

Abstract
Prostate cancer is a major cause of male death in the Western world, but few frequent genetic alterations that drive prostate cancer initiation and progression have been identified. β-Catenin is essential for many developmental processes and has been implicated in tumorigenesis in many tissues, including prostate cancer. However, expression studies on human prostate cancer samples are unclear on the role this protein plays in this disease. We have used in vivo genetic studies in the embryo and adult to extend our understanding of the role of β-Catenin in the normal and neoplastic prostate. Our gene deletion analysis revealed that prostate epithelial β-Catenin is required for embryonic prostate growth and branching but is dispensable in the normal adult organ. During development, β-Catenin controls the number of progenitors in the epithelial buds and regulates a discrete network of genes, including c-Myc and Nkx3.1. Deletion of β-Catenin in a Pten deleted model of castration-resistant prostate cancer demonstrated it is dispensable for disease progression in this setting. Complementary overexpression experiments, through in vivo protein stabilization, showed that β-Catenin promotes the formation of squamous epithelia during prostate development, even in the absence of androgens. β-Catenin overexpression in combination with Pten loss was able to drive progression to invasive carcinoma together with squamous metaplasia. These studies demonstrate that β-Catenin is essential for prostate development and that an inherent property of high levels of this protein in prostate epithelia is to drive squamous fate differentiation. In addition, they show that β-Catenin overexpression can promote invasive prostate cancer in a clinically relevant model of this disease. These data provide novel information on cancer progression pathways that give rise to lethal prostate disease in humans.
Author Summary
Prostate cancer is a major cause of male death in the Western world, but few genes involved in this disease have been identified. We have undertaken an in-depth in vivo analysis in the prostate of the β-Catenin protein, which has been shown to be important in many processes during embryogenesis and has been implicated in tumorigenesis. Our studies demonstrate that β-Catenin is essential for prostate development but is dispensable in the normal adult organ. Analysis of a mouse model of a frequently mutated human prostate tumour suppressor, Pten loss, revealed that β-Catenin is not required for neoplastic formation in this model, even in castrated conditions. However, increased β-Catenin levels can cooperate with Pten loss to promote the progression of aggressive invasive prostate cancer together with squamous metaplasia. These data uncover the role of β-Catenin in the prostate and provide new insights on how pathways interact to drive human prostate cancer.
Introduction
Prostate cancer is the most common male cancer in the developed world, and a leading cause of cancer-related death in men. To date, few common genes that promote prostate cancer progression have been identified, including the tumour suppressor PTEN (phosphatase and tensin homolog deleted on chromosome 10), the gene rearrangement TMPRSS2-ERG, and the oncogene C-MYC [1]. There is a need to determine additional drivers of prostate cancer that could be the targets of new therapies, and to determine the genetic and molecular interaction between these signalling pathways. One approach successfully used to identify and study the function of candidate cancer genes has been to investigate genes during embryonic development of the organ. This is a useful method as many genes and pathways involved in development are also reactivated in cancer initiation and progression [2]. Examples of genes that have been found to be important during both prostate development and tumorigenesis include Sox9, Hoxb13 and Nkx3.1 [3], [4], [5]
The prostate develops from the endodermally derived urogenital sinus (UGS) in response to androgens that signal through the androgen receptor (AR) [6]. This occurs at embryonic day (E) 16.5–17.5 in the mouse, when the epithelium buds out and grows into the surrounding mesenchyme. This process requires complex epithelial-mesenchymal interactions and involves signalling pathways such as androgens, FGF and SHH [6]. The buds then elongate, branch, canalize and undergo cytodifferentiation to become secretory after birth. The adult prostate epithelium is composed of p63 expressing basal cells, AR positive luminal cells and rare neuroendocrine cells [7]. The adult normal and neoplastic prostate remains responsive to androgens and castration is a first line of therapy for patients with prostate cancer. However, after an initial response to androgen withdrawal, the tumour grows in a castration-resistant phase [8].
The cytoplasmic protein β-Catenin (encoded by the CTNNB1 gene) is crucial in many steps of embryogenisis and is involved in a number of cancers [9]. β-Catenin forms part of the adherens junction with E-Cadherin and is also a component of canonical WNT signalling [9]. In the absence of WNT ligand, a destruction complex of Axin, APC, GSK3β and CK1α promotes the phosphorylation and subsequent degradation of β-Catenin via the ubiquitin pathway. When WNT ligand binds to the Frizzled/LRP receptor complex, the destruction complex is destabilized and GSK3β is unable to phosphorylate β-Catenin. This leads to an accumulation of β-Catenin that translocates to the nucleus and interacts with the transcription factors TCF/LEF to activate target genes.
Currently, the function of β-Catenin in human prostate cancer is unclear [10]. CTNNB1 mutations in prostate cancer occur rarely, in only 5% of cases [11]. It has been observed that β-Catenin expression and localization change during human prostate cancer progression, however, results are inconsistent. Several studies have seen an increase in β-Catenin expression and nuclear localization in late stage cancer samples, while others have reported a loss in nuclear expression in advanced tumours [12], [13], [14], [15], [16]. In addition to its role in WNT signalling, β-Catenin can act as a co-factor with AR, suggesting it has a role in castration-resistant disease. In prostate cancer cell lines, β-Catenin enhances androgen-stimulated AR transcriptional activation and increases sensitivity to low levels of androgens and to non-androgen ligands [17], [18], [19], [20], [21], [22]. Nuclear localization of β-Catenin may also result in increased complexes between AR and β-Catenin in prostate cancer cells, changing target gene activation [18], [23]. Furthermore, castration-resistant growth of a prostate cancer xenograft model results in increased β-Catenin and AR expression, co-localization in the nucleus and physical interaction of the proteins [24]. Mouse models have demonstrated that activating β-Catenin, through generation of a non-degradable form of this protein, leads to prostate neoplasia and squamous transdifferentiation [25], [26], [27]. Mouse prostates expressing this stabilized form of β-Catenin in combination with either the SV40 large T-antigen (LPB-Tag), which inactivates p53 and Rb, or with a mutated K-ras form invasive carcinoma [28], [29].
PTEN is frequently altered in prostate cancer, with mutations and/or deletions found in 30% of primary cancers and 63% of metastatic prostate tumours [30], [31]. PTEN is a phosphatase that negatively regulates the phosphatidylinositol-3-kinase/Akt (PI3K/Akt) pathway [32]. PTEN loss promotes phosphorylation of Akt through PI3K, which in turn phosphorylates multiple targets including GSK3β. Activation of this pathway results in an increase in cell proliferation, cell survival and protein synthesis [32]. Evidence suggests that β-Catenin can interact with the PI3K/Akt pathway following PTEN loss, through the inactivation of GSK3β and stabilization of β-Catenin. PTEN null prostate cancer cells have increased nuclear β-Catenin expression, TCF promoter activity and expression of the β-Catenin regulated gene Cyclin D1, which are suppressed upon re-expression of wild type PTEN [33], [34].
To better define the function of β-Catenin in the normal and neoplastic prostate we have used in vivo gene deletion and activation (through generation of a stabilized form of the protein) in the embryonic and adult mouse. Loss of Ctnnb1 in epithelia during mouse prostate development results in failure of bud outgrowth and branching, while β-Catenin activation leads to squamous transdifferentiation through an androgen-independent mechanism. Surprisingly, deletion of Ctnnb1 in the adult prostate had no effect on normal prostate homeostasis. To study its function in prostate cancer we analysed the role of β-Catenin in the context of the frequently deleted PTEN gene. We demonstrate that β-Catenin expression is elevated after PTEN loss but plays no function in prostate cancer in intact or castrated animals. However, increasing the level of β-Catenin in combination with PTEN deletion leads to highly invasive prostate tumours with persistent squamous metaplasia.
Results
β-Catenin is required for prostate bud growth
To determine the function of β-Catenin in the prostate during development, we used a conditional gene deletion approach to specifically delete Ctnnb1 in prostate epithelia. We used the Nkx3.1:Cre mouse line to drive the expression of Cre recombinase in epithelial cells at E17.5 just after bud induction, which in combination with a loxP containing Ctnnb1 allele (β-Cat) results in the excision of exons 2 to 6 and the production of a non-functional protein [35]. As the Nkx3.1:Cre mouse line is a knock-in allele and, therefore, heterozygous for Nkx3.1, β-Cat;Nkx3.1:Cre heterozygous animals were used as controls. At E18.5, β-Catenin is expressed at the membrane and in the cytoplasm of epithelial cells throughout the buds of control prostates and is lost in most cells of β-Cat;Nkx3.1:Cre mutant buds, although a small number of cells retain expression (Figure 1A). At this stage, LEF1, a direct transcriptional target of β-Catenin, has strong nuclear expression at the tips of developing control buds, which is lost from most epithelial cells in β-Cat;Nkx3.1:Cre prostates (Figure 1B) [36].
