

Abstract
The dopamine transporter (DAT) regulates the temporal and spatial actions of dopamine by reuptaking this neurotransmitter into the presynaptic neurons. We recently generated transgenic mice overexpressing DAT (DAT-tg) that have a 3-fold increase in DAT protein levels which results in a 40% reduction of the extracellular DA concentration in the striatum. The aim of this study was to examine the effect of this reduction in dopaminergic tone on postsynaptic responses mediated by dopamine receptors. We report here that DAT-tg mice have increased levels of striatal D1 (30%) and D2 (~60%) dopamine receptors with D1 receptor signaling components not significantly altered as evidenced by unaffected basal or stimulated levels of phospho-GluR1 (Ser845) and phospho-ERK2. However, the novel D2 mediated Akt signaling is markedly altered in DAT-tg animals. In particular, there is a 300% increase in the basal levels of phospho-Akt in the striatum of DAT-tg, reflecting the reduced extracellular dopamine tone in these animals. This increase in basal pAkt levels can be pharmacologically recapitulated by partial dopamine depletion in WT mice treated with the selective tyrosine hydroxylase inhibitor alpha-methyl-para-tyrosine (α-MPT). Behaviorally, DAT-tg animals demonstrate an augmented synergistic interaction between up-regulated D1 and D2 receptors which results in increased climbing behavior in transgenic mice after stimulation with either apomorphine or a co-administration of selective D1 and D2 receptor agonists. In sum, our study reveals that hypodopaminegia caused by up-regulation of DAT results in significant alterations at postsynaptic receptor function with most notable dysregulation at the level of D2 receptor signaling.
Keywords: Dopamine transporter (DAT), BAC Transgenic mice, hypodopaminergia, dopamine receptors, Akt, apomorphine
1. INTRODUCTION
The neurotransmitter dopamine (DA) has been shown to regulate a wide array of physiological functions including neuroendocrine secretion, control of movements, cognitive processes and motivated behaviors such as affect and reward mechanisms [1–3]. Dysfunctions in the DA systems have been proposed to be implicated in several neurological and psychiatric conditions including Parkinson’s disease (PD), attention deficit hyperactivity disorder (ADHD), drug addiction, depression and schizophrenia [4–6]. Major dopaminergic cell groups originating from substantia nigra and ventral tegmental area project their axons to the striatum and nucleus accumbens where the effects of secreted dopamine are mediated through its interaction with five distinct G protein-coupled seven transmembrane receptors (GPCRs). These receptors have been classically divided into two subfamilies: the D1-like family (D1 and D5 receptors) and the D2-like family (D2L, D2S D3 and D4 receptors) based on their ability to rapidly stimulate or inhibit adenylate cyclase respectively [7]. More recently D2 DA receptors have been shown to signal also through G protein-independent mechanisms involving Akt/GSK3 signaling cascade that is associated with slower and more sustained responses [8, 9].
A fundamental role in determining the intensity and duration of DA signalling at synapses is played by the plasma membrane DA transporter (DAT), a member of the Na+/Cl− dependent family of neurotransmitter transporters which, among others, includes the transporters for norepinephrine (NET) and serotonin (SERT) [10, 11]. DAT is specifically expressed in dopaminergic neurons [12, 13] and drives the re-uptake of DA back into the presynaptic terminals, a primary mechanism for the regulation of extracellular DA concentrations that efficiently controls the action of this neurotransmitter at pre- and postsynaptic receptors [14]. The functional importance of DAT in maintaining the homeostasis of the dopaminergic neurotransmission has been highlighted during the last decade through the study of mice with genetically altered DAT functions. Mice lacking the dopamine transporter (DAT-KO) display a 300-fold increase in the extracellular lifetime of DA and 5-fold elevation in steady-state extracellular dopamine levels in the striatum, hence representing a unique model to understand consequences of persistent hyperdopaminergia [15]. Some of the phenotypes of DAT-KO animals are also recapitulated in other mutants expressing 10% (DAT-KD mice) or 60% (DAT-siRNA treated mice) of DAT levels as compared to wild-type animals [16, 17].
Recently, mice carrying four additional copies of DAT gene (DAT-tg) were generated using a bacterial artificial chromosome (BAC) transgenesis technique. These animals display a 3-fold increase in the expression levels of DAT resulting in a more efficient uptake and a 40% reduction of extracellular striatal DA representing therefore a model of persistent hypodopaminergia [18]. While other authors have reported diminished basal locomotor activity in a different model of mice overexpressing DAT [19], the DAT-tg mice utilized in this study do not display any differences in basal locomotor behavior, hinting to a compensatory mechanism potentially involving DA receptors.
Here we evaluated whether the decreased extracellular levels of DA occurring in the striatum of DAT-tg mice can determine postsynaptic adaptations through the two major dopamine receptors (D1 and D2) and whether these two receptors or their associated signaling pathways might be regulated differently.
2. MATHERIAL AND METHODS
2.1. Animals
Experiments were carried out using age, gender and genetic background-matched WT, DAT-KO [20] and DAT-tg [18] 3–5 months old C57BL/6J mice. Animals, provided with food and water ad libitum, were housed 4 to 5 per cage in a humidity- and temperature-controlled room with 12-hour/12-hour light/dark cycle. All experiments were performed during the light cycle using every time experimentally naïve animals and were conducted in accordance with the National Institutes of Health (NIH) guidelines utilizing an approved animal protocol from the Duke University Animal Care and Use Committee.
2.2. Reagents
Radioactive compounds [3H]-SCH23390 (85 Ci/mmol), [3H]-Spiperone (16.5 Ci/mmol), [3H]-Raclopride (60.1 Ci/mmol), [35S]-GTPγS (1250 Ci/mmol) were obtained from PerkinElmer Life Sciences (Boston, MA). Ketanserin, flupenthixol, haloperidol, GDP, GTPγS, SKF 81297, quinpirole, apomorphine alpha-methyl paratyrosine (α-MPT) and protease inhibitor cocktail were purchased from Sigma-Aldrich (St. Louis, MO). Anti-phospho-GluR1-Ser-845 and anti-total-GluR1 antibodies were purchased from Chemicon International (Temecula, CA). Anti-phospho-ERK, anti-total-ERK, anti-phospho-Akt-Thr-308 and anti-total-Akt were obtained by Cell Signaling Technology (Beverly, MA). All protein extracts were quantified using a DC-protein assay from Bio-Rad (Hercules, CA).
