP MAb or goat anti-TIA-1 PAb and rabbit anti-NS2B PAb, followed by AF488-conjugated anti-mouse IgG or AF488-conjugated anti-goat IgG and AF594-conjugated anti-rabbit IgG, respectively. Cell nuclei were stained with DAPI (blue). (F) Cellular localization of SG-associated proteins (USP10, Caprin-1, TIA-1, hnRNP Q, eIF3A, and eIF4B) (green, AF488-conjugated secondary antibody) and JEV NS2B/NS3 (red, AF-594-conjugate secondary antibody) in Huh7 cells infected with JEV was determined by immunoblotting at 24 h postinfection. Cell nuclei were stained with DAPI (blue). (G) Numbers of G3BP-positive foci in 30 cells prepared as described in panel A were counted for each experimental condition. Lines, boxes, and error bars indicate the means, 25th to 75th percentiles, and 95th percentiles, respectively. The significance of differences between the means was determined by a Student's t test.*, P < 0.01; ND, no significant difference.
Fig 2.

Each SG-associated factor forms SGs under oxidative stress. After treatment with 1.0 mM sodium arsenite for 30 min at 37°C, Huh7 cells were subjected to immunofluorescence analysis with the indicated primary antibodies, followed by AF488-conjugated anti-goat IgG and AF594-conjugated anti-mouse or rabbit IgG. Cell nuclei were stained with DAPI (blue).
It has been shown previously that infection with WNV or DENV confers resistance to SG formation induced by sodium arsenite (15). To determine the effect of JEV infection on the SG formation induced by sodium arsenite, JEV-infected cells were treated with 0.5 mM sodium arsenite for 30 min at 24 h postinfection. Although many G3BP-positive foci were observed in mock-infected cells by the treatment with sodium arsenite, accumulation of G3BP in the perinuclear region was observed in the JEV-infected cells (Fig. 1A, right), and the numbers of G3BP-positive foci in the JEV-infected cells were less than those in the mock-infected cells (Fig. 1G). Although it has been reported that a significant reduction of the phosphorylation at Ser51 of eIF2α in cells treated with arsenite was induced by infection with WNV (15), the phosphorylation of eIF2α was slightly suppressed in the JEV-infected cells (Fig. 1C). Furthermore, while previous studies reported that Caprin-1 and TIA-1 were colocalized with dsRNA in cells infected with DENV (15, 26), no colocalization of G3BP or TIA-1 with the DENV core protein was observed in the present study (Fig. 3), suggesting that the mechanisms of the viral circumvention of SG formation in cells infected with JEV are different from those in cells infected with WNV and DENV.
Fig 3.

Subcellular localizations of the SG-associated proteins during DENV infection. Cellular localizations of G3BP, Caprin-1, and TIA-1 (green, AF488-conjugated secondary antibody) and viral components (core protein and dsRNA) (red, AF-594-conjugate secondary antibody) in Huh7 cells infected with DENV were determined by immunofluorescence analysis using the appropriate antibodies at 48 h postinfection. Cell nuclei were stained with DAPI (blue).
JEV core protein suppresses SG formation induced by sodium arsenite.
To elucidate the molecular mechanisms of suppression of SG formation induced by sodium arsenite during JEV infection, we tried to identify which viral protein(s) is responsible for the SG inhibition. Since G3BP was colocalized with JEV core protein, we first examined the involvement of the core protein in the perinuclear accumulation of G3BP and in the suppression of SG formation. The expression of JEV core protein alone induced the accumulation of G3BP in the perinuclear region (Fig. 4A, left panel) and suppressed sodium arsenite-induced SG formation (Fig. 4A, upper right cell in the right panel), similarly to JEV infection. In contrast, inhibition of SG formation induced by sodium arsenite was not observed in cells expressing other JEV proteins (Fig. 4B). These results suggest that JEV core protein is responsible for the circumvention of the SG formation observed in cells infected with JEV.
Fig 4.

Inhibition of the arsenite-induced SG formation by the expression of JEV proteins. (A) Huh7 cells transfected with a plasmid, pCAGPM-HA-Core, were treated with or without 1.0 mM sodium arsenite for 30 min at 37°C, and the cellular localizations of G3BP and HA-Core were determined at 24 h posttransfection by immunofluorescence analysis with mouse anti-G3BP MAb and rat anti-HA MAb, followed by AF488-conjugated anti-mouse IgG and AF594-conjugated anti-rat IgG, respectively. Cell nuclei were stained with DAPI (blue). (B) Huh7 cells, which were separately transfected with a plasmid expressing an individual viral protein (pCAGPM-HA-JEV protein) as indicated in the figure, were treated with 1.0 mM sodium arsenite for 30 min at 37°C and subjected to an immunofluorescence assay using mouse anti-G3BP MAb and rat anti-HA MAb, followed by AF488-conjugated anti-mouse IgG and AF594-conjugated anti-rat IgG, respectively. Cell nuclei were stained with DAPI (blue).
JEV core protein directly interacts with Caprin-1, an SG-associated cellular factor.
