

Abstract
Hypothermia is a powerful neuroprotective method when induced following cardiac arrest, stroke, and traumatic brain injury. The physiological effects of hypothermia are multifaceted and therefore a better knowledge of its therapeutic targets will be central to developing innovative combination therapies to augment the protective benefits of hypothermia. Altered neuronal calcium dynamics have been implicated following stroke, status epilepticus and traumatic brain injury. This study was therefore initiated to evaluate the effect of hypothermia on various modes of calcium entry into a neuron. Here, we utilized various pharmacological agents to stimulate major routes of calcium entry in primary cultured hippocampal neurons. Fluorescent calcium indicator Fura-2AM was used to compare calcium ratio under normothermic (37°C) and hypothermic (31°C) conditions. The results of this study indicate that hypothermia preferentially reduces calcium entry through N-methyl-D-aspartate receptors and ryanodine receptors. Hypothermia, on the other hand, did not have a significant effect on calcium entry through the voltage-dependent calcium channels or the inositol tri-phosphate receptors. The ability of hypothermia to selectively affect both N-methyl-D-aspartate receptors and ryanodine receptors-mediated calcium systems makes it an attractive intervention for alleviating calcium elevations that are present following many neurological injuries.
Keywords: Calcium, hypothermia, ryanodine receptor, NMDA receptor
1. Introduction
The neuroprotective benefits of hypothermia have long been recognized, but we are only now beginning to understand how hypothermia exerts its effects. It was initially believed that deep hypothermia was needed to achieve neuroprotection. However, deep hypothermia was associated with arrhythmias and other serious side effects and was therefore abandoned clinically (Polderman, 2008; 2009). Interest in hypothermia was rekindled in the 1980’s when animal studies showed that mild to moderate levels of hypothermia were neuroprotective without the detrimental side effects associated with deep hypothermia (Busto et al., 1987). Moderate hypothermia is currently used in a variety of clinical settings such as cardiac arrest (Zeiner et al., 2000), stroke (Yenari and Hemmen, 2010), neonatal hypoxia-ischemia encephalopathy (Jacobs et al., 2011), traumatic brain injury (Foundation, 2007), acute spinal cord injury (Bernard and Buist, 2003) and refractory status epilepticus (Shorvon, 2011).
The mechanism for hypothermia’s action was initially believed to be its ability to decrease cerebral blood flow and oxygen consumption (Rosomoff and Holaday, 1954). However, recent studies have demonstrated other mechanistic aspects of this intervention including decreased inflammation (Aibiki et al., 1999; Dietrich et al., 2004; Kimura et al., 2002; Suehiro et al., 2004), prevention of apoptosis (Adachi et al., 2001; Liou et al., 2003; Raghupathi et al., 2000; Xu et al., 2002) and mitochondrial dysfunction (Ning et al., 2002), and reduction in extracellular levels of excitatory neurotransmitters (Illievich et al., 1994; Okuda et al., 1986; Ooboshi et al., 2000). A better understanding of the molecular mechanisms underlying hypothermia is essential for developing treatments to augment neuroprotection following hypothermia. This study was initiated to study the effects of hypothermia on Ca2+ homeostasis as a mechanism of action of hypothermia-induced neuroprotection.
Calcium (Ca2+) is a ubiquitous second messenger involved in major signaling pathways. Ca2+ homeostasis is a tightly regulated system using a variety of channels and sequestration mechanisms. Brief and controlled elevations of intracellular Ca2+ levels are necessary for normal physiological processes such as neurotransmitter release and long-term potentiation. However, prolonged elevations, as seen in glutamate excitotoxicity, trigger several Ca2+-dependent degradative pathways leading to cell death (Choi, 1988). Altered Ca2+ dynamics have been implicated following several neurological injuries such as stroke (Sun et al., 2004), status epilepticus (SE; (Raza et al., 2004), and traumatic brain injury (TBI; (Sun et al., 2008), and the alterations in Ca2+ homeostasis are involved in the maintenance of neuronal plasticity seen in these pathologies (Raza et al., 2004). The most common neurological injuries where hypothermia is indicated, such as stroke and TBI, are associated with increased levels of glutamate following the injury. Increased levels of glutamate lead to neuronal death via Ca2+ mediated excitotoxicity. However, the effect of hypothermia on Ca2+ dynamics is unknown.
This study was initiated to evaluate the effect of moderate hypothermia on various modes of neuronal Ca2+ entry. Exploring how hypothermia affects Ca2+ dynamics in the cell can provide valuable insight into hypothermia’s neuroprotective mechanisms. Furthermore, understanding of hypothermia’s mechanisms of action may lead to further clinical use for more neurological insults.
2. Materials and Methods
2.1. Materials
All reagents were purchased from Sigma Aldrich (St. Louis, MO, USA) unless otherwise specified. TCN-213 (N-(Cyclohexylmethyl)-2-[(5-[(phenylmethyl)amino]-1,3,4-thiadiazol-2-yl}thio]acetamide) and Ro 8-4304 (4-[3-[4-(4-Fluorophenyl)-1,2,3,6-tetrahydro-1(2H)-pyridinyl]-2-hydroxypropoxy]benzamide hydrochloride) were purchased from Tocris Biosciences (Minneapolis, MN, USA). Chemicals were solubilized in physiological basal recording solution (pBRS: see composition below). Cell culture media were purchased from Invitrogen (Carlsbad, CA, USA). All animal use procedures were in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by Virginia Commonwealth University’s Institutional Animal Care and Use Committee.
2.2. Hippocampal Neuronal Cultures
Experiments were performed using primary hippocampal neuronal cultures prepared as previously described (Sombati and Delorenzo, 1995). Briefly, hippocampal cells were prepared from 2-day postnatal Sprague–Dawley rats (Harlan, Frederick, MD) and plated at a density of 2.0 × 104 cells/cm2 onto a glial support layer previously plated onto poly-L-lysine (0.05 mg/ml) coated Lab-Tek two-well cover glass chambers (Nunc, Naperville, IL). Cultures were maintained at 37°C in a 5% CO2/95% air atmosphere and fed twice weekly with MEM enriched with N3 supplement containing 25 mM HEPES buffer (pH 7.4), 2 mM L-glutamine, 3 mM glucose, 100 mg/ml transferrin, 5 mg/ml insulin, 100 mM putrescine, 3 nM sodium selenite, 200 nM progesterone, 1 mM sodium pyruvate, 0.1% ovalbumin, 0.2 ng/ml triiodothyroxine, 0.4 ng/ml corticosterone and supplemented with a glial bed-condition media (20%). These mixed cultures were used for experiments between 15–21 days in vitro following neuronal plating.
