

Abstract
Increases in intracellular Mg2+ (Mg2+i), as observed in transient cardiac ischemia, decrease L-type Ca2+ current of mammalian ventricular myocytes (VMs). However, cardiac ischemia is associated with an increase in sympathetic tone, which could stimulate L-type Ca2+ current. Therefore, the effect of Mg2+i on L-type Ca2+ current in the context of increased sympathetic tone was unclear. We tested the impact of increased Mg2+i on the β-adrenergic stimulation of L-type Ca2+ current. Exposure of acutely dissociated adult VMs to higher Mg2+i concentrations decreased isoproterenol stimulation of the L-type Ca2+ current from 75 ± 13% with 0.8 mM Mg2+i to 20 ± 8% with 2.4 mM Mg2+i. We activated this signaling cascade at different steps to determine the site or sites of Mg2+i action. Exposure of VMs to increased Mg2+i attenuated the stimulation of L-type Ca2+ current induced by activation of adenylyl cyclase with forskolin, inhibition of cyclic nucleotide phosphodiesterases with isobutylmethylxanthine, and inhibition of phosphoprotein phosphatases I and IIA with calyculin A. These experiments ruled out significant effects of Mg2+i on these upstream steps in the signaling cascade and suggested that Mg2+i acts directly on CaV1.2 channels. One possible site of action is the EF-hand in the proximal C-terminal domain, just downstream in the signaling cascade from the site of regulation of CaV1.2 channels by protein phosphorylation on the C terminus. Consistent with this hypothesis, Mg2+i had no effect on enhancement of CaV1.2 channel activity by the dihydropyridine agonist (S)-BayK8644, which activates CaV1.2 channels by binding to a site formed by the transmembrane domains of the channel. Collectively, our results suggest that, in transient ischemia, increased Mg2+i reduces stimulation of L-type Ca2+ current by the β-adrenergic receptor by directly acting on CaV1.2 channels in a cell-autonomous manner, effectively decreasing the metabolic stress imposed on VMs until blood flow can be reestablished.
INTRODUCTION
Transient cardiac ischemia is associated with increased intracellular Mg2+ (Mg2+i; Murphy et al., 1989; Headrick and Willis, 1991) and subsequently with increased sympathetic tone (Remme, 1998). During transient ischemia, Mg-ATP is hydrolyzed and free Mg2+i levels rise (Murphy et al., 1989). Mg2+i reduces the amplitude (White and Hartzell, 1988; Wang et al., 2004; Brunet et al., 2005) and increases the voltage-dependent inactivation of L-type Ca2+ current (ICa,L) in ventricular myocytes (VMs; Hartzell and White, 1989; Brunet et al., 2009). ICa,L in VMs is conducted by CaV1.2 channels consisting of a pore-forming α11.2-subunit in association with β- and α2δ-subunits (Catterall, 2000). The α1-subunits are composed of four homologous domains (I–IV) with six transmembrane segments (S1–S6) and a reentrant pore loop in each. Multiple regulatory sites are located in the large C-terminal domain (De Jongh et al., 1996; Peterson et al., 1999; Zühlke et al., 1999; Hulme et al., 2003), which is subject to in vivo proteolytic processing near its center (De Jongh et al., 1991; De Jongh et al., 1996; Hulme et al., 2005). An IQ motif in the proximal C terminus is implicated in Ca2+/calmodulin-dependent inactivation (Peterson et al., 1999; Zühlke et al., 1999). Noncovalent interaction of the distal C terminus with the proximal C-terminal domain has an autoinhibitory effect by reducing coupling efficiency of gating charge movement to channel opening (Hulme et al., 2006b). The proximal C-terminal domain contains an EF-hand motif that mediates inhibition of ICa,L by Mg2+i in the same concentration range that is reached in transient ischemia (Brunet et al., 2005, 2009).
In mammalian heart, activation of β-adrenergic receptors (β-ARs) increases contractility and heart rate (Osterrieder et al., 1982). Epinephrine or norepinephrine binding to β-AR leads to activation of the stimulatory guanine nucleotide–binding protein Gs by promoting the exchange of GDP for GTP and dissociation from Gβγ-subunits. GTP-bound Gsα binds to and stimulates adenylyl cyclase (AC), which converts ATP to cAMP (Taussig and Gilman, 1995). Binding of cAMP to the regulatory subunits of PKA results in liberation of catalytic subunits (Krebs and Beavo, 1979), which increase the amplitude of ICa,L (Tsien et al., 1972; Reuter, 1983; Kameyama et al., 1985, 1986; Catterall, 2000) by phosphorylation of a specific serine residue at the interface of the distal and proximal C-terminal domains of CaV1.2 channels (Fuller et al., 2010). The β-AR/AC/PKA cascade is negatively regulated at multiple sites, including dephosphorylation of CaV1.2 channels by phosphoprotein phosphatase 2A (PP2A; Verde et al., 1999; Hall et al., 2006), degradation of cAMP by cyclic nucleotide phosphodiesterases (PDE4 and PDE3; Verde et al., 1999; Leroy et al., 2008), reduction of AC activity by increased intracellular Ca2+ (Ishikawa and Homcy, 1997; Beazely and Watts, 2006), and hydrolysis of GTP by the intrinsic GTPase activity of the Gsα (Morris and Malbon, 1999).
Increases in Mg2+i, as observed in transient cardiac ischemia (Murphy et al., 1989), decrease ICa,L of mammalian VMs (White and Hartzell, 1988; Wang et al., 2004; Brunet et al., 2005). Transient cardiac ischemia leads to an increase in sympathetic tone (Remme, 1998), which could stimulate ICa,L via the PKA signaling cascade. Therefore, the effect of Mg2+i on ICa,L in the context of increased sympathetic tone was unclear. We observed that higher [Mg2+]i attenuated the stimulatory effect of the β-AR cascade on ICa,L. Mg2+i could affect the β-AR/AC/PKA signaling cascade at Gs proteins (Alvarez and Bruno, 1977), AC V (Cech et al., 1980; Iyengar and Birnbaumer, 1982), PDE (Alvarez et al., 1995), or CaV1.2 channels (Brunet et al., 2005, 2009). We found that Mg2+i reduces the stimulation of ICa,L by activation of PKA signaling at each step in this cascade, consistent with direct inhibition of the CaV1.2 α1-subunit in VMs by binding of Mg2+ to the proximal C-terminal EF-hand motif.
MATERIALS AND METHODS
Materials
Isoproterenol, forskolin, isobutylmethylxanthine (IBMX), and (S)-BayK8644 were purchased from Sigma-Aldrich. (R)-rolipram and cilostamide were from obtained from Tocris Bioscience. PKA peptide inhibitor (14–22 amide, myristoylated), PKC peptide inhibitor (20–28 amide, myristoylated), and Calyculin A were obtained from EMD.