Since JEV core protein was suggested to participate in the inhibition of SG formation, we tried to identify cellular factors associated with the core protein by LC-MS/MS analysis, as shown in Fig. 5A. Among the 30 factors with the best scores, two SG-associated proteins, PKR (Mascot search score, 206) and Caprin-1 (Mascot search score, 153), were identified as binding partners of JEV core protein (Fig. 5B). Although PABP1, hnRNP Q, Staufen, G3BP, and eIF4G were also identified, their scores were lower than those of PKR and Caprin-1. Because the results shown in Fig. 1B suggest that the inhibition of SG formation takes place downstream of eIF2α phosphorylation, we focused on Caprin-1 as a key factor involved in the inhibition of SG formation in cells infected with JEV. To confirm the specific interaction of JEV core protein with Caprin-1, FLAG-JEV core protein and HA-Caprin-1 were coexpressed and immunoprecipitated with anti-HA or anti-FLAG antibody in the presence or absence of nuclease. FLAG-JEV core protein was coprecipitated with HA-Caprin-1 irrespective of nuclease treatment (Fig. 5C and D), suggesting that the interaction between JEV core protein and Caprin-1 is a protein-protein interaction. On the other hand, FLAG-DENV core protein was not coprecipitated with HA-Caprin-1 (Fig. 5E), indicating that the interaction with Caprin-1 was specific for JEV core protein. Next, the direct interaction between JEV core protein and Caprin-1 was examined by a GST-pulldown assay using purified proteins expressed in bacteria. The His-tagged core protein was coprecipitated with GST-tagged Caprin-1, suggesting that JEV core protein directly interacts with Caprin-1 (Fig. 5F).
Fig 5.

JEV core protein directly interacts with Caprin-1, an SG-associated cellular factor. (A) Identification of host cellular proteins associated with JEV core protein by FOS-tagged purification and LC-MS/MS analysis. Overview of the FOS-tagged purification of cellular proteins associated with JEV core protein. (B) The 30 candidate proteins as binding partners of JEV core protein exhibiting high scores are listed. PKR and Caprin-1 are indicated in red. (C and D) FLAG-JEV core protein and HA-Caprin-1 were coexpressed in 293T cells, and the cell lysates harvested at 24 h posttransfection were treated with or without micrococcal nuclease for 30 min at 37°C and immunoprecipitated (IP) with anti-HA (αHA) or anti-FLAG (αFLAG) antibody, as indicated. The precipitates were subjected to immunoblotting (IB) to detect coprecipitated counterparts. (E) FLAG-DENV core protein was coexpressed with HA-Caprin-1 in 293T cells, immunoprecipitated with anti-HA antibody, and immunoblotted with anti-HA or anti-FLAG antibody. (F) His-tagged JEV core protein was incubated with either GST-fused Caprin-1 or GST for 2 h at 4°C, and the precipitates obtained by GST pulldown assay were subjected to CBB staining and immunoblotting with anti-His antibody.
To further determine the cellular localization of Caprin-1 in JEV-infected cells, Caprin-1 fused with AcGFP (Caprin-1-AcGFP) was lentivirally expressed in Huh7 cells. The levels of expression and recruitment of Caprin-1-AcGFP into SGs were determined by immunoblotting and immunofluorescence analysis, respectively (Fig. 6A and B). In cells infected with JEV, Caprin-1-AcGFP was concentrated in the perinuclear region and colocalized with core protein and G3BP, while no colocalization of the proteins was observed in cells infected with DENV (Fig. 6C), suggesting that Caprin-1 directly interacts with JEV core protein in the perinuclear region of the infected cells.
Fig 6.

Caprin-1 is colocalized with the JEV core protein in the perinuclear region. (A) Expression of Caprin-1 fused with AcGFP (Caprin-1-AcGFP), Caprin-1, actin, or AcGFP in lentivirally transduced Huh7 cells was determined by immunoblotting using the appropriate antibodies. (B) Subcellular localization of Caprin-1-AcGFP or AcGFP (green) and endogenous Caprin-1 (red) in cells treated with/without 1.0 mM sodium arsenite for 30 min at 37°C was determined by immunofluorescence assay with rabbit anti-Caprin-1 PAb and AF594-conjugated anti-rabbit IgG. Cell nuclei were stained with DAPI (blue). (C) Huh7/Caprin-1-AcGFP cells were infected with either JEV or DENV at an MOI of 0.5, and the cellular localizations of JEV and DENV core (red) with Caprin-1-AcGFP and G3BP (blue) were determined at 24 h and 48 h postinfection, respectively. Cells were stained with mouse anti-G3BP MAb and rabbit anti-JEV or DENV core protein PAb, followed by AF633-conjugated anti-mouse IgG and AF594-conjugated anti-rabbit IgG, respectively, and examined by immunofluorescence analysis.
Knockdown of Caprin-1 cancels SG inhibition during JEV infection and suppresses viral propagation.
To assess the biological significance of the interaction of JEV core protein with Caprin-1 in JEV propagation, the expression of Caprin-1 was suppressed by using Caprin-1-specific siRNAs (siCaprin-1). Transfection of siCaprin-1 efficiently and specifically knocked down the expression of Caprin-1 with a slight increase of cell viability and decreased the production of infectious particles in the culture supernatants of cells infected with JEV, in comparison with those treated with a control siRNA (siNC) (Fig. 7A). Furthermore, immunofluorescence analyses revealed that knockdown of Caprin-1 increased the number of G3BP-positive granules colocalized with SG-associated factors, including TIA-1 and eIF4B, and inhibited the G3BP concentration in the perinuclear region (Fig. 7B and C). These results suggest that knockdown of Caprin-1 suppresses JEV propagation through the induction of SG formation.
Fig 7.