2.3. Calcium microfluorometry
Changes in neuronal Ca2+ were measured using the ratiometric fluorescent Ca2+ indicator Fura-2-acetoxymethyl ester (AM) (Molecular Probes, Invitrogen, Eugene, OR, USA). Hippocampal neurons were loaded with 1 μM fura-2-AM dissolved 0.1% DMSO and incubated for 20 minutes (min) at 37°C in 5% CO2/95% air. The dye loading was terminated with three washes with physiological basal recording solution pBRS containing (in mM): 145 NaCl, 2.5 KCl, 10 HEPES, 2 CaCl2, 10 glucose, 1 MgCl2, and 0.002 glycine, pH 7.3, and osmolarity adjusted to 325 ± 5 mOsm with sucrose composition. The neurons were incubated for an additional 15 minutes to allow for the complete cleavage of the AM moiety from Fura-2. At this point, neurons were transferred to a temperature controlled stage (Harvard Apparatus, Holliston, MA) which maintained the temperature of the cultures at either 31°C or 37°C prior to Ca2+ imaging. Neurons were visualized on an inverted microscope (Olympus IX 70, Olympus America, Melville, NY, USA) using a 20x, 0.7 numerical aperture, fluorite water-immersion objective (Olympus America). Fura-2 ratios were obtained from a neuronal population within a single field. Approximately 5 to 10 neurons were imaged at one time within a certain field. Pyramidal neurons appeared phase-bright, had distinct pyramidal shape, and were distinguishable from glia as they were visible above the focal plane of the glial cell layer. The region of interest was restricted to neuronal soma for the purposes of this paper. Fura-2 was excited with a 75-W xenon arc lamp (Olympus America) with alternating wavelengths of 340 and 380 nm filtered through a Sutter Filter Wheel (Sutter Instruments Co., Novato, CA USA). Fluorescent emission at 510 nm was captured through a Fura filter cube (Olympus America) with a dichroic mirror at 400 nm using a Hamamatsu ORCA-ER camera (Hamamatsu Photonics, Japan). The resulting 340/380 ratios correspond directly to the total [Ca2+]i. Utilizing the ratio is important because it accounts for confounding variables such as unequal Fura-2 loading and variable cell thickness. MetaFluor (Molecular Devices, Downington, PA, USA) was used to control image acquisition and processing. Image pairs were captured and corrected for non-specific background fluorescence by subtracting images acquired from non-indicator-loaded plates. Drugs were dissolved in pBRS. High potassium solution was prepared by equimolar replacement of NaCl and was composed of 105 mM NaCl, 40 mM KCl, 10 mM HEPES, 10 mM glucose, 2 mM CaCl2, 1 mM MgCl2, and 0.002 mM glycine.
Fura-2 ratios were captured every 5 s for 1 min to obtain baseline values. At this point, the pBRS was removed and replaced with the pharmacological agent of interest. Fura-2 ratios were captured every 5 s following stimulation.
2.4. Data analyses
A sample size (n) of at least 6 plates per treatment group was used with a total of 40–60 neurons per group. Experiments were performed over several weeks so that results were representative of multiple cultures. Individual neurons from multiple experiments were pooled to calculate average and standard error of the mean (S.E.M.). Data is presented as mean ± S.E.M. To determine statistical significance between treatment groups, one-way analysis of variance (ANOVA) was used followed by Tukey or Dunnett post-hoc analysis wherever applicable. A p-value of less than 0.05 (P<0.05) was considered significant. Statistical analysis was performed and graphs were drawn using SigmaPlot (6.0, Systat Software, San Jose, CA). Image processing for representative figures was carried out with Adobe Photoshop software (CS3, Adobe Systems Inc., San Jose, CA).
3. Results
3.1. Hypothermia did not affect Ca2+ entry through voltage-dependent Ca2+ channels
Calcium entry through voltage-dependent Ca2+ channels (VDCC) was stimulated using a high potassium solution (40 mM KCl; see methods). The high potassium solution depolarizes the neuronal membrane, thus allowing an influx of Ca2+ through VDCCs. As demonstrated in Fig 1, the baseline ratio values for neurons in the 31°C group and 37°C group were not significantly different from each other at 0.27±0.01 and 0.27±0.02, respectively (P=0.998). When neurons were stimulated with 37°C high potassium, 340/380 ratio values peaked to 0.54±0.02. Neurons stimulated with high potassium at 31°C exhibited a similar peak of 0.51±0.01. The peak ratio values following stimulation were not significantly different from each other (P=0.377). In contrast, the peaks of both groups were significantly higher when compared to 37°C baseline values (One way ANOVA, P<0.001). Fig 1B further illustrates the similar rises in [Ca2+]i upon stimulation with 31°C and 37°C high potassium solution with the use of pseudocolor images. These results show that high potassium caused a significant rise in [Ca2+]i and that hypothermia did not affect Ca2+ entry through VDCCs.
Fig. 1.

Hypothermia did not affect Ca2+ entry through VDCCs. (A) Prior to stimulation of VDCCs with high potassium solution, hippocampal neurons from both treatment groups displayed similar 340/380 baseline ratios of 0.27±0.01 in the 31°C group and 0.27±0.02 in the 37°C group (black bars). When stimulated with 37°C high potassium solution 340/380 ratios peaked to 0.54±0.02. Stimulation with 31°C high potassium resulted in a similar 340/380 peak ratio of 0.51±0.01 (grey bars). *P<0.001 compared to 37°C baseline, one way ANOVA followed by post-hoc Tukey test, n=6 plates per condition. (B) Representative pseudocolor images obtained from baseline neuron (left panel), neuron stimulated with 37°C high potassium (top right panel), and neuron stimulated with 31°C high potassium (bottom right panel). Neurons from both groups exhibited elevated [Ca2+]i upon 37°C and 31°C stimulation.
3.2. Hypothermia reduced NMDA receptor-dependent Ca2+ entry
To determine if hypothermia affected N-methyl-D-aspartate (NMDA) receptor-mediated Ca2+ entry, hippocampal neurons were stimulated with glutamate (50 μM, 31°C or 37°C). Baseline 340/380 ratio values before stimulation were not significantly different between the two groups with values of 0.30±0.01 for neurons in the 31°C group and 0.31±0.02 for neurons in the 37°C group (P=0.932). Upon stimulation with 37°C glutamate solution, the ratio values peaked to 0.636±0.04. However, stimulation with 31°C glutamate solution resulted in a diminished response, with ratio values only peaking to 0.38±0.01 which was not significantly different from control (Fig 2A). The peak Ca2+ ratio between the two temperature groups were significantly different (P<0.001, one way ANOVA). Pseudocolor images further illustrate that 37°C glutamate solution causes a dramatic increase in [Ca2+]i compared to 31°C glutamate solution (Fig 2B). These results indicate that hypothermia significantly reduced Ca2+ entry through the NMDA receptor.