Isolation of VMs
Left VMs were isolated from female adult 8–12-wk-old C57BL/6 mice and maintained at 37°C until use, as previously described (Brunet et al., 2004). All protocols were approved by the University of Washington Institutional Animal Care and Use Committee.
Electrophysiology
The electrophysiological recordings were obtained as previously published (Brunet et al., 2009). In brief, patch pipettes (2.5–3.5 MΩ) were pulled from micropipette glass (VWR Scientific) and fire polished. Currents were recorded with an Axopatch 200B amplifier (Molecular Devices) and sampled at 5 kHz after anti-alias filtering at 2 kHz. Data acquisition and command potentials were controlled by Pulse (Pulse 8.50; HEKA), and data were stored for offline analysis. Voltage protocols were delivered at 10-s intervals, and leak and capacitive transients were subtracted using a P/4 protocol. Approximately 80% of series resistance was compensated with the voltage-clamp amplifier circuitry.
For whole-cell voltage-clamp recordings of VM ICa,L with Ca2+ or Ba2+ as charge carrier (ICa,L or IBa,L), the extracellular solution contained (in mM): 1.8 CaCl2 (or BaCl2), 140 TEA, 2 MgCl2, 10 d-glucose, and 10 HEPES, pH 7.3 with CsOH. The normal Mg2+ intracellular solution (0.8 mM Mg2+i) contained (in mM): 100 CsCl, 20 TEA, 10 EGTA, 10 HEPES, 5 MgATP, and 1 MgCl2 titrated to pH 7.3 with CsOH (Brunet et al., 2009). Mg2+i concentration was altered by changing the amount of MgCl2 added. Free Mg2+i was calculated by the Maxchelator program (Bers et al., 1994).
Data analysis
Voltage-clamp data were compiled and analyzed using IGOR Pro (WaveMetrics Inc.) and Excel (Microsoft). Peak ICa,L and IBa,L were measured during 300-ms depolarization to potentials between −50 and 70 mV. ICa,L density (pA/pF) was defined as the peak current elicited by the voltage depolarization normalized to the whole-cell membrane capacitance (within the same myocyte). Voltage shifts were calculated from individual I-V relationships to determine the voltage at which peak current density was observed.
All data are presented as mean ± SEM. Where no error bars are shown, errors are smaller than the symbols. The statistical significance of differences between the various experimental groups was evaluated using the Student’s t test, one-way ANOVA, or Newman–Keuls test; p-values are presented in the text.
RESULTS
[Mg2+]i reduces isoproterenol stimulation of L-type Ca2+ current
[Mg2+]i is an important regulator of ICa,L, but it is not known whether this regulation would also affect β-AR stimulation of ICa,L via the cAMP–PKA signaling pathway. Consistent with previous results (White and Hartzell, 1988; Wang et al., 2004; Brunet et al., 2005), increasing [Mg2+]i from 0.8 to 2.4 mM decreased basal unstimulated ICa,L ∼37%, from −7.9 ± 0.6 (n = 11) to −5.0 ± 0.3 pA/pF (n = 7; P < 0.01). Treatment with 1 µM isoproterenol increased ICa,L with an intracellular solution of 0.8 mM [Mg2+]i (Fig. 1 A). The peak amplitude of ICa,L increased, and the current-voltage (I-V) relationship shifted to more negative values (approximately −10 mV; Fig. 1 C). This increase was much reduced by 2.4 mM [Mg2+]i, a pathophysiologically relevant Mg2+i concentration in ischemia (Fig. 1, B and E; Murphy et al., 1989). Increasing [Mg2+]i from 0.8 to 2.4 mM decreased the isoproterenol stimulation of ICa,L from 0.75 ± 0.13 (n = 11) to 0.20 ± 0.08 (n = 7, P < 0.01; Fig. 1 D). However, increasing [Mg2+]i did not prevent the isoproterenol-induced negative shift of the I-V relationship of ICa,L (Fig. 1 F). Similar results were observed with 7.2 mM [Mg2+]i (not depicted).
Figure 1.

Effects of Mg2+i on the β-AR stimulation of ICa,L. (A) Effect of 1 µM isoproterenol (Iso) on peak ICa,L (black ICa,L traces are before isoproterenol perfusion) with 0.8 mM Mg2+i. Calibration bar: 2.5 pA/pF, 50 ms. (B) Effect of 1 µM isoproterenol on peak ICa,L (black ICa,L traces are before isoproterenol perfusion) with 2.4 mM Mg2+i. (C) Mean I-V relationship for experiments with 0.8 mM Mg2+i as described in A (n = 11). (D) Mean I-V relationship for experiments with 2.4 mM Mg2+i as described in B (n = 7). (E) Effect of increased [Mg2+]i on isoproterenol stimulation of peak ICa,L. (F) Effect of increased [Mg2+]i on the isoproterenol-induced negative shift in the I-V relationship of ICa,L. The dotted lines in A and B represent zero current level. Data are presented as mean ± SEM (some errors are smaller than the symbols; *, P < 0.01).
Ca2+ flowing through L-type Ca2+ channels enhances Ca2+-dependent inactivation and activates other Ca2+-dependent regulatory processes (Kamp and Hell, 2000). To determine whether the effect of Mg2+i on β-AR regulation requires Ca2+ entry, we substituted Ba2+ for Ca2+ as charge carrier in the recording solution and recorded IBa,L (Fig. 2). As with Ca2+ as charge carrier, isoproterenol increased IBa,L amplitude and caused a negative shift in the I-V relation (Fig. 2, A and C; Nguemo et al., 2009). Increasing Mg2+i from 0.8 to 2.4 mM decreased IBa,L ∼32%, from −7.2 ± 0.4 (n = 48) to −4.9 ± 0.4 pA/pF (n = 21; P < 0.01), in agreement with previous work (Hartzell and White, 1989). Increased Mg2+i also prevented the isoproterenol stimulation of IBa,L amplitude (Fig. 2, B, D, and E). However, increasing Mg2+i reduced, but did not completely prevent, the isoproterenol-stimulated negative shift in the I-V relation of IBa,L (Fig. 2 F). Similar results were observed with 7.2 mM Mg2+i (not depicted).
Figure 2.

