

Abstract
L-type voltage dependent Ca2+ channels (L-VDCCs; Cav1.2) are crucial in cardiovascular physiology. In heart and smooth muscle, hormones and transmitters operating via Gq enhance L-VDCC currents via essential protein kinase C (PKC) involvement. Heterologous reconstitution studies in Xenopus oocytes suggested that PKC and Gq-coupled receptors increased L-VDCC currents only in cardiac long N-terminus (NT) isoforms of α1C, whereas known smooth muscle short-NT isoforms were inhibited by PKC and Gq activators. We report a novel regulation of the long-NT α1C isoform by Gβγ. Gβγ inhibited whereas a Gβγ scavenger protein augmented the Gq- but not phorbol ester-mediated enhancement of channel activity, suggesting that Gβγ acts upstream from PKC. In vitro binding experiments reveal binding of both Gβγ and PKC to α1C-NT. However, PKC modulation was not altered by mutations of multiple potential phosphorylation sites in the NT, and was attenuated by a mutation of C-terminally located serine S1928. The insertion of exon 9a in intracellular loop 1 rendered the short-NT α1C sensitive to PKC stimulation and to Gβγ scavenging. Our results suggest a complex antagonistic interplay between Gq-activated PKC and Gβγ in regulation of L-VDCC, in which multiple cytosolic segments of α1C are involved.
Keywords: Gβγ, calcium channel, isoform, protein kinase C
Introduction
L-type voltage-dependent calcium channels (L-VDCC; CaV1.2) play a critical role in excitation-contraction coupling in cardiac, skeletal and smooth muscle.1-3 These channels are known to be modulated by a variety of hormones and transmitters, operating via GPCRs and second messengers, thereby profoundly affecting target tissues.4 A prominent modulatory pathway in the cardiovascular system is the enhancement of L-type Ca2+ currents by protein kinase C (PKC). Constitutive activity of PKC may underlie a tonic Ca2+ influx via L-VDCC in some smooth muscle cells,5 and activation of PKC is believed to critically participate in the effects of Gq-coupled GPCRs and other modulators of Cav1.2. For instance, vasoconstrictors such as angiotensin II and acetylcholine (ACh), operating mainly via Gq-coupled GPCRs in smooth muscle, induce release of Ca2+ from intracellular stores and enhance L-VDCC currents.6,7 As part of this signaling cascade, protein kinase C (PKC) is activated and was shown to be essential for Ca2+ current enhancement (discussed in refs. 5, 8–10). A less prominent inhibitory effect of PKC activators, that occasionally follows the enhancement, has been reported in cardiac and some smooth muscle cells.11-13 The enhancing effect of PKC and Gq-activating GPCRs on L-VDCC has been heterologously reconstituted only in Xenopus oocytes,10,14,15 enabling a detailed study of molecular mechanisms of these modulations. Therefore, Xenopus oocytes continued to be the heterologous expression system of choice in the current study.
The Gβγ dimer was also implicated as part of signaling cascades affecting the L-type channel. A complex and incompletely understood synergistic interaction between Gβγ, phosphoinositide 3 (PI3) kinase, PKC and often Src occurs in angiotensin, muscarinic m2 or β-adrenergic receptor-induced enhancements of L-type Ca2+ currents in some smooth muscle cells.9,16-18 The N- and C- termini (NT and CT, respectively) of the pore-forming α1 subunit of L-VDCC, Cav1.2α (α1C), contain binding sites for Gβγ, and coexpression of Gβγ with the channel in Xenopus oocytes resulted in a dual effect: a tonic Ca2+- and CaM-dependent inhibitory effect, and an enhancement when Ca2+ was chelated or when NT and/or CT gating-regulating segments were removed.19 The Gβγ-dependent, Ca2+-independent enhancement is in line with known effects of purified Gβγ on L-VDCC in smooth muscle,16,20,21 yet the physiological role of the inhibitory effect of Gβγ is unknown.
Neuronal VDCCs are inhibited by GPCRs in processes that are voltage independent (and mediated by several second messengers)22-25 and voltage dependent (VD). The VD process is mediated by Gβγ. It is fast, membrane delimited and occurs in members of the Cav2 group.26-28 Several intracellular segments of α1 subunits of neuronal VDCCs were shown to bind Gβγ; mainly the loop connecting the domains I and II, L1 (see Fig. 1A) and parts of the CT.29-31 The NT was clearly identified as another molecular determinant critical for the Gβγ modulation. It binds L1 in a Gβγ-dependent manner, thereby rendering the channel sensitive to the Gβγ inhibition.32-36 Furthermore, there is a crosstalk between the Gβγ and PKC pathways in the modulation of Cav2.2 (N-type) channels. PKC activation was shown to relieve the tonic Gβγ-mediated inhibition, suggesting a role for PKC phosphorylation of these channels.31,37-39 However, no such crosstalk was ever observed or investigated in the L-type channel; L1 of α1C does not bind Gβγ.19,29
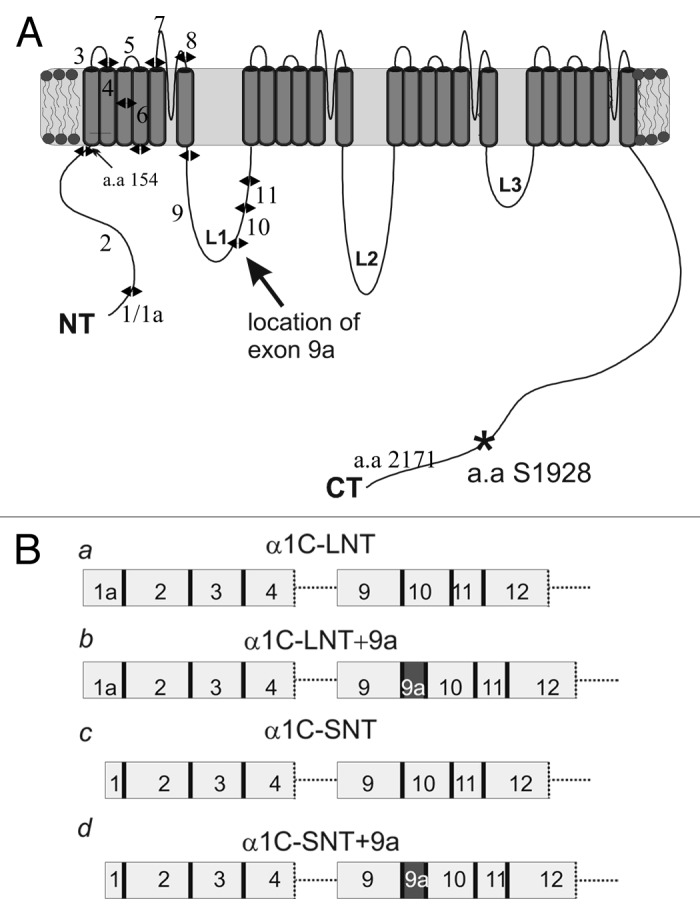
Figure 1. Exon-intron structure of four α1C constructs. (A) α1C protein structure and the location of the protein segment encoded by exon 9a, between exon 9 and 10. (B) Partial exon-intron structure of the four α1C isoforms used. These differ by the length of the NT (encoded by exon 1a, long-NT; exon 1, short-NT) and L1 (absence or presence of exon 9a).
A crucial factor determining the elaborate PKC signaling paradigm of L-VDCC is which isoform of α1C is expressed. We have previously demonstrated the reconstitution of the modulation of L-type calcium channel by PKC and by Gq-coupled GPCRs in Xenopus oocytes, and found it to be dependent on the isoform of α1C used. Only a long-NT (α1C-LNT; cardiac) isoform was upregulated by these agents, while only a decrease in the current was observed in short NT containing α1C (α1C-SNT; smooth muscle/brain type).10,40 The exact molecular mechanism underlying the current enhancement was not completely resolved. Moreover, despite the established role of PKC in the regulation of smooth muscle L-VDCCs, only a short NT isoform is known to be expressed in these cells.40,41 It was recently shown that the Cav1.2 gene undergoes extensive alternative splicing, often depending, in the smooth muscle, on culturing or pathologic conditions.42 Thus, there are two plausible explanations: (1) a unique mechanism which enhances α1C-SNT in smooth muscle in response to Gq activation must exist; or (2) one of the short-NT isoforms expressed in smooth muscle differs from the short NT α1C studied so far in a manner that renders it PKC-sensitive.
Here we report the involvement of the Gβγ dimer in the PKC-mediated, Gq-induced modulation of L-VDCC by opposing the effects of Gq-activating GPCRs but not of β-phorbol myristate acetate (PMA), a potent direct PKC activator. We characterize the effects of activation of the Gq-coupled receptor (m3R) by ACh, as well as involvement of Gβγ, on distinct isoforms of α1C, differing by the length of their NT as well as their L1 loop. Finally, we provide further insight to the mechanism of PKC-induced current enhancement by mutational analysis of putative phosphorylation sites.