2.3. D1 and D2 saturation binding assay on striatal membranes
Striatal tissues from wild-type, DAT-KO and DAT-tg mice were rapidly dissected and immediately homogenized, using a Teflonglass homogenizer, in 2 ml of lysis buffer containing 50 mM Tris-HCl (pH 7.4), 120 mM NaCl, 1 mM EDTA and a cocktail of protease inhibitors at 1:1,000 dilution. The homogenate was centrifuged at 1,000 rpm for 10 min at 4º C to remove tissue debris and nuclei and the resulting supernatant centrifuged twice at 40,000 g for 20 min at 4º C. The final pellet was suspended in assay buffer containing 50 mM Tris-HCl (pH 7.4), 120 mM NaCl, 5 mM KCl, 2mM CaCl2, 1 mM MgCl2. For D1 receptor saturation experiments, membranes (50 μl; 1.2 μg/μl final proteins concentration) were incubated with six increasing concentrations of [3H]-SCH23390 (50 μl; range: 0.5–16 nM) and 50 μl of ketanserin (100 nM), a specific antagonist of serotonin receptor, in order to prevent the binding of [3H]-SCH23390 to these receptors. For D2 receptors saturation experiments both [3H]-Spiperone and [3H]-Raclopride selective D2 antagonists were used. In the case of [3H]-Spiperone, membranes (150 μl; 0.5 μg/μl final proteins concentration) were incubated with six increasing doses of this radioactive ligand (50 μl; range: 0.03-1 nM), while for [3H]-Raclopride, membranes (50 μl; 1.2 μg/μl final proteins concentration) were incubated with six increasing doses of [3H]-Raclopride (100 μl; range: 1.8–60 nM). The different reactions were carried out at RT for 1 hr in the case of [3H]-SCH23390 and [3H]-Raclopride in a total volume of 200 μl of assay buffer and at RT for 2 hr in the case of [3H]-Spiperone in a total volume of 250 μl of assay buffer. Nonspecific binding was measured using non-radioactive flupenthixol (10 μM) for [3H]-SCH23390 binding or Haloperidol (6 μM for [3H]-Spiperone binding, 50 μM for [3H]-Raclopride binding) in parallel assay tubes and was subtracted from total binding to obtain respectively specific [3H]-SCH23390 or [3H]-Spiperone/[3H]-Raclopride binding. The incubations were terminated by rapid filtration over Brandel GF/C glass fiber filters washed with ice-cold assay buffer. The filters were incubated overnight in 5ml high flash point scintillation cocktail (Lefko-Fluor) before their radioactivity content was counted by a liquid scintillation counter.
2.4. Quinpirole-stimulated [35S]-GTPγS binding on striatal membranes
The coupling of D2/D3 dopamine receptors to Gi/Go proteins was assessed as previously described [21]. Briefly, striatal tissues of wild-type and DAT-tg mice were rapidly dissected and immediately homogenized, using a Polytron homogenizer, in 5 ml of lysis buffer containing 20 mM Hepes (pH 7.4), 100 mM NaCl, 2 mM MgCl2, 1 mM EDTA, 1 mM DTT and a cocktail of protease inhibitors at 1:1,000 dilution. The homogenate was centrifuged at 1,000 rpm for 10 min at 4º C to remove tissue debris and nuclei and the resulted supernatant centrifuged at 13,000 rpm for 20 min at 4º C. The pellet was suspended in assay buffer containing 20 mM Hepes (pH 7.4), 150 mM NaCl, 2 mM MgCl2, GDP 100 μM. Membrane fractions (20 μg) were incubated at 30ºC for 1 hour with [35S]-GTPγS (0.2 nM) and ten different concentrations of quinpirole in a range between 10−8 M and 10−3 M. Incubation mixture was filtered through Brandel GF/B glass filters, washed with 10 mM phosphate buffer and bound radioactivity determined by liquid scintillation spectrometry. Basal activity was determined in the absence of the agonist, while non-specific binding was estimated in the presence of 10 μM unlabeled GTPγS.
2.5. Western Blot analysis
Mice were sacrificed by cervical dislocation and their striatum rapidly dissected out (within 50 sec) on an ice-cold surface and frozen in liquid nitrogen before protein extraction. Preparation of the tissue samples were carried out as previously described [22]. Protein extracts (50 μg), prepared from the striatum of a single mouse, were separated by 10% SDS/PAGE and transferred to nitrocellulose membranes. The immunostaining of blots were performed overnight at 4°C with the following primary antibodies: anti-phospho-GluR1-Ser-845 (1:500), anti-total-GluR1 (1:1,000), anti-phospho-ERK (1:500), anti-total-ERK (1:1,000); anti-phospho-Akt-Thr-308 (1:100) and anti-Akt (1:2,000). Appropriate peroxidase-conjugated secondary antibodies (Jackson Immuno-Research, West Grove, PA) followed by exposure to a chemiluminescent reagent (SuperSignal West-Pico, Pierce, Rockford, IL) were utilized to reveal the immune complexes. Densitometric analysis was carried out within the linear range by using Imagequant V1.1 (Amersham Pharmacia Bioscience). Total protein signal was used as loading control for phospho-proteins and results were normalized to respective control conditions. Representative western blots show results obtained from two different mice for each group.
2.6. Behavioral responses to DA agonists
Horizontal and vertical activity of WT and DAT-tg mice were measured as previously described by Beaulieu et al. (2004). Briefly, mice were placed in an Omnitech Digiscan apparatus (AccuScan Instruments, Columbus, OH) and first habituated for 60 min. Subsequently, the animals were injected with different drugs (SKF 81297, quinpirole, apomorphine) or saline and immediately placed back into the apparatus. Their activity was then monitored continuously and tabulated at 5 min intervals for the following 60 min and cumulative counts were taken for data analysis. In all experiments, WT littermates served as control for mutant mice.
2.7. Statistical analyses
Data are reported as means ± SEM. N represents the number of animals used for each experiment. Saturation and GTP-γS binding curves where analyzed using GraphPad Prism software. Statistical significance was evaluated by two tailed t-test, one way or two ways ANOVA when appropriate.