Fig. 2.
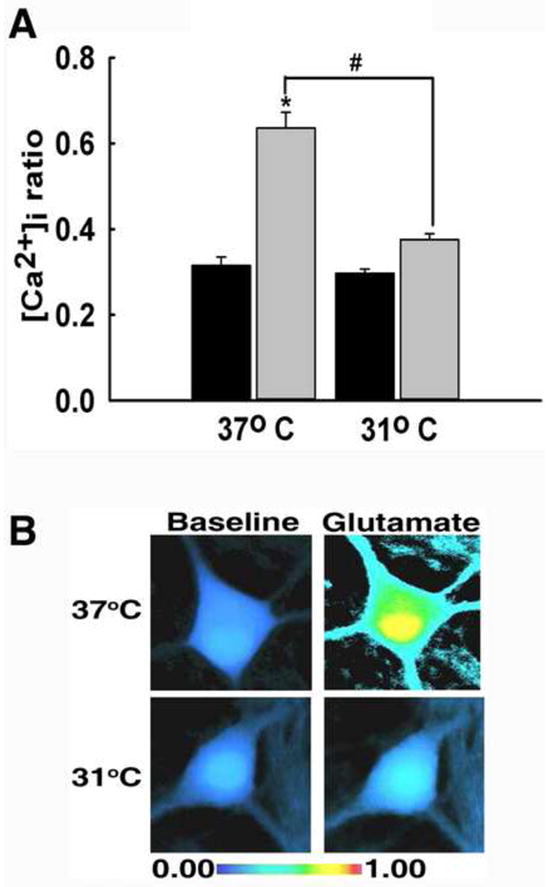
Hypothermia reduced NMDA receptor-dependent Ca2+ entry. (A) Prior to stimulation with 50 μM glutamate, hippocampal neurons exhibited similar 340/380 baseline ratios of 0.30±0.01 for neurons in the 31°C group and 0.31±0.02 for neurons in the 37°C group (black bars). Upon stimulation with 37°C glutamate, 340/380 ratios peaked to 0.636±0.04, which is significantly elevated compared to baseline. When neurons were stimulated with 31°C glutamate, 340/380 ratios peaked to 0.38±0.01 (grey bars). *P<0.001 compared to baseline; #P<0.001 between 31°C and 37°C peaks, one way ANOVA followed by post-hoc Tukey test, n=7 plates per condition. (B) Representative pseudocolor images obtained from baseline neuron (left panel), neuron stimulated with 37°C glutamate (top right panel), and neuron stimulated with 31°C glutamate (bottom right panel). Neurons exhibited elevated [Ca2+]i upon 37°C stimulation and a diminished response to 31°C stimulation.
To further investigate contribution of NMDA receptor subtypes to the elevated [Ca2+]i following glutamate stimulation, we performed these experiments in the presence of GluN2A and GluN2B subtype specific antagonists. Prior to glutamate stimulation, the average baseline ratio value was 0.38±0.01. Upon glutamate (50 μM, 37°C) stimulation, ratio values increased significantly to 1.09±0.09 (P<0.001). When neurons were stimulated with glutamate in the presence of the GluN2B selective antagonist Ro 8-4304 (10 μM), 340/380 ratios peaked to 0.94±0.08, which were not significantly different from glutamate alone condition (P= 0.19). However, when neurons were stimulated with glutamate in the presence of the GluN2A selective antagonist TCN-213 (10 μM), 340/380 ratio values increased only to 0.52±0.04, which were significantly lower than glutamate alone condition (P<0.001, one-way ANOVA). These results summarized in Fig. 3 suggest following glutamate stimulation the GluN2A receptor subtype mediate the majority of Ca2+ influx.
Fig. 3.

The role of NMDA receptor subtypes on [Ca2+]i upon glutamate stimulation. Prior to glutamate stimulation, the average baseline ratio value was 0.38±0.01. When stimulated with 50 μM glutamate, ratio values increased to 1.09±0.09. When neurons were stimulated with glutamate (50 μM) in the presence of GluN2B selective antagonist Ro 8-4304 (10 μM) 340/380 ratios increased to 0.94±0.08, which were not significantly different from glutamate alone conditions. When stimulated with GluN2A selective antagonist TCN-213, 340/380 ratios increased to 0.52±0.04, which were significantly lower than control glutamate levels. *P<0.001 compared to glutamate, one-way ANOVA followed by post-hoc Dunnett’s test, n=7 plates per condition.
3.3. Hypothermia did not affect IP3 receptor-mediated Ca2+ entry
In order to investigate how inositol trisphosphate (IP3) receptor-mediated Ca2+ release is affected by hypothermia, hippocampal neurons were stimulated with either 31°C or 37°C bradykinin (1 μM) (Osugi et al., 1986). Before stimulation with bradykinin, similar baseline ratio values of 0.27±0.01 in 31°C group and 0.27±0.01 in the 37°C group were observed (P=0.999), as shown in Fig 3. Upon stimulation with 37°C bradykinin solution, neurons exhibited a significant rise in [Ca2+]i with ratio values increasing to 0.65±0.03. When stimulated with 31°C bradykinin solution, neurons also exhibited a significant increase in [Ca2+]i with ratio values increasing to 0.60±0.01 (Fig 4A). Both peak values were significantly elevated compared to baseline ratio values (P<0.001). However, there was no significant difference between the peak values of the two treatment groups (P=0.066). Pseudocolor images illustrate the similar rises in [Ca2+]i following stimulation with 31°C and 37°C bradykinin (Fig 4B).
Fig. 4.

Hypothermia did not affect IP3 receptor-mediated Ca2+ entry. (A) Prior to bradykinin-mediated stimulation of IP3 receptors, neurons from 31°C and 37°C treatment groups displayed similar 340/380 baseline ratios of 0.27±0.01 and 0.27±0.01, respectively (black bars). Stimulation with 37°C bradykinin resulted in a peak 340/380 ratio of 0.65±0.03. Similarly, when stimulated with 31°C bradykinin, 340/380 ratios peaked to 0.60±0.01 (grey bars). *P<0.001 compared to 37°C baseline, one way ANOVA followed by post-hoc Tukey test, n=5 plates per condition. (B) Representative pseudocolor images obtained from baseline neuron (left panel), neuron stimulated with 37°C bradykinin (top right panel), and neuron stimulated with 31°C bradykinin (bottom right panel). Neurons in both groups exhibited elevated [Ca2+]i upon stimulation with 37°C and 31°C bradykinin.