Results
Gβγ hinders the enhancement of IBa following activation of Gq-coupled receptors
Modulation L-VDCC by ACh was studied in Xenopus oocytes expressing full subunit composition (α1C, β2b, α2δ) of the cardiac L-type calcium channel (α1C-LNT; see Fig. 1A and Ba) and Gq-coupled muscarinic receptors m3R or m1R. Ca2+ channel currents were studied with 40 mM Ba2+ as charge carrier. Typical whole-cell Ba2+ currents (IBa) and a current-voltage (I-V) curve in a representative oocyte expressing α1C-LNT are shown in Figure 2A. Application of ACh resulted, as previously shown, in upregulation of the current followed by a decline.10Figure 2B illustrates the time course of this effect (“control,” filled circles), and Figure 2C exemplifies typical shapes of IBa recorded at +20 mV before application of ACh (t = 0) and at the peak of ACh-induced current increase, 5 min after addition of ACh (t = 5).

Figure 2. Gβγ negates upregulation of IBa by Gq-coupled muscarinic receptors but not by direct activation of PKC by PMA. (A) a. net Ba2+ currents (obtained by subtracting currents remaining after block with 200 μM Cd2+, as described in43 recorded at 10 mV steps from -70 mV to +50 mV. b. Representative current-voltage curve. Peak amplitudes measured at each voltage step were used. (B) Time course of changes in IBa in response to ACh in oocytes expressing rabbit long-NT with m3R and m-cβARK or Gβγ. IBa was measured by 200 ms steps from -80 to 20 mV. After allowing the current to stabilize, ACh was added (as indicated). ACh was washed out after 6 min and IBa was monitored every 30 sec for additional 5 min. (C) Representative traces depicting IBa before (t = 0) and 5 min after addition of ACh (t = 5 min). (D) Summary of effects of ACh in oocytes expressed rabbit long-NT α1C and m3R with m-cβARK or Gβγ. Black bars represent the enhanced portion of the modulation measured, in each cell, at the peak of the enhancement. The gray bars represent the declining phase measured 10 min after the addition of ACh or PMA. (E) Summary of the effects of ACh in oocytes expressing rabbit long-NT α1C and m1R. (F) Coexpression of m-cβARK in oocytes expressing α1CΔN2–139 was without effect. (G) a. Summary of the effects of application of PMA to oocytes expressing rabbit long-NT α1C without (control) or with m-cβARK or Gβγ. b. representative traces of the effects of PMA on initial basal current (t = 0 min) and current recorded at t = 8 min. Statistics: asterisks (*) indicate significant differences from the control group; pound signs (#) indicate significant differences between various groups as indicated by the connecting brackets. All tests were performed by one-way ANOVA (see Experimental Procedures). ** or ##, p < 0.01; *** or ### p < 0.001; ###, p < 0.001. The number of cells tested is indicated on or above the bar.
We have previously found that the activation of PKC is crucially involved in mediating the increase in IBa. However, when the Gβγ scavenger protein, m-cβARK (a myristoylated C-terminus of β-adrenergic kinase 1), was coexpressed, the ACh-induced increase in IBa was significantly greater than in control. This was observed when either m1R or m3R were expressed to mediate the ACh action via Gq (Fig. 2B, D and E). The effect of m-cβARK was statistically significant, p < 0.01 (summarized in Fig. 2D). Bis-indolylmaleimide (Bis; a specific PKC inhibitor) significantly attenuated the ACh-induced increase in IBa, supporting the role of PKC in the current enhancement (see Fig. 2E).10 Bis also attenuated the ACh-induced increase in IBa when m-cβARK was coexpressed, but a residual increase could still be observed (Fig. 2E). The potentiating action of m-cβARK was the first indication of an involvement of Gβγ in the ACh-induced modulation. In order to further substantiate the involvement of the Gβγ dimer, we have coexpressed Gβ1γ2 along with the channel and the muscarinic receptor. This completely abolished the ACh-induced increase in IBa (Fig. 2B−D).
Coexpression of m-cβARK did not alter the modulation by ACh of the NT deletion mutant, α1CΔN2–139 (Fig. 2F). This channel variant lacks the “inhibitory module” segment of the NT (the first 20 a.a.) which is crucial for the modulation of the cardiac-type α1C by ACh and PKC.10,15,40,43 Both in the absence and presence of m-cβARK, there was only a decline in the current. The decline was quantitatively similar with or without m-cβARK. Thus, m-cβARK affects only the ACh-induced increase and does not operate by reducing or eliminating the decline in IBa. In support, overexpression of Gβγ did not alter the ACh-induced decline in IBa in α1C-SNT (data not shown), indicating that Gβγ operates on the enhancing effect of Gq-activation. To exclude a possible effect of coexpressed Gβγ or m-cβARK on the amount of receptor expressed on the plasma membrane, which may account for the observed changes in modulation, we measured total m1R protein level on the plasma membrane by biotinylation. No significant changes in m1R expression were noted in oocytes expressing channel alone, with Gβγ, or with m-cβARK (data not shown). Further, the persistence of the ACh-induced decrease in IBa in oocytes coexpressing Gβγ or m-cβARK, does not support a role for changes in plasma membrane receptor levels. While not in the focus of this study, this decrease is a genuine response to the activation of these GPCRs
To further understand the mode of action of Gβγ in PKC modulation, we activated PKC directly (as opposed to via a receptor) with the phorbol ester PMA and studied the effects of coexpressed Gβγ or m-cβARK. Both were without effect; the increase in IBa by PMA was not significantly different than in the channel expressed alone (Fig. 2G). Compartmentalization of PKC signaling in cardiac T-tubules was previously shown, where PMA had opposing effects on myocytes as compared with receptor activated PKC.44 Nevertheless, PMA was used here to robustly and directly activate PKC as means to decipher the molecular mechanism underlying PKC modulation and compare those to receptor activated PKC.
Activation of Gq in Xenopus oocytes characteristically results in typical chloride currents.45 These currents develop since calcium is being released from intracellular stores during the GPCR-Gq-PLC-Ins(1,4,5)P3 activation cascade, and the oocytes contain specific calcium-dependent chloride channels that open consequently. Indeed, activation of m1R by ACh in oocytes expressing only the receptor yielded these typical Cl- currents. Coexpression of Gβγ abolished these Cl- currents, while chelation of Gβγ (by coexpressing m-cβARK) significantly augmented Cl- currents (Fig. 3A and B). Thus, it appears that Gβγ probably acts on an event in the GPCR-Gq signaling pathway that is unrelated to PKC-induced phosphorylation (which is not involved in the activation of Ca2+-dependent Cl- channels).
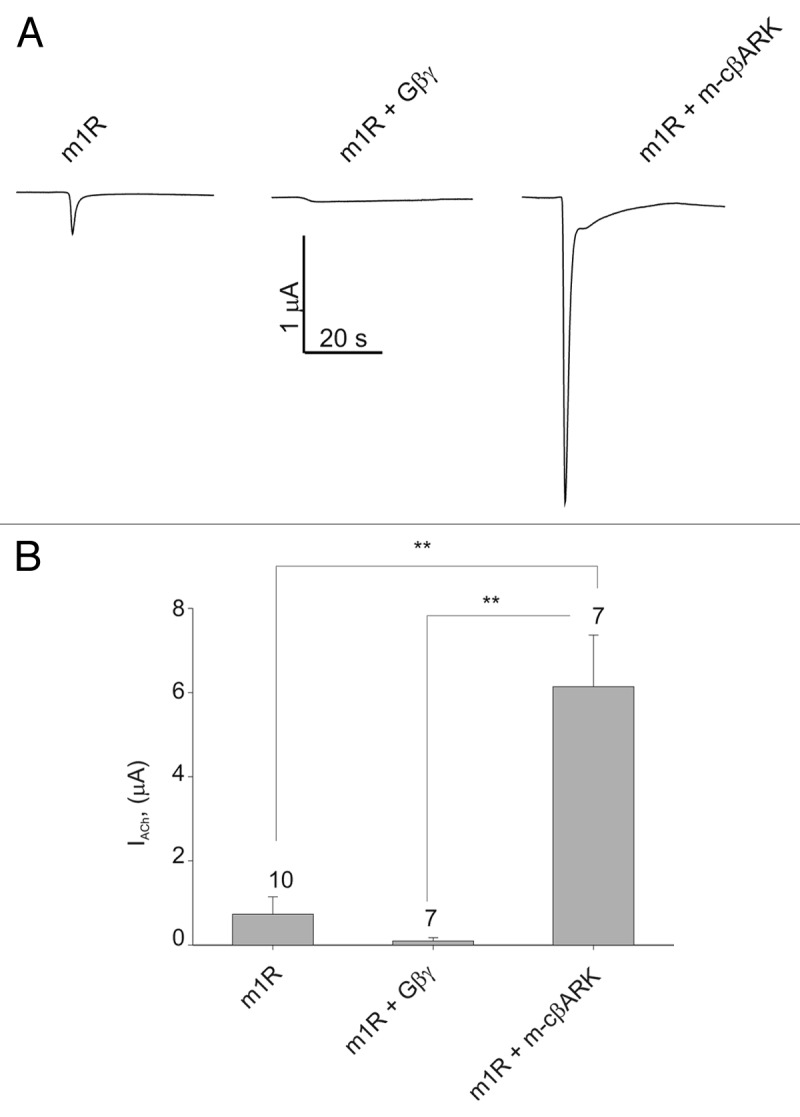
Figure 3. Gβγ abolishes typical chloride currents following Gq-mediated PKC activation. (A) Representative traces of ACh-induced chloride currents in Xenopus oocytes expressing m1R alone, with Gβγ or m-cβARK. (B) Summary of chloride currents magnitude in oocytes expressing m1R alone, with Gβγ or m-cβARK. **, p < 0.01 by t-test.
