

Abstract
Colorectal cancer (CRC) cells express renin and chymase through which they can activate angiotensin. Renin expression is induced by hyperglycemic conditions. As angiotensinogen is produced in the liver, CRC cells that can activate angiotensin have an enhanced ability to metastasize to this organ. In human CRC cases, patients with diabetes have higher activities of rennin and angiotensin-II in primary tumors, and on average, have a more progressed disease stage, especially with respect to liver metastasis. These patients exhibit a stronger association with Hemoglobin A1c levels and metastasis compared to patients without diabetes. In a combined diabetes/CRC liver metastasis mouse model, concurrent treatment with anti-angiotensin and hypoglycemic agents shows a synergic effect in terms of reduced liver metastasis and improved survival. The effect of anti-angiotensin treatment and blood sugar control as a baseline management for colon cancer patients with diabetes needs to be examined in clinical trials to establish whether it can prevent liver metastasis.
Keywords: Angiotensin, Renin, Angiotensin II receptor blocker, Diabetes, Liver metastasis
INTRODUCTION
Various factors participate in colorectal cancer (CRC) progression, such as growth factors, angiogenic factors, and cytokines. Angiotensin is a well-known hypertensive hormone and also possesses protumoral functions. Angiotensin II (A-II), an active form of angiotensin, induces angiogenesis, cell growth, invasion via activation of the specific receptor, A-II type 1 receptor (ATR1) to enhance development and progression of CRC. Some colorectal cancer cells possess the angiotensin-activating mechanism employing renin and chymase. Importantly, renin expression is upregulated by hyperglycemic condition. The data examining the importance of hyperglycemia/diabetes-induced angiotensin activation in the liver metastasis of colorectal cancer are described in this article.
PROTUMORAL EFFECT OF ANGIOTENSIN
Colorectal cancer is the fourth leading cause of cancer-related deaths in Japan, and cancer mortality continues to increase as the Western lifestyle gains popularity among the Japanese population[1]. Approximately 24% of CRC cases involving invasion beyond the submucosal layer showed liver metastasis during and/or after the operation to excise the tumor[2]. One-third of CRC patients died of liver metastasis[3], and only one-third or fewer CRC patients with liver metastasis respond to systemic chemotherapy, although even in these cases long-term survival was rare[3]. The 5-year survival rate of CRC patients with liver metastasis is reported to be less than 20%[1]. The early detection and control of liver metastasis is therefore an important goal for the successful treatment of CRCs.
A-II has multiple physiological effects mediated by the activation of the ATR1. These include vasoconstriction, inflammation, and proliferation in cardiovascular and neoplastic tissues[4]. ATR1 intracellular signaling pathways produce diverse effects, including activation of protein kinase C, angiopoietin 2, vascular endothelial growth factor (VEGF), VEGF receptors, fibroblast growth factor, platelet-derived growth factor, transforming growth factor-beta, epidermal growth factor, nitric oxide synthase, and metalloproteinase[4,5]. These properties enhance colon carcinogenesis and disease progression.
We have experimentally confirmed the protumor effect of angiotensin by studying the effects of A-II on cell growth, invasion, and apoptosis in several CRC-derived cell lines[6]. A-II enhances cell growth and in vitro invasion into type IV collagen and reduces apoptosis in a dose-dependent manner. Reduction in hepatic angiotensinogen (ATG) production by knockdown with cholesterol-conjugated antisense S-oligodeoxynucleotide (S-ODN) suppressed liver metastasis of HT29 cells in a nude mouse liver metastasis model. ATG-knockdown mice had fewer and smaller metastatic foci with a lower Ki-67 labeling index and reduced microvessel density, compared to the control mice. Knockdown of renin or chymase in HT29 cells also resulted in smaller and fewer metastatic foci in the liver compared to the control. Furthermore, examination of 121 CRC patients showed that the serum A-II concentration is significantly associated with advanced disease stage, especially liver metastasis.
