

Abstract
Diabetes is a major risk factor for stroke. However, the molecular mechanisms involved in cerebral artery dysfunction found in the diabetic patients are not completely elucidated. In cerebral artery smooth muscle cells (CASMCs), spontaneous and local increases of intracellular Ca2+ due to the opening of ryanodine receptors (Ca2+ sparks) activate large conductance Ca2+-activated K+ (BK) channels that generate spontaneous transient outward currents (STOCs). STOCs have a key participation in the control of vascular myogenic tone and blood pressure. Our goal was to investigate whether alterations in Ca2+ spark and STOC activities, measured by confocal microscopy and patch-clamp technique, respectively, occur in isolated CASMCs of an experimental model of type-2 diabetes (db/db mouse). We found that mean Ca2+ spark amplitude, duration, size and rate-of-rise were significantly smaller in Fluo-3 loaded db/db compared to control CASMCs, with a subsequent decrease in the total amount of Ca2+ released through Ca2+ sparks in db/db CASMCs, though Ca2+ spark frequency remained. Interestingly, the frequency of large-amplitude Ca2+ sparks was also significantly reduced in db/db cells. In addition, the frequency and amplitude of STOCs were markedly reduced at all voltages tested (from −50 to 0 mV) in db/db CASMCs. The latter correlates with decreased BK channel β1/α subunit ratio found in db/db vascular tissues. Taken together, Ca2+ spark alterations lead to inappropriate BK channels activation in CASMCs of db/db mice and this condition is aggravated by the decrease in the BK β1 subunit/α subunit ratio which underlies the significant reduction of Ca2+ spark/STOC coupling in CASMCs of diabetic animals.
Introduction
More than 65% of patients with diabetes die from cardiovascular disease or stroke [1]. When considering age-adjusted incidence rates, type-2 diabetic patients are two- to five times as likely to suffer cerebral vascular disease or stroke compared with non-diabetic patients, a disparity that is seen in multiple racial/geographic groups [2]–[6] and may result from abnormal cerebral artery tissue function. Interestingly, the incidence of stroke in type-2 diabetic patients is not associated with the duration of disease, smoking, fasting blood glucose, total cholesterol, lipoprotein concentrations, or hypertension [3], [4], [7]. Cerebral blood flow disturbances, impaired cerebral vascular reactivity, transient ischemic attacks and oxidative damage of cerebral vessels have been found in both type-2 diabetic patients [3] and experimental models [8]–[10] that could account for the higher incidence of diabetes-related stroke events [1]–[7]; however, the molecular mechanisms involved in cerebral artery dysfunction are not completely elucidated.
The db/db mouse, a genetic model of non-insulin dependent type-2 diabetes exhibits cerebral vascular dysfunction [9] that exacerbates brain damage, edema and inflammation after induced experimental stroke [11]–[13]. In addition to diabetes-related alterations found in cerebral vessels, vascular dysfunction is also present in mesenteric arteries [14]–[16], coronary arterioles [17], gracilis muscle arterioles [18], [19], and aorta [20]–[22] of db/db mice. Cerebral arterioles of db/db mice show impaired response to vasodilators and reduced baseline arteriolar diameter [9]. Mesenteric arteries and gracilis muscle arterioles of db/db mice show impaired response to vasodilators, enhanced response to vasoconstrictors and enhanced basal myogenic tone [14]–[16], [18], [23]. Consistent with the observations of augmented vascular tone, the myogenic pressure-diameter of arteries harvested from db/db mice are smaller than the diameters of corresponding control arteries [14], [18], [19] and are not improved by the removal of endothelium [14], [19]. Moreover, investigators have demonstrated impairment of endothelium-independent dilation in the presence of nitric oxide donors: in coronary arterioles and aorta of db/db mice in the presence of sodium nitroprusside [17], [20], and in arteries of type 2 diabetic patients after administration of glycerin trinitrate [24], [25]. All these data suggest that smooth muscle-dependent mechanisms are also responsible for the vascular dysfunction associated with type-2 diabetes. Furthermore, the disease appears to alter functional responses of resistance arteries not only at endothelial level but also in active smooth muscle layers.
In cerebral artery smooth muscle cells (CASMCs), spontaneous and local increases of intracellular Ca2+ due to the opening of Ryanodine Receptors (RyRs), visualized as Ca2+ sparks, activate large conductance Ca2+ sensitive K+ channels (BK channels) that generate spontaneous transient outward currents (STOCs) [26], [27]. STOCs have a key role in the control of arterial tone by shifting the membrane potential towards less positive values, which in turn limits Ca2+ influx through L-type Ca2+ channels, diminishes global intracellular Ca2+ concentration ([Ca2+]i), and opposes vasoconstriction [28]–[30]. Therefore, RyRs through Ca2+ sparks and BK channels, by producing STOCs regulates arterial tone favoring vasorelaxation [29]–[31]. In addition, Ca2+ spark generation is also regulated by Ca2+ influx due to an indirect coupling between L-type Ca2+ channels and RyRs [32]. The sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) participates in this indirect coupling by redirecting the Ca2+ coming from the extracellular medium towards its luminal stores, located mainly inside the sarcoplasmic reticulum (SR) of CASMCs. Navedo and collaborators [33] demonstrated that L-type Ca2+ currents and Ca2+sparklet activity –brief plasma membrane Ca2+ fluxes due to the activation of clusters of L-type Ca2+ channels– are enhanced in CASMCs of db/db mice; suggesting that the mechanisms that participate in the closure and tight regulation of L-type Ca2+ channels could be impaired in diabetic CASMCs. Therefore, we sought to investigate whether BK channels and their functional pair, the RyRs, might contribute to worsening the condition of diabetic CASMCs.
Our data represent the first demonstration of altered Ca2+ spark and STOC in CASMCs from db/db mice due to Ca2+ spark alterations which lead to inappropriate BK channels activation, and the latter is aggravated by the decrease in the BK β1 subunit/α subunit ratio also found in CASMCs of db/db mice.
Results
Reduced Spatiotemporal Properties of Ca2+ Spark in CASMCs of Diabetic Mice
Ca2+ sparks of CASMCs represent the spontaneous and coordinated opening of an undefined number of RyRs within a cluster [26], [30]. Their frequency and properties –i.e. amplitude, duration, size and mean rise rate– are used as markers of in situ RyR activity [34]. Since impaired function of RyRs might contribute to the vascular alterations in the cerebral arteries of diabetic db/db mice [8], [9], [33], our first aim was to study the in situ activity of vascular RyR in single, freshly isolated CASMCs of diabetic mice.