PKC appears to regulate α1C by phosphorylating a C-terminal, but not N-terminal, sites
Although PKC does not phosphorylate the initial 20 or 46 a.a. of long-NT which are crucial for the PKC-induced enhancement of the channel’s current,46 other putative phosphorylation sites exist on α1C, including in proximal and distal NT. We have explored most potential sites of PKC phosphorylation in the NT of the α1C-LNT isoform by mutating Ser or Thr residues to Ala (Fig. S1). The first α1C variant comprised four such mutations proximal to a.a. 75; these mutations also included T27, a site previously reported as potentially phosphorylated by PKC.47 The second variant comprised five mutations distal to aa. 75. Both mutated channel variants were still potentiated by ACh (Fig. 4A), suggesting that phosphorylation of NT is not involved in PKC-induced enhancement of L-VDCC activity. At present we cannot exclude the less likely possibility that one of the remaining non-mutated Ser or Thr in the NT may be a non-conventional PKC phosphorylation site, or that simultaneous mutation of all serines and threonines in the NT might produce a different result.

Figure 4. The S1928A mutation in α1C is involved in modulation by PKC. (A) Ser/Thr mutations in NT do not affect upregulation of IBa by ACh. Oocytes were injected with wt α1C or NT mutants 1 or 2 (see Fig. S1), NT mut I or NT mut II, α2δ, β2b and m3R. ACh was applied following current stabilization. The upregulation induced by ACh was not prevented by the mutations of putative PKC phosphorylation sites. (B) The effect of S1928A mutation of α1C-LNT on the effect of PMA on IBa. a. Time-course of the experiments in oocytes expressing wt (closed circles) or S1928A (open circles) α1C and α2δ, β2b. b. Summary of the enhancement of IBa caused by PMA, at t = 10 min. The current was significantly less augmented in the S1928A mutant as compared with wt. **, p < 0.01 by t-test. (C) The effect of S1928A mutation of α1C-LNT on the effect of ACh on IBa. a. Time-course of the experiments in oocytes co-expressing m3R and wt (closed circles) or S1928A (open circles) α1C and α2δ, β2b. b. Summary of the effect of Ach on IBa, at t = 5 min. The current was significantly less augmented in the S1928A mutant as compared with wt. ***, p < 0.001 by t-test.
An important phosphorylation site in α1C is Ser1928 on distal CT. Ser1928 was found to be phosphorylated by protein kinase A (PKA), and48-51 also by PKC in vitro.52,53 Application of PMA (10 nM) to oocytes expressing the mutated α1CS1928A with α2δ and β2b produced a significantly smaller enhancement of IBa compared with wt α1C (28% vs. 82%; Fig. 4B). Similarly, the S1928A mutation eliminated the increase in IBa caused by ACh via m3R, yet left a prominent ACh-induced decrease. This suggests that the C-terminal S1928 may be one of several sites whose phosphorylation by PKC enhances the activity of L-VDCC, whereas NT does not appear to be a target for PKC phosphorylation.
α1C NT contains a PKC binding site
Gβγ was previously shown to bind the NT of the L-type channel.19 A more detailed scan of shorter GST-fused segments of the NT revealed that in vitro synthesized Gβ1γ2 binds to the distal GST-fused segments of the NT, NT 95–140 and NT 95–154 (Fig. 5A). This part of NT is encoded by exon 2 and is conserved among all known isoforms of α1C (see Fig. 1). Similar binding experiments with in vitro synthesized PKCα and GST-fused NT sections resulted in a similar binding pattern (Fig. 5B). IBa upregulation via Gq was previously shown to be Ca2+ dependent (abolished when Ca2+ was strongly chelated) as well as PLC- (hence also diacylglycerol-) dependent.10 Thus, it appears that a conventional PKC isoform is activated via this cascade. PKCα, a conventional PKC, was shown to be abundant in both cardiac and smooth muscle preparations.54-56 This isoform was therefore selected for the in-vitro binding experiments. PKCα bound to GST fused full-length NT (1–154) and to smaller segments: 47–154 and 95–154, but not to the beginning or middle part of NT (segments 1–47, 40–87, 60–120 and 84–120), suggesting that PKC probably binds to the distal part of NT close to the plasma membrane boundary (Fig. 5B and C). These results are at odds with the report that only CT but not NT binds PKCα 53, but the autoradiography used here for the detection of bound PKCα is a more sensitive method than the western blotting used in the above work. In all, our data support the existence of a macromolecular signaling complex between α1C and PKCα as proposed by Yang et al.53 where NT may be an anchoring site for PKC.

Figure 5. Both Gβγ and PKCα bind distal segments of α1C-NT. (A) Binding of [35S]-labeled Gβ1γ2 to different GST-fused fragments of the NT. Immobilized GST fusion proteins were incubated with Gβ1γ2. After washing, Gβ1γ2-bound proteins were eluted and resolved by SDS-PAGE. Gβ1γ2 was found to bind the distal third of the NT. Similar data was obtained in six more experiments. (B) Binding of [35S]-labeled PKCα to different GST-fused fragments of the NT. PKCα was found to bind the distal half of the NT. (C) [35S]PKCα binding to segments of the NT was repeated in the presence of 1.5 μg purified Gβ1γ2 . It did not alter PKC binding to the NT, suggesting no binding competition for the same site.
To inquire whether Gβγ attenuates the effect of PKC by directly competing for interaction with the NT, binding experiments with PKCα and NT were repeated in the presence of 1.5 μg purified Gβγ. The addition of Gβγ did not alter the binding of PKCα to GST-fused segments of the NT (Fig. 5C). These results suggest that Gβγ does not interfere directly with the binding of PKC to NT.
Searching for PKC-regulated short-NT α1C isoforms
Despite the use of m-cβARK, activation of Gq-coupled muscarinic receptors still failed to enhance IBa via the previously described short-NT isoform, α1C-SNT, and PMA also did not upregulate this isoform (see Fig. 7). Yet, PKC is known to enhance L-type currents in smooth muscle (see Introduction). We envisaged that unique smooth muscle isoforms might exist that are affected by ACh and PKC, possibly in a Gβγ-dependent manner.

Figure 7. Isoforms of α1C containing the L1 insertion encoded by exon 9a show a greater sensitivity to PKC activation. (A) IBa was recorded at +20 mV in oocytes expressing an α1C isoform (as indicated), α2δ, β2b and PKCα. Following stabilization of the current, PMA (10 nM) was applied for 12–15 min. Bars depict the maximal increase in IBa evoked by PMA. *, p < 0.05 by t-test. (B−C) Oocytes were injected with α1C, α2δ, β2b and m3R with or without m-cβARK. IBa was recorded at +20 mV every 30 sec. Following current stabilization, ACh applied for 6 min and further monitored for additional 5 min after ACh washout. Black bars represent the enhanced portion of the current; gray bars represent the declining phase of the current. (B) Coexpression of m-cβARK enhanced the increase in IBa to a similar extent in both long-NT isoform. (C) Coexpression of m-cβARK enhanced IBa in both short NT isoforms. IBa increased to slightly above baseline levels in oocytes coexpressing m-cβARK and α1C-SNT, +9a, while only a decrease was present in α1C-SNT oocytes. *, p < 0.05; **, p < 0.01 by t-test (as compared with channel alone without m-cβARK).
Two isoforms of human α1C with differences in the cytosolic loop L1 have been described. The corresponding mRNAs differ in the absence or presence of a dispensable 75 nucleotide-long exon 9a, downstream of the DNA segment encoding the AID (α1C interaction domain with β subunit) (Figs. 1 and 6A; Fig. S2).57 Specific antibodies raised against protein sequences encoded by exon 9a demonstrated its presence in human smooth muscle cells in arteries.58 A detailed study of alternative splicing of human SNT smooth muscle variant also supports the 9a insertion.42 A notable rabbit α1C smooth muscle variant, referred to as rabbit lung isoform, also contains a 75 nucleotide long insertion in L159 which is absent in the well-studied cardiac α1C-LNT isoform originally cloned form a heart cDNA library.60 The a.a. composition of both short L1 (“-9a isoform”) and long L1 (“+9a isoform,” with the insertion encoded by the 75 nucleotides) is highly homologous among human and rabbit α1C (Fig. S2). Since L1 is intimately involved in PKC regulation in the N-type channels,31 we hypothesized that L1 isoforms of L-type channel may also differ in PKC regulation.

Figure 6. Exon 9a transcripts found in human RNA samples by RT-PCR. (A) Proposed exon-intron structure and the various primers used. (B) Table summarizing the expected yield of PCR amplicons using the different primers. (C) Representative 2% agarose gels of RT-PCR products. -9a and +9a transcripts were found predominantly in human bladder and aorta RNA, while -9a transcripts alone were predominantly found in human heart RNA.