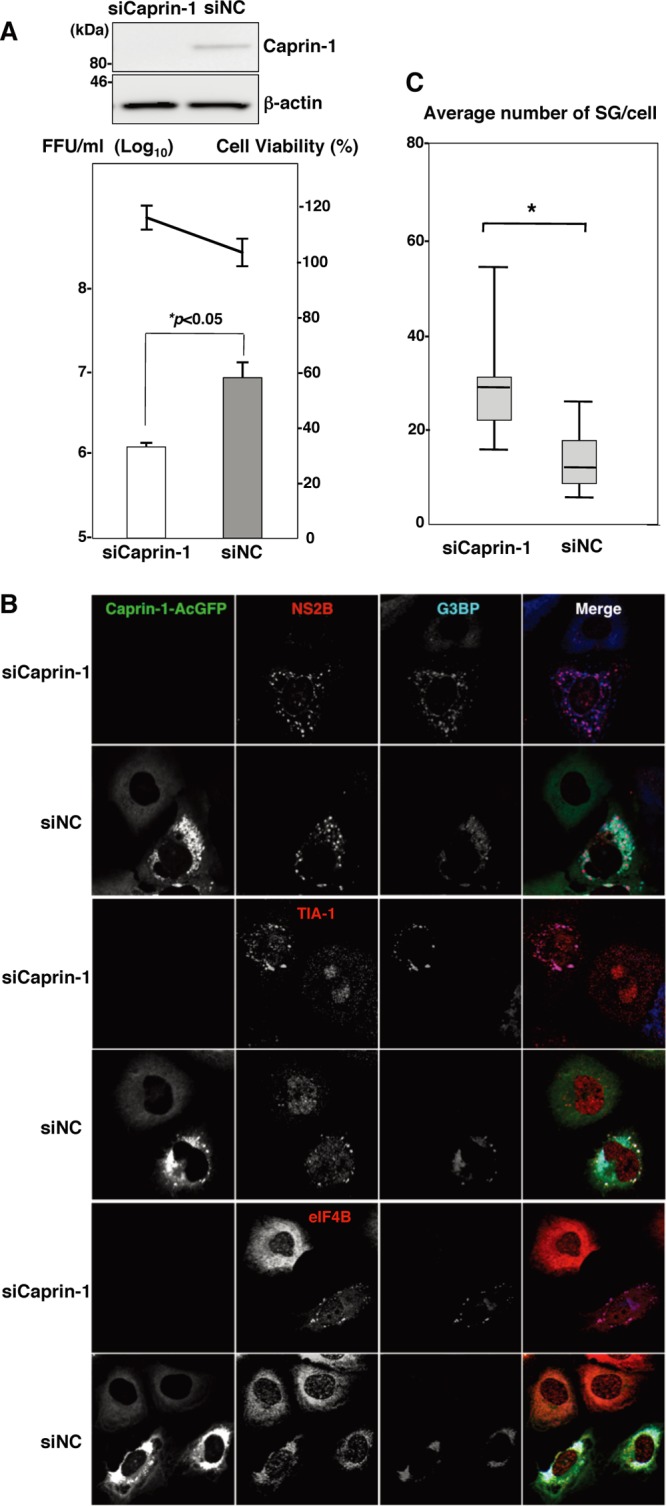
Knockdown of Caprin-1 cancels SG inhibition during JEV infection and suppresses viral propagation. (A) (Upper) The levels of expression of Caprin-1 in cells transfected with either siCaprin-1 or siNC was determined by immunoblotting using anti-Caprin-1 and anti-β-actin antibodies at 72 h posttransfection (top panel). At 48 h posttransfection with either siCaprin-1 or siNC, Huh7/Caprin-1-AcGFP cells were inoculated with JEV at an MOI of 0.5. At 24 h postinfection (72 h posttransfection), the infectious titers in the supernatants were determined by focus-forming assay in Vero cells (bottom panel, bar graph). Cell viability was determined at 72 h posttransfection and calculated as a percentage of the viability of cells treated with siNC (bottom panel, line graph). The results shown are from three independent assays, with the error bars representing the standard deviations. (B) At 48 h posttransfection with either siCaprin-1 or siNC, Huh7/Caprin-1-AcGFP cells were inoculated with JEV at an MOI of 0.5. The cellular localizations of SG-associated factors and JEV NS2B were determined at 24 h postinfection (72 h posttransfection) by immunofluorescence analysis with mouse anti-G3BP MAb and rabbit anti-NS2B PAb, rabbit anti-eIF4B PAb, or goat anti-TIA-1 PAb, followed by AF633-conjugated anti-mouse IgG and AF594-conjugated anti-rabbit IgG or AF594-conjugated anti-goat IgG, respectively. (C) Numbers of G3BP-positive foci in 30 cells prepared as described in panel B were counted. Lines, boxes, and error bars indicate the means, 25th to 75th percentiles, and 95th percentiles, respectively. The significance of differences between the means was determined by a Student's t test. *, P < 0.01.
Lys97 and Arg98 in the JEV core protein are crucial residues for the interaction with Caprin-1.