ANGIOTENSIN ACTIVATION IN CRC CELLS
As described above, the ability of angiotensin activation might be a prominent feature of cancer cell to obtain the powerful tool for progression. The mechanism of angiotensin activation in cancer is examined using colorectal cancer cell lines[6]. ATG is an inactive precursor of A-II (Figure 1), and has no effect on cancer cells prior to conversion to A-II. Effects of ATG on cell growth, invasion, and apoptosis were examined in HT29 cells. Interestingly, ATG enhanced cell growth, in vitro invasion, and anti-apoptotic survival in HT29 cells in a dose-dependent manner in a similar fashion to A-II. This finding suggests that the HT29 cells have an endogenous angiotensin-activating mechanism.
Figure 1.
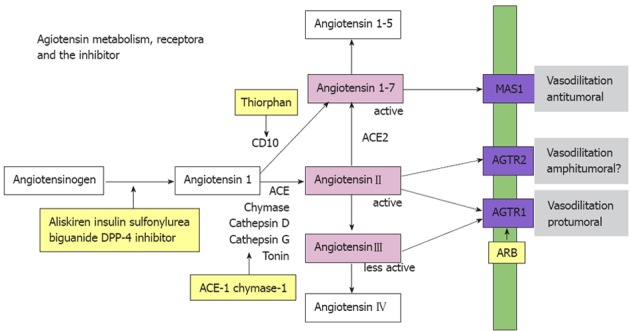
Aniotensinogen is activated to become a bioactive form, angiotensin II by some enzymes, such as ACE. In angiotensin receptors (blue boxed), angiotensin II type 1 receptor is pro-tumoral; however, angiotensin II type 2 receptor is not definitive. Angiotensin 1-7, a degraded product of angiotensin IIactivates MAS1 receptor to provide inhibitory effects. Substances marked with yellow boxes are possible inhibitors for angiotensin effects.
We then examined the expression of angiotensin-associated genes in HT29 cells[6]. They express ATR1, but not ATG or angiotensin I (A-I)-converting enzyme (ACE). However, they express chymase, which possesses an ACE-like activity. Renin is also expressed in HT29 cells, and a renin inhibitor abrogates the production of both A-I and A-II. A chymase inhibitor suppresses the production of A-II but not that of A-I. In contrast, an ACE inhibitor does not affect the production of A-I or A-II in HT29 cells. Thus, A-II is produced from ATG by renin and chymase in HT29 cells. Chymase, tonin, and cathepsin G all possess an ACE-like activity, which can be used as a substitute for ACE (Figure 1)[7]. Cathepsin D is responsible for producing A-I from ATG in cardiac myocytes, fibroblasts, and vascular smooth muscle cells in place of renin[8]. However, the CRC cells in this study did not express cathepsin D. Chymase expression is associated with hypoxia or ischemia in the human left cardiac ventricle[9]; it is not associated with hyperglycemia in CRC cells[6].
MAS1
MAS1 is a receptor of an A-II derivative produced by ACE 2-angiotensin 1-7 (A1-7), and has similar physiological effects to anti-A-II, including vessel dilation and reduction of blood pressure (Figure 1)[10]. We examined the role of MAS1 in CRC and in invasive ductal carcinoma (IDC) of the breast[11]. MAS1 was not detected by immunohistochemistry in either CRCs or the normal colon mucosa. In contrast, normal mammary lobules and ducts expressed MAS1 at high levels, although MAS1 expression was attenuated in all IDCs. Of particular note was the greatly reduced MAS1 expression in scirrhous-type IDCs compared to other types. The decrease in MAS1 expression was associated with lymph node metastasis but not T factor, grade, or the status of the estrogen or progesterone receptor. The decrease in MAS1 expression was inversely associated with human epidermal growth factor receptor 2 (HER2) expression. Using a mouse breast cancer cell line, BALB-MC, which expresses MAS1, cell growth and in vitro invasion were examined. A1-7 treatment inhibited growth and invasion of BALB-MC cells, which were abrogated by MAS1 knockdown. MAS1 intracellular signaling involves Akt phosphorylation, protein kinase C activation, and mitogen-activated protein kinase inhibition[12]. These findings suggest that MAS1 might act as an inhibitory regulator of both normal breast tissue and breast cancer.