We further investigated the presence of exon 9a-encoded insertion in human tissues that express α1C-using RT-PCR on human RNA samples. We were able to detect exon 9a in human heart, bladder and aorta (Fig. 6). Interestingly, both +9a and -9a isoforms were detected mainly in human bladder and aorta, indicating the existence of both transcripts, while the human heart RNA contained mostly the -9a isoform of the channel. There was a weak band corresponding to +9a isoform when using primers 2+5 on human heart RNA, but the main and stronger band was -9a, and the +9a band was missing in a separate experiment using the same set of primers. Furthermore, primers 11+2 yielded only -9a transcripts in the human heart RNA (Fig. 6C, “2+5,” “11+2”). We can thus conclude that human heart RNA mainly contains the -9a transcript.
In order to study the effects of 9a insertion on the channel, we have constructed two chimeras of the rabbit α1C-LNT and α1C-SNT into which we inserted the exon 9a sequence into L1 (see Fig. 1B), and compared them to the previously characterized α1C counterparts (without 9a). Oocytes were injected with equimolar concentrations of RNA corresponding to the α1C variant used and auxiliary subunit(s) α2/δ, with or without β2b. Currents were measured with Ba2+ as the charge carrier, at ascending voltage steps from -70 to +50 mV. The activation parameters in all four α1C variants were similar in the absence of β2b. As expected, expression of β2b shifted activation to hyperpolarized potentials in all chimeras (Fig. S3, Table S1). It should be noted that the half-activation voltage (Va) of α1C-SNT,+9a was shifted to hyperpolarized potentials by ~3 mV as compared with α1C-SNT, when expressed without β2b. Similar observations were made by Liao et al. in HEK cells, however the shift was much more pronounced, by ~11 mV.58 Although such differences may be attributed to the heterologous expression system used, i.e., Xenopus oocytes vs. HEK 293 cells, the difference is not negligible and may require further investigation. Inactivation kinetics of IBa were measured using a 10 sec long test pulse (from -10 mV to +40 mV) and were found to be similar in all four α1C variants (Fig. S4, Table S1). It is noteworthy that the increase in Gmax caused by coexpression of β2b appeared less prominent in +9a than in -9a isoforms, especially in α1C-SNT (Table S1). While this observation needs further study, it may indicate a novel role for the exon 9a insertion in trafficking from the ER to the plasma membrane or in the regulation of maximal open probability.43
Enhanced effect of PKC and ACh on long-L1 (+9a) isoforms
In order to study the effect of PKC on the long-L1 isoforms, we expressed all four isoforms in Xenopus oocytes and measured Ba2+ currents at +20 mV before and during application of PMA (10 nM). PMA induced a greater increase in the current in the two long-loop isoforms (+9a); the difference reached statistical significance only in the α1C-SNT isoforms (Fig. 7A). The enhancement of IBa in α1C-SNT,+9a was not much above basal levels, but remember that normally only a decrease is observed in α1C-SNT. The increase in IBa is actually underestimated since it overlaps the decrease. Moreover, the physiological relevance of the decrease in IBa induced by activation of Gq seen in Xenopus oocytes is not clear, it may be a “side effect” in this model system.10 These results strengthen the role of exon 9a in α1C-SNT,+9a as participating in the enhancing effect of PKC observed in native smooth muscle tissue.
Following the partial restoration of the increase in IBa by PMA in the α1C-SNT,+9a isoform, we set out to examine the effects of ACh, via m3R, in all four isoforms. For the most part, the two long-L1 (+9a) isoforms demonstrated similar responses to application of ACh as their controls; i.e., an increase in IBa in α1C-LNT,+9a isoform, and only a decrease in α1C-SNT,+9a isoform (Fig. 7B and C). Nevertheless, when m-cβARK was coexpressed, in the α1C-SNT,+9a isoform there was an increase in the current to slightly above basal level. The increase was especially significant when compared with the decrease seen in the control conditions in the previously characterized α1C-SNT (Fig. 7C). Again, exon 9a in α1C-SNT,+9a is shown to be important for the Gq-mediated increased current as observed in native tissues. It also points to the differences between direct activation of PKC by PMA and activation of PKC via the Gq signaling cascade.
Discussion
PKC-regulation of four Cav1.2α isoforms, differing in the length of the initial segment of their NT and the presence of an insertion in cytosolic loop 1 (L1) was studied. Utilizing Xenopus oocytes, the only heterologous system in which the enhancement of α1C by PKC has been reconstituted,10 we identified an α1C isoform with a short-NT and long L1 as a candidate PKC-upregulated isoform in the smooth muscle. PKC also plays a crucial role in mediating the regulation of α1C by Gq-activating GPCRs, such as muscarinic m1 and m3 receptors. We further investigated the molecular mechanism of Gq-mediated and PKC-induced modulation of α1C. We found a novel modulation of Gq effect by Gβγ; the Gq-mediated GPCR effects are tonically attenuated by Gβγ. In contrast, direct activation of PKC by a phorbol ester is insensitive to Gβγ, suggesting that Gβγ acts upstream from PKC. Further, we identified the NT as a possible anchoring site for PKC, where it may serve as part of a modulatory scaffold, possibly also involving the distal CT. Despite the crucial importance of the NT for PKC regulation, its phosphorylation does not appear to mediate the effect of PKC. We found that the C-terminal serine 1928, a known target for both PKA and PKC, is a functionally important site for PKC effect. Our findings imply a complex antagonistic interplay between Gq-activated PKC and Gβγ in regulation of L-VDCC, in which NT, CT, and L1 are involved.
Gβγ and PKC bind to NT of α1C and oppositely regulate the L-VDCC function
Gβγ was previously shown to directly bind to NT and CT of α1C and dually regulate the cardiac isoform of this channel.19 Nonetheless, its role in modulation of this channel remains ambiguous. Here we show that the Gq-mediated upregulation of L-VDCC, reconstituted in Xenopus oocytes, shows a distinct regulation profile by Gβγ that resembles the well-characterized antagonistic regulation of the N-type channels, where Gβγ inhibits whereas PKC enhances channel activity61 (however, this resemblance does not necessarily imply similarity of molecular mechanisms). When coexpressed with the channel and m3R, Gβγ abolished ACh-induced current enhancement performed by the Gq signaling pathway. Moreover, Gβγ sequestration by m-cβARK further augmented the Gq-mediated increase in IBa. The latter finding suggests that endogenous free Gβγ, which appears to be relatively high in Xenopus oocytes,62,63 may exert a tonic inhibitory control upon cardiac L-type (α1C-LNT) channels. Taken together, these results propose a role for Gβγ in the modulation of the cardiac L-type calcium channel by Gq-activating GPCRs.
The inhibitory Gβγ control of L-VDCC observed in oocytes appears to be at odds with the synergistic or consequential modulation of α1C by Gβγ and PKC observed in smooth muscle in response to Gs- or Gi-activating GPCRs such as β-adrenergic and muscarinic m2 receptors. In these cases Gβγ enhances the L-type currents acting indirectly, via complex mechanisms that involve activation of PI3 kinase, PKC, and PKA or Src.17,18,20,21,64 These modulations have not been so far heterologously reconstituted, hindering the study of the underlying molecular mechanisms. The controversy between these and our results may be apparent, arising from the experimental conditions used. The electrophysiological experiments in the above studies were usually performed in whole cell mode with 10 mM EGTA in the pipette (strong Ca2+ chelation). The inhibitory effect of coexpressed Gβγ on α1C-LNT in Xenopus oocytes is Ca2+- and calmodulin-dependent, and it is replaced by an enhancement under the conditions of Ca2+ chelation.19 In the experiments described in this report, no or mild Ca2+ chelation conditions were used; the calculated intracellular concentration of Ca2+ chelators used (EGTA and BAPTA) was about 2 mM, which may favor the inhibitory action of Gβγ. Other than experimental conditions, different smooth muscle channel isoforms may be modulated by Gs or Gi. The inhibitory effects of Gβγ described here are specifically exerted upon Gq-mediated modulation of L-VDCC. It remains to be investigated whether Gs or Gi mediated regulation of smooth muscle VDCC, when and if successfully reconstituted in a heterologous system, will be modulated differently by Gβγ.
Though not directly phosphorylated by PKC,46 the initial 20 a.a. segment of α1C-LNT is crucial for the upregulation of channel activity by PMA and Gq-coupled GPCRs.10,46 This segment, missing in α1C-SNT, was identified as an important regulatory module that exerts a tonic control over the gating, reducing the channel’s open probability.15,43 We have previously proposed that the action of PKC on α1C-LNT relies upon the relief of this inhibitory control, possibly by phosphorylation of the channel elsewhere.15,46 Our new data offer new insights into the role of NT in PKC regulation of α1C. Using direct in vitro protein-protein interaction measurements, we identify the universally conserved part of NT of α1C, encoded by exon 2, as a binding site for PKCα (between a.a. 95–154). Thus, the NT physically interacts with PKCα; this may be how PKC is anchored to the channel and how it exerts its action.