Effects of Mg2+i on the β-AR stimulation of IBa,L. (A) Effect of 1 µM isoproterenol (Iso) on peak IBa,L (black traces are before isoproterenol perfusion) with 0.8 mM Mg2+i. Calibration bar: 2.5 pA/pF, 50 ms. (B) Effect of isoproterenol on peak IBa,L with 2.4 mM Mg2+i. (C) Mean I-V relationship for experiments with 0.8 mM Mg2+i as described in A (n = 6). (D) Mean I-V relationship for experiments with 2.4 mM Mg2+i (n = 4). (E) Effect of increased [Mg2+]i on isoproterenol stimulation of peak IBa,L. (F) Effect of increased [Mg2+]i on isoproterenol shift in the I-V relationship of IBa,L. The dotted lines in A and B represent zero current level. Data are presented as mean ± SEM (some errors are smaller than the symbols; *, P < 0.05).
Site of action of Mg2+i in the β-AR/Gs/AC/PKA cascade
Several mechanisms could explain the [Mg2+]i modulation of the β-AR stimulation of ICa,L. Multiple steps of the β-AR/Gs/AC/PKA cascade are Mg2+i dependent, including β-AR/Gs (White and Hartzell, 1989), AC (Cech et al., 1980), PDE (PDE4D3; Alvarez et al., 1995), PP2C (Mumby and Walter, 1993), and the Ca2+ channel itself (White and Hartzell, 1988; Brunet et al., 2005, 2009). We stimulated this signaling cascade at different steps and determined the effect of Mg2+i on ICa,L stimulation.
Treatment with forskolin, an AC activator, increased IBa,L amplitude and caused a negative shift in the I-V relationship (Fig. 3, A and C), as expected (Lemke et al., 2008). Increased [Mg2+]i prevented forskolin stimulation of ICa,L (Fig. 3, B, D, and E) but did not completely prevent the forskolin-induced negative shift in the I-V relationship of ICa,L (Fig. 3 F). These results show that the site of action of Mg2+i is downstream from β-AR and AC.
Figure 3.

Effects of Mg2+i on adenylate cyclase stimulation of IBa,L. (A) Effect of 10 µM forskolin (FSK) on peak IBa,L (black IBa,L traces are before FSK perfusion). Calibration bar: 2.5 pA/pF, 50 ms. (B) Effect of forskolin on peak IBa,L with 2.4 mM Mg2+i. (C) Mean I-V relationship from experiments with 0.8 mM Mg2+i as described in A (n = 10). (D) Mean peak stimulation of IBa,L with forskolin. (E) Mean I-V relationship with 2.4 mM Mg2+i as in B. (F) I-V relationship from E normalized to maximum conductance values at V = 10–30 mV. The dotted lines in A and B represent zero current level. Data are presented as mean ± SEM (some errors are smaller than the symbols; *, P < 0.01).
Increased phosphodiesterase activity could lead to decreased cAMP levels (Alvarez et al., 1995) as a result of increased Mg2+i. PDE3 and PDE4 are the main phosphodiesterase subtypes expressed in mammalian heart, and their inhibition with IBMX, a nonselective PDE inhibitor, leads to increased ICa,L amplitude (Leroy et al., 2008). In mammalian heart, blockade of PDE activity leads to an increase in cAMP level because AC has significant basal activity (Verde et al., 1999; Leroy et al., 2008). Treatment with 100 µM IBMX increased IBa,L amplitude and caused a negative shift in the I-V relation (Fig. 4, A and C). Increased [Mg2+]i prevented the IBMX stimulation of IBa,L (Fig. 4, B, D, and E), but a small negative shift in the I-V relationship remained (Fig. 4 F). These results suggest that the main site of Mg2+i action is not the PDE and is downstream from cAMP formation and/or degradation.
Figure 4.

Effects of Mg2+i on the stimulation of IBa,L by inhibition of cyclic nucleotide phosphodiesterases. (A) Effect of 100 µM IBMX on peak IBa,L with 0.8 mM Mg2+i (n = 8; black IBa,L traces are before IBMX perfusion). Calibration bar: 2.5 pA/pF, 50 ms. (B) Effect of 100 µM IBMX on peak IBa,L with 2.4 mM Mg2+i. (C) Mean I-V relationship from experiments with 0.8 mM Mg2+i as described in A (n = 8). (D) Mean I-V relationship from experiments with 2.4 mM Mg2+i (n = 4). (E) Effect of increased [Mg2+]i on IBMX stimulation of peak IBa,L. (F) Effect of increased [Mg2+]i on the IBMX-induced negative shift in the I-V relationship of IBa,L. The dotted lines in A and B represent zero current level. Data are presented as mean ± SEM (some errors are smaller than the symbols; *, P < 0.01).
To determine which PDEs were involved in mediating the effects of IBMX on IBa,L in adult mouse VMs, we used specific inhibitors directed at PDE3 and PDE4, the dominant PDEs expressed in the murine heart (Leroy et al., 2008). The stimulatory effect of IBMX on ICa,L was recapitulated with combined inhibition of PDE4 (R-rolipram) and PDE3 (cilostamide; Fig. 5 A). Treatment with R-rolipram alone did not stimulate IBa,L (Fig. 5 B). Cilostamide application resulted in a substantial increase in IBa,L at −20 mV (P < 0.05) and a leftward shift of the I-V relation (Fig. 5 C). The increase in IBa,L observed with the combination of R-rolipram and cilostamide (Fig. 5 A) is greater than the sum of the effects of the two individual drugs (Fig. 5, B and C), suggesting that these two isoforms can compensate for each other and therefore that their combined inhibition is synergistic. In contrast, comparison of the effects of these inhibitors on the voltage dependence of activation reveals that the effect of cilostamide is dominant and addition of R-rolipram has at most a small effect (Fig. 5 D). Our results are similar to those obtained from adult rat VMs (Verde et al., 1999; Leroy et al., 2008) and suggest that similar PDEs are involved in modulating ICa,L in rodent hearts. Increased Mg2+i inhibits the increase in IBa,L caused by inhibition of these two PDEs, indicating that it acts downstream of cAMP in the PKA signaling cascade.
Figure 5.

Effect of PDE3 and PDE4 inhibition on IBa,L. (A) Mean I-V relationship for effect of combined inhibition of PDE3 (1 µM cilostamide [Cilo]) and PDE4 (10 µM R-rolipram [Rol]) on IBa,L (n = 4; *, P < 0.01). (B) Mean I-V relationships for effect of PDE4 inhibition (10 µM R-rolipram) on IBa,L (n = 5). (C) Mean I-V relationships for effect of PDE3 inhibition (1 µM cilostamide) on IBa,L (n = 7; *, P < 0.05). (D) Effects of PDE inhibition on the negative voltage shift in the I-V relationship of IBa,L (*, P < 0.05). Data are presented as mean ± SEM (some errors are smaller than the symbols).