CD10
CD10, also known as common acute lymphoblastic leukemia antigen (CALLA), is a characteristic marker of various subgroups of B-cell type acute lymphocytic leukemia[13,14]. It is a zinc-dependent membrane metalloendopeptidase (also referred to as neutral endopeptidase (EC 3.4.24.11), enkephalinase, or neprilysin)[14]. CD10 is expressed in CRC and is associated with CRC metastases, especially liver metastasis[2,15,16]. Met-enkephalin (MENK) is a high-affinity substrate of CD10[17,18]. It is produced by hepatocytes under conditions of cellular stress, such as hepatitis, bile stasis, and liver metastasis[19-21]. MENK inhibits tumor growth and the establishment of metastatic foci[22]. CD10-positive CRC cells degrade MENK and escape from MENK-induced tumor suppression[22]. CD10 possesses a weak affinity to A-I[23]; however, CD10 shows A-I-degrading activity but not the A-I-converting activity. The degradation of A-I produces A1-7, a MAS1 ligand. As discussed above, MAS1 is not expressed in CRCs. CD10-induced A1-7 does not affect CRC progression.
DIABETES AND THE RENIN/ANGIOTENSIN SYSTEM
Diabetes mellitus is a common problem in countries adopting the Western lifestyle. Several epidemiological studies have shown an association between type 2 diabetes and the risk of colorectal, pancreatic, breast, liver, gastric, and endometrial cancer[24]. The risk of malignancies is increased at earlier stages in cases of abnormalities in glucose metabolism, and there is a linear relationship between cancer risk and plasma insulin levels[24]. With regard to CRCs, a meta-analysis of 15 studies, involving a total of 2 593 935 participants, showed that diabetes is associated with an increased risk of CRC [relative risk, 1.30; 95% confidence interval (CI), 1.20-1.40]. Diabetes is also associated with CRC mortality (relative risk, 1.26; 95% CI, 1.05-1.50)[25]. High glycated hemoglobin (HbA1c) levels are also associated with an increased risk of CRC (odds ratio, 1.57; 95% CI, 0.94-2.60)[26]. Several studies have demonstrated that hyperinsulinemia, elevated levels of C-peptide, elevated body mass index, high levels of insulin growth factor-1, low levels of insulin growth factor binding protein-3, high leptin levels, and low adiponectin levels are involved in carcinogenesis[27]. Increased blood concentrations of insulin and insulin-like growth factor are particularly important in enhancing the risk of CRC[28]. However, a detailed understanding of how diabetes might increase the risk of CRC is still lacking.
We examined the expression of renin in HT29 and CT26 cells in association with changing glucose concentration. When the medium contained 100 mg/dL glucose, renin protein was detected in HT29 cells but not in CT26 cells. When the medium contained glucose at 200 mg/dL or more, the expression of renin increased with increasing glucose concentration in a dose-dependent manner in both cell lines. CT26 cells also expressed chymase but not ACE, in a similar manner to HT29 cells. These findings suggest then that the CRC cells activate angiotensin when exposed to high glucose concentrations.
In the hyperglycemic mice fed with a high-glucose diet, the size, number, Ki67 labeling index, and microvessel density of the liver metastatic foci were greater than those in the normoglycemic mice fed with the control diet. Clinical studies have suggested that a similar situation exists in patients. In the examination of 121 CRC patients, the tumor renin concentration correlated with HbA1c levels and the tumor A-II concentration correlated with the tumor renin concentration. Moreover, high blood HbA1c levels are associated with a higher incidence of liver metastasis in diabetes cases but not non-diabetes cases. In cardiac fibroblasts, a high concentration of glucose significantly increases intracellular A-II levels by increasing the intracellular levels of renin[29].