How does Gβγ oppose the effect of Gq-coupled GPCRs, m1R and m3R? A straightforward interpretation of the results of Figures 2 and 3 (no effect of Gβγ and m-cβARK on PMA-induced increase in IBa) and the fact that membrane receptor level is non-significantly affected by coespressed Gβγ or m-cβARK, is that Gβγ acts upstream of PKC, e.g., by obstructing the activation of Gq, or the activation of phospholipase C by Gαq-GTP. In support, despite the apparent proximity of N-terminal PKC and Gβγ binding sites, Gβγ does not interfere with the binding of PKC to the NT. The new findings explain why in the oocytes PMA usually induces a greater increase in IBa than Gq-activating GPCRs (e.g., Figs. 2 and 7): free Gβγ released upon activation of Gq would be expected to attenuate further activation of Gq itself or of phospholipase C (a negative feedback mechanism).65
S1928 is important for the functional effect of PKC
The decrease in Ba2+ current via rabbit long-NT channels reported by McHugh et al.47 in HEK cells was eliminated by mutating two threonines at positions 27 and 31, and the authors speculated that these two residues are phosphorylated by PKC and that this phosphorylation underlies the observed decrease. In contrast, in oocytes the PMA- and ACh-induced decrease persists in constructs lacking the first 46 a.a. that include the aforementioned threonines.10,15 It appears that PMA-induced decreases in IBa in Xenopus oocytes are not mediated by phosphorylation of the NT.
Whatever the mechanism of decrease in IBa, the main and the most important effect of PKC on cardiac and smooth muscle L-VDCCs is the enhancement of the current (see Introduction). The NT plays a crucial role in relaying the effect of PKC to channel gating in long NT isoforms of α1C, and also contains a PKC-anchoring module as discussed above. However, none of the putative PKC phosphorylation sites in the NT appear to participate in the PKC-induced enhancement of IBa. In contrast, we find that PKC effect is greatly reduced by mutation of the major PKA phosphorylation site in the distal CT, S1928. This is in line with phosphorylation of this residue by PKC in vitro.53 Furthermore, this part of the channel is an autoinhibitory module which is normally truncated in cardiac cells but remains attached to the C-terminus due to non-covalent binding.66,67 The participation of both NT and CT in PKC regulation supports the hypothesis19 that channel gating is tightly regulated by interactions between NT and CT (“NT-CT scaffold”), possibly together with L1 (as corroborated by PKC modulation of α1C-SNT,+9a); PKC, Gβγ and other regulators such as calmodulin act on L-VDCC by conformationally modulating this scaffold.
A short-NT, long-L1 α1C isoform is a candidate L-VDCC positively regulated by PKC in smooth muscle
Previously studied rabbit and human short NT α1C were not upregulated by PKC in heterologous expression systems,10,15 yet a short NT isoform is the one expressed in smooth muscle.68,69 Both in human and rabbit, an α1C isoform containing a 75 bp-long insertion in L1, encoded (in humans) by exon 9* or 9a, has been detected.58,70 Cardiac isoform of α1C contains a long NT (encoded by exon 1a),40 but, according to our RT PCR results, probably lacks exon 9a. This is in line with the findings of Liao et al. who reported lower levels of exon 9a in rat heart than in rat aorta and, using a specific antibody, found this insertion present in human smooth muscle arteries.58
We have inserted exon 9a to the wt rabbit α1C-LNT and α1C-SNT (that we used throughout our study). Remarkably, there was a notable increase in the current to above basal levels in α1C-SNT,+9a, following PMA application. Further, when both m3R and m-cβARK were expressed, following application of ACh an increase in the current to above baseline levels was observed in oocytes expressing the α1C-SNT,+9a. The differences in treatments that induce current enhancement in the α1C-SNT,+9a isoform (application of PMA vs. ACh) probably reside in the way PKC is activated/Application of PMA results in a massive mobilization of PKC to the plasma membrane.71 Nonetheless, these findings clearly distinguish this isoform from the wt α1C-SNT isoform in terms of its PKC and Gq-induced modulation. Aorta and bladder contain the short NT and, as we find, either long or short L1 (with or without exon 9a-encoded part). These are not the only isoforms of the channel present in smooth muscle, since it undergoes robust alternative splicing resulting in vast diversity in response to culturing conditions and the surrounding stimuli in vivo. Thus, it is plausible that other isoforms may be upregulated by PKC in smooth muscle; yet up to date only the one containing exon 9a behaved as such. α1C-SNT is not upregulated by PMA or ACh unless it contains exon 9a, as we have shown here.
Materials and Methods
Oocyte culture
All the experiments were performed in accordance with the Tel Aviv University Institutional Animal Care and Use Committee (permits no. 11-99-47 and 11-05-064). Xenopus laevis frogs were maintained and operated, and oocytes were collected, defolliculated, and injected with RNA as described.72 Female frogs, maintained at 20 ± 2°C on an 11 h light/13 h dark cycle, were anesthetized in a 0.15% solution of procaine methanesulfonate (MS222), and portions of ovary were removed through a small incision on the abdomen. The incision was sutured, and the animal was returned to a separate tank until it had fully recovered from the anesthesia, and afterwards was returned to a large tank where, together with the other postoperational animals, it was allowed to recover for at least 4 weeks until the next surgery. The animals did not show any signs of postoperational distress.
Oocytes were injected with equal amounts (by weight; 2.5 ng or 1 ng) of the mRNAs of CaV1.2α isoforms (original long-NT isoform: accession number X15539) or its mutants with α2/δ (accession number M21948), with or without β2b (previously referred to as rabbit heart β2A; accession number X64297), with or without 1 ng of m1R or m3R, and incubated for 3–5 d at 20–22°C in NDE96 solution (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 2.5 mM Na pyruvate, 50 μg/ml gentamycine, 5 mM HEPES, pH 7.5).
cDNA constructs and mRNA
cDNAs of α1C, α2/δ and β2b were as described.73 The rabbit heart α1C mutants used here were prepared in our laboratory as described.15 Rabbit α1C short-NT was prepared as described,43 resulting in an exchange of exon 1a of α1C-LNT with exon 1 of α1C-SNT. Rat m1R cDNA is in pGEM2. Rat m3R and rabbit PKCα are in pGEM-HJ. m-cβARK is in myr-pGEM-HE.63,74 The RNAs were prepared using a standard procedure previously described, which ensures capping of the 5′ end of the RNA and preferential inclusion of non-capped GTP in the rest of the RNA.72
Electrophysiology
Whole cell currents were recorded using the Gene Clamp 500 amplifier (Axon Instruments) using the two-electrode voltage clamp technique in a solution containing 40 mM Ba(OH)2, 50 mM NaOH, 2 mM KOH and 5 mM HEPES, titrated to pH 7.5 with methanesulfonic acid.75 Stock solution of ACh (1 M) was stored in 10–20 μl aliquots at –20°C and added to the recoding solution at a final concentration of 10 μM. Ba2+ currents were measured by 200 ms steps to +20 mV from a holding potential of -80 mV, every 30 sec. Bis-indolylmaleimide (Bis) was dissolved in water to 5 mM and stored in aliquots in -20°C. Oocytes were injected with 50 nl of 300 μM Bis and additionally incubated in the presence of 5 μM Bis for 2–4 h before measurement. Stock solution of β-phorbol-12-myristate 13-acetate (PMA; 10 mM) were prepared in DMSO and stored, protected from light, at -80°C. PMA at a final concentration of 10 nM was added to Ba2+ solution. In most experiments, all oocytes were injected with 25 nl of 50 mM BAPTA or EGTA, 30 min or 2–4 h before measurement, respectively, unless otherwise stated. All organic reagents were purchased from Sigma.
Current-voltage relations of the IBa were obtained by stepping the membrane potential from the holding potential (-80 mV) to various voltages in 10 mV steps. Voltage pulses were delivered every 10 sec. In each cell, the net IBa was obtained by subtraction of the residual currents recorded with the same protocols after applying 200 µM Cd2+. All experiments were performed at room temperature (20–22°C).
I-V curves were fitted to standard Boltzmann equation in the form of IBa = Gmax(Vm-Vrev)/(1+exp(-(Vm-Va)/Ka))), where Ka is the slope factor, Va is the voltage that causes half maximal activation, Gmax is the maximal conductance, Vm is membrane voltage, IBa is the current measured at the same voltage, and Vrev is the reversal potential of IBa. The obtained parameters of Gmax and Vrev were then used to calculate fractional conductance at each Vm, G/Gmax, using the equation: G/Gmax = I/(Gmax(Vm-Vrev)), where G is the total macroscopic conductance at Vm. The conductance-voltage (G-V) curves were plotted with the values of Va and Ka obtained from the fit of the I-V curves, using the following form of the Boltzmann equation: G/Gmax = 1/(1+exp((-(Vm-Va)/Ka))).
The waveform of decay of IBa was fitted to a two exponential equation by Levenberg-Marquardt method on Clampfit version 9 (Axon Instruments Inc.) or by a least mean square procedure in SigmaPlot in the form: f (t) = Afaste-t/τfast +Aslowe-t/τslow +C, where A is the contribution of each kinetic component (fast or slow), τfast and τslow are the respective time constants, and C is the non-inactivating current.