Figure 1A shows representative confocal images of Ca2+sparks (right images) recorded in intact control and diabetic (db/db) CASMCs that conserve their typical relaxed spindle shape (left images). We found that the db/db CASMCs produced similar Ca2+ spark frequency to control cells (in Hz: 0.8±0.1 vs. 0.8±0.1; or in events·s−1·µm−1: 0.053±0.008 vs. 0.044±0.006, in 41 db/db cells and 43 control cells, respectively (Fig.1B ), with no further modifications in the number of firing sites per cell, eager site probability and the maximum number of events within a same site (Figure S1). The maintained Ca2+ spark frequency in CASMCs contrasts with the decrease found in cardiac myocytes of the same mouse model [35]. However, db/db CASMCs presented a significant reduction in average Ca2+ spark amplitude (Fig. 1C), full duration at half maximum (FDHM, Fig. 1D), full width at half maximum (FWHM, Fig. 1E) and mean rising rate (d(F/F0)/dt: 0.181±0.006 vs. 0.163±0.005 in 460 Ca2+ sparks from 43 control CASMCs vs. 423 Ca2+ sparks from 41 db/db CASMCs, P<0.05).
Figure 1. Reduced properties of spontaneous Ca2+ sparks in CASMCs of diabetic mice. A.

Confocal images of Fluo-3 loaded CASMCs (left) and normalized representative line scan images (1.92ms/line, right) showing spontaneous Ca2+ sparks recorded in control (top images) and db/db (db/db; bottom images) cells. Intact CASMCs are relaxed and show their typical spindle shape. Scale bar = 15 µm. B. Bar graph of Ca2+ spark frequency –measured as number of Ca2+events observed per second per µm– in 43 intact control (white bars) and 41 db/db (orange bars) CASMCs. C-E Bar graphs of averaged Ca2+ spark amplitude (C, F/F0); duration (D, FDHM or full duration at half maximum in ms); and width (E, FWHM, full width at half maximum in µm). F-H. Probability density function (PDf) of Ca2+ spark amplitudes (F; F/F0), durations (G; ms), and widths (H; µm) in control (white circles; n = 460) and db/db (orange circles; n = 423 events) CASMCs. Curves represent mixed gaussian functions fitted to the data.
Similar to previous observations in cardiac cells [35], histogram distributions of Ca2+ spark parameters –plotted as a function of probability density (PDf)– were asymmetrical for both control and diabetic events (Fig 1F–H ) and were fitted to a bimodal Gaussian function [35]. The amplitude distributions (Fig 1F ) showed 2 peaks: one around 1.25 F/F0 and another of around 1.6 F/F0; however, the population of events at larger amplitudes was drastically reduced in db/db compared to control CASMCs (43.7% in control cells to 5.9% in db/db cells, Table 1), which accounted for the decrease in the average Ca2+ spark amplitude. In addition, the distribution of Ca2+ spark durations showed 2 peaks that in db/db cells were shifted towards shorter durations (in ms: first peak from 41.80 to 34.11 and second peak from 86.16 to 62.37, Table 1) with modified proportions (in %: first peak from 69.3 to 43.9 and second peak from 30.0 to 49.6). On average, these effects decreased the population of longer Ca2+ sparks in db/db CASMCs (Figure1G ). In the case of size (FWHM) distribution (Fig 1H ) the population of events changed from one peak distribution (mean at 2.77 µm) in controls to two peaks distribution (in µm: first peak at 2.03, second peak at 4.69, Table 1). Taken together, these results show a shift of the population to weaker, shorter and smaller Ca2+ sparks in db/db CASMCs. Since Ca2+ spark amplitude, duration and mean rising rate are good indicators of local release flux [34], we can conclude that although the total occurrence of Ca2+ sparks are similar between db/db and control CASMCs, the total Ca2+ mass (calculated by amplitude*duration*width) released through Ca2+ sparks was decreased in db/db cells (in F/F0·ms·µm: 317.0±18.2 in 423 db/db events vs. 472.1±30.8 in 460 control events, P<0.001). This could be due to a decreased number of RyR opening simultaneously in a cluster. We estimated RyR expression by western blots in aorta homogenates and found a decrease in db/db mice (Figure 2).
Table 1. Mean (µ), SE(σ), proportion (p in %) and correlation coefficient (r 2) of Gaussian Fits to Ca2+ spark parameters.
| Amplitude (F/F0) | |||||||
| µ1 | SE1 | p 1 | µ2 | SE2 | p 2 | r2 | |
| control | 1.19 | 0.08 | 51.8 | 1.61 | 0.77 | 43.7 | 0.99 |
| db/db | 1.28 | 0.01 | 94.1 | 2.03 | 0.08 | 5.9 | 0.99 |
| Duration (ms) | |||||||
| µ1 | SE1 | p 1 | µ2 | SE2 | p 2 | r2 | |
| control | 41.80 | 5.27 | 69.3 | 86.16 | 162.0 | 30.0 | 0.95 |
| db/db | 34.11 | 1.32 | 43.9 | 62.37 | 21.18 | 49.6 | 0.99 |
| Width (µm) | |||||||
| µ1 | SE1 | p 1 | µ2 | SE2 | p 2 | r2 | |
| control | 2.77 | 0.22 | 100 | 0.0 | 0.0 | 0.0 | 0.97 |
| db/db | 2.03 | 3.36 | 86.2 | 4.69 | 32.8 | 13.8 | 0.98 |
Figure 2. Decreased Ryanodine Receptor expression in vascular tissue homogenates of db/db mice. A.

. Representative Western Blots of RyR obtained from control and db/db vascular tissue homogenates. Samples containing 30 µg of protein were run on 15% acrylamide gels and probed with anti-RyR antibody (1∶5000) and anti-actin (1∶8000). B. Bar graph represents normalized RyR/actin densitometric ratios (n = 3 for each group).