To determine amino acid residues of the core protein that are required for the interaction with Caprin-1, we constructed a putative model based on the structural information of the DENV core protein previously resolved by nuclear magnetic resonance (NMR) (27), as shown in Fig. 8A. Based on this model, we selected hydrophobic amino acids, which were located on the solvent-exposed side in the α1 and α4 helices, as amino acid residues responsible for the binding to host proteins. Amino acid substitutions in each of the α-helices shown in Fig. 8B were designed in the context of FLAG-Core (Mα1 and Mα4), and the interaction of FLAG-Core mutants with Caprin-1 was examined by immunoprecipitation analysis. WT and Mα1, but not Mα4, core proteins were immunoprecipitated with Caprin-1 (Fig. 8B). To determine the amino acids responsible for interaction with Caprin-1, further alanine substitutions were introduced in the α4 helix, and the interaction was examined by immunoprecipitation assay. As shown in Fig. 8C, double replacing both Lys97 and Arg98 with Ala (9798A) completely abrogated the interaction with Caprin-1. The importance of these two amino acids in the interaction with Caprin-1 was also confirmed by GST pulldown assay (Fig. 8D). These results indicate that Lys97 and Arg98 in the JEV core protein are crucial for the interaction with Caprin-1. Since G3BP has been reported to be one of the key molecules for SG formation and interacts with several SG component molecules including Caprin-1 and USP10 (28, 29), interactions of the core protein with SG components were examined by immunoprecipitation assay. The wild-type but not mutant 9798A core protein was associated with G3BP1 and USP10 (Fig. 8E). In addition, the knockdown of Caprin-1 weakened the interactions of core protein with G3BP1 or USP10 (Fig. 8F). These findings indicate that JEV core protein associates with several SG component molecules, such as G3BP1 and USP10, through the interaction with Caprin-1. Next, the role of the interaction between JEV core protein and Caprin-1 in the suppression of SG formation was examined by immunofluorescence analysis. Although the expression of the wild-type JEV core protein suppressed the SG formation induced by sodium arsenite treatment, as shown above, expression of the 9798A mutant did not (Fig. 8G), suggesting that the interaction of JEV core protein with Caprin-1 through Lys97 and Arg98 plays a crucial role in the inhibition of SG formation.
Fig 8.

Lys97 and Arg98 in the JEV core protein are crucial residues for the interaction with Caprin-1. (A) Putative structural model of the core protein homodimer of JEV deduced from that of DENV obtained from the Protein Data Bank (accession number 1R6R) by using PyMOL software. The two α helices (α1 and α4) are indicated. (B) FLAG-Core mutants in which the hydrophobic amino acid residues in the α1 helix (Mα1) or α4 helix (Mα4) were replaced with alanine were coexpressed with HA-Caprin-1 in 293T cells, immunoprecipitated (IP) with anti-HA antibody, and examined by immunoblotting (IB) with anti-HA or anti-FLAG antibody. (C) FLAG-Core mutants in which the Met78, Lys79, Lys85, Arg86, Ile92, and Asp93 (7893A) or Lys97 and Arg98 (9798A) in the α4 helix domain were replaced with alanine were coexpressed with HA-Caprin-1 in 293T cells and examined as described in panel B. (D) The His-tagged JEV core protein (WT or 9798A) was incubated with GST-fused Caprin-1 for 2 h at 4°C, and the precipitates obtained by GST pulldown assay were subjected to immunoblotting with anti-His antibody. (E) FLAG-Core (WT or 9798A) was coexpressed with HA-G3BP1 or HA-USP10 in 293T cells, immunoprecipitated with anti-HA antibody, and immunoblotted with anti-HA and anti-FLAG antibodies. (F) FLAG-JEV Core was coexpressed with HA-G3BP1 or HA-USP10 in 293T cells transfected with either siCaprin-1 or siNC at 72 h posttransfection, immunoprecipitated with anti-FLAG antibody, and immunoblotted with anti-HA and anti-FLAG antibodies. The cell lysates were also subjected to immunoblotting with anti-Caprin-1 and anti-β-actin antibodies to evaluate the knockdown efficiency of Caprin-1. (G) The cellular localizations of G3BP and FLAG-Core (WT or 9798A) were determined at 24 h posttransfection after treatment with 1.0 mM sodium arsenite for 30 min at 37°C by immunofluorescence analysis with mouse anti-G3BP MAb and rabbit anti-FLAG PAb, followed by AF488-conjugated anti-mouse IgG and AF594-conjugated anti-rabbit IgG, respectively. Cell nuclei were stained with DAPI (blue).
Interaction of the JEV core protein with Caprin-1 plays crucial roles not only in viral propagation in vitro but also in the pathogenesis in mice through the suppression of SG formation.
To further examine the biological significance of the interaction between the JEV core protein and Caprin-1 in viral replication, we generated a mutant infectious cDNA clone (pMWJEAT/9798AA) of JEV encoding a mutant core protein deficient in the binding to Caprin-1 based on pMWJEAT. First, the cellular localization of the core protein in the 9798A mutant JEV-infected cells was examined by immunofluorescence analysis. The 9798A mutant core protein, as well as the wild-type core protein, was localized in the nucleus and the perinuclear region (Fig. 9A). However, the 9798A mutant core protein was not colocalized with Caprin-1, in contrast to the wild-type core protein. The sizes of infectious foci in Vero cells infected with the 9798A mutant were significantly smaller than those infected with the wild-type JEV (Fig. 9B). Furthermore, the infectious titers in C6/36 and Vero cells infected with the 9798A mutant were 6.1- and 12.6-fold lower than those infected with wild-type JEV at 48 h postinfection, respectively (Fig. 9C), suggesting that interaction of the JEV core protein with Caprin-1 plays crucial roles in the propagation of JEV in both insect and mammalian cells. Cells infected with the 9798A mutant induced SGs containing both G3BP and Caprin-1, in contrast to the accumulation of G3BP in the perinuclear region observed in those infected with the wild-type JEV (Fig. 9D). The numbers of foci in cells infected with the 9798A mutant were higher than those in cells infected with the wild-type JEV (Fig. 9E), indicating that the interaction of the JEV core protein with Caprin-1 is crucial for the suppression of SG formation. Finally, we examined the biological relevance of the interaction of JEV core protein with Caprin-1 in viral replication in vivo. Infectious particles were recovered from the cerebrums of ICR mice inoculated with wild-type JEV but not from those inoculated with the 9798A mutant (Fig. 9F). In addition, all 10 mice had died by 12 days postinoculation with the wild-type JEV, while only 1 mouse had died at day 10 postinoculation with the 9798A mutant (Fig. 9G). Collectively, these results suggest that the interaction of JEV core protein with Caprin-1 plays crucial roles not only in viral replication in vitro but also in pathogenesis in mice through the suppression of SG formation.