3. RESULTS
3.1. DAT-tg mice show increased levels of both D1 and D2 postsynaptic receptors
Previous studies have shown that the mRNA levels of the two major DA receptors (D1 and D2) are down-regulated in DAT-KO mice [20, 23]. This observation suggested that receptor down-regulation serves as a mechanism to compensate for the characteristic hyperdopaminergia in this model. Given this, DAT-tg mice, which overexpress DAT and have a 40% decrease in the level of extracellular DA, would be expected to show an up-regulation for both D1 and D2 receptors compared to wild-type (WT) littermates. To address this point, we measured the levels of striatal D1 and D2 receptors, using a saturation binding approach, with selective radioactive antagonists. Specifically, [3H]-SCH23390 was utilized to detect the levels of D1 receptors, while [3H]-Spiperone and [3H]-Raclopride were used to quantify D2 receptors. The data obtained from these experiments show that DAT-tg mice have a 30% increase in the levels of D1 receptors when compared to wild-type littermates, while the DAT-KO mice have a converse 60% decrease in D1 receptor binding (Fig. 1A). In addition, D2 saturation binding experiments conducted utilizing [3H]-Spiperone as radioligand, revealed a 64% increase in the levels of D2 receptors in DAT-tg mice and a 40% decrease in DAT-KO mice, when compared to wild-type animals (Fig. 1B). These latter results were confirmed using the radioligand [3H]-Raclopride where in DAT-tg mice we found the levels of D2 to be increased by 52%, while in DAT-KO mice the levels were decreased by 40% (Fig. 1B). Importantly, the Kd values for all the radioligands tested were not significantly changed between genotypes, confirming that D1 and D2 receptors maintain the same affinity for the antagonists tested.
Figure 1. D1 and D2 dopamine receptor levels in striatal membranes from WT, DAT-KO and DAT-tg mice determined by saturation binding experiments.
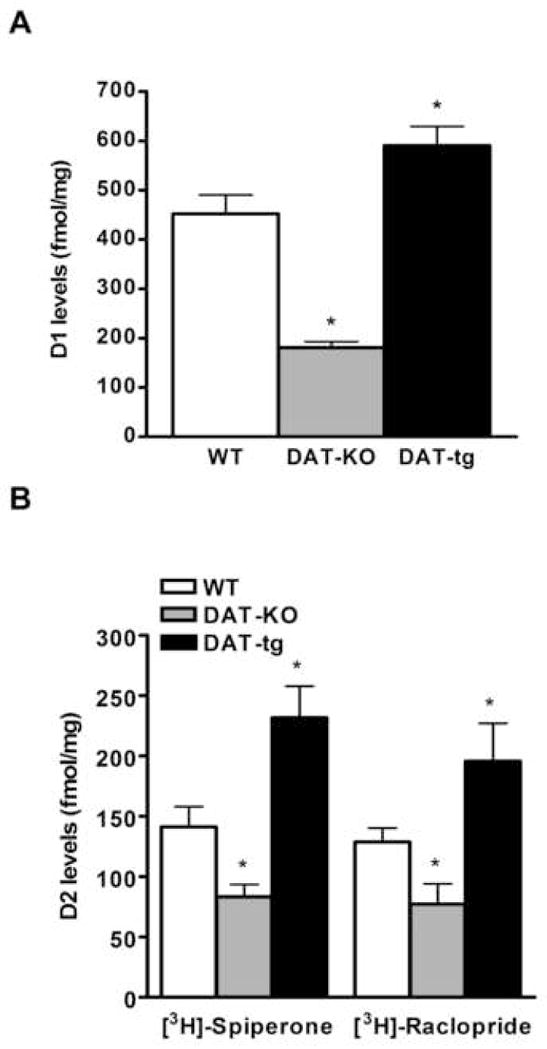
The radioligands [3H]-SCH23390 (A), [3H]-Spiperone and [3H]-Raclopride (B) were utilized in different saturation binding experiments to quantify D1 (A) and D2 (B) dopamine receptors. Levels expressed in fmol of DA receptors per mg of protein extracts utilized. Data were analyzed by GraphPad Prism software using one site binding (hyperbola) equation. Kd values (nM): [3H]-SCH23390 (WT = 1.6 ±0.3; DAT-KO = 1.2 ±0.4; DAT-tg = 1.4 ±0.6); [3H]-Spiperone (WT = 0.3 ±0.04; DAT-KO = 0.3 ±0.02; DAT-tg = 0.4 ±0.1); [3H]-Raclopride (WT = 13.3 ±1.6; DAT-KO = 13.4 ±1.2; DAT-tg = 15.5 ±1.10). N = 6–8 mice per group; data average are ±SEM. *, P ≤ 0.05.
3.2. D2-like postsynaptic receptors are functionally coupled to G proteins in DAT- tg mice
The results obtained from the saturation binding experiments show that the expression of D2 dopamine receptors is increased by ~60% in the striatum of DAT-tg mice compared to WT animals. Next we verified if these receptors were also functional and therefore able to couple to Gi/Go proteins. To address this point [35S]-GTPγS binding experiments on striatal membranes of wild-type and DAT-tg mice were performed. Analysis of the coupling of D2-like dopamine receptors to Gi/Go proteins under basal conditions revealed no differences between WT and DAT-tg mice, thus excluding the hypothesis of enhanced D2-like basal activation in transgenic animals (Fig. 2A). Instead, we observed higher levels of [35S]-GTPγS binding in the DAT-tg mice (60%) compared to their wild-type littermates (25%) after stimulation with different doses of the D2/D3 dopamine receptor agonist quinpirole (Fig. 2B). This observation indicates that the overexpression of D2 receptors in our model is followed by a concomitant increased coupling of the same receptors to Gi/Go proteins, indicating that their intrinsic activity is preserved. Moreover, the logEC50 for quinpirole did not significantly differ between genotypes (WT, 5.9 ± 0.26 M; DAT-tg, 5.5 ± 0.20 M) suggesting that these receptors are not supersensitive.
Figure 2. [35S]-GTPγS binding affinity of D2-like receptors to G proteins.