RT-PCR analysis
Five µg of total human heart, bladder and aorta RNA purchased from Ambion, Inc. (catalog no. 7966, lot 110P43B; cat. no. 7990, lot 103P010802046A; cat. no. 6844, lot 053P010802003A, respectively) were reverse-transcribed with SuperScript II reverse transcriptase (Invitrogen) with primer #1 (see text below and Fig. 4A). Each PCR reaction (50 µl) contained 2.4 µl of the product of RT reaction, 1 µl of 10 mM dNTPs, 20–50 pmol of primers, 5 µl of 10 × PCR buffer, 2 µl of Mg2+ (2 mM), and 1 µl of Taq DNA polymerase (Promega). PCR was performed under the following conditions: 95°C for 1 min, 49°C for 1 min, and 72°C for 2 min, repeated 35 times. The final elongation was performed at 72°C for 5 min. The PCR products were analyzed on a 2% agarose gel.
The primers used for RT and PCR were: #1, CTTGGACTTCTGTGAGCC (end of exon 12); #2, GAGAGTTTTCCAAAGAGA (end of exon 9); #3, TGAGCATGCCCACCAGTG (beginning of exon 10); #4, CCTCAATACACCGCAGTA (intron between exon 11 and exon 12); #5, CTGAACTTTGACTTGGAG (end of exon 11); #6, GAGGCACTCCGGCGGGCA (beginning of the exon 9a); #7, TCTGTGGAGTGACTAAAC (end of exon 9a); #8, CTGGAGTAATTCCTTCTC (intron between exon 9 and exon 9a); #9, GCCAGCACTGCCCAGAGG (intron between exon 9a and exon 10); #10, CAAAGAGAGGGAGAAGGC (10 nucl. after the beginning of exon 9a); #11, CACCAGCCAGTAGAAGAC (starting 80 nucl. from the end of exon 12); #12, TGCCCTGCCCCTCCTCTCA (intron between exon 8 and exon 9).
Interaction between GST fusion proteins and in vitro synthesized Gβγ and PKCα
The procedures were essentially as described.46 In brief, [35S]Met/Cys-labeled Gβ1 and PKCα were translated on the template of in vitro synthesized RNAs using a rabbit reticulocyte translation kit (Promega). The GST fusion proteins were synthesized and extracted from Escherichia coli using the Amersham Pharmacia Biotech kit. Purified GST fusion proteins (5–10 µg) or purified GST (10 µg) were incubated with 5 µl of the lysate, containing the 35S-labeled proteins in 500 µl of high K+ buffer (150 mM KCl, 50 mM Tris, 5 mM MgCl2, 1 mM EDTA, pH 7.0) with 0.5% CHAPS or 0.01% Lubrol (as indicated), for 2 h at room temperature, with gentle rocking. In some experiments the incubation was done in the presence of purified 1.5 μg Gβγ (kind gift of C. W. Dessauer, University of Texas, Houston). Then 30 µl of glutathione-Sepharose beads (Amersham Pharmacia Biotech) were added, and the mixture was incubated for 30 min at 4°C and washed four times in 1 ml of the same buffer. Following washing, GST fusion proteins were eluted with 30 µl of 20 mM reduced glutathione in elution buffer (120 mM NaCl, 100 mM TRIS-HCl, pH 8). 35S-labeled proteins were analyzed on 12% SDS-polyacrylamide gels. The labeled products were identified and quantified by autoradiography using PhosphorImager (Molecular Dynamics) as described.76
Statistics and data presentation
The data are presented as mean ± SEM, n = number of cells tested. To overcome the problem of batch-to-batch variability in current amplitudes, the results were normalized as follows: in each oocyte, IBa was normalized to the amplitude measured before application of an agonist. These normalized values were averaged across all oocyte batches tested. Comparisons between two groups (e.g., control and receptor expressing groups) were tested for statistically significant differences (p < 0.05 or better) using two-tailed unpaired t-test. Comparisons of amplitudes of IBa at different times in the same group were done using paired t-test. Comparison between several groups was done using one-way analysis of variance (ANOVA) followed by Tukey’s test, using the SigmaStat software (SPSS Corp.).
Supplementary Material
Acknowledgments
This paper was supported by the German-Israel Foundation (grant 930-220.2/2006). We thank Dr Carmen W. Dessauer (University of Texas, Houston) for kindly providing us with purified Gβγ. We thank W.A. Catterall (University of Washington, Seattle) for insightful scientific discussions.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental material may be found here: http://www.landesbioscience.com/journals/channels/article/22016/
Footnotes
Previously published online: www.landesbioscience.com/journals/channels/article/22016
References
- 1.Hughes AD. Calcium channels in vascular smooth muscle cells. J Vasc Res. 1995;32:353–70. doi: 10.1159/000159111. [DOI] [PubMed] [Google Scholar]
- 2.Reuter H. Calcium channel modulation by neurotransmitters, enzymes and drugs. Nature. 1983;301:569–74. doi: 10.1038/301569a0. [DOI] [PubMed] [Google Scholar]
- 3.Xiong Z, Sperelakis N. Regulation of L-type calcium channels of vascular smooth muscle cells. J Mol Cell Cardiol. 1995;27:75–91. doi: 10.1016/S0022-2828(08)80009-0. [DOI] [PubMed] [Google Scholar]
- 4.Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–55. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- 5.Navedo MF, Amberg GC, Votaw VS, Santana LF. Constitutively active L-type Ca2+ channels. Proc Natl Acad Sci U S A. 2005;102:11112–7. doi: 10.1073/pnas.0500360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huster M, Frei E, Hofmann F, Wegener JW. A complex of Ca(V)1.2/PKC is involved in muscarinic signaling in smooth muscle. FASEB J. 2010;24:2651–9. doi: 10.1096/fj.09-149856. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y, Yan T, Wang Q, Wang W, Xu J, Wu X, et al. PKC-dependent extracellular signal-regulated kinase 1/2 pathway is involved in the inhibition of Ib on AngiotensinII-induced proliferation of vascular smooth muscle cells. Biochem Biophys Res Commun. 2008;375:151–5. doi: 10.1016/j.bbrc.2008.07.137. [DOI] [PubMed] [Google Scholar]
- 8.Fish RD, Sperti G, Colucci WS, Clapham DE. Phorbol ester increases the dihydropyridine-sensitive calcium conductance in a vascular smooth muscle cell line. Circ Res. 1988;62:1049–54. doi: 10.1161/01.RES.62.5.1049. [DOI] [PubMed] [Google Scholar]
- 9.Callaghan B, Zhong J, Keef KD. Signaling pathway underlying stimulation of L-type Ca2+ channels in rabbit portal vein myocytes by recombinant Gbetagamma subunits. Am J Physiol Heart Circ Physiol. 2006;291:H2541–6. doi: 10.1152/ajpheart.00420.2006. [DOI] [PubMed] [Google Scholar]
- 10.Weiss S, Doan T, Bernstein KE, Dascal N. Modulation of cardiac Ca2+ channel by Gq-activating neurotransmitters reconstituted in Xenopus oocytes. J Biol Chem. 2004;279:12503–10. doi: 10.1074/jbc.M310196200. [DOI] [PubMed] [Google Scholar]
- 11.Lacerda AE, Rampe D, Brown AM. Effects of protein kinase C activators on cardiac Ca2+ channels. Nature. 1988;335:249–51. doi: 10.1038/335249a0. [DOI] [PubMed] [Google Scholar]
- 12.Tseng GN, Boyden PA. Different effects of intracellular Ca and protein kinase C on cardiac T and L Ca currents. Am J Physiol. 1991;261:H364–79. doi: 10.1152/ajpheart.1991.261.2.H364. [DOI] [PubMed] [Google Scholar]
- 13.Boixel C, Tessier S, Pansard Y, Lang-Lazdunski L, Mercadier JJ, Hatem SN. Tyrosine kinase and protein kinase C regulate L-type Ca(2+) current cooperatively in human atrial myocytes. Am J Physiol Heart Circ Physiol. 2000;278:H670–6. doi: 10.1152/ajpheart.2000.278.2.H670. [DOI] [PubMed] [Google Scholar]
- 14.Singer-Lahat D, Gershon E, Lotan I, Hullin R, Biel M, Flockerzi V, et al. Modulation of cardiac Ca2+ channels in Xenopus oocytes by protein kinase C. FEBS Lett. 1992;306:113–8. doi: 10.1016/0014-5793(92)80980-U. [DOI] [PubMed] [Google Scholar]
- 15.Shistik E, Ivanina T, Blumenstein Y, Dascal N. Crucial role of N terminus in function of cardiac L-type Ca2+ channel and its modulation by protein kinase C. J Biol Chem. 1998;273:17901–9. doi: 10.1074/jbc.273.28.17901. [DOI] [PubMed] [Google Scholar]
- 16.Viard P, Exner T, Maier U, Mironneau J, Nürnberg B, Macrez N. Gbetagamma dimers stimulate vascular L-type Ca2+ channels via phosphoinositide 3-kinase. FASEB J. 1999;13:685–94. doi: 10.1096/fasebj.13.6.685. [DOI] [PubMed] [Google Scholar]