Figure 1. β-Catenin is required for prostate growth and branching.

(A) immunohistochemistry (IHC) on sections of β-Cat;Nkx3.1:Cre mutant and control E18.5 UGS with a β-Catenin antibody. Adjacent panels show high magnification of buds. Note the strong staining in the buds of control prostates that is lost in most cells of mutant prostates. Far right panel is high magnification of a bud that has not lost β-Catenin, indicated with a black arrow. (B) double IHC for β-Catenin and LEF1 on β-Cat;Nkx3.1:Cre and control E18.5 UGS sections. Strong nuclear LEF1 expression is observed in control prostatic buds, but lost in mutant buds. Adjacent panels show buds at high magnification. White outline indicates bud. White cells are autofluorescence from red blood cells. (C) X-gal staining of ROSA-LacZ β-Cat;Nkx3.1:Cre and control prostate organ cultures grown for 5 days. (D) quantitative analysis shows a significant decrease in the total number of prostatic ducts in β-Cat;Nkx3.1:Cre mutants (p = 0.0068, n = 5). Error bars represent standard deviation.
All β-Cat;Nkx3.1:Cre mutant pups died at birth and showed defects in vertebrae formation, likely a result of Nkx3.1 expression in paraxial mesoderm [37]. To overcome this, prostates were grown in ex vivo organ culture. Prostates were dissected at E18.5, placed on filters and grown such that individual lobes could be assessed. To evaluate epithelial bud development, mutant animals were mated to ROSA26-LacZ reporter mice that express LacZ upon Cre mediated recombination [38]. After 5–6 days in culture, control anterior and ventral lobes grew thick buds with multiple branches and dorsal-lateral lobes grew long buds with branches (Figure 1C). In contrast, β-Cat;Nkx3.1:Cre anterior lobes grew primary buds that were thinner and lacked branches, while ventral and dorsal-lateral lobes grew small buds with no branches. Quantification of prostate duct tips revealed a significant decrease in β-Cat;Nkx3.1:Cre mutants, demonstrating a reduction in branching (Figure 1D). After 5 days of culture, most epithelial cells have lost β-Catenin but a small number of cells still retain protein expression (Figure 2A and 2B). To determine if the smaller mutant prostates are due to a decrease in cellular proliferation the marker Ki-67 was analysed. At E18.5 there is no significant difference in the proliferation of cells in mutant buds (p = 0.095, n = 5) (Figure 2B and 2C). In contrast, after being grown for 5 days in culture, there is a significant decrease in dividing cells in mutant prostates (13%) compared to control prostates (37%) (p = 0.0016, n = 5) (Figure 2B and 2C). At this stage, there is a concomitant decrease in the number p63 positive cells at the tip of β-Cat;Nkx3.1:Cre mutant buds (Figure 2D). Control buds have multiple layers of p63 expressing cells at the tip of the bud with many co-expressing LEF1 (Figure 2E). In contrast, β-Cat;Nkx3.1:Cre buds have lost epithelial LEF1 expression and a single layer of p63 expressing cells is present. These data show that β-Catenin is required for prostate growth and branching and regulates the number of p63 positive cells at the tip of the buds.
Figure 2. β-Catenin regulates proliferation of prostatic duct progenitors.

(A) IHC for β-Catenin on sections of β-Cat;Nkx3.1:Cre and control E18.5 UGS that have been grown in organ culture for 5 days. Black arrows indicate buds that have lost β-Catenin expression. (B) double IHC for β-Catenin and Ki-67 on β-Cat;Nkx3.1:Cre and control E18.5 UGS sections (top panel) and β-Cat;Nkx3.1:Cre and control prostate organ culture sections (bottom panels). White circles indicate mutant buds that have lost β-Catenin. White arrowhead indicates an epithelial bud that has not lost β-Catenin. (C) quantitative analysis of Ki-67 shows there is no significant decrease in proliferation in mutant prostate ducts at E18.5. A significant decrease in proliferation is evident after 5 days in culture, compared to control prostates (E18.5 p = 0.095, 5 days in culture p = 0.0016). (D) β-Catenin and p63 IHC on serial sections of β-Cat;Nkx3.1:Cre and control prostate organ cultures. Arrows indicates multiple p63 expressing cells at the tips of control buds that are absent from mutant buds. (E) double IHC for LEF1 and p63 on β-Cat;Nkx3.1:Cre and control E18.5 UGS that have been grown in organ culture for 5 days. White boxes indicate position of high magnification images shown below. Error bars represent standard deviation.
To identify molecular targets of β-Catenin in prostate bud growth, we analysed the expression of established targets and genes known to be involved in prostate development by whole-mount in situ hybridization (WISH) on prostates dissected at E18.5 and grown in organ culture. Two well-described targets of WNT/β-Catenin signalling are Axin2, a negative regulator of the pathway, and c-Myc, a transcription factor that regulates multiple processes including cell proliferation and differentiation. Axin2 is expressed throughout the buds of control prostates with higher levels in the tips, while c-Myc is expressed in the bud tips (Figure 3). Expression of both genes are lost in the buds of β-Cat;Nkx3.1:Cre mutant prostates (n = 4), with only a few small remaining patches of c-Myc expression, presumably a result of epithelial cells that have not deleted β-Catenin. Interestingly, expression analysis of Nkx3.1, a gene important in prostate duct morphology [5], showed a dramatic loss of expression in mutant prostates (n = 5), while mutant buds still expressed Sox9 (n = 5), another key regulator of prostate development (Figure 3) [39]. In addition, Fgfr2 expression is downregulated in β-Cat;Nkx3.1:Cre mutant buds (n = 5), while Shh is expressed at similar levels to control prostates (n = 4) (Figure 3). This demonstrates that β-Catenin controls a discrete network of transcription factors and signalling pathways, including Nkx3.1 and Fgfr2, to promote prostate bud development.
Figure 3. β-Catenin regulates a discrete network of genes during prostate development.

WISH analysis of β-Cat;Nkx3.1:Cre mutant and control prostate organ cultures grown for 5 days shows expression of Axin2, c-Myc, Nkx3.1 and Fgfr2 are lost in mutant buds, while Sox9 and Shh expression are still present. VP is the ventral lobe, AP is the anterior lobe and DLP is the dorsal lateral lobe.
Stabilized β-Catenin causes transdifferentiation of embryonic prostate epithelium
In a complementary approach to our β-Catenin deletion model, we used Nkx3.1:Cre to express a stabilized form of the protein (Actβ-Cat) in the developing mouse prostate epithelium, Actβ-Cat;Nkx3.1:Cre [40]. The Actβ-Cat allele has loxP sites flanking exon 3 and Cre-mediated deletion results in the loss of the GSK3β regulatory phosphorylation sites, leading to stable expression of β-Catenin. At E18.5, the epithelial buds of Actβ-Cat;Nkx3.1:Cre mutants are larger than control prostates (Actβ-Cat animals with no Cre) and the morphology of the bud tip is irregular (Figure 4A). The bigger Actβ-Cat;Nkx3.1:Cre buds express high levels of β-Catenin, contain many p63 expressing cells and have levels of proliferation comparable to control buds (Actβ-Cat;Nkx3.1:Cre buds 75% Ki-67 positive cells, control buds 58% Ki-67 positive cells, p = 0.12) (Figure 4A). Similar to β-Cat;Nkx3.1:Cre mutant pups, Actβ-Cat;Nkx3.1:Cre pups die at birth, so prostates were again dissected and grown in culture to assess the phenotype at later stages of development. After 5–6 days in culture, abnormal round structures developed in the place of normal prostate bud growth (Figure 4B). β-Catenin and haematoxylin and eosin (H&E) stained sections demonstrate that the epithelium of these structures has a stratified squamous morphology (Figure 4B). The normally round epithelial cells are now multi-layered and flat. Detailed analysis shows a correlation of high β-Catenin expression and the change in cell morphology, shown clearly by p63 expression (Figure S1A). Areas of squamous epithelia have lower levels of proliferation as marked by Ki-67 when compared to control prostates (Figure 4B). Furthermore, they express the squamous cell marker CK10 (Figure 4B). The abnormal structures and squamous epithelia form in Actβ-Cat;Nkx3.1:Cre mutants even when grown in the absence of dihydrotestosterone (DHT) (Figure S1B). Nkx3.1 and Sox9 expression was reduced in Actβ-Cat;Nkx3.1:Cre organ cultures (n = 4), while Fgfr2 continues to be expressed in the novel structures of these mutants (n = 4) (Figure 4C). Loss of expression of these markers suggests that these cells have lost prostate identity. Overall, these data demonstrate that overexpression of β-Catenin in the developing prostate epithelia causes an androgen-independent transdifferentiation to squamous cells.