Effect of Mg2+i on the increase in CaV1.2 channel activity by PPs
The action of protein kinases on the CaV1.2 complex is counteracted by PPs. Treatment with calyculin A, a selective inhibitor of PP1 and PP2A, stimulates ICa,L of VMs by relieving PP reversal of the action of protein kinases (Hartzell et al., 1995; duBell and Rogers, 2004). We tested whether the effect of Mg2+i on β-AR stimulation of L-type Ca2+ current was mediated by activation of PP1 and PP2A by using calyculin A to inhibit them. In agreement with previous reports, exposure of VMs to calyculin A increased IBa,L (Fig. 6, A and B; Hartzell et al., 1995; duBell and Rogers, 2004). Dialysis of cells with 7.2 mM Mg2+i reduced basal IBa,L (−4.3 ± 0.7 pA/pF with 0.8 mM Mg2+i (n = 6); −3.4 ± 0.3 pA/pF (n = 8) with 7.2 mM Mg2+i) and produced a strong inhibition of IBa,L that had been prestimulated with calyculin A (Fig. 6 C). The degree of inhibition indicated that Mg2+i both prevented up-regulation of IBa,L and inhibited the basal level of IBa,L. Thus, Mg2+i does not act by increasing the activity of PP1 and PP2A.
Figure 6.

Effect of Mg2+i on increased IBa,L induced by calyculin A. (A) Effect of 100 nM calyculin A on peak IBa,L (black trace is before calyculin A application). Calibration bar: 2.5 pA/pF, 50 ms. (B) Mean I-V relationship for experiments as described in A (control, n = 10; calyculin A, n = 16). (C) Effect of increased [Mg2+]i (7.2 mM; n = 6) on the increase in peak IBa,L induced by calyculin A (n = 16). (D) Effect of PKA inhibition and PKC inhibition on peak IBa,L prestimulated by calyculin A. PKA was inhibited by the preincubation of PKA inhibitor (PKI)14–22 amide, 5 µM myristoylated peptide (n = 12). PKC was inhibited by the preincubation of PKC inhibitor 20–28 amide, 24 µM myristoylated peptide (n = 8), and 1 µM Ro 31–8220 (n = 5). Myristoyl peptide inhibitors were incubated with cells for 10 min in standard extracellular solution before beginning recordings. The dotted line in A represents zero current level. Data are presented as mean ± SEM (some errors are smaller than the symbols; *, P < 0.01).
We investigated the role of PKA and PKC in calyculin A stimulation of IBa,L. In agreement with previous work, calyculin A stimulation of IBa,L is not altered by inhibition of PKA (Fig. 6 D; Hartzell et al., 1995; duBell and Rogers, 2004). The kinase inhibitor PKI (PKA inhibitor 14–22 myristoylated peptide) blocked isoproterenol stimulation of L-type Ca2+ current at 5 µM (Fig. 7). However, PKI did not prevent calyculin A stimulation of IBa,L (Fig. 6 D). The PKC inhibitor myristoyl PKCI[20–28] also did not impact calyculin A stimulation of IBa,L (Fig. 6 D), in agreement with previous work (Hartzell et al., 1995). These results show that the increase in channel activity caused by calyculin-enhanced phosphorylation of CaV1.2 channels or associated regulatory proteins by protein kinases other than PKA is effectively inhibited by Mg2+i. The ability of Mg2+i to both inhibit CaV1.2 channel activity stimulated by PKA in response to treatment with isoproterenol and to inhibit stimulation by other protein kinases whose phosphorylation of the channel is increased in the presence of calyculin A suggests that Mg2+i acts downstream of these protein phosphorylation reactions in the regulatory cascade, directly on the CaV1.2 channel protein itself.
Figure 7.
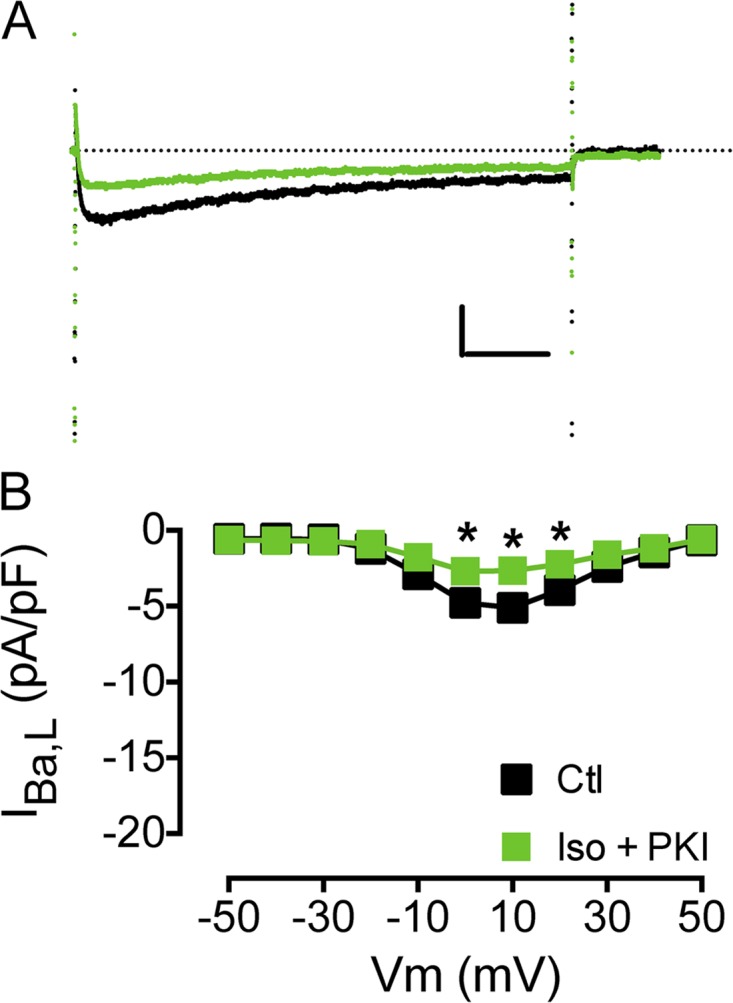
Effects of PKA inhibition on isoproterenol stimulation of IBa,L. (A and B) Effect of PKI perfusion on isoproterenol (Iso) stimulation of peak IBa,L before (black) and after 1 µM isoproterenol and PKI (PKA inhibitor 14–22 amide, 5 µM myristoylated peptide [n = 5]; green; A) and I-V relationship (*, P < 0.05; B). Calibration bar: 2.5 pA/pF, 50 ms. The dotted line in A represents zero current level. Data are presented as mean ± SEM (some errors are smaller than the symbols).