A-II AND LIVER METASTASIS OF COLORECTAL CANCER
The A-II precursor, ATG is mainly produced in hepatocytes[30]. We confirmed that CRC cells possessing angiotensin-activating ability establish liver metastasis because they can produce abundant A-II from AGT in the liver[6]. To examine the prometastatic effect, CRC cells with angiotensin-activating ability were used in the mouse liver metastasis model. We suppressed AGT production in the mouse liver by using pro-AGT antisense S-ODN, which significantly suppressed the liver metastasis of CRC cells. Thus, CRC cells with angiotensin-activating ability are more likely to form liver metastasis. In CRC cases, A-II is associated with renin concentration in primary tumors[6]. Thus, high levels of A-II in primary CRC tissues, which indicates the potential to activate angiotensin in CRC cells, was associated with a high frequency of liver metastasis. The A-II concentration in primary CRC tissues may be a good marker for liver metastasis.
ANGIOTENSIN-TARGETING THERAPY FOR COLORECTAL CANCER
The renin/angiotensin-activating system is recognized as an important molecular target for CRC prevention and treatment. Several inhibitors of the renin/angiotensin-activating system suppress cancer development, cancer cell growth, angiogenesis, and metastasis[4,5,31-34]. Inhibitors of the renin-angiotensin system are widely used to treat hypertension. We have shown that some anti-angiotensin agents, inhibitors of renin and chymase, suppress liver metastasis of CRCs[6,35]. In addition, ACE inhibitors and/or A-II receptor blocker (ARB) have been reported to improve disease prognosis or progression in pancreatic and urogenital cancer[36,37].
We have also examined the combined effect of anti-angiotensin treatment and hypoglycemic treatment[35]. In a streptozotocin-induced BALB/c mouse diabetes model fed a high-calorie diet, the blood sugar level increased and was associated with increasing size and number of CT26 cell liver metastases. In this diabetes mouse model, we examined the effect of concurrent hypoglycemic and anti-angiotensin treatments[35]. Insulin and gliclazide (sulfonylurea) were administered with or without a renin inhibitor (aliskiren) to the liver metastasis mouse model fed a high-calorie diet and treated with streptozotocin injection. Treatment with insulin and gliclazide resulted in lower blood sugar levels compared to that in the untreated mice. The mice treated with insulin or gliclazide showed a decrease in the number of metastatic foci and improved survival compared to the untreated mice. Concurrent treatment with anti-angiotensin using aliskiren or captopril ARB and hypoglycemic agents (insulin or gliclazide) resulted in lower serum A-II concentration, fewer metastatic foci, and longer survival compared to the untreated mice or the mice treated with hypoglycemic agents alone. Combined treatment with anti-angiotensin and hypoglycemic agents showed a synergistic inhibitory effect on liver metastasis. The mice treated with the combination showed suppression of liver metastasis and improved survival, equal to that of the control mice.
Given that the association between hyperglycemia and liver metastasis in colon cancer is a result of renin upregulation, diabetes status is likely to be a risk factor for liver metastasis. Control of blood sugar could, therefore, be important in preventing liver metastasis in colon cancer patients. The use of anti-angiotensin treatment and blood sugar control as a baseline management for colon cancer patients with diabetes deserves to be examined in clinical trials in order to establish whether it helps in the prevention of liver metastasis (Figure 1).
CONCLUSION
As described above, the angiotensin activation is a pivotal feature of colorectal cancer for disease progression and liver metastasis. The angiotensin blockade and blood sugar control are hopeful tools for suppressing the liver metastasis of colorectal cancer. These treatments are provided commonly to patients of the hypertension and diabetes in an effective and safe manner. For CRC patients, especially, in their postoperative status, the angiotensin blockade and blood sugar control are relevant to prevent the liver metastasis in addition to the anti-cancer chemotherapy.