Figure 4. Stabilized β-Catenin transdifferentiates the developing prostate buds into squamous epithelium.

(A) IHC on Actβ-Cat;Nkx3.1:Cre mutant and control (Actβ-Cat) E18.5 UGS sections with β-Catenin, p63 and Ki-67 antibodies. Adjacent panels show high magnification of buds. (B) H&E stain and IHC for β-Catenin, p63, Ki-67 and CK10 on sections of Actβ-Cat;Nkx3.1:Cre and control prostate organ cultures grown for 5 days. High magnification of CK10 is shown in the lower panels. (C) WISH analysis of Actβ-Cat;Nkx3.1:Cre mutant and control prostate organ cultures grown for 5 days shows expression of Nkx3.1 and Sox9 are lost in mutants, while Fgfr2 is still present. VP is the ventral lobe, AP is the anterior lobe and DLP is the dorsal lateral lobe.
β-Catenin is not required in the adult prostate
To test whether β-Catenin is required for normal adult prostate homeostasis, we deleted it using the loss of function allele described (β-Cat) and mice with the PBCre transgene, which drives expression of Cre recombinase in mouse prostate epithelia from 2 weeks of age (β-Cat;PBCre). H&E stained sections of β-Cat;PBCre prostates 6 months, 9 months and 19 months of age show no observable phenotype compared to control littermates (Figure 5 and Figure S2). Analysis of β-Catenin expression in mutant prostates confirmed that it is deleted in adult prostate epithelial cells (Figure 5). In β-Cat;PBCre prostates E-Cadherin is correctly localized at the cell membrane suggesting the adherens junctions form (Figure 5). As our developmental studies revealed that β-Catenin regulates the number of p63 positive cells, we analysed p63 expressing basal cells in β-Cat;PBCre mutant and control prostates. Identical to control prostates, p63 is restricted to expression in basal cells of β-Cat;PBCre prostates (Figure 5). Although β-Catenin is deleted in a subpopulation of basal cell due to the mosaic expression of PBCre, quantification shows there are a similar number of basal cells in mutant prostates compared to controls (β-Cat;PBCre 23% p63 positive cells, control prostates 21% p63 positive cells, p = 0.107) (Figure 5B). In addition, β-Cat;PBCre prostates have similar expression of c-Myc, LEF1, AR and Probasin compared to control prostates, and mutant animals are fertile (Figure S2). These data indicate that β-Catenin has no role in maintaining normal morphology of the adult prostate.
Figure 5. β-Catenin is not required for adult prostate homeostasis.
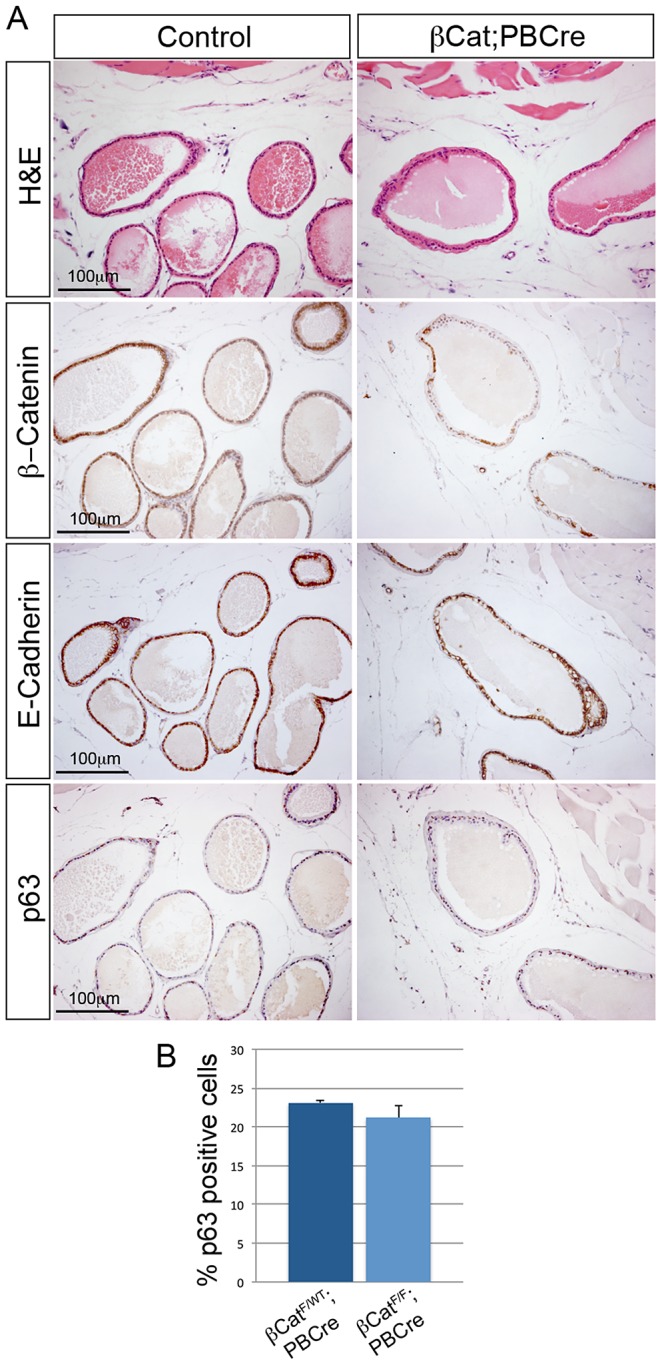
(A) H&E stain and IHC for β-Catenin, E-Cadherin and p63 on sections of β-Cat;PBCre mutants and control prostates. Sections are cut through the dorsal-lateral lobe from 6 months old animals. (B) quantification of p63 expressing cells shows a similar number of basal cells in β-Cat;PBCre mutants as controls (p = 0.107). Error bars represent standard deviation.
Pten regulated β-Catenin is not required for prostate tumorigenesis
To better understand the role of β-Catenin in human prostate cancer, we wanted to explore its relationship with the frequently mutated PTEN gene. To do this we used the well-defined Pten murine prostate cancer model [41]. In this model, Pten deletion in mice results in the formation of hyperplasia, prostatic intraepithelial neoplasia (PIN) and carcinoma. Pten null prostates were generated using a loxP containing Pten allele and the PBCre transgene (PtenF/F;PBCre). We first analysed the expression of β-Catenin protein to determine if β-Catenin levels are altered in these animals, as studies in prostate cancer cell lines have shown that Pten loss leads to β-Catenin accumulation [33], [34]. Immunohistochemistry shows that PtenF/F;PBCre prostates have a dramatic increase in the level of β-Catenin compared to controls, which is confirmed by Western blot (Figure 6A and Figure S3A). In addition, in Pten null prostate tumours there is an increase in the active non-phosphorylated stabilized form of β-Catenin, which is thought to be involved in the transcription of target genes, although we could not detect an induction of LEF1 (Figure S3B). To test whether this increase in β-Catenin expression has a role in Pten loss-driven prostate cancer, we deleted Ctnnb1 in the Pten null model using the loxP alleles and the PBCre transgene described. Detailed histopathological analysis of Pten mutant (PtenF/F;PBCre and β-CatF/WT;PtenF/F;PBCre), Pten;Ctnnb1 double mutant (β-CatF/F;PtenF/F;PBCre) and control animals (β-CatF/F;PtenF/F) aged 3 months demonstrates a similar progression to PIN in Pten null mutants and in β-CatF/F;PtenF/F;PBCre mutants (Figure 6B). pAKT levels remain high and E-Cadherin expression remain unaltered in both β-CatF/WT;PtenF/F;PBCre mutant prostates and in β-CatF/F;PtenF/F;PBCre mutant prostates (Figure 6B). Consistent with this, the percentage of Ki-67 positive cells in Pten null prostates, which is comparable to previous reports, is not significantly different to prostates that have lost Pten and Ctnnb1 (Figure 6C) [41], [42]. This shows there is no difference in the progression to PIN in prostates that have lost Pten and those that have lost both Pten and Ctnnb1, suggesting that β-Catenin does not play a role in Pten loss-driven prostate cancer.
Figure 6. Pten regulated β-Catenin is not required for prostate tumorigenesis.

