

Abstract
Of the eight catalytic transglutaminases (TGs) transglutaminase 2 (TG2) has been the most comprehensively studied due to its ubiquitous expression in multiple cell types. Despite the observed critical role for this enzyme in multiple biological processes in vitro, TG2 knock-out mouse models have shown no severe developmental phenotypes, suggesting compensation by other TGs. To begin characterization of the compensating mechanisms, we analyzed total transamidating activity and expression patterns of all catalytically active TGs in seven different tissues/organs from wild-type and TG2 knock-out mice. Inhibitory analysis with TG2-specific inhibitor KCC-009 suggests that relative contribution of TG2 in total transamidating activity differs in various tissues. Accordingly, our data indicate tissue-specific mechanisms of compensation for the loss of TG2, including transcriptional compensation in heart and liver versus functional compensation in aorta, kidney and skeletal/cartiagenous tissues. On the contrary, no compensation has been detected in skeletal muscle, suggesting a limited role for the TG2-mediated transamidation in normal development of this tissue.
Keywords: Transglutaminase, Inhibitors, Cartilage, Heart, Aorta, Liver, Kidney, Muscle, Compensation
Introduction
The mammalian transglutaminase (TG) protein family consists of nine proteins with eight zymogens/enzymes, designated Factor XIIIa (FXIIIa) and TG1-7 in addition to a structural protein, protein 4.2, which lacks catalytic activity. TG-mediated reactions are essential for multiple biological processes ranging from blood coagulation to skin barrier formation and extracellular matrix assembly (reviewed in Griffin et al., 2002; Lorand and Graham 2003). These enzymes function in a wide range of biological processes by catalyzing three types of posttranslational modifications: transamidation, esterification, and hydrolysis (Iismaa et al., 2009). In addition, TG2, TG4 and TG5 can bind and hydrolyze GTP, which inhibits their transamidase catalytic activity (Iismaa et al., 1997; Spina et al., 1999; Candi et al., 2004). Interestingly, while these distinct enzymes are able to recognize the same protein substrate, they generally exhibit remarkable substrate specificity.
TG2 is considered the most fascinating and comprehensively studied of this diverse enzyme family. It is constitutively expressed in many cell types, including but not limited to, endothelial cells, vascular smooth muscle cells and fibroblasts (Thomazy and Fesus, 1989). Additionally, its expression correlates with cell differentiation in some cell lineages, such as the osteochondrogenic lineage (Thomazy and Fesus, 1989; Aeshlimann et al., 1993; Nurminsky et al., 2011). In addition to the traditional TG activities, TG2 has been reported to act as a protein kinase (Mishra et al., 2004) and a protein disulfide isomerase (Hasegawa et al., 2003), as well as to facilitate cell-matrix interaction independently from its enzymatic activity (Akimov et al., 2000; Xu et al., 2006; Dardik et al., 2006). TG2 is localized to both the extracellular matrix and multiple cellular compartments, with ample in vitro studies showing a wide range of TG2 functions from cell adhesion to cell death (Griffin et al., 2002; Fesus et al., 2005; Iismaa et al., 2009; Nadalutti et al., 2011).
Two mouse knockout models for TG2 were developed simultaneously by different groups to evaluate its in vivo function (De Laurenzi et al., 2001; Nanda et al., 2001). These models were based on disrupting mouse Tgm2 gene around exon 5 (which encodes part of the catalytic core domain) and both showed absence of TG2 protein in homozygote progeny. However, no obvious developmental phenotype was observed in either of these mouse models despite the previously demonstrated in vitro role for TG2 in multiple biological processes. These phenotypes suggest the common biological phenomenon of backup compensation, which occurs when functionally overlapping proteins compensate for the loss of each other. For example, such rescuing/compensation mechanisms have been described for the family of small leucine-rich proteoglycans (Ameye et al., 2002). In TG2 null (TG2−/−) chondrocytes the compensatory activation of FXIIIa has been observed, resulting in an unchanged level of total transamidase activity (Nurminskaya and Kaartinen 2006; Tanaka et al., 2007). In this study, we analyzed the relative contribution of the TG2-mediated catalytic activity in seven different wild-type (WT) mouse tissues. Next we examined enzymatic activity in the TG2−/− tissues and analyzed expression of the eight TGs in the TG2−/− versus wild-type tissues to identify possible tissue-specific compensation mechanisms supporting the TG2−/− phenotype.