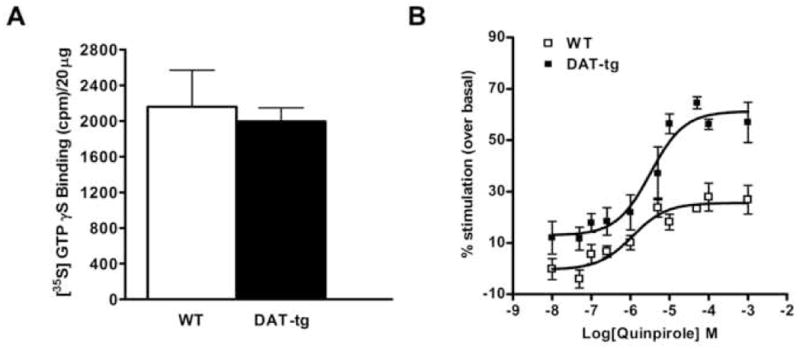
(A) Basal [35S]-GTPγS binding to D2/D3 receptors in striatal membranes from WT and DAT-tg mice as determined by quantity of radioactivity bound expressed in counts per minute (cpm) per 20ug (B) Percent stimulated [35S]-GTPγS binding after stimulation with increasing concentration of quinpirole was calculated by dividing the quantity of radioactivity at each concentration of quinpirole tested by the basal radioactivity in absence of quinpirole. The data were analyzed by GraphPad Prism software using a sigmoidal dose-response equation. The logEC50 parameters (WT, 5.9 ± 0.26 M; DAT-tg, 5.5 ± 0.20 M) did not significantly differ between genotypes. Each point represents the mean of N = 4 mice where every experiment was performed in triplicate.
3.3. D1 dopamine receptor signaling downstream targets pGluR1 (Ser845) and pERK2 are indistinguishable between WT and DAT-tg mice
Considering the increased expression of D1 dopamine receptors observed in the striatum of DAT-tg, we examined whether this augmentation could alter the phosphorylation state of specific D1 downstream targets, thus affecting their signal transduction mechanisms. It has previously been shown that stimulation of D1 receptors in vivo leads to increases in phospho-GluR1 (Ser 845) [24] and phospho-ERK2 levels [25-28]. The phosphorylation state of these two proteins was used to monitor D1 receptor activation in both WT and DAT-tg mice under basal and stimulated conditions. After the animals were injected with saline or two different doses of the selective D1 agonist SKF 81297 (2 and 3 mg/kg, i.p) their striatum was rapidly dissected and utilized to perform western blot analysis. The basal levels of both phopsho-GluR1 and phopho-ERK2 in transgenic mice were found to be similar to WT controls (Fig. 3A and B). Treatments with SKF 81297 (2 mg/kg) for 15 minutes significantly increased the levels of phospho-GluR1 (Ser 845) to 180% in WT and 175% in DAT-tg mice, as compared to the saline’s levels for the same genotype (Fig. 3C and D). Under the same conditions the levels of phopsho-ERK2 were not significantly changed in either of the two genotypes (Fig. 3E and F). When we increased the dose of SKF 81297 to 3 mg/kg a significant augmentation in the phosphorylation levels of both GluR1 (WT 190%, DAT-tg 260%) and ERK2 (WT 218%, DAT-tg 224%) was observed, indicating a dose depend stimulation of these D1 receptor downstream targets (Fig. 3C- E ). Although at the dose of 3mg/kg treatment with SKF 81297 highlighted a tendency for phospho-GluR1 to be increased in DAT-tg mice, the extent of the phosphorylation was not significantly different compared to the one observed in WT animals, revealing that, under these conditions, D1 receptor signaling for these two pathways is indistinguishable among the two genotypes.
Figure 3.
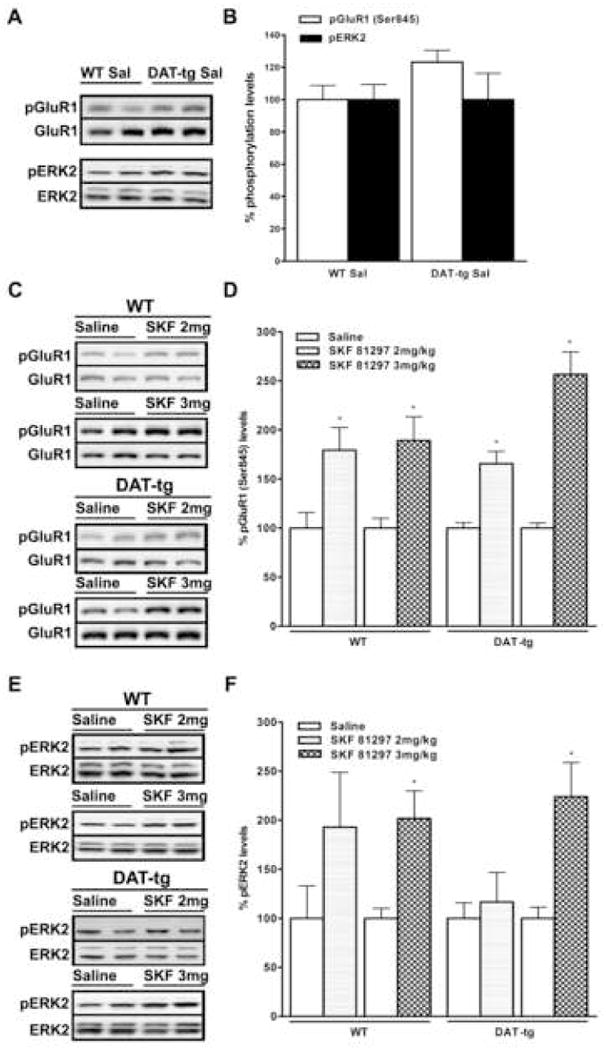
Phospho-GluR1 (Ser845) and phospho-ERK2 levels in the striatum of wild- type and DAT-tg mice under basal or stimulated (SKF 81297) conditions. (A and B) Basal levels of phospho-GluR1 (Ser845) and phospho-ERK2 in WT and DAT-tg mice injected with saline. (C and D) Effects of SKF 81297 (2 and 3mg/kg) on phospho-GluR1 (Ser845) levels in WT and DAT-tg mice. (E and F) Levels of phospho-ERK2 in WT and DAT-tg injected with SKF 81297 (2 and 3mg/kg). Representative western blots (A, C, E) show results obtained from two separate striatal extracts prepared from different mice. Analyses were conducted at 15 min after injection. Results of densitometric analysis (B, D, F) are presented as percentage normalized to saline-treated mice of the same genotype. N = 5–8 mice per group; data average are ±SEM. *, P≤0.03.