(A) IHC of β-Catenin on sections of PtenF/F and PtenF/F;PBCre prostates showing upregulation of β-Catenin after Pten loss. (B) H&E stain and IHC for β-Catenin, pAKT and E-Cadherin on sections of 3-month-old control (no Cre), β-CatF/WT;PtenF/F;PBCre and β-CatF/F;PtenF/F;PBCre prostates. (C) double IHC for β-Catenin and Ki-67 on β-CatF/WT;PtenF/F;PBCre and β-CatF/F;PtenF/F;PBCre prostate sections. Right, quantitative analysis of Ki-67 shows no significant difference in proliferation in β-CatF/F;PtenF/F;PBCre prostates compared to β-CatF/WT;PtenF/F;PBCre prostates (p = 0.196). (D) H&E stain and IHC for β-Catenin, AR and Ki-67 on prostates sections of 3-month-old β-CatF/WT;PtenF/F;PBCre and β-CatF/F;PtenF/F;PBCre animals that were castrated for one month. Black arrows highlight proliferating cells and white arrows indicate highly proliferative foci. (E) double IHC for β-Catenin and Ki-67 on prostates sections of 3-month-old β-CatF/WT;PtenF/F;PBCre and β-CatF/F;PtenF/F;PBCre animals that were castrated for one month. Below left, quantitative analysis of Ki-67 shows no significant difference in proliferation in castrated β-CatF/F;PtenF/F;PBCre prostates compared to β-CatF/WT;PtenF/F;PBCre prostates (p = 0.695). Below right, quantitative analysis of Ki-67 foci shows no significant difference between β-CatF/F;PtenF/F;PBCre and β-CatF/WT;PtenF/F;PBCre prostates (p = 0.562). Error bars represent standard deviation.
Castration of mice with Pten null prostates results in regression of PIN, but cells continue to proliferate [41]. As β-Catenin has been implicated in the progression of CRPC, we investigated if β-Catenin has a role in the continued growth of Pten deficient prostates. To do this, we surgically castrated 3 month old β-CatF/WT;PtenF/F;PBCre and β-CatF/F;PtenF/F;PBCre mice and analysed the prostates 1 month later. H&E and β-Catenin stained sections demonstrate that both Pten null and β-CatF/F;PtenF/F;PBCre mutant prostates regress (Figure 6D). Castration of animals with Pten null prostates resulted in AR protein being retained in the cytoplasm and a more diffuse expression pattern [41]. This response to androgen withdrawal is also seen in β-CatF/F;PtenF/F;PBCre mutant prostates and suggests a similar effect on AR in both models. Analysis of Ki-67 expression in β-CatF/WT;PtenF/F;PBCre and β-CatF/F;PtenF/F;PB4Cre prostates shows that cellular proliferation continues after castration (Figure 6D). In addition, there were often foci of many Ki-67 positive cells in animals of both genotypes (more than 20 positive cells) (Figure 6D). In β-CatF/F;PtenF/F;PBCre mutant prostates, Ki-67 foci were present in areas of epithelia that has lost β-Catenin as well as those that had retained expression. Quantification of Ki-67 shows there is no significant difference in total proliferation or in Ki-67 foci between Pten null prostates and β-CatF/F;PtenF/F;PBCre mutant prostates after castration (Figure 6E). Taken together, this work demonstrates that Pten loss results in increased levels of β-Catenin in the prostate, but this is not required for tumour growth in intact or castrated animals.
Stabilized β-Catenin cooperates with Pten loss to drive prostate cancer progression
Pten null animals form high-grade PIN but do not readily progress to invasive carcinoma [42]. Although we found the higher β-Catenin levels after Pten deletion did not have a function in prostate cancer initiation, we wanted to test whether increasing levels of β-Catenin further would affect progression. To do this, we crossed the Pten loxP (PtenF/F) animals with mice with the stabilized form of β-Catenin (Actβ-Cat) and the PBCre transgene. This allowed the comparison of prostates that are Pten null (PtenF/F;PBCre), have stabilized β-Catenin (Actβ-Cat;PBCre), Pten null in combination with stabilized β-Catenin (Actβ-Cat;PtenF/F;PBCre) and controls (Actβ-Cat;PtenF/F). Actβ-Cat;PtenF/F;PBCre prostates have higher levels of active non-phosphorylated β-Catenin than Pten null prostates and show a clear induction of LEF1, indicating these tumours have an increase in β-Catenin mediated transcription (Figure S3B). At 2 months of age, Actβ-Cat;PtenF/F;PBCre prostates display high-grade PIN and focally invasive epithelial cells in the stroma (Figure S4A). By 3 months of age, Actβ-Cat;PtenF/F;PBCre prostates are larger and weigh significantly more than PtenF/F;PB4Cre or Actβ-Cat;PBCre prostates (Figure 7A and 7B). H&E staining shows that Actβ-Cat;PtenF/F;PBCre prostates have large areas of high-grade PIN and invasive adenocarcinoma with extensive infiltrative growths of atypical epithelial cells invading into the surrounding fibromuscular stroma and connective tissue, which are not present in PtenF/F;PBCre or Actβ-Cat;PBCre prostates (Figure 6B and Figure 7C). These invasive areas express high levels of β-Catenin, LEF1 and pAKT, indicating that these pathways cooperate to promote the invasive phenotype (Figure 7C and Figure S4B). In double mutant prostates, smooth-muscle actin staining is broken-up and irregular, confirming that epithelial cells have spread into the stroma (Figure 7C). In addition, Actβ-Cat;PtenF/F;PBCre lesions are highly proliferative and have a significant increase in the number of proliferating cells compared to Actβ-Cat;PBCre prostates (Actβ-Cat;PBCre 17% Ki-67 cells, Actβ-Cat;PtenF/F;PBCre 26% Ki-67 cells, p = 0.039) (Fig. 7C and 7D). Comparable with embryonic prostates expressing stabilized β-Catenin, Actβ-Cat;PBCre and Actβ-Cat;PtenF/F;PBCre prostates display areas of squamous differentiation and keratinization that express the squamous cell markers CK10 and CK1 and that frequently associate with many p63 positive cells (Figure 7C and Figure S4B). These data demonstrate that β-Catenin can interact with Pten loss to form highly invasive prostate cancer and squamous metaplasia.
Figure 7. Stabilized β-Catenin cooperates with Pten loss to drive prostate cancer progression.

(A) brightfield images of mouse prostates with epithelial Pten deletion and stabilized β-Catenin, as indicated. The dorsal-lateral-ventral lobes (DLVP) and anterior lobes (AP) from individual animals of each genotype are shown. (B) wet weights of prostates with epithelial Pten deletion and stabilized β-Catenin. (C) H&E stain and IHC for β-Catenin, pAKT, smooth muscle actin (SMA), LEF1, CK10, CK1, p63 and Ki-67 on sections of 3-month-old Actβ-Cat;PBCre and Actβ-Cat;PtenF/F;PBCre prostates. (D) quantitative analysis of Ki-67 shows a significant increase in proliferation in Actβ-Cat;PtenF/F;PBCre prostates compared to Actβ-Cat;PBCre (p = 0.039). Black arrows indicate epithelial cells invading into the stroma. White arrows indicate areas of squamous metaplasia. Black arrowhead indicates loss of SMA. Error bars represent standard deviation.
Discussion
The role of β-Catenin in prostate development is not known and its function in prostate cancer is not clearly defined. We have undertaken an in depth genetic study in the embryonic and adult mouse to identify the role β-Catenin plays in these processes. Our developmental genetic studies demonstrate that epithelial β-Catenin is essential for early growth of the prostatic buds but that high levels of β-Catenin lead to the formation of squamous epithelia. Deletion of Ctnnb1 in the adult prostate has no effect on normal prostate homeostasis or a Pten model of prostate cancer. However, the functional significance of β-Catenin in prostate cancer is highlighted as increased levels can cooperate with Pten loss to drive progression to invasive carcinoma together with squamous transdifferentiation. The ability of β-Catenin to differentiate prostate epithelia into squamous cells in the embryo and adult suggests that this protein is playing a similar role in both developmental stages. These studies further highlight the use of developmental systems to inform on the function of genes and pathways in the adult.
Our studies show that during prostate development β-Catenin controls the proliferation of cells at the tip of the epithelial buds and regulates the number of p63 positive cells in this region. This lack of proliferation leads to a defect in prostatic bud growth and branching in mutant animals, consistent with other studies that have shown that the tips of the buds have high numbers of proliferating cells and are important in the formation of branches [43]. Our data reveal that β-Catenin regulates the expression of LEF1 and c-Myc at the tip of the developing prostate bud. c-Myc is a well-known regulator of proliferation and it is therefore likely that β-Catenin controls cell number in prostate buds through this gene [44], [45]. We were unable to determine if β-Catenin is required for prostate induction and bud initiation as the Nkx3.1:Cre mouse line is first expressed after this has begun at E17.5. A recent study using an inducible system to delete β-Catenin prior to prostate formation has demonstrated that this protein is required for bud initiation [46]. Together, these data demonstrate that β-Catenin is required for multiple steps of prostate development, including prostatic epithelial progenitor cell proliferation at the tip of the buds and for ductal branching.