Materials and Methods
Animals and tissue dissection
Animals used were CB57/B6 and TG2−/− mice (a kind gift from Robert Graham, Victor Chang Cardiovascular Institute, New South Wales, Australia). All procedures were approved by the institutional animal care and use committee at the University of Maryland Medical School and were conducted in compliance with NIH guidelines for the care and use of laboratory animals. Two 3–4 weeks old mice of each genotype were used to dissect sternum (designated as non-hypertrophic cartilage), knee joint (designated as ossifying cartilage), skeletal muscle from the limb, aorta, heart, kidney, and liver. Tissues from both animals were pooled together and total RNA was isolated with Trizol reagent (Invitrogen).
Real-Time PCR
Primers for TGs were designed using NCBI primer design software. The real-time PCR was run using first-strand synthesized cDNA as a template on a BIO RAD CFX96 Real-Time System following the manufactures instructions for heat activation, amplification and melting curves for 45 cycles. Expression levels were normalized to RLP-19 mRNA with anything showing expression after 35 cycles being disregarded for analysis.
TG Activity Assay
Total TG cross-linking activity in mouse tissue was assayed by incorporation of the biotinylated pentylamine Ez-link Pentylamine-Biotin (Pierce, IL) into N,N′-Dimethylcasein (Sigma-Aldrich, MO) in the ELISA-like assay as previously described (Trigwell et al., 2004). The 96-well microtiter plates (Maxisorp NUNC, UK) were incubated overnight with 250μl of 1mg/ml N,N′-Dimethylcasein (Sigma-Aldrich, MO) in 5mM Sodium Carbonate (pH 9.8), and blocked with 200μL of 0.1% bovine serum albumin (BSA) (HyClone, Ut) in 5mM Sodium Carbonate (pH 9.8) for one hour at 37°C. Mouse tissue was lysed in 5mM Tris-HCl pH 7.5, 0.25M Sucrose, 0.2mM MgSO4, 2mM DTT, 0.4mM PMSF, 5μg/ml Leupeptine and 0.4% Triton X-100 (lysis buffer), centrifuged and TG-containing supernatant was used for further assays. Purified guinea pig liver transglutaminase 2 (gplTG2) (Sigma-Aldrich, MO) was used as a standard for activity tests. For inhibitory studies, mouse lysates (20μg total protein) or purified gplTG2 (75ng purified protein) were pre-incubated with 30μM inhibitors for one hour at 37°C. Reaction was carried out in 100mM Tris-HCl pH 8.5, 6.7mM CaCl2, 13.3mM DTT and 2.5mM Ez-link Pentylamine-Biotin (Pierce, IL) for one hour at 37°C. Incorporated Ez-link Pentylamine-Biotin was detected with 1:5000 ExtrAvidin-Peroxidase (Sigma, MO) and Super AquaBlue ELISA Substrate (eBioscience, CA) followed by reading the absorbance at 405nm on a Polarstar Optima plate reader.
Data and Statistical Analysis
Statistical significance was calculated by the student’s T-test (*P≤0.05; **P≤0.005) and error bars demonstrate the standard error mean.
Results and Discussion
Tissue-specific expression of TG family
The expression pattern of all catalytic TG enzymes was analyzed in wild type (WT) mouse tissues that were chosen based on previously implicated roles for TG2 in development and pathology. Earlier studies have reported expression of these proteins in various cell types and tissues, however, to our knowledge a comparative analysis of their expression in various tissues has been limited. The summary of the results is shown in Fig. 1. We were unable to detect expression of TG7 and TG4 in any of the tissues, in agreement with the previous studies identifying restricted expression of TG4 protein to the prostate (Ho et al., 1992). Expression of FXIIIa varies between the analyzed tissues with lowest expression of FXIIIa observed in the liver and kidney where the regulatory/carrier B subunit of the heterotetrameric plasma coagulation Factor XIII is expressed. Despite FXIIIa’s historical identification as a “plasma” transglutaminase, our results combined with previous reports (www.ncbi.nlm.nih.gov/UniGene) demonstrate its expression in a vast variety of tissues.
Fig. 1.