3.4. Hypothermia reduced ryanodine receptor-mediated Ca2+ release
Caffeine (10 mM) was used to stimulate ryanodine receptor-mediated Ca2+ release (McPherson et al., 1991). Prior to caffeine stimulation, both 31°C and 37°C groups exhibited similar 340/380 ratio values of 0.28±0.01 and 0.34±0.01, respectively (P=0.649). Upon stimulation with a 37°C caffeine solution, neurons exhibited a marked increased in [Ca2+]i with peak ratio values of 0.72±0.05 (Fig 5A). When stimulated with 31°C caffeine solution, neurons exhibited a diminished response with a peak ratio value of 0.44±0.04. Stimulation with 31°C caffeine solution results in a diminished response compared to the peak in [Ca2+]i observed following 37°C caffeine stimulation. The peak value in the hypothermia condition is significantly lower than the peak response in the 37°C group (P<0.001). There was also a significant difference between the peak values of both treatment groups compared to 37°C baseline (P<0.001). Fig 5B illustrates the differences in peak [Ca2+]i through pseudocolor images. These results suggest that hypothermia reduces the release of Ca2+ from intracellular stores by inhibiting the ryanodine receptor.
Fig. 5.

Hypothermia reduced ryanodine receptor-mediated Ca2+ induced Ca2+ release. (A) Prior to caffeine-mediated RyR stimulation, baseline 340/380 ratio values from both treatment groups were not significantly different with values of 0.28±0.01 in the 31°C group and 0.34±0.01 in the 37°C group (black bars). Upon stimulation with 37°C caffeine, 340/380 ratios peaked to 0.72±0.05. When stimulated with 31°C caffeine, neurons exhibited a diminished response with a peak ratio of 0.44±0.04 (grey bars). *P<0.001 compared to 37°C baseline, **P<0.05 compared to 37°C baseline, #P<0.001 between 31°C and 37°C peaks, one way ANOVA followed by post-hoc Tukey test, n=10 plates per group. (B) Representative pseudocolor images obtained from baseline neuron (left panel), neuron stimulated with 37°C caffeine (top right panel), and neuron stimulated with 31°C caffeine (bottom right panel). Neurons exhibited elevated [Ca2+]i upon 37°C stimulation and a diminished response to 31°C stimulation.
4. Discussion
Understanding the mechanisms by which hypothermia protects brain tissue may lead to advances in treatment of neurological injuries. Moderate hypothermia, which ranges from 30°C to 33°C, is being increasingly used clinically to reduce mortality and improve patient outcome following ischemic injuries (Polderman, 2009). The role of Ca2+ signaling in producing excitotoxic injuries is well documented. However, the effects of hypothermia on Ca2+ transmission are not fully understood. To better understand how hypothermia affects Ca2+ dynamics, this study utilized pharmacological agents to stimulate various modes of Ca2+ entry under normothermic and moderate hypothermic conditions. The results of this study indicate that moderate hypothermia specifically targets Ca2+ entry via the NMDA receptors and ryanodine receptors in the hippocampal neuronal cultures. The ability of hypothermia to selectively affect two major sources of Ca2+ elevations lends further support to its use following ischemic injuries.
Calcium influx through VDCCs occurs in response to changes in the membrane potential and triggers various physiological events such as neuronal excitability and neurotransmitter release. While many animal models have found VDCC blockers to be useful following neuronal injuries, their use in clinical settings has met limited success and is associated with deleterious side effects (Rauck et al., 2006). To investigate if hypothermic intervention affects Ca2+ entry through VDCCs, a high potassium solution, which depolarized the neuronal membrane, thus allowing an influx of Ca2+ was used. Under hypothermic conditions, Ca2+ influx following stimulation of VDCC was not significantly reduced compared to normothermic controls, indicating that hypothermia does not significantly affect Ca2+ influx through VDCCs.
NMDA receptor-mediated Ca2+ entry has been shown to play a major role in excitotoxic neuronal injury and death. We have previously shown that following glutamate exposure or in vitro status epilepticus (SE) which induces glutamate release and activates NMDA receptors, there is an increase in [Ca2+]i that is sensitive to inhibition by both competitive NMDA receptor antagonist APV and non-competitive NMDA receptor antagonist MK-801 (DeLorenzo et al., 1998; Sun et al., 2002). Other researchers have also published that following glutamate stimulation in cultured hippocampal neurons, the majority of Ca2+ influx occurs through NMDA receptors and can be blocked by APV or MK-801 (Choi et al., 1988; Michaels and Rothman, 1990; Rothman et al., 1987). In stroke models, NMDA receptor antagonists reduce the ischemic infarct volume (Gill et al., 1992) and protect neurons from excitotoxicity (Choi, 1988; Moudy et al., 1994). In models of SE, NMDA antagonists administered prior to injury provide neuroprotection (Deshpande et al., 2008; Raza et al., 2004). However, the effect of hypothermia on extracellular glutamate concentrations is controversial. Several studies have shown that hypothermia reduces extracellular glutamate concentrations (Busto et al., 1989; Kvrivishvili, 2002; Winfree et al., 1996), thereby reducing to NMDA receptor activation and concomitant Ca2+ entry. Other studies have demonstrated that hypothermia has no effect on glutamate concentrations (Asai et al., 2000; Hua et al., 2010). The results of this study demonstrate that hypothermia significantly reduces Ca2+ entry via NMDA receptors. Hippocampal neurons stimulated under hypothermic conditions exhibited a markedly diminished response to glutamate. This provides evidence that hypothermia reduces Ca2+ entry through NMDA receptors despite the presence of extracellular glutamate.
We also investigated the relative contributions of GluN2A and GluN2B containing NMDA receptors to the rise in [Ca2+]i following glutamate stimulation. NMDA subunit expression is affected developmentally. GluN2B is widely expressed prenatally and during the first two weeks following birth. Around P10 levels of GluN2A start to increase dramatically, peaking around P14 and replacing GluN2B as the primary subunit in the hippocampus and cortex (Sanz-Clemente et al., 2012; Wenzel et al., 1997). We use cultured hippocampal neurons between days 15–21. This suggests that in our neuronal culture preparation GluN2A containing NMDA receptors are predominantly expressed. Indeed, under normothermic conditions a significant inhibition of Ca2+ entry is observed only in the presence of GluN2A antagonist TCN-213 but not when GluN2B antagonist Ro 8-4304 is applied.