Prostate development is regulated by a set of transcription factors and signalling networks that are poorly defined. Two of the earliest markers of prostate identity are the transcription factors Nkx3.1 and Sox9. These, together with FGF signalling, through Fgfr2, and the Hedgehog signalling pathway, through Shh, are required for normal prostate development [6]. Our work has uncovered a novel divergence in the pathways that control prostate development, with β-Catenin regulating the expression of Nkx3.1 and Fgfr2 but having no effect on Sox9 and Shh expression. This suggests a complex gene network during prostate development with different pathway branches regulating prostate identity in parallel, and β-Catenin regulating Nkx3.1 and Fgfr2 in one of these branches. FGF signalling has been implicated in the early formation of prostate buds and β-Catenin could be acting through this pathway [47]. However, the prostate phenotype in mice where Fgfr2 was deleted using the Nkx3.1:Cre strain was not the same as in β-Cat;Nkx3.1:Cre mutants, suggesting that Fgfr2 is not the only factor responsible for the defects in growth and branching in the latter animals [47].
Our studies indicate that β-Catenin acts through the canonical WNT pathway to regulate prostate epithelial cell proliferation, as we observe the loss of expression of classical downstream genes of this pathway after Ctnnb1 deletion, including LEF1, c-Myc and Axin2. In addition, several members of the WNT family have been shown to be expressed in the epithelia and mesenchyme of the developing prostate [48]. Our results are consistent with published studies where addition of WNT ligand to rat prostate cultures was shown to increase the number of p63 positive cells, while the WNT signalling inhibitor Dkk had the opposite effect, and led to disrupted prostatic branching [49]. From our gene deletion experiments, it may have been expected that expressing stabilized β-Catenin during embryogenesis would result in an expansion of the prostatic ducts. Although we observe this at E18.5, at later stages the epithelium becomes squamous-like. Our studies suggest that WNT signalling needs to be tightly regulated during prostate development and negative regulators of the pathway, such as Dkk1, may be required to control the level of β-Catenin and prevent prostate epithelia transdifferentiating to a squamous phenotype.
Our data demonstrate that β-Catenin regulates the number of p63 positive progenitor cells in the developing prostate. Interestingly, WNT/β-Catenin signalling has been shown to control self-renewal of stem/progenitor cells of primary adult prostate epithelial cells and prostate cancer cell lines in prostate sphere assays [50], [51]. These studies have shown that Bmi-1 is necessary for β-Catenin self-renewal activity. Bmi-1 can induce the self-renewal activity of prostate spheres and results in the expansion of p63 positive cells. In this assay, the p63 expressing cells have a higher capacity for self-renewal, indicating they contain adult prostatic progenitor cells [52]. Together, these data suggest that pathways that regulate progenitor cells during embryonic prostate development also control stem/progenitor cells in the adult prostate.
We have shown that during prostate development β-Catenin overexpression results in squamous epithelia differentiation, even in the absence of androgens. Similarly, squamous cells form when stabilized β-Catenin is expressed in the adult prostate, even after Pten loss. This suggests that an intrinsic property of high levels of β-Catenin in prostate epithelia is to drive squamous differentiation. Squamous metaplasia is also seen after exogenous administration of estrogen to male mice. Estrogen has been shown to act through estrogen receptor alpha (ERα) in the epithelium to promote proliferation of p63 expressing basal cells and the formation of squamous metaplasia [53], [54], [55]. Interestingly, we found an increase in ERα expression in Actβ-Cat;PBCre prostates, however, this was not the case for Actβ-Cat;PtenF/F;PBCre mice, suggesting that high ERα is not a requisite for squamous cell formation driven by high levels of β-Catenin (data not shown). Ectopic high levels of p63 expression were able to induce transdifferentiation and squamous metaplasia of mouse lung epithelium [56]. In addition, high p63 levels have been associated with squamous cell carcinoma and β-Catenin has been shown to regulate this protein in this disease [57], [58]. Therefore our studies suggest that stabilized β-Catenin acts to induce high levels of p63, which drives the transdifferentiation process towards a squamous fate.
β-Catenin has been implicated in CRPC and has been shown to act as an AR co-factor in low androgen conditions [17], [18], [19], [20], [21], [22], [24]. Pten mutant mice show resistant properties with proliferation of epithelial cells in the prostates of castrated animals [41]. Our data demonstrate that although β-Catenin is upregulated after Pten loss, it is not essential in neoplastic growth of Pten null cancer after androgen withdrawal. This data suggests that, in this CRPC model, β-Catenin does not act as a co-factor with AR or in an AR-independent manner to regulate neoplastic proliferation. The increase in β-Catenin expression we observe after Pten deletion is predominantly localized at the membrane and in the cytoplasm, with little transcriptionally active protein accumulating. This low level of active β-Catenin may not induce transcription of targets, such as LEF1, and have no affect on Pten-loss driven PIN. This is in contrast to our Actβ-Cat;PtenF/F;PBCre model that has high levels of nuclear β-Catenin, increased levels of LEF1 expression and progression to an aggressive phenotype. Alternatively, the growth of Pten null prostate lesions deficient in β-Catenin may be due to compensation by the multiple downstream effectors of the PI3K/Akt pathway that are affected by Pten loss [32].
Our study, and work by others, has shown that activating β-Catenin through a stabilized form of the protein in the prostate leads to PIN and squamous metaplasia, with lesions that are highly proliferative but not invasive [27]. Interestingly, our data demonstrates that increased β-Catenin levels are able to drive Pten deficient prostate epithelia, a frequent genetic lesion in human prostate cancer, to invasive carcinoma suggesting that these pathways could interact during human prostate tumorigenesis. Consistent with this, recent exome sequencing has shown that members of the WNT pathway, including APC, are frequently mutated in aggressive prostate tumours, and these cancers are enriched for mutations in a PTEN interacting network [59]. β-Catenin has also been shown to cooperate with mutant K-ras and SV40 large T-antigen to drive tumour progression to invasive carcinoma [28], [29]. Mice expressing active β-Catenin and mutant K-ras in prostate epithelial cells have a loss of p63 expressing cells and do not display squamous metaplasia, consistent with our hypothesis that p63 expression is involved in transdifferentiation to squamous cells [29]. The combination of active β-Catenin and SV40 large T-antigen results in epithelial cell invasion into the stroma [28]. Similar to our model, nuclear β-Catenin and expression of downstream targets were found in the epithelial cells within the stroma suggesting that this pathway can act synergistically with multiple signalling pathways to promote the formation of invasive aggressive prostate cancer.
The ability of β-Catenin to cooperate with other oncogenic events such as Pten deletion, mutant K-ras and SV40 large T-antigen to drive tumour progression suggests that it may be a general drug target in prostate cancer [28], [29]. Changes in β-Catenin expression are typically a late event in human prostate cancer and detailed sequencing studies have shown that mutations in the WNT pathway occur in late stage lethal CRPC [59], [60]. This suggests that inhibitors of this pathway, such as tankyrase inhibitors, could be used in the clinic to treat patients with these mutations [61]. Squamous metaplasia was also observed in these mice, which is rarely seen in human prostate cancer. Our data therefore suggests that other events need to take place to allow β-Catenin to drive disease progression without squamous formation. In the model we used, Pten and Actβ-Cat undergo Cre-mediated recombination at the same time within luminal cells, although some basal cells are also targeted. Therefore genetic lesions may need to occur in a step-wise fashion in patients so that β-Catenin expression is altered at a later stage in tumour formation. Alternatively, β-Catenin may need to interact with other genetic lesions or be upregulated in a different cell type to give rise to tumours that do not exhibit squamous differentiation. These studies provide novel information on the necessary steps in tumour development that are required for progression to the invasive phenotype seen in human patients with lethal prostate cancer.
Materials and Methods
Mouse strains
Mice containing the Ctnnb1 conditional allele (β-Cat) have been described previously [35]. Mice containing the stabilized Ctnnb1 conditional allele (Actβ-Cat) were kindly provided by M.M. Taketo [40]. The ARR2PBCre transgenic mice, PBCre, have been described previously [62]. The Nkx3.1:Cre allele was created by introducing the Cre gene by homologous recombination into the Nkx3.1 locus and was kindly provided by Michael Shen. Mice with the conditional allele of Pten were obtained from The Jackson Laboratory [63]. These animals were bred on a mixed genetic background. We define midday of the day of plug as E0.5 and most pregnant females in this study gave birth on E19. All procedures were in accordance with UK Home Office legislation.