Footnotes
Supported by Grant-in-Aid for Scientific Research from Japan Society for the Promotion of Science, Japan
Peer reviewers: Antonio Araujo, MD, PhD, Medical Oncology Department, Portuguese Institute of Oncology – Porto Centre, R Dr Antonio Bernardino Almeida, 4200 Porto, Portugal; Bin Wu, MD, PhD, Professor, Chairman, Department of Gastroenterology, the Third Affiliated Hospital of Sun Yat-Sen University, Guangzhou 510630, Guangdong Province, China
S-Editor Jiang L L-Editor A E-Editor Lu YJ
References
- 1.Cancer Statistics in Japan, 2008. 2008 ed. Tokyo: National Cancer Center; 2008. Available from: http: //ganjoho.jp/public/statistics/pub/statistics01.html. [Google Scholar]
- 2.Fujimoto Y, Nakanishi Y, Sekine S, Yoshimura K, Akasu T, Moriya Y, Shimoda T. CD10 expression in colorectal carcinoma correlates with liver metastasis. Dis Colon Rectum. 2005;48:1883–1889. doi: 10.1007/s10350-005-0141-6. [DOI] [PubMed] [Google Scholar]
- 3.Fong Y, Kemeny N, Paty P, Blumgart LH, Cohen AM. Treatment of colorectal cancer: hepatic metastasis. Semin Surg Oncol. 1996;12:219–252. doi: 10.1002/(SICI)1098-2388(199607/08)12:4<219::AID-SSU3>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 4.Escobar E, Rodríguez-Reyna TS, Arrieta O, Sotelo J. Angiotensin II, cell proliferation and angiogenesis regulator: biologic and therapeutic implications in cancer. Curr Vasc Pharmacol. 2004;2:385–399. doi: 10.2174/1570161043385556. [DOI] [PubMed] [Google Scholar]
- 5.Wu XZ. New strategy of antiangiogenic therapy for hepatocellular carcinoma. Neoplasma. 2008;55:472–481. [PubMed] [Google Scholar]
- 6.Shimomoto T, Ohmori H, Luo Y, Chihara Y, Denda A, Sasahira T, Tatsumoto N, Fujii K, Kuniyasu H. Diabetes-associated angiotensin activation enhances liver metastasis of colon cancer. Clin Exp Metastasis. 2012;29:915–925. doi: 10.1007/s10585-012-9480-6. [DOI] [PubMed] [Google Scholar]
- 7.Belova LA. Angiotensin II-generating enzymes. Biochemistry ( Mosc) 2000;65:1337–1345. doi: 10.1023/a:1002848402911. [DOI] [PubMed] [Google Scholar]
- 8.Kumar R, Singh VP, Baker KM. The intracellular renin-angiotensin system: implications in cardiovascular remodeling. Curr Opin Nephrol Hypertens. 2008;17:168–173. doi: 10.1097/MNH.0b013e3282f521a8. [DOI] [PubMed] [Google Scholar]
- 9.Nishimura H, Hoffmann S, Baltatu O, Sugimura K, Ganten D, Urata H. Angiotensin I converting enzyme and chymase in cardiovascular tissues. Kidney Int Suppl. 1996;55:S18–S23. [PubMed] [Google Scholar]
- 10.Ferreira AJ, Bader M, Santos RA. Therapeutic targeting of the angiotensin-converting enzyme 2/Angiotensin-(1-7)/Mas cascade in the renin-angiotensin system: a patent review. Expert Opin Ther Pat. 2012;22:567–574. doi: 10.1517/13543776.2012.682572. [DOI] [PubMed] [Google Scholar]
- 11.Kuniyasu H. Establish of anti-liver metastasis therapy of colorectal cancer-targeting of angiotensin activation system. Vol. 24601. Japan: Proceeding of Grant-in-Aid for Scientific Research from Japan Society for the Promotion of Science; 2011. p. 20590349. [Google Scholar]