The GluN2A and GluN2B containing receptors have distinct distribution and function (Erreger et al., 2004). The GluN2B receptors are extrasynaptically located, are predominant in developing neurons and responsible for long term depression (Hrabetova et al., 2000; Liu et al., 2004). In contrast, GluN2A receptors are synaptically located, are predominant in mature neurons and responsible for long term potentiation (Hrabetova et al., 2000; Liu et al., 2004). Compared to GluN2B containing NMDA receptors, synaptically located GluN2A containing NMDA receptors have 2–5x higher probability of opening upon activation (Erreger et al., 2005). NMDA receptor subunits also have opposing roles in mediating excitotoxic neuronal death. Stimulation of GluN2B containing receptors is thought to be excitotoxic, while activation of GluN2A containing receptors is thought to be neuroprotective (Liu et al., 2007). In our model bath application of glutamate will activate both synaptic and extrasynaptic NMDA receptors. Even though GluN2A containing NMDA receptors are mediating majority of Ca2+ influx, hypothermia will likely reduce Ca2+ entry via both the GluN2A and GluN2B containing NMDA receptors. Such an action could probably explain the effect of hypothermia on not only lowering elevated [Ca2+]i following glutamate stimulation but also the reported neuroprotective effects following excitotoxic brain injuries (Polderman, 2009). It will insightful to further explore these possibilities in future experiments.
Another mechanism for cytosolic Ca2+ entry is by Ca2+-induced Ca2+ release (CICR). Activation of IP3 receptors and ryanodine receptors present on the endoplasmic reticulum (ER) surface releases Ca2+ from intracellular stores into the cytosol, thereby leading to an elevation of [Ca2+]i. This study used bradykinin to stimluate IP3 receptor-mediated Ca2+ release (Osugi et al., 1986). Binding of bradykinin to its receptor activates phospholipase C, which cleaves phosphatidylinositol 4,5-bisphosphate, producing diacylglycerol and IP3. IP3 then binds to and activates the IP3 receptor on the ER membrane. In this study, hypothermia did not significantly reduce Ca2+ entry through bradykinin-mediated IP3 receptor stimulation. To investigate the effects of hypothermia on ryanodine receptor-mediated Ca2+ entry, caffeine was employed to activate ryanodine receptors. Caffeine is an established pharmacological tool for activating ryanodine receptor-mediated Ca2+ release (Angehagen et al., 2003; Endo, 1977; Greene et al., 1985; Jenden and Fairhurst, 1969; McPherson et al., 1991; Uneyama et al., 1993). Non-physiological concentrations of caffeine have been shown to function similarly to nanomolar concentrations of ryanodine with the advantage of having faster kinetics and rapid reversibility upon washout (McPherson et al., 1991). Caffeine at concentrations greater than 5 mM leads to activation of the ryanodine receptor that is independent of cytosolic Ca2+ (Koulen and Thrower, 2001). Thus, this study employed 10 mM caffeine to study the effects of hypothermia on CICR. Hypothermia significantly reduced the height of the Ca2+ transient upon caffeine induced-ryanodine receptor stimulation, thereby reducing CICR and providing novel evidence that hypothermia reduces Ca2+ release via ryanodine receptors.
Following acute neurological injuries such as stroke, SE and traumatic brain injury (TBI), significant and prolonged increases in [Ca2+]i have been reported (Pal et al., 1999; Pal et al., 2001; Raza et al., 2001; Sun et al., 2008; Sun et al., 2004). This is thought to underlie neuronal loss, neuronal plasticity changes resulting in cognitive impairments and the expression of seizures. Preventing these sustained increases in [Ca2+]i (“Ca2+ plateau”) has the potential to ameliorate the neurological deficits. Calcium entry via NMDA receptors is thought to underlie the generation of this Ca2+ plateau. Despite the large body of evidence supporting the neuroprotective effects of NMDA receptor antagonists in animal models, clinical trials in humans have been unsuccessful due to their poor tolerance, toxicity, and CNS-adverse effects (Ikonomidou and Turski, 2002; Muir, 2006). Therefore finding mechanisms for alleviating the Ca2+ plateau beyond the NMDA receptor would be clinically relevant. Our observation that hypothermia lowers Ca2+ entry through the NMDA receptor and the clinical data that suggest neuroprotective role of hypothermia makes this intervention an attractive therapeutic option for treatment of excitotoxic brain injuries.
We recently reported that CICR plays a vital role in maintaining elevated [Ca2+]i following SE. In fact, dantrolene, a ryanodine receptor antagonist, administered following SE reduced intracellular Ca2+, was neuroprotective, and remarkably prevented development of seizures (antiepileptogenic) (Nagarkatti et al., 2010). There is evidence that dantrolene is neuroprotective in animal and cell culture models of various neurological injuries including SE, ischemia, and TBI (Lei et al., 1992; Nagarkatti et al., 2010; Niebauer and Gruenthal, 1999; Yoon et al., 1996; Zhang et al., 1993). However, the use of dantrolene clinically is limited by its high lipid solubility and poor permeability across the blood brain barrier (Wuis et al., 1989). The ability of hypothermia to selectively affect both NMDA receptor and ryanodine receptor-mediated Ca2+ systems makes it an extremely attractive intervention for alleviating Ca2+ elevations following neurological injuries. It will be interesting to study the role of hypothermia when used as a therapeutic intervention either alone or in combination with existing therapies in animal models and whether this intervention is associated with limiting side-effects. Better knowledge of therapeutic targets of hypothermia will help improve protection using hypothermia and be central to developing innovative combination therapies to augment the protective benefits of hypothermia.