Sarcoplasmic Reticulum Ca2+ Loading CASMCs from Diabetic Animals
Sarcoplasmic reticulum (SR) Ca2+ load is a major determinant of Ca2+ release regulating RyR opening and, by consequence, Ca2+ spark properties and activity. It is plausible, therefore, that a reduced SR Ca2+ load could contribute to the reduction in Ca2+ spark properties described from CASMCs of diabetic mice. Thus, we measured global SR Ca2+ content by rapid application of caffeine (10 mmol/L). Global [Ca2+]i responses were measured in CASMCs from control and db/db mice using fura-2 based Ca2+ microfluorometry. Examples of such traces are shown in Fig. 3A , left traces. No significant differences in the peak amplitude of the caffeine-evoked [Ca2+]i transients were observed (in nmol/L: 858.6±81.6, n = 28 control cells vs. 731.4±99.5, n = 18 db/db cells) (Fig. 3B , left bars) thus ruling out a difference on the SR Ca2+ load at rest between db/db and control CASMCs underlying the weaker Ca2+ sparks. To observe the time dependence of luminal Ca2+ replenishment [36], we allowed 5 min of recovery and applied caffeine again. In this case, the peak amplitude of the second [Ca2+]i transient was significantly reduced in the CASMCs of diabetic animals (in nmol/L: 675.9±78.2, n = 16 control cells vs. 443.1±41.9, n = 13 db/db cells, P<0.05) (Fig. 3B ). The normalized SR Ca2+ load recovery with respect to the first caffeine-induced [Ca+]i transient was of 62.6±0.08% for db/db CASMCs, vs. 78.4±0.1% for control CASMCs. Thus the time dependence of SR Ca2+ load recovery is impaired in the vascular cells of diabetic mice, with no alterations in basal [Ca2+] (74.5±5.1 nmol/L, n = 23 control vs. 76.4±6.9 nmol/L, n = 21 db/db CASMCs. Fig. 3C ), suggesting that SERCA pump function might be compromised.
Figure 3. SR Ca2+ load in CASMCs of diabetic mouse. A.

. Representative caffeine-induced Ca2+ transients in Fura-2 loaded CASMCs from control (upper graph) and db/db mice (bottom graph); bottom traces indicate the start and end of caffeine applications (10 mmol/L). Breaks in x-axis indicate a 5-min interval between the first and the second caffeine application to allow the recovery of SR Ca2+ stores. B. Bar graph of averaged caffeine-induced Ca2+ transients. *P<0.05 C. Bar graph of averaged basal [Ca2+]i concentrations in CASMCs from control (open bar, n = 50 cells) and db/db mice (fill bar, n = 62 cells).
Decreased STOC Activity in CASMCs from Diabetic Mice
In CASMCs, Ca2+sparks activate large conductance K+ (BK) channels, which play a critical role in controlling vascular tone [26], [30]. We aimed to investigate whether the smaller Ca2+ sparks in diabetic CASMCs had an impact on BK channel activity by analyzing spontaneous transient outward currents (STOCs). Intact CASMCs were patch-clamped and progressively depolarized from a holding potential of −50 mV to 0 mV in 10 mV voltage increments (Fig. 4A ). STOC frequency was significantly decreased in db/db CASMCs compared to control cells across a range of voltages from −40 mV to 0 mV (Fig. 4B ). However, sigmoidal fitting of the data revealed that the voltage dependence of STOC frequency was unaffected (voltage of half maximum activation, V50: −20.1±0.9, and −17.9±2.9 mV, in 9 control and 8 db/db CASMCs, respectively), suggesting that voltage sensitivity of BK channel activation is unaltered in type 2 diabetes. Amplitude was also significantly reduced in db/db CASMCs as compared to control cells, with statistical difference from −30 mV to 0 mV. However, linear fitting of these data demonstrated that the voltage dependence of STOC amplitude was diminished 0.55 fold in db/db CASMCs (slope: 0.40±0.01, vs. 0.22±0.02 pA/mV; Fig. 4C ). The diminished STOC amplitude in the diabetic CASMCs was not due to a change in cell size, since there was no significant difference in membrane capacitance (an indicator of cell surface) between diabetic and non-diabetic CASMCs (9.5±2.4 pF for db/db n = 10 cells vs. 10.0±1.5 pF for control n = 15 cells) nor to a change in zero current potential (−33.5±2.1 mV in 7 WT cells, vs. −33.1±3.4 mV in 5 db/db cells). Further analysis of STOC kinetics revealed that both STOC time-to-rise (Fig. 5A ) and time-to-decay (Fig. 5B ) had shorter durations in db/db CASMCs compared to control. Furthermore, STOC area was significantly diminished in db/db CASMCs vs control cells (Fig. 5C ).
Figure 4. Decreased STOC frequency and amplitude in CASMCs of db/db mice. A.

. Representative traces of STOC activity at different holding potentials (−50, −30 and 0 mV) obtained from freshly isolated control (upper traces) and db/db (bottom traces) CASMCs. Voltage dependence of STOC frequency (B) and amplitude (C); cells were voltage clamped at different holding potentials (from −50 to 0 mV) and the STOC activity was recorded. The plots show STOC frequencies and amplitudes (M ± SEM) from 9 controls (open circles, dash line) and 8 db/db (filled circles, solid line) CASMCs at each holding potential. *P<0.05 and #P<0.01; control vs. db/db cells.
Figure 5. Reduced properties of STOCs from diabetic CASMCs.

Voltage dependence of STOC time-to-rise (A), time-to-decay (B) and area (C). Cells were voltage clamped at different holding potentials (from −50 to 0 mV) and the STOC activity was recorded. The plots show STOC properties (M ± SEM) of 9 controls (open circles, dash lines) and 8 diabetic (filled circles, solid lines) CASMCs at each holding potential. *P<0.05 and #P<0.01; control vs. diabetic cells.