Expression pattern of TGs in mouse tissues. Quantitative real-time PCR was employed to analyze expression of each enzyme and the data was normalized to expression of the housekeeping gene RPL-19. Tissue-specific levels of expression for each enzyme were compared to its average expression between all analyzed tissues. With crosses indicating no expression detected above the determined threshold. (p<0.05)
TG1 and TG3 are expressed at similar levels in all analyzed internal tissues which were cleared of skin, where expression of these enzymes is required for stabilization of the cornified cell envelope (Kuramoto et al., 2002; Candi et al., 2002). TG1 identified in the skeletal muscle, aorta and skeletal tissue of mice may function to stabilize adherent junctions, similar to its previously proposed role in the lung epithelium, liver, kidney and endothelium of the myocardial microvasculature (Hiigari et al., 1999; Baumgartner et al., 2004). However, the biological role of TG3 in these skinless tissues remains largely unknown.
The highest level of TG2 expression was detected in the aorta, correlating with its previously proposed role in vascular remodeling (Bakker et al., 2006). Similarly, the highest expression of TG5 was also detected in aorta. Expression of both TG2 and TG5 in liver was significantly lower than that of the other analyzed tissues, while in skeletal muscle and joint expression of TG5 was undetectable. Despite this low level of expression, TG2 still contributed almost 80% of total transamidating activity in liver as detected by using the TG2-specific inhibitor KCC-009 (Choi et al., 2005) (Table 1). The tissue-specific expression pattern of TG3 is similar to that of TG2, possibly implicating a common long-range regulatory mechanism for these genes localized on the same chromosome (Grenard et al., 2001). Expression of TG6 was detected in the cardiovascular tissues, both heart and aorta, and also in kidney, but absent from skeletal muscle and joint, adding new sites of expression to the previously described skin, eyes and neurons (www.ncbi.nlm.nih.gov/UniGene; Hadjivassiliou et al., 2008). Thus, TG6 seems to be an isoform with a wider distribution than previously believed.
Table 1.
Percent inhibition of TG activity by KCC-009 in wild-type mouse tissues
| Organ/Tissue | Activity (U/mg Protein) | Activity (U/mg Protein) with 30μM KCC-009 Treatment | Percent Inhibition with KCC-009 |
|---|---|---|---|
| Skeletal Muscle | 7.39 ± 0.34 | 2.85 ± 0.60 | 61.37% |
| Liver | 46.74 ± 0.19 | 10.05 ± 0.84 | 78.50% |
| Heart | 29.00 ± 2.52 | 12.74 ± 0.70 | 56.06% |
| Aorta | 8.11 ± N.A. | 6.16 ± N.A. | 23.94% |
| Sternum | 4.53 ± 1.07 | Not Detectable | 100% |
| Joint/ossifying cartilage | 19.80 ± 0.87 | 7.33 ± 0.55 | 62.98% |
| Kidney | 20.99 ± 1.46 | 11.80 ± 0.78 | 43.78% |
In conclusion, our data revealed wide and varying patterns of expression for many TGs, implicating an additional level of complexity in the biological functions of TGs and potentially different compensation mechanisms for the loss of TG2 in various tissues.
Compensation for TG2 loss in skeletal muscle
Expression of three TGs was identified in the WT skeletal muscle with–TG2 and FXIIIa expressed at relatively high levels and TG1 at much lower levels (Fig. 2a). The TG2 inhibitor KCC-009 inhibited approximately 60% of the total transamidating activity (Table 1), attributing this activity to TG2. Accordingly, genetic ablation of TG2 resulted in a 60% reduction in total transamidating activity in the skeletal muscle (Fig. 2b), corresponding to the portion of TG activity attributed to endogenous TG2. This result indicates a lack of compensation for the loss of TG2 by other TGs, suggesting that TG2 functions in the skeletal muscle independently from its catalytic activity. In agreement with this conclusion, expression of TG1 did not change significantly and expression of FXIIIa was significantly reduced in the TG2−/− versus wild-type muscle (Fig. 2c).
Fig. 2.