Effect of Mg2+i on activation of CaV1.2 channels by (S)-BayK8644
The increase of CaV1.2 current by the β-AR/Gs/AC/PKA cascade is thought to be mediated by the phosphorylation of Ser1700 in the proximal C-terminal domain of CaV1.2 channels and consequent relief of the autoinhibition exerted by the interaction of the proteolytically cleaved distal C terminus with the proximal C terminus (Bünemann et al., 1999; Hulme et al., 2006a,b; Fuller et al., 2010). The inhibitory effect of the distal C-terminal domain requires binding of Mg2+i to the EF-hand in the proximal C-terminal domain (Brunet et al., 2009). Thus, we proposed that the inhibitory effect of the distal C-terminal domain is propagated to the gating apparatus in the transmembrane segments of the channel through coupled conformation changes involving the EF-hand structural element (Brunet et al., 2009). If this model is correct, stimulation of the activity of the CaV1.2 channel by a direct action on the transmembrane domains of the channel should be independent of binding of Mg2+i to the EF-hand in the proximal C-terminal domain. We have tested this idea by examining the effect of Mg2+i on the enhancement of channel activity by the dihydropyridine agonist (S)-BayK8644, which binds to a receptor site formed by nine amino acid residues in the IIIS5, IIIS6, and IVS6 transmembrane segments (Striessnig et al., 1991; Grabner et al., 1996; Hockerman et al., 1997). When VMs were dialyzed with 0.8 mM Mg2+i, subsequent exposure to 0.3 µM (S)-BayK8644 significantly increased IBa,L (Fig. 8, A and C). In contrast to β-AR/Gs/AC/PKA cascade stimulation, when myocytes were dialyzed with 2.4 mM Mg2+i or 7.2 mM Mg2+i (not depicted) and then exposed to (S)-BayK8644, a similar stimulation as in 0.8 mM Mg2+i was observed (Fig. 8, B, D, and E). In addition, when 0.8 mM Mg2+i or 2.4 mM Mg2+i was dialyzed, (S)-BayK8644 exposure caused a similar negative shift in the I-V relation (Fig. 8 F). The most likely interpretation of our results is that Mg2+i acts upstream from the site of action of (S)-BayK8644 on the L-type Ca2+ channel α-subunit but downstream of the site of phosphorylation stimulated by the PKA cascade, consistent with binding at the EF-hand motif in the proximal C-terminal domain.
Figure 8.

Effects of Mg2+i on Ca2+ channel agonist stimulation of IBa,L. (A) Effect of 0.3 µM (S)-BayK8644 on peak IBa,L (black traces are before (S)-BayK8644) with 0.8 mM Mg2+i. Calibration bar: 2.5 pA/pF, 50 ms. (B) Effect of 0.3 µM (S)-BayK8644 on peak IBa,L with 2.4 mM Mg2+i. (C) Mean I-V relationship for experiments as described in A with 0.8 mM Mg2+i (n = 8). (D) Mean I-V relationship for experiments as described in B with 2.4 mM Mg2+i (n = 6). (E) Effect of increased [Mg2+]i on (S)-BayK8644 stimulation of peak IBa,L. (F) Effect of increased [Mg2+]i on the negative shift of voltage dependence induced by (S)-BayK8644. The dotted lines in A and B represent zero current level. Data are presented as mean ± SEM (some errors are smaller than the symbols; *, P < 0.01).
DISCUSSION
Our results show that increases in Mg2+i significantly attenuate β-AR stimulation of ICa,L of VMs. Based on electrophysiological and pharmacological experiments, we propose that Mg2+i acts directly on the CaV1.2 channel complex, most likely on the EF-hand in the proximal C-terminal domain, to attenuate the β-adrenergic stimulation of ICa,L. This inhibition of CaV1.2 channels by Mg2+ is likely to have pathophysiological and therapeutic significance in the context of ischemia as discussed below.
Mg2+i attenuates β-AR stimulation of L-type Ca2+ current
Transient ischemia is associated with both an increase in free [Mg2+]i and subsequently with an increase in sympathetic tone (Murphy et al., 1989; Remme, 1998). Increases in [Mg2+]i decrease ICa,L in VMs (White and Hartzell, 1988; Wang et al., 2004; Brunet et al., 2005, 2009). In contrast, β-AR stimulation increases ICa,L. We found that increased Mg2+i attenuated isoproterenol stimulation of ICa,L and IBa,L. This finding was not anticipated from prior studies, which suggested that PKA phosphorylation could either increase or decrease the potency of Mg2+i to inhibit ICa,L (White and Hartzell, 1988; Yamaoka and Seyama, 1998; Pelzer et al., 2001; Wang et al., 2004). These investigators first activated PKA by increasing cAMP concentration and then tested the effect of increases in Mg2+i concentration subsequent to PKA activation and channel phosphorylation (White and Hartzell, 1988; Yamaoka and Seyama, 1998; Pelzer et al., 2001; Wang et al., 2004). In contrast, we exposed VMs to increased [Mg2+]i and then stimulated them with isoproterenol to more closely mimic the sequence of events observed in transient ischemia (Murphy et al., 1989; Remme, 1998). Under these conditions, increased [Mg2+]i effectively inhibits β-AR stimulation of ICa,L. These results predict that the activation of β-AR signaling cascade during transient ischemia would be opposed by increased [Mg2+]i, but only in cells whose ATP concentration is decreased, thereby providing a cell-autonomous effect to prevent increased entry of Ca2+ in cells with impaired metabolic status. This effect would add to the cell-autonomous inhibition of basal activity of CaV1.2 channels by [Mg2+]i, which we demonstrated in previous work (Brunet et al., 2005, 2009). Together, these two parallel actions could have an important cardioprotective effect on cardiac myocytes experiencing a decrease in intracellular ATP concentration as a result of ischemia.
Site of Mg2+i action in the β-AR/Gs/AC/PKA cascade
Where does Mg2+i act in the β-AR/AC/PKA signaling cascade? To determine the sites of action of Mg2+i in the β-AR/AC/PKA cascade, we stimulated this cascade at multiple steps. We found that increased [Mg2+]i inhibits β-AR stimulation no matter where the signaling cascade is activated. Our data show that the site of Mg2+i action is downstream from β-AR/Gs/AC activation, cAMP formation or degradation, and dephosphorylation by PPs but upstream of the dihydropyridine agonist (S)-BayK8644 that acts directly on a receptor site in the transmembrane core of the CaV1.2 channel. In vitro experiments have shown that the catalytic (C) subunit of PKA can bind two Mg2+ (Zheng et al., 1993a,b) at a high affinity site and a low affinity site (Shaffer and Adams, 1999; Zimmermann et al., 2008). At resting Mg2+i and ATP concentrations, both Mg2+-binding sites of the C-subunit of PKA are occupied. Binding of Mg2+ at the high affinity site is important for enzyme activity, whereas binding of Mg2+ at the low affinity site is important for the binding of the type I PKA regulatory subunit and PKI to maintain the C-subunit in an inactive state in resting conditions (Zimmermann et al., 2008). As these sites are both occupied by Mg2+i under resting conditions, it is unlikely that they would be involved in inhibition of the β-AR stimulation of ICa,L as Mg2+i increases.