Fig 9.

Interaction of JEV core protein with Caprin-1 plays crucial roles not only in viral replication in vitro but also in pathogenesis in mice through the suppression of SG formation. (A) Huh7/Caprin-1-AcGFP cells were infected with JEV (WT or 9798A mutant) at an MOI of 1.0, and the cellular localizations of Caprin-1-AcGFP and JEV core protein were determined at 24 h postinfection by immunofluorescence analysis with rabbit anti-core PAb and AF594-conjugated anti-rabbit IgG. Cell nuclei were stained with DAPI (blue). (B) Focus formation of JEV (WT or 9798A mutant) in Vero cells incubated in methylcellulose overlay medium at 48 h postinfection. The infectious foci were immunostained as described previously (20). (C) Growth kinetics of JEV (WT or 9798A mutant) in C6/36 and Vero cells infected at an MOI of 0.1. Infectious titers in the culture supernatants harvested at the indicated times were determined by focus-forming assays in Vero cells. Means of three experiments are indicated. (D) Huh7/Caprin-1-AcGFP cells were infected with either WT or 9798A at an MOI of 0.5, and cellular localizations of Caprin-1-AcGFP, G3BP (blue), and JEV NS2B (red) were determined at 24 h postinfection by immunofluorescence analysis with mouse anti-G3BP MAb and rabbit anti-NS2B PAb, followed by AF633-conjugated anti-mouse IgG and AF594-conjugated anti-rabbit IgG, respectively. (E) Numbers >of G3BP-positive foci in 30 cells prepared as described in panel D were counted. Lines, boxes, and error bars indicate the means, 25th to 75th percentiles, and 95th percentiles, respectively. The significance of differences between the means was determined by Student's t test. *, P < 0.01. (F) Infectious titers in the cerebrums of mice at 7 days postintraperitoneal inoculation with 5 × 104 FFU/100 μl of either WT or 9798A virus were determined in Vero cells. The means of titers in the homogenates of the cerebrums from three mice are indicated. The detection limit is 102 FFU/g of cerebrum. (G) Percentages of surviving mice (n = 10) after intraperitoneal inoculation with 5 × 104 FFU of either WT or 9798A virus. Mock, inoculation with DMEM.
DISCUSSION
Viruses are obligatory intracellular parasites, and their life cycles rely on host cellular functions. Many viruses have evolved to inhibit SG formation and thereby evade the host translation shutoff mechanism and facilitate viral replication (6, 30), while some viruses co-opt molecules regulating SG formation for viral replication (11, 31). The vaccinia virus subverts SG components to generate aggregates containing G3BP, Caprin-1, eIF4G, eIF4E, and mRNA of the virus, but not of the host, in order to stimulate viral translation (11). Replication, translation, and assembly of transmissible gastroenteritis coronavirus, a member of the Coronaviridae family, are regulated by the interaction of polypyrimidine tract-binding protein and TIA-1 with viral RNA (31). HIV-1 utilizes Staufen1, which is a principal component of SG, in the viral RNA selection to form ribonucleoproteins (RNPs) through interaction with Gag protein, instead of SG translation silencing (8). In the case of flaviviruses, TIA-1 and TIAR bind to the 3′ untranslated region (UTR) of the negative-stranded RNA of WNV to facilitate viral replication (16), and G3BP1, Caprin-1, and USP10 interact with DENV RNA, although the biological significance of these interactions remains unknown (26). In this study, we have shown that JEV infection suppresses SG formation by the recruitment of several effector molecules promoting SG assembly, including G3BP and USP10, to the perinuclear region through the interaction of JEV core protein with Caprin-1. Furthermore, a mutant JEV carrying a core protein incapable of binding to Caprin-1 exhibited reduced replication in vitro and attenuated pathogenicity in mice.
G3BP is one of the key molecules involved in the SG aggregation process and self-oligomerizes in a phosphorylation-dependent manner to sequester mRNA in SGs (4). Therefore, G3BP knocked down cells (6) and G3BP knockout mouse embryonic fibroblast cells are deficient in the SG formation. In addition, G3BP sequestration inhibits SG formation in response to arsenite treatment (32). Caprin-1, known as RNA granule protein 105 or p137 (33), also participates in SG formation through phosphorylation of eIF2α (28) and is ubiquitously expressed in the cytoplasm. Caprin-1 regulates the transport and translation of mRNAs of proteins involved in the synaptic plasticity in neurons (34) and cellular proliferation and migration in multiple cell types (28) through an interaction with G3BP. USP10, another SG-associated molecule, also interacts with G3BP and forms the G3BP/USP10 complex (29), suggesting that several SG-associated RBPs participate in the formation of a protein-protein network. In this study, the JEV core protein was shown to directly interact with Caprin-1, to sequester several key molecule complexes involved in SG formation to the perinuclear region in cells infected with JEV, and to facilitate viral propagation through the suppression of SG formation.