Acknowledgments
This research is supported by the CounterACT Program, National Institutes of Health Office of the Director (NIH OD), and the National Institute of Neurological Disorders and Stroke (NINDS), Grant Number 2U01NS058213-06 and 1R21NS072061-01A1 to RJD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi M, Sohma O, Tsuneishi S, Takada S, Nakamura H. Combination effect of systemic hypothermia and caspase inhibitor administration against hypoxic-ischemic brain damage in neonatal rats. Pediatr Res. 2001;50:590–595. doi: 10.1203/00006450-200111000-00010. [DOI] [PubMed] [Google Scholar]
- Aibiki M, Maekawa S, Ogura S, Kinoshita Y, Kawai N, Yokono S. Effect of moderate hypothermia on systemic and internal jugular plasma IL-6 levels after traumatic brain injury in humans. J Neurotrauma. 1999;16:225–232. doi: 10.1089/neu.1999.16.225. [DOI] [PubMed] [Google Scholar]
- Angehagen M, Margineanu DG, Ben-Menachem E, Ronnback L, Hansson E, Klitgaard H. Levetiracetam reduces caffeine-induced Ca2+ transients and epileptiform potentials in hippocampal neurons. Neuroreport. 2003;14:471–475. doi: 10.1097/00001756-200303030-00035. [DOI] [PubMed] [Google Scholar]
- Asai S, Zhao H, Kohno T, Takahashi Y, Nagata T, Ishikawa K. Quantitative evaluation of extracellular glutamate concentration in postischemic glutamate re-uptake, dependent on brain temperature, in the rat following severe global brain ischemia. Brain Res. 2000;864:60–68. doi: 10.1016/s0006-8993(00)02151-x. [DOI] [PubMed] [Google Scholar]
- Bernard SA, Buist M. Induced hypothermia in critical care medicine: a review. Crit Care Med. 2003;31:2041–2051. doi: 10.1097/01.CCM.0000069731.18472.61. [DOI] [PubMed] [Google Scholar]
- Busto R, Dietrich WD, Globus MY, Valdes I, Scheinberg P, Ginsberg MD. Small differences in intraischemic brain temperature critically determine the extent of ischemic neuronal injury. J Cereb Blood Flow Metab. 1987;7:729–738. doi: 10.1038/jcbfm.1987.127. [DOI] [PubMed] [Google Scholar]
- Busto R, Globus MY, Dietrich WD, Martinez E, Valdes I, Ginsberg MD. Effect of mild hypothermia on ischemia-induced release of neurotransmitters and free fatty acids in rat brain. Stroke. 1989;20:904–910. doi: 10.1161/01.str.20.7.904. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Choi DW, Koh JY, Peters S. Pharmacology of glutamate neurotoxicity in cortical cell culture: attenuation by NMDA antagonists. J Neurosci. 1988;8:185–196. doi: 10.1523/JNEUROSCI.08-01-00185.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLorenzo RJ, Pal S, Sombati S. Prolonged activation of the N-methyl-D-aspartate receptor-Ca2+ transduction pathway causes spontaneous recurrent epileptiform discharges in hippocampal neurons in culture. Proc Natl Acad Sci U S A. 1998;95:14482–14487. doi: 10.1073/pnas.95.24.14482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande LS, Lou JK, Mian A, Blair RE, Sombati S, Attkisson E, DeLorenzo RJ. Time course and mechanism of hippocampal neuronal death in an in vitro model of status epilepticus: role of NMDA receptor activation and NMDA dependent calcium entry. Eur J Pharmacol. 2008;583:73–83. doi: 10.1016/j.ejphar.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich WD, Chatzipanteli K, Vitarbo E, Wada K, Kinoshita K. The role of inflammatory processes in the pathophysiology and treatment of brain and spinal cord trauma. Acta Neurochir Suppl. 2004;89:69–74. doi: 10.1007/978-3-7091-0603-7_9. [DOI] [PubMed] [Google Scholar]
- Endo M. Calcium release from the sarcoplasmic reticulum. Physiol Rev. 1977;57:71–108. doi: 10.1152/physrev.1977.57.1.71. [DOI] [PubMed] [Google Scholar]
- Erreger K, Chen PE, Wyllie DJ, Traynelis SF. Glutamate receptor gating. Crit Rev Neurobiol. 2004;16:187–224. doi: 10.1615/critrevneurobiol.v16.i3.10. [DOI] [PubMed] [Google Scholar]
- Erreger K, Dravid SM, Banke TG, Wyllie DJ, Traynelis SF. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol. 2005;563:345–358. doi: 10.1113/jphysiol.2004.080028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foundation BT. Guidelines for the management of severe traumatic brain injury. J Neurotrauma. 2007;24(Suppl 1):S1–106. doi: 10.1089/neu.2007.9999. [DOI] [PubMed] [Google Scholar]
- Gill R, Andine P, Hillered L, Persson L, Hagberg H. The effect of MK-801 on cortical spreading depression in the penumbral zone following focal ischaemia in the rat. J Cereb Blood Flow Metab. 1992;12:371–379. doi: 10.1038/jcbfm.1992.54. [DOI] [PubMed] [Google Scholar]
- Greene RW, Haas HL, Hermann A. Effects of caffeine on hippocampal pyramidal cells in vitro. Br J Pharmacol. 1985;85:163–169. doi: 10.1111/j.1476-5381.1985.tb08843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrabetova S, Serrano P, Blace N, Tse HW, Skifter DA, Jane DE, Monaghan DT, Sacktor TC. Distinct NMDA receptor subpopulations contribute to long-term potentiation and long-term depression induction. J Neurosci. 2000;20:RC81. doi: 10.1523/JNEUROSCI.20-12-j0002.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Hisano K, Morimoto Y. Effect of mild and moderate hypothermia on hypoxic injury in nearly pure neuronal culture. J Anesth. 2010;24:726–732. doi: 10.1007/s00540-010-0999-x. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002;1:383–386. doi: 10.1016/s1474-4422(02)00164-3. [DOI] [PubMed] [Google Scholar]
- Illievich UM, Zornow MH, Choi KT, Scheller MS, Strnat MA. Effects of hypothermic metabolic suppression on hippocampal glutamate concentrations after transient global cerebral ischemia. Anesth Analg. 1994;78:905–911. doi: 10.1213/00000539-199405000-00012. [DOI] [PubMed] [Google Scholar]