- 12.Iwai M, Horiuchi M. Devil and angel in the renin-angiotensin system: ACE-angiotensin II-AT1 receptor axis vs. ACE2-angiotensin-(1-7)-Mas receptor axis. Hypertens Res. 2009;32:533–536. doi: 10.1038/hr.2009.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patey G, De La Baume S, Schwartz JC, Gros C, Roques B, Fournie-Zaluski MC, Soroca-Lucas E. Selective protection of methionine enkephalin released from brain slices by enkephalinase inhibition. Science. 1981;212:1153–1155. doi: 10.1126/science.7015510. [DOI] [PubMed] [Google Scholar]
- 14.Shipp MA, Vijayaraghavan J, Schmidt EV, Masteller EL, D’Adamio L, Hersh LB, Reinherz EL. Common acute lymphoblastic leukemia antigen (CALLA) is active neutral endopeptidase 24.11 (“enkephalinase”): direct evidence by cDNA transfection analysis. Proc Natl Acad Sci USA. 1989;86:297–301. doi: 10.1073/pnas.86.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yao T, Takata M, Tustsumi S, Nishiyama K, Taguchi K, Nagai E, Tsuneyoshi M. Phenotypic expression of gastrointestinal differentiation markers in colorectal adenocarcinomas with liver metastasis. Pathology. 2002;34:556–560. doi: 10.1080/0031302021000035965-4. [DOI] [PubMed] [Google Scholar]
- 16.Ohji Y, Yao T, Eguchi T, Yamada T, Hirahashi M, Iida M, Tsuneyoshi M. Evaluation of risk of liver metastasis in colorectal adenocarcinoma based on the combination of risk factors including CD10 expression: multivariate analysis of clinicopathological and immunohistochemical factors. Oncol Rep. 2007;17:525–530. [PubMed] [Google Scholar]
- 17.Sumitomo M, Shen R, Nanus DM. Involvement of neutral endopeptidase in neoplastic progression. Biochim Biophys Acta. 2005;1751:52–59. doi: 10.1016/j.bbapap.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 18.Ogasawara M, Murata J, Ayukawa K, Saimi I. Differential effect of intestinal neuropeptides on invasion and migration of colon carcinoma cells in vitro. Cancer Lett. 1997;116:111–116. doi: 10.1016/s0304-3835(97)00167-5. [DOI] [PubMed] [Google Scholar]
- 19.Boyella VD, Nicastri AD, Bergasa NV. Human hepatic met-enkephalin and delta opioid receptor-1 immunoreactivities in viral and autoimmune hepatitis. Ann Hepatol. 2008;7:221–226. [PubMed] [Google Scholar]
- 20.Spivey JR, Jorgensen RA, Gores GJ, Lindor KD. Methionine-enkephalin concentrations correlate with stage of disease but not pruritus in patients with primary biliary cirrhosis. Am J Gastroenterol. 1994;89:2028–2032. [PubMed] [Google Scholar]
- 21.Nicoll J, Axiotis CA, Bergasa NV. The delta opioid receptor 1 is expressed by proliferating bile ductules in rats with cholestasis: implications for the study of liver regeneration and malignant transformation of biliary epithelium. Med Hypotheses. 2005;65:1099–1105. doi: 10.1016/j.mehy.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 22.Kuniyasu H, Luo Y, Fujii K, Sasahira T, Moriwaka Y, Tatsumoto N, Sasaki T, Yamashita Y, Ohmori H. CD10 enhances metastasis of colorectal cancer by abrogating the anti-tumoural effect of methionine-enkephalin in the liver. Gut. 2010;59:348–356. doi: 10.1136/gut.2009.178376. [DOI] [PubMed] [Google Scholar]
- 23.Skidgel RA, Erdös EG. Angiotensin converting enzyme (ACE) and neprilysin hydrolyze neuropeptides: a brief history, the beginning and follow-ups to early studies. Peptides. 2004;25:521–525. doi: 10.1016/j.peptides.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 24.Nicolucci A. Epidemiological aspects of neoplasms in diabetes. Acta Diabetol. 2010;47:87–95. doi: 10.1007/s00592-010-0187-3. [DOI] [PubMed] [Google Scholar]