3.4. Decreased striatal extracellular concentration of DA leads to an increase in phospho-Akt (Thr308) levels
Previous work conducted in our laboratory has demonstrated that treatment with the non selective dopamine receptor agonist apomorphine leads to a decrease in the phosphorylation of Akt (Thr308). This effect has been shown to be mediated by D2-like receptors and to be involved in the expression of certain DA-related behaviors [22, 29]. Taking into account the marked increase in the levels of D2 dopamine receptors observed in DAT-tg, we investigated the phospho-Akt levels under basal and stimulated conditions. Basal levels of phospho-Akt (Thr308) were found to be markedly increased (300%) in DAT-tg mice as compared to their wild-type littermates (Fig. 4A and B). This was an unexpected result, since a decrease in the basal phosphorylation state of Akt, with increased functional D2 receptors, would be anticipated. For this reason we evaluated whether the condition of hypodopaminergia characterizing our model might play a role itself in affecting the phosphorylation state of Akt. Wild-type mice were treated for 3 hours with α-MPT (250 mg/kg), an inhibitor of tyrosine hydroxylase and therefore DA synthesis. It has been shown that this treatment reduces the striatal extracellular concentration of dopamine by 50% [30], thus reproducing the state of hypodopaminergia characterizing our DAT-tg mice. After treatment with α-MPT the levels of phospho-Akt in the striatum of these animals were measured by western blot and compared to the ones obtained from wild-type mice injected with saline. This treatment resulted in a significant increase in the levels of phospho-Akt (Thr308) (~220%) similar to the one observed in DAT-tg (Fig. 4A and B). Moreover, D2 receptor levels in WT animals treated with α-MPT were found not to be significantly changed compared to WT saline controls (data not shown). This suggests that diminished extracellular DA levels, rather than augmented D2 receptors, may be responsible for the changes in the basal phosphorylation state of Akt observed in DAT-tg animals.
Figure 4. Phospho-Akt (Thr308) levels are affected by diminished extracellular DA in the striatum.
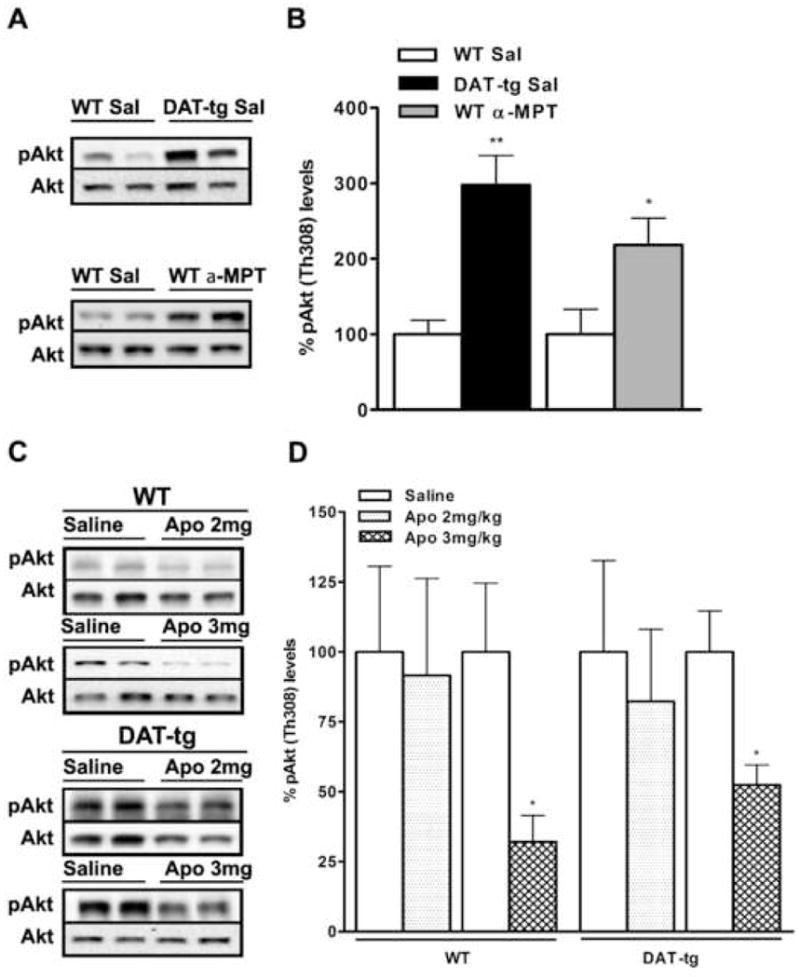
(A and B) Levels of phospho-Akt (Thr308) in WT and DAT-tg mice injected with saline as compared to WT animals treated with α-MTP (250 mg/kg). (C and D) Effects of apomorphine (2 and 3 mg/kg) on phospho-Akt (Thr308) levels in WT and DAT-tg mice. Representative western blots (A and C) show results obtained from two separate striatal extracts prepared from different mice. Analyses were conducted at 60 min (saline and apomorphine) or 180 min (saline and α-MTP) after injection. Results of densitometric analysis (B and D) are presented as percentage normalized to saline-treated mice of the same genotype. N = 5–8 mice per group; data average are ±SEM. *, P≤0.03, **, P≤0.001.
Finally, treatments with apomorphine (3 mg/kg but not 2 mg/kg) for 60 minutes led to a decrease in the phosphorylation state of Akt in both WT (65%) and DAT-tg animals (50%) as compared to saline treatments for the same genotypes (Fig. 4C and D). Because of differences in the basal levels of phospho-Akt between WT and DAT-tg mice, the absolute decrease in phosphorylation of Akt in DAT-tg mice is considerably greater than in WT striatum, suggesting a conserved effectiveness of D2R signaling under these experimental conditions.