- Jacobs SE, Morley CJ, Inder TE, Stewart MJ, Smith KR, McNamara PJ, Wright IM, Kirpalani HM, Darlow BA, Doyle LW. Whole-body hypothermia for term and near-term newborns with hypoxic-ischemic encephalopathy: a randomized controlled trial. Arch Pediatr Adolesc Med. 2011;165:692–700. doi: 10.1001/archpediatrics.2011.43. [DOI] [PubMed] [Google Scholar]
- Jenden DJ, Fairhurst AS. The pharmacology of ryanodine. Pharmacol Rev. 1969;21:1–25. [PubMed] [Google Scholar]
- Kimura A, Sakurada S, Ohkuni H, Todome Y, Kurata K. Moderate hypothermia delays proinflammatory cytokine production of human peripheral blood mononuclear cells. Crit Care Med. 2002;30:1499–1502. doi: 10.1097/00003246-200207000-00017. [DOI] [PubMed] [Google Scholar]
- Koulen P, Thrower EC. Pharmacological modulation of intracellular Ca(2+) channels at the single-channel level. Mol Neurobiol. 2001;24:65–86. doi: 10.1385/MN:24:1-3:065. [DOI] [PubMed] [Google Scholar]
- Kvrivishvili G. Glycine and neuroprotective effect of hypothermia in hypoxic-ischemic brain damage. Neuroreport. 2002;13:1995–2000. doi: 10.1097/00001756-200211150-00001. [DOI] [PubMed] [Google Scholar]
- Lei SZ, Zhang D, Abele AE, Lipton SA. Blockade of NMDA receptor-mediated mobilization of intracellular Ca2+ prevents neurotoxicity. Brain Res. 1992;598:196–202. doi: 10.1016/0006-8993(92)90183-a. [DOI] [PubMed] [Google Scholar]
- Liou AK, Clark RS, Henshall DC, Yin XM, Chen J. To die or not to die for neurons in ischemia, traumatic brain injury and epilepsy: a review on the stress-activated signaling pathways and apoptotic pathways. Prog Neurobiol. 2003;69:103–142. doi: 10.1016/s0301-0082(03)00005-4. [DOI] [PubMed] [Google Scholar]
- Liu XB, Murray KD, Jones EG. Switching of NMDA receptor 2A and 2B subunits at thalamic and cortical synapses during early postnatal development. J Neurosci. 2004;24:8885–8895. doi: 10.1523/JNEUROSCI.2476-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, Wang YT. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson PS, Kim YK, Valdivia H, Knudson CM, Takekura H, Franzini-Armstrong C, Coronado R, Campbell KP. The brain ryanodine receptor: a caffeine-sensitive calcium release channel. Neuron. 1991;7:17–25. doi: 10.1016/0896-6273(91)90070-g. [DOI] [PubMed] [Google Scholar]
- Michaels RL, Rothman SM. Glutamate neurotoxicity in vitro: antagonist pharmacology and intracellular calcium concentrations. J Neurosci. 1990;10:283–292. doi: 10.1523/JNEUROSCI.10-01-00283.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moudy AM, Yamada KA, Rothman SM. Rapid desensitization determines the pharmacology of glutamate neurotoxicity. Neuropharmacology. 1994;33:953–962. doi: 10.1016/0028-3908(94)90153-8. [DOI] [PubMed] [Google Scholar]
- Muir KW. Glutamate-based therapeutic approaches: clinical trials with NMDA antagonists. Curr Opin Pharmacol. 2006;6:53–60. doi: 10.1016/j.coph.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Nagarkatti N, Deshpande LS, Carter DS, DeLorenzo RJ. Dantrolene inhibits the calcium plateau and prevents the development of spontaneous recurrent epileptiform discharges following in vitro status epilepticus. Eur J Neurosci. 2010;32:80–88. doi: 10.1111/j.1460-9568.2010.07262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niebauer M, Gruenthal M. Neuroprotective effects of early vs. late administration of dantrolene in experimental status epilepticus. Neuropharmacology. 1999;38:1343–1348. doi: 10.1016/s0028-3908(99)00059-3. [DOI] [PubMed] [Google Scholar]
- Ning XH, Chen SH, Xu CS, Li L, Yao LY, Qian K, Krueger JJ, Hyyti OM, Portman MA. Hypothermic protection of the ischemic heart via alterations in apoptotic pathways as assessed by gene array analysis. J Appl Physiol. 2002;92:2200–2207. doi: 10.1152/japplphysiol.01035.2001. [DOI] [PubMed] [Google Scholar]
- Okuda C, Saito A, Miyazaki M, Kuriyama K. Alteration of the turnover of dopamine and 5-hydroxytryptamine in rat brain associated with hypothermia. Pharmacol Biochem Behav. 1986;24:79–83. doi: 10.1016/0091-3057(86)90048-1. [DOI] [PubMed] [Google Scholar]
- Ooboshi H, Ibayashi S, Takano K, Sadoshima S, Kondo A, Uchimura H, Fujishima M. Hypothermia inhibits ischemia-induced efflux of amino acids and neuronal damage in the hippocampus of aged rats. Brain Res. 2000;884:23–30. doi: 10.1016/s0006-8993(00)02861-4. [DOI] [PubMed] [Google Scholar]
- Osugi T, Uchida S, Imaizumi T, Yoshida H. Bradykinin-induced intracellular Ca2+ elevation in neuroblastoma X glioma hybrid NG108-15 cells; relationship to the action of inositol phospholipids metabolites. Brain Res. 1986;379:84–89. doi: 10.1016/0006-8993(86)90258-1. [DOI] [PubMed] [Google Scholar]
- Pal S, Sombati S, Limbrick DD, Jr, DeLorenzo RJ. In vitro status epilepticus causes sustained elevation of intracellular calcium levels in hippocampal neurons. Brain Res. 1999;851:20–31. doi: 10.1016/s0006-8993(99)02035-1. [DOI] [PubMed] [Google Scholar]
- Pal S, Sun D, Limbrick D, Rafiq A, DeLorenzo RJ. Epileptogenesis induces long-term alterations in intracellular calcium release and sequestration mechanisms in the hippocampal neuronal culture model of epilepsy. Cell Calcium. 2001;30:285–296. doi: 10.1054/ceca.2001.0236. [DOI] [PubMed] [Google Scholar]
- Polderman KH. Hypothermia and neurological outcome after cardiac arrest: state of the art. Eur J Anaesthesiol Suppl. 2008;42:23–30. doi: 10.1017/S026502150700333X. [DOI] [PubMed] [Google Scholar]
- Polderman KH. Mechanisms of action, physiological effects, and complications of hypothermia. Crit Care Med. 2009;37:S186–202. doi: 10.1097/CCM.0b013e3181aa5241. [DOI] [PubMed] [Google Scholar]