Direct action of Mg2+i on CaV1.2 channels
Overall, our results are most consistent with a direct action of [Mg2+]i on the EF-hand in the proximal C-terminal domain, as described previously (Brunet et al., 2005, 2009). Full Mg2+i occupancy of the EF-hand of CaV1.2 α-subunit could prevent transduction of the effect of PKA phosphorylation to the transmembrane body of the CaV1.2 channel. This EF-hand motif is positioned between the site of regulation by the distal C-terminal domain and PKA phosphorylation, which are located more distally in the amino acid sequence of the C terminus (Brunet et al., 2009; Fuller et al., 2010), and the site of action of (S)-BayK8644 in the transmembrane core of the channel (Striessnig et al., 1991; Grabner et al., 1996; Hockerman et al., 1997). Therefore, binding of Mg2+i to the proximal C-terminal EF-hand enhances the autoinhibitory effect of the noncovalently associated distal C terminus, as we showed previously (Brunet et al., 2009), and also prevents PKA phosphorylation from reversing the autoinhibitory effect of the distal C terminus when the β-AR signaling cascade is activated. Interestingly, Ca2+/calmodulin interacting with the C-terminal IQ motif, adjacent to the EF-hand, was also proposed to be important in regulating β-AR stimulation of L-type Ca2+ current (Walsh and Cheng, 2004). We propose that the effects of phosphorylation of the CaV1.2 α-subunit on its C-terminal phosphorylation sites are transmitted through the more proximal regulatory structural elements, including the IQ motif and EF-hand, which are positioned between the distal C terminus and the transmembrane core of the channel. Further experiments involving structure–function analysis in transfected cells will be required to test this hypothesis.
In a previous study, we found that PKA regulation of CaV1.2 channels can be reconstituted in nonmuscle cells by expression of CaV1.2Δ1800, the separate distal C-terminal domain CaV1.2(1801–2171), and A-Kinase Anchoring Protein 15 (Fuller et al., 2010). Phosphorylation of Ser1700 at the interface between the distal and proximal regions of the C terminus was both necessary and sufficient for the increase in peak current and in coupling of gating charge movement to channel opening (Fuller et al., 2010), but the negative shift in voltage dependence observed in cardiac myocytes was not fully reconstituted in this system and may require additional regulatory mechanisms. In light of this apparent difference in mechanism of regulation, it is not surprising that our experiments here show that Mg2+i reduces peak CaV1.2 current more completely than it prevents the negative shift in voltage dependence and that different mechanisms of activating the PKA signaling cascade have quantitatively different effects on the negative shift in voltage dependence. Further work on the basic mechanism of regulation of CaV1.2 channels by PKA and other protein kinases will be required to fully understand the relationship between regulation of peak Ca2+ currents and the voltage dependence of activation.
Therapeutic implications
We suggest that inhibition of CaV1.2 channels by transient increases in Mg2+i is an integral component of a cell autonomous “stress response” for VMs during transient ischemia, designed to keep intracellular Ca2+ at low levels to decrease the VM contractile function and related metabolic needs until the stressful episode subsides. Therefore, patients at risk of cardiac stress should have deficiencies in free Mg2+i corrected. MagT1 and TUSC3 are important Mg2+ transporters in vertebrates (Zhou and Clapham, 2009), and their regulation by physiological events and/or pharmacological agents may be significant in controlling cellular response to ischemia. Overall our study suggests that increased Mg2+i, as observed in transient ischemia, is important because it acts in a cell-autonomous manner to maintain physiological intracellular Ca2+ concentration at a low level by reducing the L-type Ca2+ current and consequently lowering energy expenditure, even in the face of increased sympathetic tone.
Acknowledgments
We would like to acknowledge Dr. Selva Baltan for constructive discussions and insightful comments.
This work was supported by the American Heart Association (National Development Award to S. Brunet) and the National Institutes of Health (grant R01 HL085372 to W.A. Catterall).
Lawrence G. Palmer served as editor.
Footnotes
Abbreviations used in this paper:
- AC
- adenylyl cyclase
- β-AR
- β-adrenergic receptor
- IBMX
- isobutylmethylxanthine
- VM
- ventricular myocyte
References
- Alvarez R., Bruno J.J. 1977. Activation of cardiac adenylate cyclase: hormonal modification of the magnesium ion requirement. Proc. Natl. Acad. Sci. USA. 74:92–95 10.1073/pnas.74.1.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez R., Sette C., Yang D., Eglen R.M., Wilhelm R., Shelton E.R., Conti M. 1995. Activation and selective inhibition of a cyclic AMP-specific phosphodiesterase, PDE-4D3. Mol. Pharmacol. 48:616–622 [PubMed] [Google Scholar]
- Beazely M.A., Watts V.J. 2006. Regulatory properties of adenylate cyclases type 5 and 6: A progress report. Eur. J. Pharmacol. 535:1–12 10.1016/j.ejphar.2006.01.054 [DOI] [PubMed] [Google Scholar]
- Bers D.M., Patton C.W., Nuccitelli R. 1994. A practical guide to the preparation of Ca2+ buffers. Methods Cell Biol. 40:3–29 10.1016/S0091-679X(08)61108-5 [DOI] [PubMed] [Google Scholar]
- Brunet S., Aimond F., Li H., Guo W., Eldstrom J., Fedida D., Yamada K.A., Nerbonne J.M. 2004. Heterogeneous expression of repolarizing, voltage-gated K+ currents in adult mouse ventricles. J. Physiol. 559:103–120 10.1113/jphysiol.2004.063347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet S., Scheuer T., Klevit R., Catterall W.A. 2005. Modulation of CaV1.2 channels by Mg2+ acting at an EF-hand motif in the COOH-terminal domain. J. Gen. Physiol. 126:311–323 10.1085/jgp.200509333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet S., Scheuer T., Catterall W.A. 2009. Cooperative regulation of Cav1.2 channels by intracellular Mg2+, the proximal C-terminal EF-hand, and the distal C-terminal domain. J. Gen. Physiol. 134:81–94 10.1085/jgp.200910209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bünemann M., Gerhardstein B.L., Gao T., Hosey M.M. 1999. Functional regulation of L-type calcium channels via protein kinase A-mediated phosphorylation of the β2 subunit. J. Biol. Chem. 274:33851–33854 10.1074/jbc.274.48.33851 [DOI] [PubMed] [Google Scholar]