- 25.Larsson SC, Orsini N, Wolk A. Diabetes mellitus and risk of colorectal cancer: a meta-analysis. J Natl Cancer Inst. 2005;97:1679–1687. doi: 10.1093/jnci/dji375. [DOI] [PubMed] [Google Scholar]
- 26.Saydah SH, Platz EA, Rifai N, Pollak MN, Brancati FL, Helzlsouer KJ. Association of markers of insulin and glucose control with subsequent colorectal cancer risk. Cancer Epidemiol Biomarkers Prev. 2003;12:412–418. [PubMed] [Google Scholar]
- 27.Pais R, Silaghi H, Silaghi AC, Rusu ML, Dumitrascu DL. Metabolic syndrome and risk of subsequent colorectal cancer. World J Gastroenterol. 2009;15:5141–5148. doi: 10.3748/wjg.15.5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giovannucci E. Metabolic syndrome, hyperinsulinemia, and colon cancer: a review. Am J Clin Nutr. 2007;86:s836–s842. doi: 10.1093/ajcn/86.3.836S. [DOI] [PubMed] [Google Scholar]
- 29.Singh VP, Baker KM, Kumar R. Activation of the intracellular renin-angiotensin system in cardiac fibroblasts by high glucose: role in extracellular matrix production. Am J Physiol Heart Circ Physiol. 2008;294:H1675–H1684. doi: 10.1152/ajpheart.91493.2007. [DOI] [PubMed] [Google Scholar]
- 30.Dzau VJ. Implications of local angiotensin production in cardiovascular physiology and pharmacology. Am J Cardiol. 1987;59:59A–65A. doi: 10.1016/0002-9149(87)90178-0. [DOI] [PubMed] [Google Scholar]
- 31.Grossman E, Messerli FH, Goldbourt U. Carcinogenicity of antihypertensive therapy. Curr Hypertens Rep. 2002;4:195–201. doi: 10.1007/s11906-002-0007-4. [DOI] [PubMed] [Google Scholar]
- 32.Attoub S, Gaben AM, Al-Salam S, Al Sultan MA, John A, Nicholls MG, Mester J, Petroianu G. Captopril as a potential inhibitor of lung tumor growth and metastasis. Ann N Y Acad Sci. 2008;1138:65–72. doi: 10.1196/annals.1414.011. [DOI] [PubMed] [Google Scholar]
- 33.Deshayes F, Nahmias C. Angiotensin receptors: a new role in cancer? Trends Endocrinol Metab. 2005;16:293–299. doi: 10.1016/j.tem.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 34.Fyhrquist F, Saijonmaa O. Renin-angiotensin system revisited. J Intern Med. 2008;264:224–236. doi: 10.1111/j.1365-2796.2008.01981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo Y, Ohmori H, Shimomoto T, Fujii K, Sasahira T, Chihara Y, Kuniyasu H. Anti-angiotensin and hypoglycemic treatments suppress liver metastasis of colon cancer cells. Pathobiology. 2011;78:285–290. doi: 10.1159/000330169. [DOI] [PubMed] [Google Scholar]
- 36.Nakai Y, Isayama H, Ijichi H, Sasaki T, Sasahira N, Hirano K, Kogure H, Kawakubo K, Yagioka H, Yashima Y, et al. Inhibition of renin-angiotensin system affects prognosis of advanced pancreatic cancer receiving gemcitabine. Br J Cancer. 2010;103:1644–1648. doi: 10.1038/sj.bjc.6605955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyajima A, Kikuchi E, Kosaka T, Oya M. Angiotensin II Type 1 Receptor Antagonist as an Angiogenic Inhibitor in Urogenital Cancer. Rev Recent Clin Trials. 2009;4:75–78. doi: 10.2174/157488709788185996. [DOI] [PubMed] [Google Scholar]