3.5. Stimulation with apomorphine or combined D1- and D2-like dopamine receptor agonists results in enhanced climbing behavior in DAT-tg mice
The striatum is involved in mediating locomotion, as well as sniffing, licking, biting and other stereotypic behaviors, known to be induced by dopaminergic drugs via stimulation of both D1- and D2-like receptor-containing neurons [31, 32]. In particular, the characteristic climbing response to high doses of the non-selective DA agonist apomorphine is a well-established approach to evaluate responsiveness of postsynaptic DA receptors in vivo [33]. In order to asses the impact of increased D1 and D2 receptors found in the striatum of transgenic mice and delineate their contribution to climbing behavior, groups of WT and DAT-tg were injected with saline, apomorphine (3 mg/kg, s.c) or a combination of D1 and D2 dopamine receptor selective agonists (SKF 81297, 3 mg/kg, i.p. plus quinpirole, 2 mg/kg, i.p.) [34, 35]. After 60 min habituation period, vertical activity counts were measured for the following 60 min post drug injection and found increased in DAT-tg mice as compared to WT treated with either apomorphine (67%) or SKF 81297 plus quinpirole (62%), thus reflecting augmented climbing behavior in mutant mice (Fig. 5). Moreover, none of the doses of SKF 81297 or quinpirole (both at 0.5, 2.5 and 5 mg/kg, i.p) tested alone produced significantly different locomotor-stimulating effects between genotypes (data not shown). Taken together our data suggest that increased climbing behavior in DAT-tg treated with mixed D1 and D2 agonists requires the concomitant activation of D1 and D2 receptors and may result from augmented D1 and D2 receptors number observed in transgenic animals as compared to WT controls.
Figure 5.
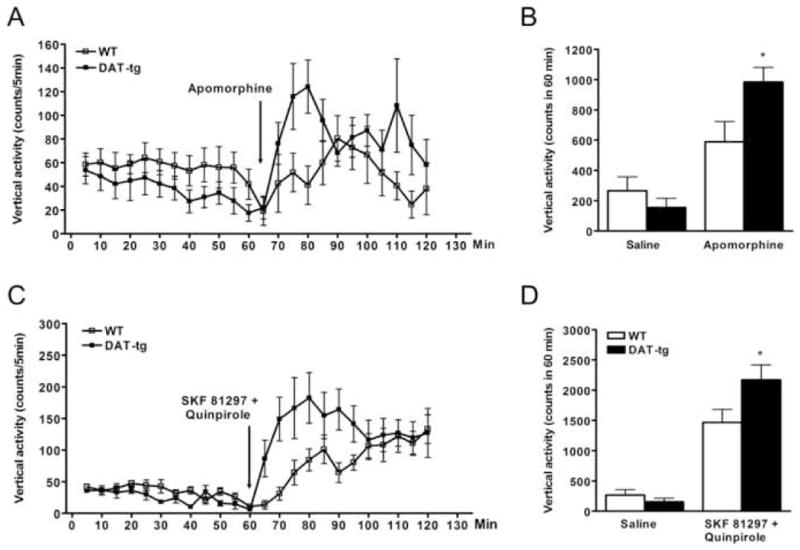
Vertical activity counts in response to injection of apomorphine or a combination of SKF 81297 and quinpirole are enhanced in DAT-tg mice. Time course on the effect of combined injection of SKF 81297 (3 mg/kg, i.p.) and quinpirole (2 mg/kg, i.p.) (A) or apomorphine (3 mg/kg, s.c.) (C) on the vertical activity of wild-type and DAT-tg mice. Quantification of vertical activity in WT and DAT-tg mice measured for 60 min after injection of combined SKF 81297 and quinpirole (B) or apomorphine (D). N = 6–8 mice for each experimental condition. Data average are ±SEM. *, P < 0.05.
4. DISCUSSION
Dopamine exerts a modulatory control on basal ganglia neurons at the pre- and postsynaptic level by acting on D2-like autoreceptors and on D1/D2 class of receptors respectively. Among other factors, the concentration and the lifetime of this neurotransmitter in the extracellular space have been shown to regulate the expression of dopamine related genes in different brain areas [36]. In particular, a significant decrease of both D1 and D2 mRNA levels was observed in the basal ganglia [20], caudate putamen and nucleus accumbens [23] of DAT-KO mice. Our radioligand binding results, demonstrating decreased levels of D1 (60%) and D2 (40%) receptors in striatal membrane of DAT-KO mice, provide further support for these observations. This down regulation could be a mechanism to compensate for the characteristic hyperdopaminergia in this model and re-equilibrate the homeostasis of this system. Our observations with D1 and D2 receptor levels in DAT-tg mice suggests that this adaptive process may function in both directions to adjust postsynaptic dopamine neurotransmission when extracellular levels are not properly regulated. Indeed, when compared to WT, DAT-tg mice show an increase in the striatal levels of D1 (30%) and D2 (~60%) receptors. In addition, the higher levels of quinpirole stimulated [35S]-GTPγS binding found in DAT-tg mice, when compared to their WT littermates, appear to be in line with the overexpression of D2 dopamine receptors in our model. In fact, the lack of significant differences in the logEC50 for quinpirole and in the basal [35S]-GTPγS binding between genotypes precludes the possibility that D2-like receptors in transgenic mice may be supersensitive or presenting an enhanced D2-like basal activation compared to WT controls. Thus, the increased coupling to Gi/Go proteins observed upon stimulation with quinpirole correlates with the overexpression of D2 dopamine receptors in DAT-tg animals, suggesting that their intrinsic activity and functionality is preserved.