- Catterall W.A. 2000. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 16:521–555 10.1146/annurev.cellbio.16.1.521 [DOI] [PubMed] [Google Scholar]
- Cech S.Y., Broaddus W.C., Maguire M.E. 1980. Adenylate cyclase: the role of magnesium and other divalent cations. Mol. Cell. Biochem. 33:67–92 10.1007/BF00224572 [DOI] [PubMed] [Google Scholar]
- De Jongh K.S., Warner C., Colvin A.A., Catterall W.A. 1991. Characterization of the two size forms of the α 1 subunit of skeletal muscle L-type calcium channels. Proc. Natl. Acad. Sci. USA. 88:10778–10782 10.1073/pnas.88.23.10778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jongh K.S., Murphy B.J., Colvin A.A., Hell J.W., Takahashi M., Catterall W.A. 1996. Specific phosphorylation of a site in the full-length form of the alpha 1 subunit of the cardiac L-type calcium channel by adenosine 3′,5′-cyclic monophosphate-dependent protein kinase. Biochemistry. 35:10392–10402 10.1021/bi953023c [DOI] [PubMed] [Google Scholar]
- duBell W.H., Rogers T.B. 2004. Protein phosphatase 1 and an opposing protein kinase regulate steady-state L-type Ca2+ current in mouse cardiac myocytes. J. Physiol. 556:79–93 10.1113/jphysiol.2003.059329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller M.D., Emrick M.A., Sadilek M., Scheuer T., Catterall W.A. 2010. Molecular mechanism of calcium channel regulation in the fight-or-flight response. Sci. Signal. 3:ra70 10.1126/scisignal.2001152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabner M., Wang Z.Y., Hering S., Striessnig J., Glossmann H. 1996. Transfer of 1,4-dihydropyridine sensitivity from L-type to class A (BI) calcium channels. Neuron. 16:207–218 10.1016/S0896-6273(00)80037-9 [DOI] [PubMed] [Google Scholar]
- Hall D.D., Feekes J.A., Arachchige Don A.S., Shi M., Hamid J., Chen L., Strack S., Zamponi G.W., Horne M.C., Hell J.W. 2006. Binding of protein phosphatase 2A to the L-type calcium channel Cav1.2 next to Ser1928, its main PKA site, is critical for Ser1928 dephosphorylation. Biochemistry. 45:3448–3459 10.1021/bi051593z [DOI] [PubMed] [Google Scholar]
- Hartzell H.C., White R.E. 1989. Effects of magnesium on inactivation of the voltage-gated calcium current in cardiac myocytes. J. Gen. Physiol. 94:745–767 10.1085/jgp.94.4.745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartzell H.C., Hirayama Y., Petit-Jacques J. 1995. Effects of protein phosphatase and kinase inhibitors on the cardiac L-type Ca current suggest two sites are phosphorylated by protein kinase A and another protein kinase. J. Gen. Physiol. 106:393–414 10.1085/jgp.106.3.393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Headrick J.P., Willis R.J. 1991. Cytosolic free magnesium in stimulated, hypoxic, and underperfused rat heart. J. Mol. Cell. Cardiol. 23:991–999 10.1016/0022-2828(91)91635-5 [DOI] [PubMed] [Google Scholar]
- Hockerman G.H., Peterson B.Z., Sharp E., Tanada T.N., Scheuer T., Catterall W.A. 1997. Construction of a high-affinity receptor site for dihydropyridine agonists and antagonists by single amino acid substitutions in a non-L-type Ca2+ channel. Proc. Natl. Acad. Sci. USA. 94:14906–14911 10.1073/pnas.94.26.14906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme J.T., Lin T.W., Westenbroek R.E., Scheuer T., Catterall W.A. 2003. β-adrenergic regulation requires direct anchoring of PKA to cardiac CaV1.2 channels via a leucine zipper interaction with A kinase-anchoring protein 15. Proc. Natl. Acad. Sci. USA. 100:13093–13098 10.1073/pnas.2135335100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme J.T., Konoki K., Lin T.W., Gritsenko M.A., Camp D.G., II, Bigelow D.J., Catterall W.A. 2005. Sites of proteolytic processing and noncovalent association of the distal C-terminal domain of CaV1.1 channels in skeletal muscle. Proc. Natl. Acad. Sci. USA. 102:5274–5279 10.1073/pnas.0409885102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme J.T., Westenbroek R.E., Scheuer T., Catterall W.A. 2006a. Phosphorylation of serine 1928 in the distal C-terminal domain of cardiac CaV1.2 channels during beta1-adrenergic regulation. Proc. Natl. Acad. Sci. USA. 103:16574–16579 10.1073/pnas.0607294103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme J.T., Yarov-Yarovoy V., Lin T.W.-C., Scheuer T., Catterall W.A. 2006b. Autoinhibitory control of the CaV1.2 channel by its proteolytically processed distal C-terminal domain. J. Physiol. 576:87–102 10.1113/jphysiol.2006.111799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa Y., Homcy C.J. 1997. The adenylyl cyclases as integrators of transmembrane signal transduction. Circ. Res. 80:297–304 10.1161/01.RES.80.3.297 [DOI] [PubMed] [Google Scholar]
- Iyengar R., Birnbaumer L. 1982. Hormone receptor modulates the regulatory component of adenylyl cyclase by reducing its requirement for Mg2+ and enhancing its extent of activation by guanine nucleotides. Proc. Natl. Acad. Sci. USA. 79:5179–5183 10.1073/pnas.79.17.5179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameyama M., Hofmann F., Trautwein W. 1985. On the mechanism of β-adrenergic regulation of the Ca channel in the guinea-pig heart. Pflugers Arch. 405:285–293 10.1007/BF00582573 [DOI] [PubMed] [Google Scholar]
- Kameyama M., Hescheler J., Hofmann F., Trautwein W. 1986. Modulation of Ca current during the phosphorylation cycle in the guinea pig heart. Pflugers Arch. 407:123–128 10.1007/BF00580662 [DOI] [PubMed] [Google Scholar]
- Kamp T.J., Hell J.W. 2000. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ. Res. 87:1095–1102 10.1161/01.RES.87.12.1095 [DOI] [PubMed] [Google Scholar]