Flaviviruses replicate at a relatively low rate in comparison with most of the other positive-stranded RNA viruses, and thus rapid shutdown of host cellular protein synthesis would be deleterious for the viral life cycle. In cells infected with JEV, several SG components were colocalized with the core protein in the perinuclear region, while in those infected with WNV or DENV, SG components were accumulated in a replication complex composed of viral RNA and nonstructural proteins. In addition, the phosphorylation of eIF2α induced by arsenite was completely canceled by the infection with WNV or DENV, whereas the suppression of the phosphorylation was limited in JEV infection (15). Incorporation of the nascent viral RNA into the membranous structure induced by viral nonstructural proteins prevents PKR activation and inhibits SG formation in cells infected with WNV (17). In cells infected with hepatitis C virus (HCV), which belongs to the genus Hepacivirus in the family Flaviviridae, induction of SG formation was observed in the early stage of infection, in contrast to the inhibition of the arsenite-induced SG formation in the late stage (35). Several SG components, such as G3BP1, PABP1, and ataxin-2, were colocalized with HCV core protein around lipid droplets (35), and G3BP1 was also associated with the NS5B protein and the 5′ terminus of the minus-strand viral RNA (36) to mediate efficient viral replication. Collectively, these data suggest that flaviviruses have evolved to regulate cellular processes involved in SG formation through various strategies.
PKR is one of the interferon-stimulated genes and plays a crucial role in antiviral defense through phosphorylation of eIF2α, which leads to host translational shutoff (37, 38). In the early stage of flavivirus infection, both positive- and negative-stranded RNAs transcribe at low levels, while genomic RNA predominantly synthesizes in the late stage of infection (39). It was shown that activation of PKR was suppressed (40) or only induced in the late stage of WNV infection (41) and impaired by the expression of HCV NS5A (42–44). Very recently, JEV NS2A was shown to suppress PKR activation through inhibition of dimerization of PKR in the early stage but not in the late stage of infection (45). In this study, we have shown that JEV core protein interacts with Caprin-1 and inhibits SG formation downstream of the phosphorylation of eIF2α in the late stage of infection, suggesting that JEV has evolved to escape from host antiviral responses in the multiple stages of viral replication by using structural and nonstructural proteins.
The flavivirus core protein is a multifunctional protein involved in many aspects of the viral life cycle. In addition to the formation of viral nucleocapsid through the interaction with viral RNA (as a structural protein) (46), flavivirus core proteins interact with various host factors, such as B23 (47), Jab1 (48), hnRNP K (49), and hnRNP A2 (23), and regulate viral replication and/or modify the host cell environment (as a nonstructural protein). Although further investigations are needed to clarify the precise mechanisms underlying the circumvention of SG formation through the interaction of JEV core protein with Caprin-1, leading to efficient propagation in vitro and pathogenicity in mice, these findings could help not only to provide new insight into strategies by which viruses escape host stress responses but also to develop novel antiviral agents for flavivirus infection.
ACKNOWLEDGMENTS
We thank M. Tomiyama for secretarial assistance. We also thank K. Saito and T. Wakita for technical advice and the infectious clone of JEV, respectively.
This work was supported in part by grants-in-aid from the Ministry of Health, Labor, and Welfare, the Ministry of Education, Culture, Sports, Science, and Technology, and the Osaka University Global Center of Excellence Program. H. Katoh is a research fellow of the Japanese Society for the Promotion of Science.