The Proportion of β1 to α Subunit Expression of BK Channels is Decreased in db/db CASMCs
We next sought to determine whether the diminished frequency and amplitude of STOCs in the diabetic CASMCs might be related to a change in BK channel composition. Vascular BK channels are composed of four pore-forming α-subunits and four accessory β-subunits, β1 being the predominant isoform expressed in vascular smooth muscle [37]. The α-subunit is the pore forming subunit, and the β1-subunit enhances the sensitivity of BK channels to Ca2+-dependent activation [37]. Downregulation or genetic disruption of β1 subunit has been implicated in the pathogenesis of diabetic vascular dysfunction [38]–[41] and arterial hypertension [42]. We determined protein expression of BK channel α- and β1-subunits in aorta homogenates of control and db/db mice. Anti-actin antibody was used to normalize the loading. Representative immunoblots (Fig. 6A ) and bar graphs (Fig. 6B ) show that BK β1/α subunit ratio is 4.2-fold decreased in the diabetic mice (normalized ratio to actin: 1.0±0.37 vs. 0.24±0.05, in n = 7 controls and n = 7 db/db tissue homogenates, P<0.05); suggesting that composition stoichiometry of BK channels is modified by the diabetic condition; with decreased proportion of BK channel β1-subunits to α-subunits. Separate analysis of BK α and β1 subunit expression levels were slightly but not significantly modified (BKα normalized ratio to actin: 1.0±0.3 in controls vs. 1.6±0.7 in diabetic samples; BKβ1 normalized ratio to actin: 1.0±0.3 in n = 7 controls vs. 0.6±0.2 in n = 7 diabetic samples).
Figure 6. Decreased BK β1/α -subunit ratio in vascular tissues of db/db mice.

A. Representative Western Blots of BK α and β1 subunits obtained from control and db/db vascular tissues. Samples containing 30 µg of protein from aorta whole homogenates were run on 15% acrylamide gels and probed with anti-BK α subunit antibody (1∶1000), anti-BK β1 subunit antibody (1∶500) and anti-actin (1∶8000). B. Bar graph represents normalized BKβ1/α densitometric ratios (n = 7 for each group). *P<0.05.
Reduced STOC/spark Coupling in CASMCs of Diabetic Mice
It has been shown that Ca2+ sparks have a vasorelaxing effect by activating STOCs, which hyperpolarizes the cell, leading to inactivation of VDCCs. The STOC/Ca2+spark coupling ratio was indirectly estimated by dividing the number of STOCs at near resting membrane potential (−40 mV) by the number of spontaneous Ca2+ sparks in single quiescent cells. Fig. 7A shows that this coupling ratio was decreased in db/db CASMCs, suggesting that many of the Ca2+ sparks produced in the diabetic CASMCs lack STOCs, contrary to what is observed in normal vascular myocytes [29]. The estimated reduced coupling ratio may have a considerable impact on the deregulation of vascular tone under the diabetic condition.
Figure 7. Reduced STOC/spark coupling as a factor contributing to an increased vascular tone in diabetic CASMCs. A.

. Estimated STOC/Spark coupling ratio in control (white bar) and db/db (orange bar) CASMCs. The estimated coupling ratio was obtained by dividing the frequency of STOCs in each cell (n = 11 control vs n = 10 db/db cells) at near resting membrane potential (−40 mV) between the average frequency of spontaneous Ca2+ sparks in single quiescent cells; *P<0.05. B. Schematic representation of single db/db CASMC where the triggering of STOCs is not tightly controlled by Ca2+ sparks. The decreased STOC/Spark coupling ratio suggests that most of the Ca2+ sparks produced in the diabetic cells are STOCless having a detrimental impact on the regulation of vascular myogenic tone. SERCA: sarco/endoplasmic reticulum Ca2+ ATPase, BK: big conductance Ca2+-activated K+ Channel, VDCC: L-type Ca2+ channel, RyR: ryanodine receptor.
Discussion
In this work we present the first compelling evidence of the RyR and BK channel functional loss in native CASMCs of db/db mice. We have demonstrated: 1) Ca2+ spark properties are significantly decreased in the CASMCs of diabetic animals; 2) STOC frequency, amplitude, kinetics and area were further reduced; and 3) diminution of the BK channel β1/α subunit ratio, resulting in an abnormal Ca2+ spark-STOC coupling in db/db CASMCs.
Taken together, our data suggests the scenario schematized in Fig. 7B where the triggering of STOCs is not tightly controlled by Ca2+ sparks in db/db CASMCs, probably due to 1) smaller and shorter Ca2+ sparks with reduced amplitude than then could be STOCless; 2) a compromised SR Ca2+ load recovery; and 3) decreased STOC frequency, amplitude, area, time-to-rise and time-to-decay. The latter is probably due to reduced Ca2+ sparks because of decreased RyRs expression and further enhanced by the altered subunit composition of BK channels. Taken together, the reduced efficacy of Ca2+ sparks to trigger STOCs might contribute to an increase in vascular tone of db/db arteries.
Previous studies have shown that the function of cerebral arteries is impaired in experimental and genetic models of type-2 diabetes [8]–[10], [41]. Specifically, the cerebral vascular dysfunction found in the db/db mice is characterized by reduced baseline arteriolar diameter, increased basal tone and reduced response to acetylcholine, in part due to enhanced Rho-kinase activity, superoxide production [9] and overall oxidative damage [8] that worsens brain damage, edema and inflammation after induced experimental stroke [11]–[13]. In the current study, we have analyzed intracellular Ca2+ handling and STOCs which might contribute to these alterations, since it has been previously shown that spontaneous Ca2+ sparks contribute to vascular relaxation by activating STOCs.
We found that the overall Ca2+ spark frequency was not significantly altered, but the Ca2+ spark amplitude, duration, width, and as a consequence Ca2+ spark mass were decreased in db/db CASMCs. To our knowledge, this is the first study of Ca2+ sparks in CASMCs of type 2 diabetes. In type 1 diabetes, Ca2+ sparks frequency has been found to be unaltered [40] or increased [39], while their amplitudes were found to be increased [39], [40]. Although both type 1 and type-2 diabetes are characterized by hyperglycemia, the mechanism of disease is quite different, which may explain the differing results in Ca2+ spark amplitude.
It has been previously established that decreasing SR Ca2+ load in vascular myocytes reduces Ca2+ spark properties [43]. We did not find a significant decrease in the SR Ca2+ load in the conditions of Ca2+ sparks recording (Fig. 3), ruling out this possibility. However, the recovery of SR Ca2+ load after 5 min of depletion was impaired in db/db mice, which could be indicative of an underlying defect in SERCA activity. In fact, it has been documented that cytokines (i.e. interleukin-1 βand γ-interferon), which are increased in the db/db mice, depress SERCA function [44]. On the other hand, reduced number of RyRs that simultaneously open in a cluster may account for the reduction in Ca2+ spark amplitude, duration, spatial spread, and mass of the Ca2+ spark. This could be supported by the reduced RyR expression in db/db arteries (Fig. 2).