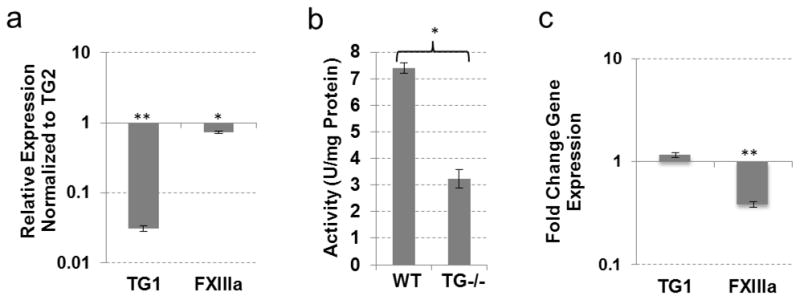
TG expression and activity in skeletal muscle tissue from wild-type and TG2 knock-out mice. (a) Real-time PCR analysis showing expression of TGs compared to TG2 expression in wild-type mouse skeletal muscle. (b) TG cross-linking activity assayed by pentylamine-biotin incorporation into N,N′-dimethylcasein. Total protein lysates from wild-type and TG2 knock-out mouse skeletal muscle were used. (c) Real-time PCR analysis showing expression of TGs in TG2 knock-out mouse skeletal muscle compared to wild-type tissue. (*P≤0.05; **P≤0.005)
Compensation for TG2 loss in liver
Similar to muscle, the same three TGs - TG1, FXIIIa and TG2–were expressed in the liver (Fig. 3a). Approximately 80% of total transmidating activity was attributed to TG2 (Table 1), however the total transamidating activity in the TG2−/− liver was reduced to 42% (Fig. 3b), indicating a possible compensation effect by other TGs. Expression analysis of the TG−/− liver revealed a 2-fold increase in FXIIIa expression (SEM 0.157, p<0.05) and a 1.6-fold increase in TG1 expression (SEM 0.076, p<0.005) (Fig. 3c). No other TGs were induced in the TG2−/− liver tissue, suggesting transcriptional compensation for the loss of TG2 via increased expression of TG1 and FXIIIa, which are expressed in the WT tissue. Interestingly, the combined activity of TG1 and FXIIIa in the liver constitutes approximately 22% of the total transamidating activity, in contrast to their 40% contribution in skeletal muscle (Table 1). This is accompanied by high levels of TG1expression in the liver (compared to TG2) while FXIIIa expression is comparable in these tissues (Fig. 2, 3). Two possible explanations can be – proposed first, that TG1 and/or FXIIIa, both requiring proteolytic activation, are activated to a higher extent in the muscle than the liver. Alternatively, TG2 in the muscle maybe less active than in liver, possibly due to regulation via the Ca2+/GTP binding balance. The significantly lower levels of total TG activity in the muscle (Table 1) favor the latter explanation.
Fig. 3.
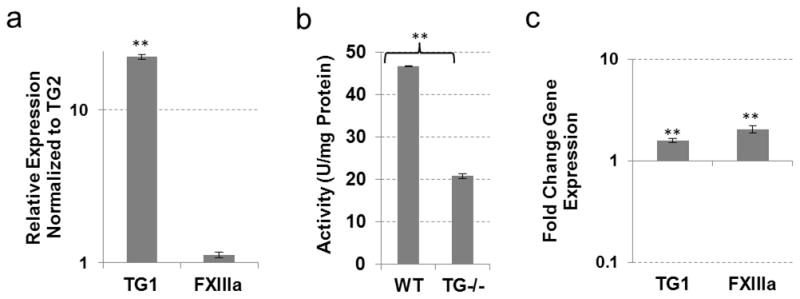
TG expression and activity in liver tissue from wild-type and TG2 knock-out mice. (a) Real-time PCR analysis showing expression of TGs compared to TG2 expression in wild-type mouse liver. (b) TG cross-linking activity assayed by pentylamine-biotin incorporation into N,N′-dimethylcasein. Total protein lysates from wild-type and TG2 knock-out mouse liver were used. (c) Real-time PCR analysis showing expression of TGs in TG2 knockout mouse liver compared to wild-type liver tissue. (*P 0≤05; **P≤0.005)
Compensation for TG2 loss in non-hypertrophic cartilage
In our previous studies, TG2 was shown to regulate the early stages of chondrogenic differentiation in mesenchymal cells (Nurminsky et al., 2011). However, the cartilaginous tissues in TG2−/− mice have been found to be phenotypically normal, suggesting a compensation mechanism by other TGs. We analyzed expression of the eight TG enzymes in the sternum cartilage, which is composed of mostly non-hypertrophic chondrocytes. Relative to TG2 expression, FXIIIa was expressed at comparable levels, while TG1 and TG3 were expressed at lower levels (Fig. 4a). The TG2 inhibitor KCC-009 dramatically decreased total transamidating activity in the wild type sternum (Table 1), suggesting that TG2 is the major active enzyme in this tissue. Unexpectedly, genetic ablation of TG2 resulted in a significant 3-fold increase in total TG activity (SEM 0.6, p<0.005) (Fig. 4b). However, no significant change in FXIIIa expression and a slight down-regulation of TG1 was observed (Fig. 4c), and expression of TG3 is reduced to almost undetectable levels (data not shown). These results implicate catalytic rather than transcriptional activation of the FXIIIa, TG1 and/or TG3 in the TG2−/− cartilage, and present an example of functional in contrast to transcriptional compensation for the loss of TG2 that was proposed for liver.