- Raghupathi R, Graham DI, McIntosh TK. Apoptosis after traumatic brain injury. J Neurotrauma. 2000;17:927–938. doi: 10.1089/neu.2000.17.927. [DOI] [PubMed] [Google Scholar]
- Rauck RL, Wallace MS, Leong MS, Minehart M, Webster LR, Charapata SG, Abraham JE, Buffington DE, Ellis D, Kartzinel R. A randomized, double-blind, placebo-controlled study of intrathecal ziconotide in adults with severe chronic pain. J Pain Symptom Manage. 2006;31:393–406. doi: 10.1016/j.jpainsymman.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Raza M, Blair RE, Sombati S, Carter DS, Deshpande LS, DeLorenzo RJ. Evidence that injury-induced changes in hippocampal neuronal calcium dynamics during epileptogenesis cause acquired epilepsy. Proc Natl Acad Sci U S A. 2004;101:17522–17527. doi: 10.1073/pnas.0408155101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raza M, Pal S, Rafiq A, DeLorenzo RJ. Long-term alteration of calcium homeostatic mechanisms in the pilocarpine model of temporal lobe epilepsy. Brain Res. 2001;903:1–12. doi: 10.1016/s0006-8993(01)02127-8. [DOI] [PubMed] [Google Scholar]
- Rosomoff HL, Holaday DA. Cerebral blood flow and cerebral oxygen consumption during hypothermia. Am J Physiol. 1954;179:85–88. doi: 10.1152/ajplegacy.1954.179.1.85. [DOI] [PubMed] [Google Scholar]
- Rothman SM, Thurston JH, Hauhart RE. Delayed neurotoxicity of excitatory amino acids in vitro. Neuroscience. 1987;22:471–480. doi: 10.1016/0306-4522(87)90347-2. [DOI] [PubMed] [Google Scholar]
- Sanz-Clemente A, Nicoll RA, Roche KW. Diversity in NMDA Receptor Composition: Many Regulators, Many Consequences. Neuroscientist. 2012 doi: 10.1177/1073858411435129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorvon S. Super-refractory status epilepticus: an approach to therapy in this difficult clinical situation. Epilepsia. 2011;52(Suppl 8):53–56. doi: 10.1111/j.1528-1167.2011.03238.x. [DOI] [PubMed] [Google Scholar]
- Sombati S, Delorenzo RJ. Recurrent spontaneous seizure activity in hippocampal neuronal networks in culture. J Neurophysiol. 1995;73:1706–1711. doi: 10.1152/jn.1995.73.4.1706. [DOI] [PubMed] [Google Scholar]
- Suehiro E, Fujisawa H, Akimura T, Ishihara H, Kajiwara K, Kato S, Fujii M, Yamashita S, Maekawa T, Suzuki M. Increased matrix metalloproteinase-9 in blood in association with activation of interleukin-6 after traumatic brain injury: influence of hypothermic therapy. J Neurotrauma. 2004;21:1706–1711. doi: 10.1089/neu.2004.21.1706. [DOI] [PubMed] [Google Scholar]
- Sun DA, Deshpande LS, Sombati S, Baranova A, Wilson MS, Hamm RJ, DeLorenzo RJ. Traumatic brain injury causes a long-lasting calcium (Ca2+)-plateau of elevated intracellular Ca levels and altered Ca2+ homeostatic mechanisms in hippocampal neurons surviving brain injury. Eur J Neurosci. 2008;27:1659–1672. doi: 10.1111/j.1460-9568.2008.06156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun DA, Sombati S, Blair RE, DeLorenzo RJ. Calcium-dependent epileptogenesis in an in vitro model of stroke-induced “epilepsy”. Epilepsia. 2002;43:1296–1305. doi: 10.1046/j.1528-1157.2002.09702.x. [DOI] [PubMed] [Google Scholar]
- Sun DA, Sombati S, Blair RE, DeLorenzo RJ. Long-lasting alterations in neuronal calcium homeostasis in an in vitro model of stroke-induced epilepsy. Cell Calcium. 2004;35:155–163. doi: 10.1016/j.ceca.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Uneyama H, Munakata M, Akaike N. Caffeine response in pyramidal neurons freshly dissociated from rat hippocampus. Brain Res. 1993;604:24–31. doi: 10.1016/0006-8993(93)90348-q. [DOI] [PubMed] [Google Scholar]
- Wenzel A, Fritschy JM, Mohler H, Benke D. NMDA receptor heterogeneity during postnatal development of the rat brain: differential expression of the NR2A, NR2B, and NR2C subunit proteins. J Neurochem. 1997;68:469–478. doi: 10.1046/j.1471-4159.1997.68020469.x. [DOI] [PubMed] [Google Scholar]
- Winfree CJ, Baker CJ, Connolly ES, Jr, Fiore AJ, Solomon RA. Mild hypothermia reduces penumbral glutamate levels in the rat permanent focal cerebral ischemia model. Neurosurgery. 1996;38:1216–1222. doi: 10.1097/00006123-199606000-00034. [DOI] [PubMed] [Google Scholar]
- Wuis EW, Rijntjes NV, Van der Kleijn E. Whole-body autoradiography of 14C-dantrolene in the marmoset monkey. Pharmacol Toxicol. 1989;64:156–158. doi: 10.1111/j.1600-0773.1989.tb00621.x. [DOI] [PubMed] [Google Scholar]
- Xu L, Yenari MA, Steinberg GK, Giffard RG. Mild hypothermia reduces apoptosis of mouse neurons in vitro early in the cascade. J Cereb Blood Flow Metab. 2002;22:21–28. doi: 10.1097/00004647-200201000-00003. [DOI] [PubMed] [Google Scholar]
- Yenari MA, Hemmen TM. Therapeutic hypothermia for brain ischemia: where have we come and where do we go? Stroke. 2010;41:S72–74. doi: 10.1161/STROKEAHA.110.595371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KW, Mitchell HL, Broder LD, Brooker RW, Delisle RK. Trauma-induced neurotoxicity in rat hippocampal neurons. Stroke. 1996;27:122–126. doi: 10.1161/01.str.27.1.122. [DOI] [PubMed] [Google Scholar]
- Zeiner A, Holzer M, Sterz F, Behringer W, Schorkhuber W, Mullner M, Frass M, Siostrzonek P, Ratheiser K, Kaff A, Laggner AN. Mild resuscitative hypothermia to improve neurological outcome after cardiac arrest. A clinical feasibility trial. Hypothermia After Cardiac Arrest (HACA) Study Group. Stroke. 2000;31:86–94. doi: 10.1161/01.str.31.1.86. [DOI] [PubMed] [Google Scholar]
- Zhang L, Andou Y, Masuda S, Mitani A, Kataoka K. Dantrolene protects against ischemic, delayed neuronal death in gerbil brain. Neurosci Lett. 1993;158:105–108. doi: 10.1016/0304-3940(93)90623-s. [DOI] [PubMed] [Google Scholar]