The ability of D1- and D2- class receptors to transmit intracellular signals was then evaluated in more details by examining the phosphorylation state of different downstream target proteins known to be selectively activated by the two different classes of receptors. Previous in vivo studies have demonstrated that stimulation of D1 receptors increases the levels of phospho-GluR1 and phospho-ERK2. This phenomenon is believed to be c-AMP dependent, mediated by D1 receptors and has been shown to peak within the first 30 minutes after drug injection [24-28]. On the other hand, apomorphine has been recently shown to induce a decrease in Akt phosphorylation. This novel G protein-independent pathway develops progressively during the first 30-60 minutes after drug administration and has been shown to be mediated by D2-like receptors [8, 29]. Our results reveal only a marginal effect on the DAT-tg genotype on basal phosphorylation of GluR1 and ERK2 as compared to their wild-type littermates, suggesting that these protein targets are not affected by decreased extracellular DA or increased D1 receptor levels characterizing our model. In striking contrast, the basal phosphorylation state of Akt is markedly increased (300%) in DAT-tg mice as compared to their WT littermates. This observation is quite interesting since, a decrease in the basal phosphorylation state of Akt, due to the increased number of functional D2 receptors, would have been expected. Our data support the hypothesis that the phosphorylation state of Akt is more sensitive to DA tone rather than D2 receptors number. Indeed, in wild-type mice treated with α-MPT, which display reduced extracellular concentration of dopamine, there is an increase in the levels of phospho-Akt comparable to that observed in transgenic littermates. The fact that treatment with α-MPT does not influence D2 receptors number further support the idea that the condition of hypodopaminergia characterizing our model is responsible for the increase in basal Akt phophorylation, while augmented D2 receptors levels may be an adaptive mechanism occurring to maintain homeostasis of the system. This interpretation is further corroborated by additional results showing reduced basal phospho-Akt levels in the hyperdopaminergic DAT-KO mice [22] or enhanced phospho-Akt levels in wild-type mice where D2-like receptors were blocked pharmacologically [37]. In addition, our data reveal a tendency for D1- and D2-mediated signaling pathways to be enhanced upon stimulation with appropriate agonists. In particular, stimulation of D1 receptors with the selective agonist SKF 81297 leads to intracellular transmission of the signal that appear to be more pronounced in transgenic mice, although not significantly, thus suggesting that the 30% increase of D1 receptors observed in the striatum of DAT-tg mice may be not sufficient to induce a profound alteration in D1-mediated signaling. Moreover, the results obtained upon stimulation with apomorphine reveal a similar extent of D2-mediated Akt dephosphorylation in WT (65%) and DAT-tg (50%) mice. However, considering that the basal levels of phospho-Akt observed in DAT-tg mice are 3 times higher than WT controls, it appears that the treatment with apomorphine results in much greater absolute changes in the phospho-Akt signals in transgenic mice, since these levels are comparable to the ones observed in non-treated WT animals.
Behaviorally, our data support the idea that simultaneous stimulation of overexpressed D1 and D2 receptors in DAT-tg mice are necessary to produce increased climbing behavior compared to WT littermates. It is in fact already well appreciated that increases in DA neurotransmission and consequent augmentation of motor stimulant effects reach its maximum when both D1- and D2-class of receptors are simultaneously stimulated. This phenomenon, known as synergism, was first observed when many effects generated by D2 receptor stimulation were found to occur only after D1 receptor stimulation was enhanced by administration of selective D1 receptor agonists [31, 32]. Indeed, the difference in climbing behavior between wild-type and transgenic mice appear evident only when the animals are treated with non selective DA agonist apomorphine or combination of selective D1 (SKF 81297) and D2 (quinpirole) agonists. Altogether our results indicate that the overexpression of DAT plays a critical role in mediating postsynaptic adaptations in the striatum. DAT-related reduction of extracellular dopamine levels appears to be selectively affecting the D2 receptor-mediated signaling pathway as evidenced by an increase in the basal phosphorylation state of Akt. At the same time, this particular hypodopaminergia causes an augmented D1 and D2 receptors number and synergistic interaction as evidenced by enhanced behavioral responses to non-selective DA agonists. Considering the different consequences of the hypodopaminergic state of DAT-tg on D2 receptor signaling versus D2 receptor levels, our results clearly demonstrate that simply measuring receptor levels is insufficient to allow any definitive conclusions with regards to the actual signaling of a studied receptor. This observation is particularly interesting when considering that in postmortem studies only receptor levels can be reliably measured and no data can be ascertained with regards to the receptor signaling.
Acknowledgments
We thank Xiu-Qin Zhang and Katherine Clark for assistance with genotyping of mice. We thank Drs. Erin Whalen, Graciela Pineyro and Laura Bohn for protocols for GTP-γS experiments. This work was supported in part by National Institutes of Health, National Institute on Drug Abuse Grants NS-19576 to M.G.C. B.M holds a European Marie-Curie Outgoing International Fellowship (FP6-2005-Mobility-6). A.J.R. holds a National Institute on Drug Abuse K01 award (DA017703). A.S. was supported by a fellowship from the Canadian Institutes of Health Research (CIHR).
Abbreviations
- ANOVA
analysis of variance
- ADHD
attention deficit hyperactivity disorder
- BAC
bacterial artificial chromosome
- cAMP
cyclic adenonine monophosphate,
- DA
dopamine
- DAT
dopamine transporter
- DAT-KD
dopamine transporter knock-down
- DAT-KO
dopamine transporter knock-out
- DAT-tg
dopamine transporter transgenic
- ERK
extracellular signal-regulated kinase