Fig. 4.

TG expression and activity in non-hypertrophic cartilage from wild-type and TG2 knock-out mice. (a) Real-time PCR analysis showing expression of TGs compared to TG2 expression in wild-type mouse non-hypertrophic cartilage. (b) TG cross-linking activity assayed by pentylamine-biotin incorporation into N,N′-dimethylcasein. Total protein lysates from wild-type and TG2 knock-out mouse non-hypertrophic cartilage were used. (c) Real-time PCR analysis showing expression of TGs in TG2 knock-out mouse non-hypertrophic cartilage compared to wild-type tissue. (*P≤0.05; **P≤0.005)
Compensation for TG2 loss in joint/ossifying cartilage
Differentiating chondrocytes of the growth plate have been shown to express both TG2 and FXIIIa (Aeschlimann 1993; Nurminskaya and Linsenmayer 1996). Here, we analyzed expression of eight TGs in the whole joint, which includes the cartilaginous growth plate, articular cartilage, periosteum and secondary ossification center. All soft tissues, including tendon, muscle, ligament, bursa and synovial sac, were carefully removed at dissection. In addition to the previously described expression of TG2 and FXIIIa, TG1 was expressed in the joint tissues although at much lower levels (Fig. 5a). Specific inhibition of TG2 with KCC-009 significantly inhibited the transamidase activity in the wild type joint tissue (Table 1). However, genetic ablation of TG2 has minor effect on the transamidase activity (Fig. 5b), indicating that enzymes other than TG2 can support the transamidase activity in the skeletal tissues, in agreement with earlier studies (Nurminskaya et al., 1998; Nurminskaya and Kaartinen, 2006; Tanaka, 2007). A novel observation of this study was the up-regulation of TG1 and induction of TG3 expression in the TG2−/− joint (Fig. 5c). In addition to the previously demonstrated proteolytic activation of the FXIIIa proenzyme (Nurminskaya et al., 1998; Tanaka et al., 2007), TG1 and TG3 may also compensate for the loss of TG2 in skeletal tissue. This finding suggests that even a double TG2/FXIIIa knock-out model may be insufficient to delineate the role of the TG-mediated protein modifications in skeletal formation.
Fig. 5.