Ca2+ sparks in vascular smooth muscle cells contribute to relaxation by activating BK channels. Diabetic CASMCs show a significant decrease in STOC frequency, amplitude, time-to-rise, time-to-decay and area (Fig. 4 and 5), as in type 1 diabetes [39], [40]. STOCs are outward (hyperpolarizing) currents mediated by BK channels. The K+ efflux brings the cell membrane to more negative potentials, limiting the activation of voltage-dependent Ca2+ channels. This in turn decreases Ca2+ influx, inducing relaxation [26]–[31]. Hence, the decrease in Ca2+ sparks properties, STOCs and Ca2+ sparks-STOCs coupling could underlie or contribute to depolarization under certain conditions in vivo.
Because STOCs are activated by Ca2+ sparks, and the occurrence of Ca2+ sparks is not significantly altered, the reduction in STOCs could be interpreted as a decreased ability of the Ca2+ sparks to activate STOCs in db/db CASMCs (Fig. 7A). There are multiple potential explanations for this uncoupling. It is possible that the smaller Ca2+ sparks are less effective in activating STOCs. Indeed, after careful analysis of Ca2+ spark amplitude, duration, and width PDf (Fig. 1F–H), we found that the global decrease in Ca2+ spark properties was related to a decrease in the population of larger Ca2+ sparks. Additionally, the reduction in STOCs could be due to the relative decrease in β1 channel subunit (Fig. 6) [38]–[41], [45], since it has been shown that the β-subunit plays a major functional role in defining both the Ca2+ dependence of activation and availability of BK channels [37]. In this regard, BK channels of coronary arteries from Zucker diabetic fatty rats showed impaired Ca2+ dependent activation attributable to a decrease in BK channel β1-subunit expression [38]. We found a decrease in the β1/α BK subunit proportion (Fig. 7), suggesting that in db/db mice, some BK channels may be devoid of β1-subunit. Additional biochemical studies of type-1 diabetic animals also support the evidence that BK channel β1-subunit relative to BK channel α-subunit protein expression is reduced in arterial tissues [39], [40]. Interestingly, similar findings have been reported in CASMCs of spontaneously hypertensive rats [42] underlying a common mechanism to decrease STOC/Ca2+spark functional coupling. Additionally, a key finding of this study is that the function of BK channels is impaired in CASMCs of type-2 diabetic mice, but that this is not attributable to an altered voltage dependence of BK channel activation. Although we cannot exclude the possibility that the dysfunction of BK channels found in diabetic CASMCs could be due to high glucose-mediated oxidative modulation [46], we suggest that Ca2+ sparks with diminished amplitude, size, duration, and mass may be less efficient in activating STOCs. Indeed, the STOC/Ca2+ spark coupling efficiency was significantly reduced in db/db cells (Fig. 7A), which suggests that spark/STOC coupling plays a key role in contributing to the previously well documented vascular dysfunction associated with this model of type 2 diabetes [9]. Further, the decreased STOC activity in db/db mice could underlie the increased activity of L-type Ca2+ channels reported by Navedo et al. [33].
In summary, we have dissected local Ca2+ signaling-STOC activity in CASMCs from db/db mice and their controls. We have shown that the alteration in RyR activity, manifested as a decrease in the larger Ca2+ spark population, is involved in reduced STOC properties, with consequent dysfunction. Thus, defective crosstalk between RyRs and BK channels may be important contributors to the vascular pathology of diabetic individuals and, as such, are potentially attractive targets for therapeutic intervention.
Materials and Methods
All experiments were carried out according to European Union Council Directives (86/609/EEC) for the care of laboratory animals. Animal Protocol was approved by the Comité Régional d’Ethique sur l’expérimentation animale of Languedoc-Roussillon on the Use and Care of Animals (authorization B34-172-16 for animal facility manager).
Mice
The diabetic (db/db) mice used in the current study were on a C57BL/KsJ genetic background. We used male animals at the age of 14 to 16 weeks. db/db mice were significantly heavier than WT controls (body weight: 55.5±0.8 g for db/db, n = 17 animals vs. 30.0±0.8 g for controls, n = 13; P<0.001).
CASM Cell Isolation
Cerebral artery smooth muscle cells (CASMCs) were enzymatically isolated from 14 to 16 week-old male C57BL/KsJ-db (db/db) mice and their controls (+/+), using a previously standardized method [47]. Mice were anesthetized by peritoneal injection of pentobarbital solution (100 mg/kg) plus heparin (4000 U/kg). The brain was removed and transferred to a Petri dish filled with oxygenated ice-cold HEPES-buffered dissection solution (HBDS, containing in mmol/L: 55 NaCl, 6 KCl, 80 Na-glutamate, 2 MgCl2, 10 glucose and 10 HEPES, pH 7.4). The cerebral basilar arteries were dissected, transferred to a 1.5 ml tube and incubated in the shaker for 16 min at 37 °C in HBDS containing in mg/ml: 1.0 papain (2X crystallized), 1.0 bovine serum albumin (BSA), and 1.0 dithiothreitol. Then, the tissue was transferred to a collagenase solution (mixture of collagenases type F and type H, 7∶3 ratio) plus 100 µmol/L CaCl2 and incubated for 8 min at 37 °C with shaking. Enzyme digestion was stopped by washing the tissue 5 times with HBDS. Digested tissue was triturated with a fire-polished glass Pasteur pipette to yield single smooth muscle cells. Myocytes used for confocal experiments were loaded with 10 µmol/L Fluo-3 AM for 30 min at room temperature, washed and kept at 4 °C in physiological saline solution (PSS, composition in mmol/L: 137 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 10 glucose and 10 HEPES, pH 7.4) to be used within the same day.