Organ culture system
E18.5 urogenital sinuses were dissected from male embryos and grown on Biopore membranes (MilliPore) with culture media (DMEM/F-12 HAM 1/1 mixture) supplemented with ITS+1 (Sigma-Aldrich I-2521) at 20 µl/ml, gentamicin (0.05 mg/ml), benzylpenicillin sodium (0.12 mg/ml), streptomycin sulphate (0.2 mg/ml), ampicillin (0.1 mg/ml) and 10−8 M dihydrotestosterone.
Whole-mount in situ hybridisation (WISH)
WISH was carried out on an in situ processor (Intavis In Situ Pro) according to standard protocols [64]. Probes for Sox9 and Nkx3.1 have been described previously [5], [65]. The Axin2, c-Myc, Fgfr2 and Shh DIG containing probes were synthesized from PCR fragments containing T7 RNA polymerase promoter sequences as described previously [64]. These fragments were derived from the amplification of mouse cDNA or genomic DNA using the following primers:
Axin2: 5′ AGGATGCTGAAGGCTCAAAGC 3′ and
5′ GTAATACGACTCACTATAGGGACTCTGGATGGTCCCCAAAG 3′
c-Myc: 5′ AAGCTGGTCTCGGAGAAGCTGG 3′ and
5′ TAATACGACTCACTATAGGGAGGTTCAGGGATCTGGTCACG 3′
Fgfr2: 5′ CGCAGGATGGACCTCTCTAC 3′ and
5′ GTAATACGACTCACTATAGGGCGACCAACTGCTTGAATGTG 3′;
Shh: 5′ GACAGCTCACAAGTCCTCAG 3′ and
5′ GTAATACGACTCACTATAGGGACGTAAGTCCTTCACCA 3′.
For each WISH probe, at least four embryos of each genotype were processed.
Mouse prostate histology
Histological phenotype of samples was assessed on H&E stained sections. Serial sections were then stained for immunohistochemical analysis. Histological assessment was based on published guidelines and assisted by the pathologists R. Cardiff, D. Berney and G. Stamp [66], [67].
Immunohistochemistry and Western blot
Antibody stains were performed on paraffin sections. Tissues were fixed overnight in 4% paraformaldehyde (PFA), dehydrated by washing through an ethanol gradient series, washed in histoclear and embedded in wax. 4 µm sections were cut, treated with histoclear and rehydrated through an ethanol gradient series. Antigen retrieval was obtained by boiling the sections in citrate buffer (0.1 M sodium citrate pH6 and 0.05% Tween) and sections were treated with 3% H2O2 to block endogenous peroxidase activity. Sections were incubated in PBS with 10% sheep serum and then incubated with primary and secondary antibodies in PBS with 1% sheep serum. Sections were counterstained with hematoxylin. The following antibodies were used for immunohistochemistry; β-Catenin (BD Biosciences, 1∶100 fluorescence, 1∶800 DAB), AR (PG-21, Upstate, 1∶250 DAB), p63 (4A4, Santa Cruz Biotechnology, 1∶50 fluorescence, 1∶200 DAB), CK10 (PRB-159P-100, Covance, 1∶1000 DAB), CK1 (PRB-165P-100, Covance, 1∶1000 DAB), LEF1 (2230, Cell Signalling Technology, 1∶100 fluorescence, 1∶1000 DAB), pAKT (Ser473) (9271, Cell Signalling Technology, 1∶100 DAB), Ki-67 (clone TEC-3, Dako, 1∶20 fluorescence, 1∶200 DAB), smooth-muscle actin (clone 1A4, Sigma, 1∶5000 DAB), E-Cadherin (BD Transduction Laboratories, 1∶250 DAB). For DAB chromogen staining the ABC vector kit was used with biotinlyated secondary antibodies (Vector Laboratories) according to manufacturer's instructions and the DAB substrate (Dako). Secondary fluorescent antibodies were obtained from Molecular Probes and were used at a 1∶500 dilution. Fluorescent images were visualized and collected on a Leica TCS-SP2 confocal microscope. For all antibody stains, sections were stained from at least four animals of each genotype. Western blotting was performed using standard protocols, and the following antibodies were used; β-Catenin 1∶500; active non-phosphorylated β-Catenin 1∶500 (Millipore), LEF1 1∶1000, HSC70 1∶15,000 (Santa Cruz Biotechnology), β-Actin 1∶500 (Santa Cruz Biotechnology).
Quantification of proliferation and basal cells
To quantify proliferating cells, Ki-67 and β-Catenin double-labelled fluorescent immunohistochemistry was performed on sections and stained with nuclear DAPI. For developmental and adult studies prostatic ducts were identified using DAPI. For control ducts and ducts expressing stabilized β-Catenin Ki-67 positive cells were counted in the β-Catenin positive epithelia. For loss-of-function β-Catenin mutant buds Ki-67 positive cells were counted in the β-Catenin negative prostate epithelia. To quantify basal cells, p63 expressing cells were counted and shown as a percentage of the total epithelial cells. For developmental studies, cells were counted from a section of five embryos of each genotype. For adult studies, cells from at least 4 high power fields were counted per animal, which totalled more than 900 cells per animal. At least three animals of each genotype were analysed. All values are significant with p<0.05 using Student t-test unless otherwise stated.
β-Galactosidase staining
Tissues were dissected and fixed in PBS with 2% PFA and 0.1% glutaraldehyde for 30 min at 37°C. After washing in PBS, tissues were stained at 37°C in staining buffer (1 mg/ml X-gal, 2 mM MgCl2, 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6 and 0.02% NP-40 in PBS) in the dark.
Supporting Information
Embryonic squamous formation of prostate epithelium by stabilized β-Catenin. (A) high magnification of β-Catenin and p63 IHC on sections of Actβ-Cat;Nkx3.1Cre mutant and control (Actβ-Cat) prostate organ cultures grown for 3 days. Arrows indicate area of high β-Catenin that has become squamous stratified epithelium. (B) H&E and IHC for β-Catenin on sections of Actβ-Cat;Nkx3.1Cre prostate organ cultures grown for 5 days with no DHT.
(TIF)
β-Catenin is not required for adult prostate homeostasis. H&E stain and IHC for AR, Probasin, c-Myc and LEF1 on sections of β-Cat;PBCre mutant and control prostates. Sections cut through the dorsal-lateral lobe.
(TIF)
Western blot analysis of β-Catenin and LEF1 in adult mouse prostate tissue. (A) β-Catenin levels increase in Pten null (PtenF/F;PBCre) prostates compared to controls. (B) undetectable levels of active non-phosphorylated β-Catenin are present in control prostates, while in PtenF/F;PBCre prostates there is an accumulation of wild type endogenous active non-phosphorylated β-Catenin, as indicated. Actβ-Cat;PtenF/F;PBCre prostates have very high levels of the smaller exon 3 deleted mutant active β-Catenin, as indicated. The anti-active β-Catenin antibody also detects a smaller unknown band in control and PtenF/F;PBCre samples. LEF1 is not detected in control or PtenF/F;PBCre prostates and is upregulated Actβ-Cat;PtenF/F;PBCre prostates.
(TIF)
Stabilized β-Catenin and Pten homozygous loss prostate cancer. (A) H&E stain on sections of 2-month-old Actβ-Cat;PtenF/F;PBCre prostates. Left panel shows areas of adenocarcinoma and squamous metaplasia. Right panel shows epithelial cells invading into the surrounding stroma. (B) H&E stain and IHC for β-Catenin and p63 on sections of 3-month-old Actβ-Cat;PtenF/F;PBCre prostates showing detail of adenocarcinoma and squamous metaplasia. Black arrows indicate mitotic figures. White arrows indicate cords of β-Catenin positive epithelial cells that have invaded the stroma. Arrowheads indicate squamous metaplasia.
(TIF)
Acknowledgments
We thank Jenny Gardiner for helpful comments when preparing the manuscript, Michael Shen for providing the Nkx3.1:Cre mice, and Beatrice Howard for the c-Myc in situ probe. We are grateful to Robert Cardiff, Dan Berney, and Gordon Stamp for assistance with histopathological analysis.