- 17.Quignard JF, Mironneau J, Carricaburu V, Fournier B, Babich A, Nurnberg B, et al. Phosphoinositide 3-kinase γ mediates angiotensin II-induced stimulation of L-type calcium channels in vascular myocytes. J Biol Chem. 2001;276:32545–51. doi: 10.1074/jbc.M102582200. [DOI] [PubMed] [Google Scholar]
- 18.Callaghan B, Koh SD, Keef KD. Muscarinic M2 receptor stimulation of Cav1.2b requires phosphatidylinositol 3-kinase, protein kinase C, and c-Src. Circ Res. 2004;94:626–33. doi: 10.1161/01.RES.0000118248.17466.B7. [DOI] [PubMed] [Google Scholar]
- 19.Ivanina T, Blumenstein Y, Shistik E, Barzilai R, Dascal N. Modulation of L-type Ca2+ channels by gbeta gamma and calmodulin via interactions with N and C termini of alpha 1C. J Biol Chem. 2000;275:39846–54. doi: 10.1074/jbc.M005881200. [DOI] [PubMed] [Google Scholar]
- 20.Zhong J, Dessauer CW, Keef KD, Hume JR. Regulation of L-type Ca2+ channels in rabbit portal vein by G protein alphas and betagamma subunits. J Physiol. 1999;517:109–20. doi: 10.1111/j.1469-7793.1999.0109z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong J, Hume JR, Keef KD. β-Adrenergic receptor stimulation of L-type Ca2+ channels in rabbit portal vein myocytes involves both alphas and betagamma G protein subunits. J Physiol. 2001;531:105–15. doi: 10.1111/j.1469-7793.2001.0105j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bernheim L, Beech DJ, Hille B. A diffusible second messenger mediates one of the pathways coupling receptors to calcium channels in rat sympathetic neurons. Neuron. 1991;6:859–67. doi: 10.1016/0896-6273(91)90226-P. [DOI] [PubMed] [Google Scholar]
- 23.Diversé-Pierluissi M, Dunlap K. Distinct, convergent second messenger pathways modulate neuronal calcium currents. Neuron. 1993;10:753–60. doi: 10.1016/0896-6273(93)90175-Q. [DOI] [PubMed] [Google Scholar]
- 24.Diversé-Pierluissi M, Goldsmith PK, Dunlap K. Transmitter-mediated inhibition of N-type calcium channels in sensory neurons involves multiple GTP-binding proteins and subunits. Neuron. 1995;14:191–200. doi: 10.1016/0896-6273(95)90254-6. [DOI] [PubMed] [Google Scholar]
- 25.Shapiro MS, Wollmuth LP, Hille B. Modulation of Ca2+ channels by PTX-sensitive G-proteins is blocked by N-ethylmaleimide in rat sympathetic neurons. J Neurosci. 1994;14:7109–16. doi: 10.1523/JNEUROSCI.14-11-07109.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dolphin AC. Mechanisms of modulation of voltage-dependent calcium channels by G proteins. J Physiol. 1998;506:3–11. doi: 10.1111/j.1469-7793.1998.003bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ikeda SR, Dunlap K. Voltage-dependent modulation of N-type calcium channels: role of G protein subunits. Adv Second Messenger Phosphoprotein Res. 1999;33:131–51. doi: 10.1016/S1040-7952(99)80008-1. [DOI] [PubMed] [Google Scholar]
- 28.Zamponi GW, Snutch TP. Modulation of voltage-dependent calcium channels by G proteins. Curr Opin Neurobiol. 1998;8:351–6. doi: 10.1016/S0959-4388(98)80060-3. [DOI] [PubMed] [Google Scholar]
- 29.De Waard M, Liu H, Walker D, Scott VE, Gurnett CA, Campbell KP. Direct binding of G-protein betagamma complex to voltage-dependent calcium channels. Nature. 1997;385:446–50. doi: 10.1038/385446a0. [DOI] [PubMed] [Google Scholar]
- 30.Qin N, Platano D, Olcese R, Stefani E, Birnbaumer L. Direct interaction of gbetagamma with a C-terminal gbetagamma-binding domain of the Ca2+ channel alpha1 subunit is responsible for channel inhibition by G protein-coupled receptors. Proc Natl Acad Sci U S A. 1997;94:8866–71. doi: 10.1073/pnas.94.16.8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel alpha1 subunit. Nature. 1997;385:442–6. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- 32.Agler HL, Evans J, Tay LH, Anderson MJ, Colecraft HM, Yue DT. G protein-gated inhibitory module of N-type (ca(v)2.2) ca2+ channels. Neuron. 2005;46:891–904. doi: 10.1016/j.neuron.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 33.Cantí C, Page KM, Stephens GJ, Dolphin AC. Identification of residues in the N terminus of alpha1B critical for inhibition of the voltage-dependent calcium channel by Gbeta gamma. J Neurosci. 1999;19:6855–64. doi: 10.1523/JNEUROSCI.19-16-06855.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Page KM, Cantí C, Stephens GJ, Berrow NS, Dolphin AC. Identification of the amino terminus of neuronal Ca2+ channel alpha1 subunits alpha1B and alpha1E as an essential determinant of G-protein modulation. J Neurosci. 1998;18:4815–24. doi: 10.1523/JNEUROSCI.18-13-04815.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simen AA, Miller RJ. Involvement of regions in domain I in the opioid receptor sensitivity of alpha1B Ca(2+) channels. Mol Pharmacol. 2000;57:1064–74. [PubMed] [Google Scholar]
- 36.Zhang JF, Ellinor PT, Aldrich RW, Tsien RW. Multiple structural elements in voltage-dependent Ca2+ channels support their inhibition by G proteins. Neuron. 1996;17:991–1003. doi: 10.1016/S0896-6273(00)80229-9. [DOI] [PubMed] [Google Scholar]
- 37.Swartz KJ. Modulation of Ca2+ channels by protein kinase C in rat central and peripheral neurons: disruption of G protein-mediated inhibition. Neuron. 1993;11:305–20. doi: 10.1016/0896-6273(93)90186-U. [DOI] [PubMed] [Google Scholar]
- 38.Barrett CF, Rittenhouse AR. Modulation of N-type calcium channel activity by G-proteins and protein kinase C. J Gen Physiol. 2000;115:277–86. doi: 10.1085/jgp.115.3.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu Y, Ikeda SR. Modulation of Ca(2+)-channel currents by protein kinase C in adult rat sympathetic neurons. J Neurophysiol. 1994;72:1549–60. doi: 10.1152/jn.1994.72.4.1549. [DOI] [PubMed] [Google Scholar]
- 40.Blumenstein Y, Kanevsky N, Sahar G, Barzilai R, Ivanina T, Dascal N. A novel long N-terminal isoform of human L-type Ca2+ channel is up-regulated by protein kinase C. J Biol Chem. 2002;277:3419–23. doi: 10.1074/jbc.C100642200. [DOI] [PubMed] [Google Scholar]
- 41.Dai B, Saada N, Echetebu C, Dettbarn C, Palade P. A new promoter for α1C subunit of human L-type cardiac calcium channel Ca(V)1.2. Biochem Biophys Res Commun. 2002;296:429–33. doi: 10.1016/S0006-291X(02)00894-X. [DOI] [PubMed] [Google Scholar]
- 42.Tiwari S, Zhang Y, Heller J, Abernethy DR, Soldatov NM. Atherosclerosis-related molecular alteration of the human CaV1.2 calcium channel alpha1C subunit. Proc Natl Acad Sci U S A. 2006;103:17024–9. doi: 10.1073/pnas.0606539103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kanevsky N, Dascal N. Regulation of maximal open probability is a separable function of Ca(v)beta subunit in L-type Ca2+ channel, dependent on NH2 terminus of alpha1C (Ca(v)1.2alpha) J Gen Physiol. 2006;128:15–36. doi: 10.1085/jgp.200609485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robu VG, Pfeiffer ES, Robia SL, Balijepalli RC, Pi Y, Kamp TJ, et al. Localization of functional endothelin receptor signaling complexes in cardiac transverse tubules. J Biol Chem. 2003;278:48154–61. doi: 10.1074/jbc.M304396200. [DOI] [PubMed] [Google Scholar]
- 45.Dascal N, Cohen S. Further characterization of the slow muscarinic responses in Xenopus oocytes. Pflugers Arch. 1987;409:512–20. doi: 10.1007/BF00583809. [DOI] [PubMed] [Google Scholar]
- 46.Shistik E, Keren-Raifman T, Idelson GH, Blumenstein Y, Dascal N, Ivanina T. The N terminus of the cardiac L-type Ca(2+) channel alpha(1C) subunit. The initial segment is ubiquitous and crucial for protein kinase C modulation, but is not directly phosphorylated. J Biol Chem. 1999;274:31145–9. doi: 10.1074/jbc.274.44.31145. [DOI] [PubMed] [Google Scholar]