Ca2+spark Recordings
Spontaneous and local Ca2+ release events were recorded as previously reported [47]. Fluo-3 loaded CASMCs (50 µl of cell suspension) were allowed to adhere to the bottom of a glass coverslip in a perfusion chamber. The cells were perfused with physiological saline solution (PSS) at room temperature before starting the experiments. Ca2+ sparks were recorded at room temperature with a laser scanning confocal microscope (Zeiss, LSM510 META) equipped with an x63 water-immersion objective (N.A. 1.2) in the line scan mode (5 images of 1000 lines at speed of 1.92 ms/line). Fluo-3 was excited at 488 nm with an Argon laser (3% intensity), and emission measured at above 510 nm. Ca2+ sparks were reconstructed by stacking consecutive line scans and performing a time-intensity plot. Ca2+ sparks were automatically detected and properties of amplitude (F/F0), duration (FDHM, Full duration at half maximum, in ms) and width (FWHM, Full width at half maximum, in µm) measured with a custom-made program running in IDL 5.5 software (Research Systems Inc.), with a detection threshold of 4.3×S.D [36]. Images of Ca2+ sparks were normalized by dividing the fluorescence intensity of each pixel (F) by the average resting fluorescence intensity (F0) to generate an F/F0 image. Mean rate of rise was determined as reported by Shen et al [34].
[Ca2+]i Transients in CASMCs
Myocytes were loaded with 2.5 µmol/L fura 2-AM. Cells were transferred to a bath perfusion chamber and continuously superfused with PSS. Two caffeine-induced [Ca2+]i transients were elicited consecutively with a 5-minute interval in between to allow refilling of intracellular Ca2+ stores. Images of Fura-2 fluorescence were captured by a cooled CCD camera (Photometrics, USA) with an oil-immersion x40 objective mounted on an inverted microscope (Axiovert, Zeiss, Germany), and acquired at 510 nm emission after excitations at 340 nm and 380 nm using a lambda-DG4 excitation system (Sutter Instrument Company, USA). Ca2+ signals were analyzed using Metafluor software (Universal Imaging Corporation, USA). [Ca2+]i (in nmol/L) was calculated from the ratio of 340/380 nm after correcting for background. Fluorescence emission ratios were converted to [Ca2+]i according to the Grynkiewicz equation [48], using a dissociation constant for fura-2 of 239 nM. Values of 0.32, 5.7, and 10.3 obtained from fura-2 in situ calibration were used for Rmin, Rmax, and β, respectively.
Electrophysiological Recordings
STOCs were recorded in the whole cell configuration of the patch clamp technique using solutions and protocol previously described [48]. The pipette solution (in mmol/L): 80 Kglutamate, 5 NaCl, 40 KCl, 1 MgCl2, 3 MgATP, 0.1 NaGTP, 0.05 KEGTA, 20 HEPES, pH 7.2 with KOH. CASMCs were superfused with PSS and clamped at −50 mV or stepped from −50 to −10 mV in 10 mV increments. Membrane capacitance (Cm) was determined from the current amplitude elicited in response to a hyperpolarizing voltage pulse from a holding potential of −80 mV (duration, 10 ms; amplitude, 10 mV). Currents were filtered at 500 Hz and digitalized at 2 kHz (500 µs/point). STOC analysis was performed off line, using the event detection tool of Clampfit 9.2 (Axon Instruments, Inc); where both STOC time-to-rise and time-to-decay were measured at half peak of STOC in ms.
Western Blots
Aorta homogenates were used because they provide more protein. They were prepared with homogenization buffer (in mmol/L: 300 sucrose, 20 HEPES, pH 7.2) plus protease inhibitors (1 µg/ml aprotinin, 500 µmol/L benzamidine, 12 µmol/L leupeptin, 100 µmol/L PMSF). Aortas were homogenized on ice with a glass homogenizer and spun at 2,000 g for 15 min at 4°C. Supernatants were fractionated on 15% SDS-PAGE gels, transferred onto nitrocellulose membranes (1 h at 100 V, Hybond-ECL, GE Healthcare Bio-Sciences Corp, NJ, USA) and probed with anti-BK α antibody (1∶1000; Alomone Labs, Jerusalem Israel), anti BK β1 antibody (1∶500; Alomone Labs, Jerusalem Israel), anti RyR (1∶5000; Thermo Scientific) and anti-actin antibody (1∶8000, Sigma-Aldrich) in phosphate-buffered saline solution containing Tween-20 (PBS-T; in mmol/L: 3 KH2PO4, 10 Na2HPO4, 150 NaCl, 0.1% Tween-20, pH 7.2–7.4). Membranes were incubated 1 h with secondary peroxidase-conjugated goat anti-rabbit (1∶15000). Signal was detected by chemiluminiscence. Densitometry was done with Kodak ID Software (v. 3.635, Molecular Imaging Systems). Actin signals were detected in the same blots. Protein concentration was assessed by the Bradford method.
STOCs and Ca2+ Spark Analyses
Data are expressed as the mean ± SE. Statistical significance was assessed using SigmaStat 3.0 software by Student’s t-tests, one way ANOVA test or by nonparametric Mann-Whitney Rank-sum test for cases in which data failed the Shapiro-Wilk normality test. Values of P<0.05 were considered statistically significant. The probability density function analysis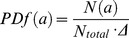 , where N(a) is the histogram distribution of Ca2+ spark parameter a, Ntotal is the total number of Ca2+ sparks, and Δ is the bin width. Bimodal Gaussian distributions were done as previously reported [35].
, where N(a) is the histogram distribution of Ca2+ spark parameter a, Ntotal is the total number of Ca2+ sparks, and Δ is the bin width. Bimodal Gaussian distributions were done as previously reported [35].
Supporting Information
A, Average of sites where Ca2+ sparks were recorded within the same cell during the recording period (9.6 seconds). Firing sites were counted as the sites where we recorded at least one Ca2+ spark. B, Ca2+ spark frequency within each firing site reported as number of events recorded within each site/s. C, Probability in each cell to present sites that fire repetitively. D, Maximum number of Ca2+sparks recorded within the same site. N = 43 for control CASMCs (white bars) and n = 41 for db/db cells (gray bars).
(DOCX)
Acknowledgments
We thank Dr. E. Michelle Capes (Department of Cellular and Regenerative Biology, School of Medicine and Public Health; University of Wisconsin-Madison) for critical reading of the manuscript.
Funding Statement
This work was funded by Conacyt (No. 80960), ICyTDF No.331/2010), Agence National de la Recherche (Geno-09-HyperEpac and Geno-09-Carythm), Région Ile de France (CODDIM: COD100256), European Union (2005 N°018802, CONTICA) and FRM (Fondation pour la Recherche médicale). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.(2011) Centers for Disease Control and Prevention. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta,GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention.