Funding Statement
This work was supported by CR-UK grant number C13590/A10228. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, et al. (2010) Integrative genomic profiling of human prostate cancer. Cancer Cell 18: 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schaeffer EM, Marchionni L, Huang Z, Simons B, Blackman A, et al. (2008) Androgen-induced programs for prostate epithelial growth and invasion arise in embryogenesis and are reactivated in cancer. Oncogene 27: 7180–7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ewing CM, Ray AM, Lange EM, Zuhlke KA, Robbins CM, et al. (2012) Germline mutations in HOXB13 and prostate-cancer risk. N Engl J Med 366: 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thomsen MK, Ambroisine L, Wynn S, Cheah KS, Foster CS, et al. (2010) SOX9 elevation in the prostate promotes proliferation and cooperates with PTEN loss to drive tumor formation. Cancer Res 70: 979–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bhatia-Gaur R, Donjacour AA, Sciavolino PJ, Kim M, Desai N, et al. (1999) Roles for Nkx3.1 in prostate development and cancer. Genes Dev 13: 966–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marker PC, Donjacour AA, Dahiya R, Cunha GR (2003) Hormonal, cellular, and molecular control of prostatic development. Dev Biol 253: 165–174. [DOI] [PubMed] [Google Scholar]
- 7. Hudson DL (2004) Epithelial stem cells in human prostate growth and disease. Prostate Cancer Prostatic Dis 7: 188–194. [DOI] [PubMed] [Google Scholar]
- 8. Yap TA, Zivi A, Omlin A, de Bono JS (2011) The changing therapeutic landscape of castration-resistant prostate cancer. Nat Rev Clin Oncol 8: 597–610. [DOI] [PubMed] [Google Scholar]
- 9. Clevers H (2006) Wnt/beta-catenin signaling in development and disease. Cell 127: 469–480. [DOI] [PubMed] [Google Scholar]
- 10. Kypta RM, Waxman J (2012) Wnt/beta-catenin signalling in prostate cancer. Nat Rev Urol [DOI] [PubMed] [Google Scholar]
- 11. Voeller HJ, Truica CI, Gelmann EP (1998) Beta-catenin mutations in human prostate cancer. Cancer Res 58: 2520–2523. [PubMed] [Google Scholar]
- 12. Szasz AM, Nyirady P, Majoros A, Szendroi A, Szucs M, et al. (2010) beta-catenin expression and claudin expression pattern as prognostic factors of prostatic cancer progression. BJU Int 105: 716–722. [DOI] [PubMed] [Google Scholar]
- 13. Whitaker HC, Girling J, Warren AY, Leung H, Mills IG, et al. (2008) Alterations in beta-catenin expression and localization in prostate cancer. Prostate 68: 1196–1205. [DOI] [PubMed] [Google Scholar]
- 14. Chen G, Shukeir N, Potti A, Sircar K, Aprikian A, et al. (2004) Up-regulation of Wnt-1 and beta-catenin production in patients with advanced metastatic prostate carcinoma: potential pathogenetic and prognostic implications. Cancer 101: 1345–1356. [DOI] [PubMed] [Google Scholar]
- 15. Morita N, Uemura H, Tsumatani K, Cho M, Hirao Y, et al. (1999) E-cadherin and alpha-, beta- and gamma-catenin expression in prostate cancers: correlation with tumour invasion. Br J Cancer 79: 1879–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de la Taille A, Rubin MA, Chen MW, Vacherot F, de Medina SG, et al. (2003) Beta-catenin-related anomalies in apoptosis-resistant and hormone-refractory prostate cancer cells. Clin Cancer Res 9: 1801–1807. [PubMed] [Google Scholar]
- 17. Masiello D, Chen SY, Xu Y, Verhoeven MC, Choi E, et al. (2004) Recruitment of beta-catenin by wild-type or mutant androgen receptors correlates with ligand-stimulated growth of prostate cancer cells. Mol Endocrinol 18: 2388–2401. [DOI] [PubMed] [Google Scholar]
- 18. Mulholland DJ, Read JT, Rennie PS, Cox ME, Nelson CC (2003) Functional localization and competition between the androgen receptor and T-cell factor for nuclear beta-catenin: a means for inhibition of the Tcf signaling axis. Oncogene 22: 5602–5613. [DOI] [PubMed] [Google Scholar]
- 19. Song LN, Coghlan M, Gelmann EP (2004) Antiandrogen effects of mifepristone on coactivator and corepressor interactions with the androgen receptor. Mol Endocrinol 18: 70–85. [DOI] [PubMed] [Google Scholar]
- 20. Truica CI, Byers S, Gelmann EP (2000) Beta-catenin affects androgen receptor transcriptional activity and ligand specificity. Cancer Res 60: 4709–4713. [PubMed] [Google Scholar]
- 21. Verras M, Brown J, Li X, Nusse R, Sun Z (2004) Wnt3a growth factor induces androgen receptor-mediated transcription and enhances cell growth in human prostate cancer cells. Cancer Res 64: 8860–8866. [DOI] [PubMed] [Google Scholar]
- 22. Song LN, Herrell R, Byers S, Shah S, Wilson EM, et al. (2003) Beta-catenin binds to the activation function 2 region of the androgen receptor and modulates the effects of the N-terminal domain and TIF2 on ligand-dependent transcription. Mol Cell Biol 23: 1674–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pawlowski JE, Ertel JR, Allen MP, Xu M, Butler C, et al. (2002) Liganded androgen receptor interaction with beta-catenin: nuclear co-localization and modulation of transcriptional activity in neuronal cells. J Biol Chem 277: 20702–20710. [DOI] [PubMed] [Google Scholar]
- 24. Wang G, Wang J, Sadar MD (2008) Crosstalk between the androgen receptor and beta-catenin in castrate-resistant prostate cancer. Cancer Res 68: 9918–9927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bierie B, Nozawa M, Renou JP, Shillingford JM, Morgan F, et al. (2003) Activation of beta-catenin in prostate epithelium induces hyperplasias and squamous transdifferentiation. Oncogene 22: 3875–3887. [DOI] [PubMed] [Google Scholar]
- 26. Gounari F, Signoretti S, Bronson R, Klein L, Sellers WR, et al. (2002) Stabilization of beta-catenin induces lesions reminiscent of prostatic intraepithelial neoplasia, but terminal squamous transdifferentiation of other secretory epithelia. Oncogene 21: 4099–4107. [DOI] [PubMed] [Google Scholar]
- 27. Yu X, Wang Y, Jiang M, Bierie B, Roy-Burman P, et al. (2009) Activation of beta-Catenin in mouse prostate causes HGPIN and continuous prostate growth after castration. Prostate 69: 249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu X, Wang Y, DeGraff DJ, Wills ML, Matusik RJ (2011) Wnt/beta-catenin activation promotes prostate tumor progression in a mouse model. Oncogene 30: 1868–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pearson HB, Phesse TJ, Clarke AR (2009) K-ras and Wnt signaling synergize to accelerate prostate tumorigenesis in the mouse. Cancer Res 69: 94–101. [DOI] [PubMed] [Google Scholar]
- 30. Dahia PL (2000) PTEN, a unique tumor suppressor gene. Endocr Relat Cancer 7: 115–129. [DOI] [PubMed] [Google Scholar]
- 31. Suzuki H, Freije D, Nusskern DR, Okami K, Cairns P, et al. (1998) Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic prostate cancer tissues. Cancer Res 58: 204–209. [PubMed] [Google Scholar]
- 32. Song MS, Salmena L, Pandolfi PP (2012) The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 13: 283–296. [DOI] [PubMed] [Google Scholar]
- 33. Persad S, Troussard AA, McPhee TR, Mulholland DJ, Dedhar S (2001) Tumor suppressor PTEN inhibits nuclear accumulation of beta-catenin and T cell/lymphoid enhancer factor 1-mediated transcriptional activation. J Cell Biol 153: 1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sharma M, Chuang WW, Sun Z (2002) Phosphatidylinositol 3-kinase/Akt stimulates androgen pathway through GSK3beta inhibition and nuclear beta-catenin accumulation. J Biol Chem 277: 30935–30941. [DOI] [PubMed] [Google Scholar]
- 35. Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, et al. (2001) Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 128: 1253–1264. [DOI] [PubMed] [Google Scholar]
- 36. Eastman Q, Grosschedl R (1999) Regulation of LEF-1/TCF transcription factors by Wnt and other signals. Curr Opin Cell Biol 11: 233–240. [DOI] [PubMed] [Google Scholar]
- 37. Herbrand H, Pabst O, Hill R, Arnold HH (2002) Transcription factors Nkx3.1 and Nkx3.2 (Bapx1) play an overlapping role in sclerotomal development of the mouse. Mech Dev 117: 217–224. [DOI] [PubMed] [Google Scholar]
- 38. Soriano P (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21: 70–71. [DOI] [PubMed] [Google Scholar]