- GPCRs
G protein-coupled receptors
- EC50
median effective concentration,
- mRNA
messenger ribonucleic acid,
- NET
norepinephrine transporter,
- PD
Parkinson’s disease
- PKB or Akt
proteine kinase B
- PP2A
protein phosphatase 2A,
- SERT
serotonin transporter
- siRNA
small interfering ribonucleic acid
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carlsson A. Science. 2001;294:1021–1024. doi: 10.1126/science.1066969. [DOI] [PubMed] [Google Scholar]
- 2.Greengard P. Science. 2001;294:1024–1030. doi: 10.1126/science.294.5544.1024. [DOI] [PubMed] [Google Scholar]
- 3.Gainetdinov RR, Sotnikova TD, Caron MG. Trends Pharmacol Sci. 2002;23:367–373. doi: 10.1016/s0165-6147(02)02044-8. [DOI] [PubMed] [Google Scholar]
- 4.Mohn AR, Yao WD, Caron MG. Neuropharmacology. 2004;47 (Suppl 1):101–110. doi: 10.1016/j.neuropharm.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 5.Bozzi Y, Borrelli E. Trends Neurosci. 2006;29:167–174. doi: 10.1016/j.tins.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 6.Iversen SD, Iversen LL. Trends Neurosci. 2007;30:188–193. doi: 10.1016/j.tins.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Physiol Rev. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- 8.Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 9.Bibb JA. Cell. 2005;122 (2):153–5. doi: 10.1016/j.cell.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 10.Masson J, Sagne C, Hamon M, El Mestikawy S. Pharmacol Rev. 1999;51:439–464. [PubMed] [Google Scholar]
- 11.Torres GE, Gainetdinov RR, Caron MG. Nat Rev Neurosci. 2003;4:13–25. doi: 10.1038/nrn1008. [DOI] [PubMed] [Google Scholar]
- 12.Ciliax BJ, Heilman C, Demchyshyn LL, Pristupa ZB, Ince E, Hersch SM, Niznik HB, Levey AI. J Neurosci. 1995;15:1714–1723. doi: 10.1523/JNEUROSCI.15-03-01714.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffman BJ, Hansson SR, Mezey E, Palkovits M. Front Neuroendocrinol. 1998;19:187–231. doi: 10.1006/frne.1998.0168. [DOI] [PubMed] [Google Scholar]
- 14.Sotnikova TD, Beaulieu JM, Gainetdinov RR, Caron MG. CNS Neurol Disord Drug Targets. 2006;5:45–56. doi: 10.2174/187152706784111579. [DOI] [PubMed] [Google Scholar]
- 15.Gainetdinov RR, Caron MG. Annu Rev Pharmacol Toxicol. 2003;43:261–284. doi: 10.1146/annurev.pharmtox.43.050802.112309. [DOI] [PubMed] [Google Scholar]
- 16.Zhuang X, Oosting RS, Jones SR, Gainetdinov RR, Miller GW, Caron MG, Hen R. Proc Natl Acad Sci U S A. 2001;98:1982–1987. doi: 10.1073/pnas.98.4.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salahpour A, Medvedev IO, Beaulieu JM, Gainetdinov RR, Caron MG. Biol Psychiatry. 2007;61:65–69. doi: 10.1016/j.biopsych.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 18.Salahpour A, Ramsey AJ, Medvedev IO, Kile B, Sotnikova TD, Holmstrand E, Ghisi V, Nicholls PJ, Wong L, Murphy K, Sesack SR, Wightman RM, Gainetdinov RR, Caron MG. Proc Natl Acad Sci U S A. 2008;105:4405–4410. doi: 10.1073/pnas.0707646105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donovan DM, Miner LL, Perry MP, Revay RS, Sharpe LG, Przedborski S, Kostic V, Philpot RM, Kirstein CL, Rothman RB, Schindler CW, Uhl GR. Brain Res Mol Brain Res. 1999;73:37–49. doi: 10.1016/s0169-328x(99)00235-1. [DOI] [PubMed] [Google Scholar]
- 20.Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- 21.Gainetdinov RR, Bohn LM, Walker JK, Laporte SA, Macrae AD, Caron MG, Lefkowitz RJ, Premont RT. Neuron. 1999;24:1029–1036. doi: 10.1016/s0896-6273(00)81048-x. [DOI] [PubMed] [Google Scholar]
- 22.Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG. Proc Natl Acad Sci U S A. 2004;101:5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fauchey V, Jaber M, Caron MG, Bloch B, Le Moine C. Eur J Neurosci. 2000;12:19–26. doi: 10.1046/j.1460-9568.2000.00876.x. [DOI] [PubMed] [Google Scholar]
- 24.Snyder GL, Allen PB, Fienberg AA, Valle CG, Huganir RL, Nairn AC, Greengard P. J Neurosci. 2000;20:4480–4488. doi: 10.1523/JNEUROSCI.20-12-04480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valjent E, Corvol JC, Pages C, Besson MJ, Maldonado R, Caboche J. J Neurosci. 2000;20:8701–8709. doi: 10.1523/JNEUROSCI.20-23-08701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol JC, Stipanovich A, Caboche J, Lombroso PJ, Nairn AC, Greengard P, Hervé D, Girault JA. Proc Natl Acad Sci U S A. 2005;102(2):491–6. doi: 10.1073/pnas.0408305102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Svenningsson P, Tzavara ET, Carruthers R, Rachleff I, Wattler S, Nehls M, McKinzie DL, Fienberg AA, Nomikos GG, Greengard P. Science. 2003;302:1412–1415. doi: 10.1126/science.1089681. [DOI] [PubMed] [Google Scholar]
- 28.Beaulieu JM, Sotnikova TD, Gainetdinov RR, Caron MG. J Biol Chem. 2006;281:32072–32080. doi: 10.1074/jbc.M606062200. [DOI] [PubMed] [Google Scholar]
- 29.Beaulieu JM, Tirotta E, Sotnikova TD, Masri B, Salahpour A, Gainetdinov RR, Borrelli E, Caron MG. J Neurosci. 2007;27:881–885. doi: 10.1523/JNEUROSCI.5074-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sotnikova TD, Beaulieu JM, Barak LS, Wetsel WC, Caron MG, Gainetdinov RR. PLoS Biol. 2005;3:e271. doi: 10.1371/journal.pbio.0030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walters JR, Bergstrom DA, Carlson JH, Chase TN, Braun AR. Science. 1987;236:719–722. doi: 10.1126/science.2953072. [DOI] [PubMed] [Google Scholar]
- 32.White FJ, Bednarz LM, Wachtel SR, Hjorth S, Brooderson RJ. Pharmacol Biochem Behav. 1988;30:189–193. doi: 10.1016/0091-3057(88)90442-x. [DOI] [PubMed] [Google Scholar]
- 33.Wilcox RE, Smith RV, Anderson JA, Riffee WH. Pharmacol Biochem Behav. 1980;12:29–33. doi: 10.1016/0091-3057(80)90411-6. [DOI] [PubMed] [Google Scholar]
- 34.Xu M, Koeltzow TE, Santiago GT, Moratalla R, Cooper DC, Hu XT, White NM, Graybiel AM, White FJ, Tonegawa S. Neuron. 1997;19:837–848. doi: 10.1016/s0896-6273(00)80965-4. [DOI] [PubMed] [Google Scholar]
- 35.Gainetdinov RR, Bohn LM, Sotnikova TD, Cyr M, Laakso A, Macrae AD, Torres GE, Kim KM, Lefkowitz RJ, Caron MG, Premont RT. Neuron. 2003;38:291–303. doi: 10.1016/s0896-6273(03)00192-2. [DOI] [PubMed] [Google Scholar]
- 36.Jaber M, Robinson SW, Missale C, Caron MG. Neuropharmacology. 1996;35:1503–1519. doi: 10.1016/s0028-3908(96)00100-1. [DOI] [PubMed] [Google Scholar]
- 37.Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA. Nat Genet. 2004;36:131–137. doi: 10.1038/ng1296. [DOI] [PubMed] [Google Scholar]