Footnotes
Published ahead of print 24 October 2012
REFERENCES
- 1. Nover L, Scharf KD, Neumann D. 1989. Cytoplasmic heat shock granules are formed from precursor particles and are associated with a specific set of mRNAs. Mol. Cell. Biol. 9:1298–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anderson P, Kedersha N. 2002. Stressful initiations. J. Cell Sci. 115:3227–3234 [DOI] [PubMed] [Google Scholar]
- 3. Gilks N, Kedersha N, Ayodele M, Shen L, Stoecklin G, Dember LM, Anderson P. 2004. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 15:5383–5398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tourriere H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Bertrand E, Tazi J. 2003. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 160:823–831 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5. Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, Golan DE, Anderson P. 2000. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J. Cell Biol. 151:1257–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. White JP, Cardenas AM, Marissen WE, Lloyd RE. 2007. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2:295–305 [DOI] [PubMed] [Google Scholar]
- 7. Khaperskyy DA, Hatchette TF, McCormick C. 2012. Influenza A virus inhibits cytoplasmic stress granule formation. FASEB J. 26:1629–1639 [DOI] [PubMed] [Google Scholar]
- 8. Abrahamyan LG, Chatel-Chaix L, Ajamian L, Milev MP, Monette A, Clement JF, Song R, Lehmann M, DesGroseillers L, Laughrea M, Boccaccio G, Mouland AJ. 2010. Novel Staufen1 ribonucleoproteins prevent formation of stress granules but favour encapsidation of HIV-1 genomic RNA. J. Cell Sci. 123:369–383 [DOI] [PubMed] [Google Scholar]
- 9. McInerney GM, Kedersha NL, Kaufman RJ, Anderson P, Liljestrom P. 2005. Importance of eIF2α phosphorylation and stress granule assembly in alphavirus translation regulation. Mol. Biol. Cell 16:3753–3763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smith JA, Schmechel SC, Raghavan A, Abelson M, Reilly C, Katze MG, Kaufman RJ, Bohjanen PR, Schiff LA. 2006. Reovirus induces and benefits from an integrated cellular stress response. J. Virol. 80:2019–2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Katsafanas GC, Moss B. 2007. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe 2:221–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Misra UK, Kalita J. 2010. Overview: Japanese encephalitis. Prog. Neurobiol. 91:108–120 [DOI] [PubMed] [Google Scholar]
- 13. Sumiyoshi H, Mori C, Fuke I, Morita K, Kuhara S, Kondou J, Kikuchi Y, Nagamatu H, Igarashi A. 1987. Complete nucleotide sequence of the Japanese encephalitis virus genome RNA. Virology 161:497–510 [DOI] [PubMed] [Google Scholar]
- 14. Murray CL, Jones CT, Rice CM. 2008. Architects of assembly: roles of Flaviviridae non-structural proteins in virion morphogenesis. Nat. Rev. Microbiol. 6:699–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Emara MM, Brinton MA. 2007. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc. Natl. Acad. Sci. U. S. A. 104:9041–9046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li W, Li Y, Kedersha N, Anderson P, Emara M, Swiderek KM, Moreno GT, Brinton MA. 2002. Cell proteins TIA-1 and TIAR interact with the 3′ stem-loop of the West Nile virus complementary minus-strand RNA and facilitate virus replication. J. Virol. 76:11989–12000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Courtney SC, Scherbik SV, Stockman BM, Brinton MA. 2012. West Nile virus infections suppress early viral RNA synthesis and avoid inducing the cell stress granule response. J. Virol. 86:3647–3657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kambara H, Tani H, Mori Y, Abe T, Katoh H, Fukuhara T, Taguwa S, Moriishi K, Matsuura Y. 2011. Involvement of cyclophilin B in the replication of Japanese encephalitis virus. Virology 412:211–219 [DOI] [PubMed] [Google Scholar]
- 19. Mori Y, Yamashita T, Tanaka Y, Tsuda Y, Abe T, Moriishi K, Matsuura Y. 2007. Processing of capsid protein by cathepsin L plays a crucial role in replication of Japanese encephalitis virus in neural and macrophage cells. J. Virol. 81:8477–8487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mori Y, Okabayashi T, Yamashita T, Zhao Z, Wakita T, Yasui K, Hasebe F, Tadano M, Konishi E, Moriishi K, Matsuura Y. 2005. Nuclear localization of Japanese encephalitis virus core protein enhances viral replication. J. Virol. 79:3448–3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kambara H, Fukuhara T, Shiokawa M, Ono C, Ohara Y, Kamitani W, Matsuura Y. 2012. Establishment of a novel permissive cell line for the propagation of hepatitis C virus by expression of microRNA miR122. J. Virol. 86:1382–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhao Z, Date T, Li Y, Kato T, Miyamoto M, Yasui K, Wakita T. 2005. Characterization of the E-138 (Glu/Lys) mutation in Japanese encephalitis virus by using a stable, full-length, infectious cDNA clone. J. Gen. Virol. 86:2209–2220 [DOI] [PubMed] [Google Scholar]
- 23. Katoh H, Mori Y, Kambara H, Abe T, Fukuhara T, Morita E, Moriishi K, Kamitani W, Matsuura Y. 2011. Heterogeneous nuclear ribonucleoprotein A2 participates in the replication of Japanese encephalitis virus through an interaction with viral proteins and RNA. J. Virol. 85:10976–10988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hamamoto I, Nishimura Y, Okamoto T, Aizaki H, Liu M, Mori Y, Abe T, Suzuki T, Lai MM, Miyamura T, Moriishi K, Matsuura Y. 2005. Human VAP-B is involved in hepatitis C virus replication through interaction with NS5A and NS5B. J. Virol. 79:13473–13482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jones CT, Ma L, Burgner JW, Groesch TD, Post CB, Kuhn RJ. 2003. Flavivirus capsid is a dimeric alpha-helical protein. J. Virol. 