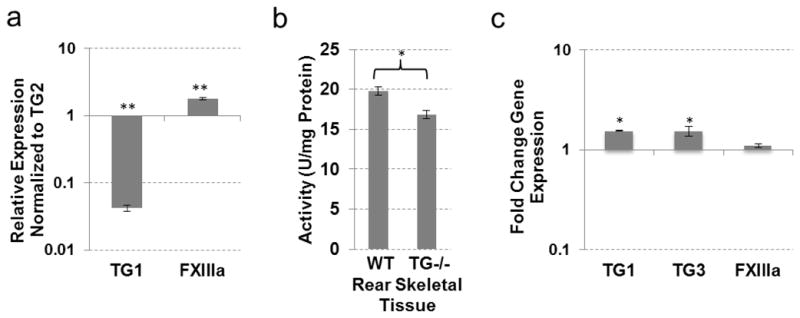
TG expression and activity in joint/ossifying cartilage from wild-type and TG2 knock-out mice. (a) Real-time PCR analysis showing expression of TGs compared to TG2 expression in wild-type mouse joint/ossifying cartilage. (b) TG cross-linking activity assayed by pentylamine-biotin incorporation into N,N′-dimethylcasein. Total protein lysates from wild-type and TG2 knock-out mouse joint/ossifying cartilage were used. (c) Real-time PCR analysis showing expression of TGs in TG2 knock-out mouse joint/ossifying cartilage compared to wild-type tissue. (*P≤0.05; **P≤0.005)
Compensation for TG2 loss in cardiovascular tissues
Aorta
High levels of TG2 expression have been detected in the aortic tissues, where different cell types express TG2 including endothelial cells, vascular smooth muscle cells and fibroblasts of the adventitia (Greenberg et al., 1991). Additionally, a significant role for TG2 has been implicated in vascular pathologies such as vascular inward remodeling (Bakker et al., 2006; Pistea et al., 2008), and medial calcification (Johnson et al 2008; our unpublished data). Nevertheless, no phenotypic abnormalities have been reported in the developing vasculature of TG2−/− mice. Endogenous TG activity in the wild-type aortic tissue was much lower than in any of the other analyzed tissues (Table 1), and expression analysis revealed dominant expression of TG2, although several other TGs were expressed at low levels as well (Fig. 6a). Surprisingly, total transamidating activity in the aortic tissue was only slightly inhibited by KCC-009 (Table 1), suggesting that TG2 is mostly present in an inactive (maybe GTP-bound) form in the aorta. However, genetic ablation of TG2 resulted in enhanced total TG activity (Fig. 6b) despite no induction in TG expression (Fig. 6c). Proteolytic activation of the pro-enzymes expressed in the TG2−/− aortic tissue offers a credible explanation for this observation, but further analysis is needed to elucidate the molecular regulation of this effect. Of note, we did not detect up-regulation of TG5 expression in the fresh TG2−/− aortic tissue which has been previously reported in the passaged TG2−/− VSMCs and maybe an artifact of cell culture (Johnson et al., 2008).
Fig. 6.

TG expression and activity in the aorta from wild-type and TG2 knock-out mice. (a) Real-time PCR analysis showing expression of TGs compared to TG2 expression in wild-type mouse aorta. (b) TG cross-linking activity assayed by pentylamine-biotin incorporation into N,N′-dimethylcasein. Total protein lysates from wild-type and TG2 knock-out mouse aorta were used. (c) Real-time PCR analysis showing expression of TGs in TG2 knock-out mouse aorta compared to wild-type tissue. (*P≤0.05; **P≤0.005)
Heart
A role for TG2 in heart biology has been suggested by the finding that its activity is down-regulated in cardiac failure (Hwang et al., 1996) and TG2-induced ventricular remodeling caused by cardiomyocyte-specific transgenic overexpression of TG2 (Small et al., 1999). In addition to TG2, heart tissue expresses FXIIIa at a level similar to TG2 along with lower levels of TG1 and TG3 (Fig. 7a). The TG2-mediated transamidation contributes to almost 60% of total activity as determined with specific inhibitor KCC-009 (Table 1). Nevertheless, in the TG2−/− heart tissue total TG activity remains practically unchanged suggesting compensation by other TGs (Fig. 7b). In this tissue, transcriptional compensation by TG3, TG5 and TG6 is suggested by the real-time PCR analysis (Fig. 7c). Further studies are needed to identify the cell origin of elevated TG3, TG5 and TG6 expression in the TG2−/− hearts.
Fig. 7.

TG expression and activity in the heart from wild-type and TG2 knock-out mice. (a) Real-time PCR analysis showing expression of TGs compared to TG2 expression in wild-type mouse heart. (b) TG cross-linking activity assayed by pentylamine-biotin incorporation into N,N′-dimethylcasein. Total protein lysates from wild-type and TG2 knock-out mouse heart were used. (c) Real-time PCR analysis showing expression of TGs in TG2 knock-out mouse heart compared to expression in the wild-type tissue. (*P≤0.05; **P≤0.005)
Compensation for TG2 loss in the kidney
TG2 has previously been shown to contribute to extracellular matrix accumulation by accelerating matrix deposition of collagens in kidneys (Fisher et al., 2009). In our studies we found TG2 to be the most abundantly expressed TG in the WT liver followed by TG1 and low levels of TG3, TG5, TG6 and FXIIIa (Fig. 8a). In the kidney, TG2 contributed to approximately 44% of the transamidase activity as shown by KCC-009 inhibition (Table 1), with only a 25% reduction in transamidase activity in the TG−/− kidney (Fig. 8b). When examining expression of seven other enzymatic TGs we found that TG1 and FXIIIa were significantly up-regulated while TG6 was further down-regulated (Fig. 8c), indicating that TG1 and FXIIIa could be functioning to compensate for the decreased transamidating activity in the TG−/− mice. However, further analysis is required to determine whether compensation is supported by an increase in transcription or proteolytic activation of TG1 and FXIIIa.