- 47.McHugh D, Sharp EM, Scheuer T, Catterall WA. Inhibition of cardiac L-type calcium channels by protein kinase C phosphorylation of two sites in the N-terminal domain. Proc Natl Acad Sci U S A. 2000;97:12334–8. doi: 10.1073/pnas.210384297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perets T, Blumenstein Y, Shistik E, Lotan I, Dascal N. A potential site of functional modulation by protein kinase A in the cardiac Ca2+ channel alpha 1C subunit. FEBS Lett. 1996;384:189–92. doi: 10.1016/0014-5793(96)00303-1. [DOI] [PubMed] [Google Scholar]
- 49.Mitterdorfer J, Froschmayr M, Grabner M, Moebius FF, Glossmann H, Striessnig J. Identification of PK-A phosphorylation sites in the carboxyl terminus of L-type calcium channel α 1 subunits. Biochemistry. 1996;35:9400–6. doi: 10.1021/bi960683o. [DOI] [PubMed] [Google Scholar]
- 50.De Jongh KS, Murphy BJ, Colvin AA, Hell JW, Takahashi M, Catterall WA. Specific phosphorylation of a site in the full-length form of the α 1 subunit of the cardiac L-type calcium channel by adenosine 3′,5′-cyclic monophosphate-dependent protein kinase. Biochemistry. 1996;35:10392–402. doi: 10.1021/bi953023c. [DOI] [PubMed] [Google Scholar]
- 51.Gao T, Yatani A, Dell’Acqua ML, Sako H, Green SA, Dascal N, et al. cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron. 1997;19:185–96. doi: 10.1016/S0896-6273(00)80358-X. [DOI] [PubMed] [Google Scholar]
- 52.Yang L, Doshi D, Morrow J, Katchman A, Chen X, Marx SO. Protein kinase C isoforms differentially phosphorylate Ca(v)1.2 alpha(1c) Biochemistry. 2009;48:6674–83. doi: 10.1021/bi900322a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang L, Liu G, Zakharov SI, Morrow JP, Rybin VO, Steinberg SF, et al. Ser1928 is a common site for Cav1.2 phosphorylation by protein kinase C isoforms. J Biol Chem. 2005;280:207–14. doi: 10.1074/jbc.M410509200. [DOI] [PubMed] [Google Scholar]
- 54.Park WS, Son YK, Kim N, Youm JB, Warda M, Ko JH, et al. Direct modulation of Ca(2+)-activated K(+) current by H-89 in rabbit coronary arterial smooth muscle cells. Vascul Pharmacol. 2007;46:105–13. doi: 10.1016/j.vph.2006.08.413. [DOI] [PubMed] [Google Scholar]
- 55.Simonis G, Briem SK, Schoen SP, Bock M, Marquetant R, Strasser RH. Protein kinase C in the human heart: differential regulation of the isoforms in aortic stenosis or dilated cardiomyopathy. Mol Cell Biochem. 2007;305:103–11. doi: 10.1007/s11010-007-9533-3. [DOI] [PubMed] [Google Scholar]
- 56.Wang Y, Wang W, Wang Q, Wu J, Xu J, Wu X. [Ca2+]i and PKC-alpha are involved in the inhibitory effects of Ib, a novel nonpeptide AngiotensinII subtype AT1 receptor antagonist, on AngiotensinII-induced vascular contraction in vitro. Biochem Biophys Res Commun. 2007;364:118–23. doi: 10.1016/j.bbrc.2007.09.101. [DOI] [PubMed] [Google Scholar]
- 57.Tang ZZ, Liang MC, Lu S, Yu D, Yu CY, Yue DT, et al. Transcript scanning reveals novel and extensive splice variations in human l-type voltage-gated calcium channel, Cav1.2 alpha1 subunit. J Biol Chem. 2004;279:44335–43. doi: 10.1074/jbc.M407023200. [DOI] [PubMed] [Google Scholar]
- 58.Liao P, Yu D, Lu S, Tang Z, Liang MC, Zeng S, et al. Smooth muscle-selective alternatively spliced exon generates functional variation in Cav1.2 calcium channels. J Biol Chem. 2004;279:50329–35. doi: 10.1074/jbc.M409436200. [DOI] [PubMed] [Google Scholar]
- 59.Biel M, Ruth P, Bosse E, Hullin R, Stühmer W, Flockerzi V, et al. Primary structure and functional expression of a high voltage activated calcium channel from rabbit lung. FEBS Lett. 1990;269:409–12. doi: 10.1016/0014-5793(90)81205-3. [DOI] [PubMed] [Google Scholar]
- 60.Mikami A, Imoto K, Tanabe T, Niidome T, Mori Y, Takeshima H, et al. Primary structure and functional expression of the cardiac dihydropyridine-sensitive calcium channel. Nature. 1989;340:230–3. doi: 10.1038/340230a0. [DOI] [PubMed] [Google Scholar]
- 61.Zamponi GW. Determinants of G protein inhibition of presynaptic calcium channels. Cell Biochem Biophys. 2001;34:79–94. doi: 10.1385/CBB:34:1:79. [DOI] [PubMed] [Google Scholar]
- 62.Lutz LB, Kim BE, Jahani D, Hammes SR. G protein β γ subunits inhibit nongenomic progesterone-induced signaling and maturation in Xenopus laevis oocytes. Evidence for a release of inhibition mechanism for cell cycle progression. J Biol Chem. 2000;275:41512–20. doi: 10.1074/jbc.M006757200. [DOI] [PubMed] [Google Scholar]
- 63.Rishal I, Porozov Y, Yakubovich D, Varon D, Dascal NG. Gbetagamma-dependent and Gbetagamma-independent basal activity of G protein-activated K+ channels. J Biol Chem. 2005;280:16685–94. doi: 10.1074/jbc.M412196200. [DOI] [PubMed] [Google Scholar]
- 64.Viard P, Macrez N, Mironneau C, Mironneau J. Involvement of both G protein alphas and β γ subunits in β-adrenergic stimulation of vascular L-type Ca(2+) channels. Br J Pharmacol. 2001;132:669–76. doi: 10.1038/sj.bjp.0703864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bonacci TM, Ghosh M, Malik S, Smrcka AV. Regulatory interactions between the amino terminus of G-protein betagamma subunits and the catalytic domain of phospholipase Cbeta2. J Biol Chem. 2005;280:10174–81. doi: 10.1074/jbc.M412514200. [DOI] [PubMed] [Google Scholar]
- 66.Gao T, Cuadra AE, Ma H, Bunemann M, Gerhardstein BL, Cheng T, et al. C-terminal fragments of the α 1C (CaV1.2) subunit associate with and regulate L-type calcium channels containing C-terminal-truncated α 1C subunits. J Biol Chem. 2001;276:21089–97. doi: 10.1074/jbc.M008000200. [DOI] [PubMed] [Google Scholar]
- 67.Hulme JT, Yarov-Yarovoy V, Lin TW, Scheuer T, Catterall WA. Autoinhibitory control of the CaV1.2 channel by its proteolytically processed distal C-terminal domain. J Physiol. 2006;576:87–102. doi: 10.1113/jphysiol.2006.111799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pang L, Koren G, Wang Z, Nattel S. Tissue-specific expression of two human Ca(v)1.2 isoforms under the control of distinct 5′ flanking regulatory elements. FEBS Lett. 2003;546:349–54. doi: 10.1016/S0014-5793(03)00629-X. [DOI] [PubMed] [Google Scholar]
- 69.Saada NI, Carrillo ED, Dai B, Wang WZ, Dettbarn C, Sanchez J, et al. Expression of multiple CaV1.2 transcripts in rat tissues mediated by different promoters. Cell Calcium. 2005;37:301–9. doi: 10.1016/j.ceca.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 70.Biel M, Hullin R, Freundner S, Singer D, Dascal N, Flockerzi V, et al. Tissue-specific expression of high-voltage-activated dihydropyridine-sensitive L-type calcium channels. Eur J Biochem. 1991;200:81–8. doi: 10.1111/j.1432-1033.1991.tb21051.x. [DOI] [PubMed] [Google Scholar]
- 71.Nishizuka Y. Studies and perspectives of protein kinase C. Science. 1986;233:305–12. doi: 10.1126/science.3014651. [DOI] [PubMed] [Google Scholar]
- 72.Dascal N, Lotan I. Expression of exogenous ion channels and neurotransmitter receptors in RNA-injected Xenopus oocytes. Protocols in Molecular Neurobiology, Chapter 13: Humana Press, Totowa, NJ, 1992:205-25. [Google Scholar]
- 73.Singer D, Biel M, Lotan I, Flockerzi V, Hofmann F, Dascal N. The roles of the subunits in the function of the calcium channel. Science. 1991;253:1553–7. doi: 10.1126/science.1716787. [DOI] [PubMed] [Google Scholar]
- 74.Jing J, Chikvashvili D, Singer-Lahat D, Thornhill WB, Reuveny E, Lotan I. Fast inactivation of a brain K+ channel composed of Kv1.1 and Kvbeta1.1 subunits modulated by G protein beta gamma subunits. EMBO J. 1999;18:1245–56. doi: 10.1093/emboj/18.5.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shistik E, Ivanina T, Puri T, Hosey M, Dascal N. Ca2+ current enhancement by alpha 2/delta and beta subunits in Xenopus oocytes: contribution of changes in channel gating and alpha 1 protein level. J Physiol. 1995;489:55–62. doi: 10.1113/jphysiol.1995.sp021029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ivanina T, Perets T, Thornhill WB, Levin G, Dascal N, Lotan I. Phosphorylation by protein kinase A of RCK1 K+ channels expressed in Xenopus oocytes. Biochemistry. 1994;33:8786–92. doi: 10.1021/bi00195a021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.