- 39. Thomsen MK, Butler CM, Shen MM, Swain A (2008) Sox9 is required for prostate development. Dev Biol 316: 302–311. [DOI] [PubMed] [Google Scholar]
- 40. Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, et al. (1999) Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J 18: 5931–5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, et al. (2003) Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 4: 209–221. [DOI] [PubMed] [Google Scholar]
- 42. Ding Z, Wu CJ, Chu GC, Xiao Y, Ho D, et al. (2011) SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature 470: 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sugimura Y, Cunha GR, Donjacour AA, Bigsby RM, Brody JR (1986) Whole-mount autoradiography study of DNA synthetic activity during postnatal development and androgen-induced regeneration in the mouse prostate. Biol Reprod 34: 985–995. [DOI] [PubMed] [Google Scholar]
- 44. Bouchard C, Staller P, Eilers M (1998) Control of cell proliferation by Myc. Trends Cell Biol 8: 202–206. [DOI] [PubMed] [Google Scholar]
- 45. Trumpp A, Refaeli Y, Oskarsson T, Gasser S, Murphy M, et al. (2001) c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 414: 768–773. [DOI] [PubMed] [Google Scholar]
- 46. Simons BW, Hurley PJ, Huang Z, Ross AE, Miller R, et al. (2012) Wnt signaling though beta-catenin is required for prostate lineage specification. Dev Biol 371: 246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lin Y, Liu G, Zhang Y, Hu YP, Yu K, et al. (2007) Fibroblast growth factor receptor 2 tyrosine kinase is required for prostatic morphogenesis and the acquisition of strict androgen dependency for adult tissue homeostasis. Development 134: 723–734. [DOI] [PubMed] [Google Scholar]
- 48. Mehta V, Abler LL, Keil KP, Schmitz CT, Joshi PS, et al. (2011) Atlas of Wnt and R-spondin gene expression in the developing male mouse lower urogenital tract. Dev Dyn 240: 2548–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang BE, Wang XD, Ernst JA, Polakis P, Gao WQ (2008) Regulation of epithelial branching morphogenesis and cancer cell growth of the prostate by Wnt signaling. PLoS ONE 3: e2186 doi:10.1371/journal.pone.0002186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lukacs RU, Memarzadeh S, Wu H, Witte ON (2010) Bmi-1 is a crucial regulator of prostate stem cell self-renewal and malignant transformation. Cell Stem Cell 7: 682–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bisson I, Prowse DM (2009) WNT signaling regulates self-renewal and differentiation of prostate cancer cells with stem cell characteristics. Cell Res 19: 683–697. [DOI] [PubMed] [Google Scholar]
- 52. Xin L, Lukacs RU, Lawson DA, Cheng D, Witte ON (2007) Self-renewal and multilineage differentiation in vitro from murine prostate stem cells. Stem Cells 25: 2760–2769. [DOI] [PubMed] [Google Scholar]
- 53. Risbridger G, Wang H, Young P, Kurita T, Wang YZ, et al. (2001) Evidence that epithelial and mesenchymal estrogen receptor-alpha mediates effects of estrogen on prostatic epithelium. Dev Biol 229: 432–442. [DOI] [PubMed] [Google Scholar]
- 54. Risbridger GP, Wang H, Frydenberg M, Cunha G (2001) The metaplastic effects of estrogen on mouse prostate epithelium: proliferation of cells with basal cell phenotype. Endocrinology 142: 2443–2450. [DOI] [PubMed] [Google Scholar]
- 55. Chen M, Yeh CR, Chang HC, Vitkus S, Wen XQ, et al. (2012) Loss of epithelial oestrogen receptor alpha inhibits oestrogen-stimulated prostate proliferation and squamous metaplasia via in vivo tissue selective knockout models. J Pathol 226: 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Romano RA, Ortt K, Birkaya B, Smalley K, Sinha S (2009) An active role of the DeltaN isoform of p63 in regulating basal keratin genes K5 and K14 and directing epidermal cell fate. PLoS ONE 4: e5623 doi:10.1371/journal.pone.0005623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hu H, Xia SH, Li AD, Xu X, Cai Y, et al. (2002) Elevated expression of p63 protein in human esophageal squamous cell carcinomas. Int J Cancer 102: 580–583. [DOI] [PubMed] [Google Scholar]
- 58. Ruptier C, De Gasperis A, Ansieau S, Granjon A, Taniere P, et al. (2011) TP63 P2 promoter functional analysis identifies beta-catenin as a key regulator of DeltaNp63 expression. Oncogene 30: 4656–4665. [DOI] [PubMed] [Google Scholar]
- 59. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, et al. (2012) The mutational landscape of lethal castration-resistant prostate cancer. Nature 487: 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kumar A, White TA, MacKenzie AP, Clegg N, Lee C, et al. (2011) Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc Natl Acad Sci U S A 108: 17087–17092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, et al. (2009) Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461: 614–620. [DOI] [PubMed] [Google Scholar]
- 62. Wu X, Wu J, Huang J, Powell WC, Zhang J, et al. (2001) Generation of a prostate epithelial cell-specific Cre transgenic mouse model for tissue-specific gene ablation. Mech Dev 101: 61–69. [DOI] [PubMed] [Google Scholar]
- 63. Lesche R, Groszer M, Gao J, Wang Y, Messing A, et al. (2002) Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis 32: 148–149. [DOI] [PubMed] [Google Scholar]
- 64. Val P, Jeays-Ward K, Swain A (2006) Identification of a novel population of adrenal-like cells in the mammalian testis. Dev Biol 299: 250–256. [DOI] [PubMed] [Google Scholar]
- 65. Swain A, Narvaez V, Burgoyne P, Camerino G, Lovell-Badge R (1998) Dax1 antagonizes Sry action in mammalian sex determination. Nature 391: 761–767. [DOI] [PubMed] [Google Scholar]
- 66. Park JH, Walls JE, Galvez JJ, Kim M, Abate-Shen C, et al. (2002) Prostatic intraepithelial neoplasia in genetically engineered mice. Am J Pathol 161: 727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shappell SB, Thomas GV, Roberts RL, Herbert R, Ittmann MM, et al. (2004) Prostate pathology of genetically engineered mice: definitions and classification. The consensus report from the Bar Harbor meeting of the Mouse Models of Human Cancer Consortium Prostate Pathology Committee. Cancer Res 64: 2270–2305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Embryonic squamous formation of prostate epithelium by stabilized β-Catenin. (A) high magnification of β-Catenin and p63 IHC on sections of Actβ-Cat;Nkx3.1Cre mutant and control (Actβ-Cat) prostate organ cultures grown for 3 days. Arrows indicate area of high β-Catenin that has become squamous stratified epithelium. (B) H&E and IHC for β-Catenin on sections of Actβ-Cat;Nkx3.1Cre prostate organ cultures grown for 5 days with no DHT.
(TIF)
β-Catenin is not required for adult prostate homeostasis. H&E stain and IHC for AR, Probasin, c-Myc and LEF1 on sections of β-Cat;PBCre mutant and control prostates. Sections cut through the dorsal-lateral lobe.
(TIF)
Western blot analysis of β-Catenin and LEF1 in adult mouse prostate tissue. (A) β-Catenin levels increase in Pten null (PtenF/F;PBCre) prostates compared to controls. (B) undetectable levels of active non-phosphorylated β-Catenin are present in control prostates, while in PtenF/F;PBCre prostates there is an accumulation of wild type endogenous active non-phosphorylated β-Catenin, as indicated. Actβ-Cat;PtenF/F;PBCre prostates have very high levels of the smaller exon 3 deleted mutant active β-Catenin, as indicated. The anti-active β-Catenin antibody also detects a smaller unknown band in control and PtenF/F;PBCre samples. LEF1 is not detected in control or PtenF/F;PBCre prostates and is upregulated Actβ-Cat;PtenF/F;PBCre prostates.
(TIF)
Stabilized β-Catenin and Pten homozygous loss prostate cancer. (A) H&E stain on sections of 2-month-old Actβ-Cat;PtenF/F;PBCre prostates. Left panel shows areas of adenocarcinoma and squamous metaplasia. Right panel shows epithelial cells invading into the surrounding stroma. (B) H&E stain and IHC for β-Catenin and p63 on sections of 3-month-old Actβ-Cat;PtenF/F;PBCre prostates showing detail of adenocarcinoma and squamous metaplasia. Black arrows indicate mitotic figures. White arrows indicate cords of β-Catenin positive epithelial cells that have invaded the stroma. Arrowheads indicate squamous metaplasia.
(TIF)