- Krebs E.G., Beavo J.A. 1979. Phosphorylation-dephosphorylation of enzymes. Annu. Rev. Biochem. 48:923–959 10.1146/annurev.bi.48.070179.004423 [DOI] [PubMed] [Google Scholar]
- Lemke T., Welling A., Christel C.J., Blaich A., Bernhard D., Lenhardt P., Hofmann F., Moosmang S. 2008. Unchanged β-adrenergic stimulation of cardiac L-type calcium channels in Cav1.2 phosphorylation site S1928A mutant mice. J. Biol. Chem. 283:34738–34744 10.1074/jbc.M804981200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy J., Abi-Gerges A., Nikolaev V.O., Richter W., Lechêne P., Mazet J.L., Conti M., Fischmeister R., Vandecasteele G. 2008. Spatiotemporal dynamics of β-adrenergic cAMP signals and L-type Ca2+ channel regulation in adult rat ventricular myocytes: role of phosphodiesterases. Circ. Res. 102:1091–1100 10.1161/CIRCRESAHA.107.167817 [DOI] [PubMed] [Google Scholar]
- Morris A.J., Malbon C.C. 1999. Physiological regulation of G protein-linked signaling. Physiol. Rev. 79:1373–1430 [DOI] [PubMed] [Google Scholar]
- Mumby M.C., Walter G. 1993. Protein serine/threonine phosphatases: structure, regulation, and functions in cell growth. Physiol. Rev. 73:673–699 [DOI] [PubMed] [Google Scholar]
- Murphy E., Steenbergen C., Levy L.A., Raju B., London R.E. 1989. Cytosolic free magnesium levels in ischemic rat heart. J. Biol. Chem. 264:5622–5627 [PubMed] [Google Scholar]
- Nguemo F., Sasse P., Fleischmann B.K., Kamanyi A., Schunkert H., Hescheler J., Reppel M. 2009. Modulation of L-type Ca2+ channel current density and inactivation by β-adrenergic stimulation during murine cardiac embryogenesis. Basic Res. Cardiol. 104:295–306 10.1007/s00395-008-0755-7 [DOI] [PubMed] [Google Scholar]
- Osterrieder W., Brum G., Hescheler J., Trautwein W., Flockerzi V., Hofmann F. 1982. Injection of subunits of cyclic AMP-dependent protein kinase into cardiac myocytes modulates Ca2+ current. Nature. 298:576–578 10.1038/298576a0 [DOI] [PubMed] [Google Scholar]
- Pelzer S., La C., Pelzer D.J. 2001. Phosphorylation-dependent modulation of cardiac calcium current by intracellular free magnesium. Am. J. Physiol. Heart Circ. Physiol. 281:H1532–H1544 [DOI] [PubMed] [Google Scholar]
- Peterson B.Z., DeMaria C.D., Adelman J.P., Yue D.T. 1999. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 22:549–558 10.1016/S0896-6273(00)80709-6 [DOI] [PubMed] [Google Scholar]
- Remme W.J. 1998. The sympathetic nervous system and ischaemic heart disease. Eur. Heart J. 19:F62–F71 [PubMed] [Google Scholar]
- Reuter H. 1983. Calcium channel modulation by neurotransmitters, enzymes and drugs. Nature. 301:569–574 10.1038/301569a0 [DOI] [PubMed] [Google Scholar]
- Shaffer J., Adams J.A. 1999. An ATP-linked structural change in protein kinase A precedes phosphoryl transfer under physiological magnesium concentrations. Biochemistry. 38:5572–5581 10.1021/bi982768q [DOI] [PubMed] [Google Scholar]
- Striessnig J., Murphy B.J., Catterall W.A. 1991. Dihydropyridine receptor of L-type Ca2+ channels: identification of binding domains for [3H](+)-PN200-110 and [3H]azidopine within the α1 subunit. Proc. Natl. Acad. Sci. USA. 88:10769–10773 10.1073/pnas.88.23.10769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taussig R., Gilman A.G. 1995. Mammalian membrane-bound adenylyl cyclases. J. Biol. Chem. 270:1–4 10.1074/jbc.270.1.1 [DOI] [PubMed] [Google Scholar]
- Tsien R.W., Giles W., Greengard P. 1972. Cyclic AMP mediates the effects of adrenaline on cardiac purkinje fibres. Nat. New Biol. 240:181–183 [DOI] [PubMed] [Google Scholar]
- Verde I., Vandecasteele G., Lezoualc’h F., Fischmeister R. 1999. Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L-type Ca2+ current in rat ventricular myocytes. Br. J. Pharmacol. 127:65–74 10.1038/sj.bjp.0702506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh K.B., Cheng Q. 2004. Intracellular Ca2+ regulates responsiveness of cardiac L-type Ca2+ current to protein kinase A: role of calmodulin. Am. J. Physiol. Heart Circ. Physiol. 286:H186–H194 10.1152/ajpheart.00272.2003 [DOI] [PubMed] [Google Scholar]
- Wang M., Tashiro M., Berlin J.R. 2004. Regulation of L-type calcium current by intracellular magnesium in rat cardiac myocytes. J. Physiol. 555:383–396 10.1113/jphysiol.2003.048538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R.E., Hartzell H.C. 1988. Effects of intracellular free magnesium on calcium current in isolated cardiac myocytes. Science. 239:778–780 10.1126/science.2448878 [DOI] [PubMed] [Google Scholar]
- White R.E., Hartzell H.C. 1989. Magnesium ions in cardiac function. Regulator of ion channels and second messengers. Biochem. Pharmacol. 38:859–867 10.1016/0006-2952(89)90272-4 [DOI] [PubMed] [Google Scholar]
- Yamaoka K., Seyama I. 1998. Phosphorylation modulates L-type Ca channels in frog ventricular myocytes by changes in sensitivity to Mg2+ block. Pflugers Arch. 435:329–337 10.1007/s004240050519 [DOI] [PubMed] [Google Scholar]
- Zheng J., Knighton D.R., ten Eyck L.F., Karlsson R., Xuong N., Taylor S.S., Sowadski J.M. 1993a. Crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MgATP and peptide inhibitor. Biochemistry. 32:2154–2161 10.1021/bi00060a005 [DOI] [PubMed] [Google Scholar]
- Zheng J., Trafny E.A., Knighton D.R., Xuong N.H., Taylor S.S., Ten Eyck L.F., Sowadski J.M. 1993b. 2.2 Å refined crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MnATP and a peptide inhibitor. Acta Crystallogr. D Biol. Crystallogr. 49:362–365 10.1107/S0907444993000423 [DOI] [PubMed] [Google Scholar]
- Zhou H., Clapham D.E. 2009. Mammalian MagT1 and TUSC3 are required for cellular magnesium uptake and vertebrate embryonic development. Proc. Natl. Acad. Sci. USA. 106:15750–15755 10.1073/pnas.0908332106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann B., Schweinsberg S., Drewianka S., Herberg F.W. 2008. Effect of metal ions on high-affinity binding of pseudosubstrate inhibitors to PKA. Biochem. J. 413:93–101 10.1042/BJ20071665 [DOI] [PubMed] [Google Scholar]
- Zühlke R.D., Pitt G.S., Deisseroth K., Tsien R.W., Reuter H. 1999. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 399:159–162 10.1038/20200 [DOI] [PubMed] [Google Scholar]