77:7143–7149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ward AM, Bidet K, Yinglin A, Ler SG, Hogue K, Blackstock W, Gunaratne J, Garcia-Blanco MA. 2011. Quantitative mass spectrometry of DENV-2 RNA-interacting proteins reveals that the DEAD-box RNA helicase DDX6 binds the DB1 and DB2 3′ UTR structures. RNA Biol. 8:1173–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ma L, Jones CT, Groesch TD, Kuhn RJ, Post CB. 2004. Solution structure of dengue virus capsid protein reveals another fold. Proc. Natl. Acad. Sci. U. S. A. 101:3414–3419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Solomon S, Xu Y, Wang B, David MD, Schubert P, Kennedy D, Schrader JW. 2007. Distinct structural features of Caprin-1 mediate its interaction with G3BP-1 and its induction of phosphorylation of eukaryotic translation initiation factor 2α, entry to cytoplasmic stress granules, and selective interaction with a subset of mRNAs. Mol. Cell. Biol. 27:2324–2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Soncini C, Berdo I, Draetta G. 2001. Ras-GAP SH3 domain binding protein (G3BP) is a modulator of USP10, a novel human ubiquitin specific protease. Oncogene 20:3869–3879 [DOI] [PubMed] [Google Scholar]
- 30. Montero H, Rojas M, Arias CF, Lopez S. 2008. Rotavirus infection induces the phosphorylation of eIF2α but prevents the formation of stress granules. J. Virol. 82:1496–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sola I, Galan C, Mateos-Gomez PA, Palacio L, Zuniga S, Cruz JL, Almazan F, Enjuanes L. 2011. The polypyrimidine tract-binding protein affects coronavirus RNA accumulation levels and relocalizes viral RNAs to novel cytoplasmic domains different from replication-transcription sites. J. Virol. 85:5136–5149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hinton SD, Myers MP, Roggero VR, Allison LA, Tonks NK. 2010. The pseudophosphatase MK-STYX interacts with G3BP and decreases stress granule formation. Biochem. J. 427:349–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Grill B, Wilson GM, Zhang KX, Wang B, Doyonnas R, Quadroni M, Schrader JW. 2004. Activation/division of lymphocytes results in increased levels of cytoplasmic activation/proliferation-associated protein-1: prototype of a new family of proteins. J. Immunol. 172:2389–2400 [DOI] [PubMed] [Google Scholar]
- 34. Shiina N, Shinkura K, Tokunaga M. 2005. A novel RNA-binding protein in neuronal RNA granules: regulatory machinery for local translation. J. Neurosci. 25:4420–4434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ariumi Y, Kuroki M, Kushima Y, Osugi K, Hijikata M, Maki M, Ikeda M, Kato N. 2011. Hepatitis C virus hijacks P-body and stress granule components around lipid droplets. J. Virol. 85:6882–6892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yi Z, Pan T, Wu X, Song W, Wang S, Xu Y, Rice CM, Macdonald MR, Yuan Z. 2011. Hepatitis C virus co-opts Ras-GTPase-activating protein-binding protein 1 for its genome replication. J. Virol. 85:6996–7004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gale M, Jr, Katze MG. 1998. Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR, the interferon-induced protein kinase. Pharmacol. Ther. 78:29–46 [DOI] [PubMed] [Google Scholar]
- 38. Pindel A, Sadler A. 2011. The role of protein kinase R in the interferon response. J. Interferon Cytokine Res. 31:59–70 [DOI] [PubMed] [Google Scholar]
- 39. Chu PW, Westaway EG. 1985. Replication strategy of Kunjin virus: evidence for recycling role of replicative form RNA as template in semiconservative and asymmetric replication. Virology 140:68–79 [DOI] [PubMed] [Google Scholar]
- 40. Elbahesh H, Scherbik SV, Brinton MA. 2011. West Nile virus infection does not induce PKR activation in rodent cells. Virology 421:51–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Samuel MA, Whitby K, Keller BC, Marri A, Barchet W, Williams BR, Silverman RH, Gale M, Jr, Diamond MS. 2006. PKR and RNase L contribute to protection against lethal West Nile Virus infection by controlling early viral spread in the periphery and replication in neurons. J. Virol. 80:7009–7019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gale M, Jr, Blakely CM, Kwieciszewski B, Tan SL, Dossett M, Tang NM, Korth MJ, Polyak SJ, Gretch DR, Katze MG. 1998. Control of PKR protein kinase by hepatitis C virus nonstructural 5A protein: molecular mechanisms of kinase regulation. Mol. Cell. Biol. 18:5208–5218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gale MJ, Jr, Korth MJ, Tang NM, Tan SL, Hopkins DA, Dever TE, Polyak SJ, Gretch DR, Katze MG. 1997. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology 230:217–227 [DOI] [PubMed] [Google Scholar]
- 44. He Y, Tan SL, Tareen SU, Vijaysri S, Langland JO, Jacobs BL, Katze MG. 2001. Regulation of mRNA translation and cellular signaling by hepatitis C virus nonstructural protein NS5A. J. Virol. 75:5090–5098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tu YC, Yu CY, Liang JJ, Lin E, Liao CL, Lin YL. 2012. Blocking dsRNA-activated protein kinase PKR by Japanese encephalitis virus nonstructural protein 2A. J. Virol. 86:10347–10358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Khromykh AA, Westaway EG. 1996. RNA binding properties of core protein of the flavivirus Kunjin. Arch. Virol. 141:685–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tsuda Y, Mori Y, Abe T, Yamashita T, Okamoto T, Ichimura T, Moriishi K, Matsuura Y. 2006. Nucleolar protein B23 interacts with Japanese encephalitis virus core protein and participates in viral replication. Microbiol. Immunol. 50:225–234 [DOI] [PubMed] [Google Scholar]
- 48. Oh W, Yang MR, Lee EW, Park KM, Pyo S, Yang JS, Lee HW, Song J. 2006. Jab1 mediates cytoplasmic localization and degradation of West Nile virus capsid protein. J. Biol. Chem. 281:30166–30174 [DOI] [PubMed] [Google Scholar]
- 49. Chang CJ, Luh HW, Wang SH, Lin HJ, Lee SC, Hu ST. 2001. The heterogeneous nuclear ribonucleoprotein K (hnRNP K) interacts with dengue virus core protein. DNA Cell Biol. 20:569–577 [DOI] [PubMed] [Google Scholar]