- 2. Jørgensen H, Nakayama H, Raaschou H, Olsen T (1994) Stroke in patients with diabetes. The Copenhagen Stroke Study. Stroke 25: 1977–1984. [DOI] [PubMed] [Google Scholar]
- 3. Mankovsky B, Metzger B, Molitch M, Biller J (1996) Cerebrovascular disorders in patients with diabetes mellitus. J Diabetes Complications 10: 228–242. [DOI] [PubMed] [Google Scholar]
- 4. Air EL, Kissela BM (2007) Diabetes, the metabolic syndrome, and ischemic stroke: epidemiology and possible mechanisms. Diabetes Care 30: 3131–3140. [DOI] [PubMed] [Google Scholar]
- 5. Janghorbani M, Hu FB, Willett WC, Li TY, Manson JE, et al. (2007) Prospective study of type 1 and type 2 diabetes and risk of stroke subtypes: the Nurses’ Health Study. Diabetes Care 30: 1730–1735. [DOI] [PubMed] [Google Scholar]
- 6. Ergul A, Li W, Elgebaly MM, Bruno A, Fagan SC (2009) Hyperglycemia, diabetes and stroke: focus on the cerebrovasculature. Vascul Pharmacol 51: 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Asfandiyarova N, Kolcheva N, Ryazantsev I, Ryazantsev V (2006) Risk factors for stroke in type 2 diabetes mellitus. Diab Vasc Dis Res 3: 57–60. [DOI] [PubMed] [Google Scholar]
- 8. Liao Y, Ueno M, Nakagawa T, Huang C, Kanenishi K, et al. (2008) Oxidative damage in cerebral vessels of diabetic db/db mice. Diabetes Metab Res Rev 21: 554–559. [DOI] [PubMed] [Google Scholar]
- 9. Didion S, Lynch C, Baumbach G, Faraci F (2005) Impaired endothelium-dependent responses and enhanced influence of Rho-kinase in cerebral arterioles in type II diabetes. Stroke 36: 342–347. [DOI] [PubMed] [Google Scholar]
- 10. Didion SP, Lynch CM, Faraci FM (2007) Cerebral vascular dysfunction in TallyHo mice: a new model of Type II diabetes. Am J Physiol Heart Circ Physiol 292: H1579–1583. [DOI] [PubMed] [Google Scholar]
- 11. Vannucci S, Willing L, Goto S, Alkayed N, Brucklacher R, et al. (2001) Experimental stroke in the female diabetic, db/db, mouse. J Cereb Blood Flow Metab 21: 52–60. [DOI] [PubMed] [Google Scholar]
- 12.Tureyen K, Bowen K, Liang J, Dempsey RJ, Vemuganti R (2010) Exacerbated Brain Damage, Edema and Inflammation in Type-2 Diabetic Mice Subjected to Focal Ischemia. J Neurochem. [DOI] [PMC free article] [PubMed]
- 13. Yeung CM, Lo AC, Cheung AK, Chung SS, Wong D, et al. (2010) More severe type 2 diabetes-associated ischemic stroke injury is alleviated in aldose reductase-deficient mice. J Neurosci Res 88: 2026–2034. [DOI] [PubMed] [Google Scholar]
- 14. Lagaud G, Masih-Khan E, Kai S, van Breemen C, Dubé G (2001) Influence of type II diabetes on arterial tone and endothelial function in murine mesenteric resistance arteries. J Vasc Res 38: 578–589. [DOI] [PubMed] [Google Scholar]
- 15. Pannirselvam M, Wiehler W, Anderson T, Triggle C (2005) Enhanced vascular reactivity of small mesenteric arteries from diabetic mice is associated with enhanced oxidative stress and cyclooxygenase products. Br J Pharmacol 144: 953–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Su J, Lucchesi PA, Gonzalez-Villalobos RA, Palen DI, Rezk BM, et al. (2008) Role of advanced glycation end products with oxidative stress in resistance artery dysfunction in type 2 diabetic mice. Arterioscler Thromb Vasc Biol 28: 1432–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bagi Z, Koller A, Kaley G (2003) Superoxide-NO interaction decreases flow- and agonist-induced dilations of coronary arterioles in Type 2 diabetes mellitus. Am J Physiol Heart Circ Physiol 285: H1404–1410. [DOI] [PubMed] [Google Scholar]
- 18. Bagi Z, Erdei N, Toth A, Li W, Hintze T, et al. (2005) Type 2 diabetic mice have increased arteriolar tone and blood pressure: enhanced release of COX-2-derived constrictor prostaglandins. Arterioscler Thromb Vasc Biol 25: 1610–1616. [DOI] [PubMed] [Google Scholar]
- 19. Erdei N, Bagi Z, Edes I, Kaley G, Koller A (2007) H2O2 increases production of constrictor prostaglandins in smooth muscle leading to enhanced arteriolar tone in Type 2 diabetic mice. Am J Physiol Heart Circ Physiol 292: H649–656. [DOI] [PubMed] [Google Scholar]
- 20. Piercy V, Taylor S (1998) A comparison of spasmogenic and relaxant responses in aortae from C57/BL/KsJ diabetic mice with those from their non-diabetic litter mates. Pharmacology 56: 267–275. [DOI] [PubMed] [Google Scholar]
- 21. Guo Z, Su W, Allen S, Pang H, Daugherty A, et al. (2005) COX-2 up-regulation and vascular smooth muscle contractile hyperreactivity in spontaneous diabetic db/db mice. Cardiovasc Res 67: 723–735. [DOI] [PubMed] [Google Scholar]
- 22. Miike T, Kunishiro K, Kanda M, Azukizawa S, Kurahashi K, et al. (2008) Impairment of endothelium-dependent ACh-induced relaxation in aorta of diabetic db/db mice–possible dysfunction of receptor and/or receptor-G protein coupling. Naunyn Schmiedebergs Arch Pharmacol 377: 401–410. [DOI] [PubMed] [Google Scholar]
- 23. Pannirselvam M, Verma S, Anderson T, Triggle C (2002) Cellular basis of endothelial dysfunction in small mesenteric arteries from spontaneously diabetic (db/db −/−) mice: role of decreased tetrahydrobiopterin bioavailability. Br J Pharmacol 136: 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McVeigh GE, Brennan GM, Johnston GD, McDermott BJ, McGrath LT, et al. (1992) Impaired endothelium-dependent and independent vasodilation in patients with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 35: 771–776. [DOI] [PubMed] [Google Scholar]
- 25. Williams SB, Cusco JA, Roddy MA, Johnstone MT, Creager MA (1996) Impaired nitric oxide-mediated vasodilation in patients with non-insulin-dependent diabetes mellitus. J Am Coll Cardiol 27: 567–574. [DOI] [PubMed] [Google Scholar]
- 26. Jaggar J, Wellman G, Heppner T, Porter V, Perez G, et al. (1998) Ca2+ channels, ryanodine receptors and Ca(2+)-activated K+ channels: a functional unit for regulating arterial tone. Acta Physiol Scand 164: 577–587. [DOI] [PubMed] [Google Scholar]
- 27. Gollasch M, Wellman G, Knot H, Jaggar J, Damon D, et al. (1998) Ontogeny of local sarcoplasmic reticulum Ca2+ signals in cerebral arteries: Ca2+ sparks as elementary physiological events. Circ Res 83: 1104–1114. [DOI] [PubMed] [Google Scholar]
- 28. Knot H, Standen N, Nelson M (1998) Ryanodine receptors regulate arterial diameter and wall [Ca2+] in cerebral arteries of rat via Ca2+-dependent K+ channels. J Physiol 508 (Pt 1): 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pérez G, Bonev A, Patlak J, Nelson M (1999) Functional coupling of ryanodine receptors to KCa channels in smooth muscle cells from rat cerebral arteries. J Gen Physiol 113: 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gollasch M, Löhn M, Fürstenau M, Nelson M, Luft F, et al. (2000) Ca2+ channels, Ca2+ sparks, and regulation of arterial smooth muscle function. Z Kardiol 89 Suppl 2: 15–19. [DOI] [PubMed] [Google Scholar]
- 31. Jaggar J, Porter V, Lederer W, Nelson M (2000) Calcium sparks in smooth muscle. Am J Physiol Cell Physiol 278: C235–256. [DOI] [PubMed] [Google Scholar]
- 32. Essin K, Welling A, Hofmann F, Luft F, Gollasch M, et al. (2007) Indirect coupling between Cav1.2 channels and ryanodine receptors to generate Ca2+ sparks in murine arterial smooth muscle cells. J Physiol 584: 205–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shen JX, Wang S, Song LS, Han T, Cheng H (2004) Polymorphism of Ca2+ sparks evoked from in-focus Ca2+ release units in cardiac myocytes. Biophys J 86: 182–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gómez A, Rueda A, Sainte-Marie Y, Pereira L, Zissimopoulos S, et al. (2009) Mineralocorticoid modulation of cardiac ryanodine receptor activity is associated with downregulation of FK506-binding proteins. Circulation 119: 2179–2187. [DOI] [PubMed] [Google Scholar]
- 36. Gómez-Viquez L, Guerrero-Serna G, García U, Guerrero-Hernández A (2003) SERCA pump optimizes Ca2+ release by a mechanism independent of store filling in smooth muscle cells. Biophys J 85: 370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brenner R, Peréz G, Bonev A, Eckman D, Kosek J, et al. (2000) Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature 407: 870–876. [DOI] [PubMed] [Google Scholar]
- 38. Lu T, Ye D, He T, Wang X, Wang H, et al. (2008) Impaired Ca2+-dependent activation of large-conductance Ca2+-activated K+ channels in the coronary artery smooth muscle cells of Zucker Diabetic Fatty rats. Biophys J 95: 5165–5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dong L, Zheng Y, Van Riper D, Rathore R, Liu Q, et al. (2008) Functional and molecular evidence for impairment of calcium-activated potassium channels in type-1 diabetic cerebral artery smooth muscle cells. J Cereb Blood Flow Metab 28: 377–386. [DOI] [PubMed] [Google Scholar]
- 40. McGahon M, Dash D, Arora A, Wall N, Dawicki J, et al. (2007) Diabetes downregulates large-conductance Ca2+-activated potassium beta 1 channel subunit in retinal arteriolar smooth muscle. Circ Res 100: 703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Y, Zhang HT, Su XL, Deng XL, Yuan BX, et al. (2010) Experimental diabetes mellitus down-regulates large-conductance Ca2+-activated K+ channels in cerebral artery smooth muscle and alters functional conductance. Curr Neurovasc Res 7: 75–84. [DOI] [PubMed] [Google Scholar]
- 42. Amberg G, Santana L (2003) Downregulation of the BK channel beta1 subunit in genetic hypertension. Circ Res 93: 965–971. [DOI] [PubMed] [Google Scholar]
- 43. Cheranov S, Jaggar J (2002) Sarcoplasmic reticulum calcium load regulates rat arterial smooth muscle calcium sparks and transient K(Ca) currents. J Physiol 544: 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cardozo AK, Ortis F, Storling J, Feng YM, Rasschaert J, et al. (2005) Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes 54: 452–461. [DOI] [PubMed] [Google Scholar]
- 45. McGahon M, Zhang X, Scholfield C, Curtis T, McGeown J (2007) Selective downregulation of the BKbeta1 subunit in diabetic arteriolar myocytes. Channels (Austin) 1: 141–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu T, He T, Katusic Z, Lee H (2006) Molecular mechanisms mediating inhibition of human large conductance Ca2+-activated K+ channels by high glucose. Circ Res 99: 607–616. [DOI] [PubMed] [Google Scholar]
- 47. Rueda A, Song M, Toro L, Stefani E, Valdivia H (2006) Sorcin modulation of Ca2+ sparks in rat vascular smooth muscle cells. J Physiol 576: 887–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rueda A, Garcia L, Soria-Jasso LE, Arias-Montano JA, Guerrero-Hernandez A (2002) The initial inositol 1,4,5-trisphosphate response induced by histamine is strongly amplified by Ca(2+) release from internal stores in smooth muscle. Cell Calcium 31: 161–173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A, Average of sites where Ca2+ sparks were recorded within the same cell during the recording period (9.6 seconds). Firing sites were counted as the sites where we recorded at least one Ca2+ spark. B, Ca2+ spark frequency within each firing site reported as number of events recorded within each site/s. C, Probability in each cell to present sites that fire repetitively. D, Maximum number of Ca2+sparks recorded within the same site. N = 43 for control CASMCs (white bars) and n = 41 for db/db cells (gray bars).
(DOCX)