Fig. 8.

TG expression and activity in the kidney from wild-type and TG2 knock-out mice. (a) Real-time PCR analysis showing expression of TGs compared to TG2 expression in wild-type mouse kidney. (b) TG cross-linking activity assayed by pentylamine-biotin incorporation into N,N′-dimethylcasein. Total protein lysates from wild-type and TG2 knock-out mouse kidney were used. (c) Real-time PCR analysis showing expression of TGs in TG2 knockout mouse kidney compared to expression in the wild-type tissue. (*P≤0.05; **P≤0.005)
Conclusion
In this study we show a comparative expression analysis of TGs in several tissues from both wild-type and TG2 knock-out mice in which TG2 has been suggested to have a role by in vitro studies. Furthermore, we determined the level of contribution by TG2 to the total transamidating activity present in each tissue. This acquired information allowed for preliminary identification of potential tissue-specific mechanisms of compensation for genetic ablation of TG2. Our data suggests translational compensation in the heart and liver, although different TGs were induced for compensation. In contrast, functional compensation in the cartilaginous tissues and aorta is suggested by the presented analysis. Additionally, we found that in skeletal muscle there was no compensation for the loss of TG2. Understanding tissue-specific compensation mechanisms may help in design and generation of double
Acknowledgments
We would like to thank Dr. Chaitan Khosla for providing us with the TG inhibitor KCC-009 and Dr. Robert Graham for providing us with the CB57/B6 and TG2−/− mice.
Funding
This work was supported by NIH grants R01R01HL093305, R56DK071920 and R03AR057126 and a grant from Maryland Stem Cell Research Fund to M. Nurminskaya.
Abbreviation List
- TG
Transglutaminase
- FXIIIa
Factor XIIIa
- TG1-7
Transglutaminase 1-7
- WT
Wild-type
- TG−/−
Transglutaminase 2 null
Contributor Information
Stephanie Deasey, Dept. of Biochemistry and Molecular Biology, University of Maryland School of Medicine, Baltimore, MD 21201.
Maria Nurminskaya, Email: mnurminskaya@som.umaryland.edu, Dept. of Biochemistry and Molecular Biology, University of Maryland School of Medicine, Baltimore, MD 21201, Phone: 410-706-7469.
References
- Aeschlimann D, Wetterwald A, Fleisch H, Paulsson M. Expression of tissue transglutaminase in skeletal tissues correlates with events of terminal differentiation of chondrocytes. J Cell Biol. 1993;120:1461–1470. doi: 10.1083/jcb.120.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimov SS, Krylov D, Fleischmann LF, Belkin AM. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J Cell Biol. 2000;148:825–838. doi: 10.1083/jcb.148.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameye L, Young MF. Mice Deficient in Small Leucine-Rich Proteoglycans: Novel In Vivo Models for Osteoporosis, Osteoarthritis, Ehlers-Danlos Syndrome, Muscular Dystrophy and Corneal Diseases. Glycobiology. 2002;12(9):107R–1167R. doi: 10.1093/glycob/cwf065. [DOI] [PubMed] [Google Scholar]
- Bakker EN, Pistea A, Spaan JA, Rolf T, de Vries CJ, van Rooijen N, Candi E, VanBavel E. Flow-Dependent Remodeling of Small Arteries in Mice Deficient for Tissue-Type Transglutaminase: Possible Compensation by Macrophage-Derived Factor XIII. Circ Res. 2006;99:86–92. doi: 10.1161/01.RES.0000229657.83816.a7. [DOI] [PubMed] [Google Scholar]
- Baumgartner W, Golenhofen N, Weth A, Hiiragi T, Saint R, Griffin M, Drenckhahn D. Role of Transglutaminase 1 in Stabilisation of Intercellular Junctions of the Vascular Endothelium. Histochem Cell Biol. 2004;122:17–25. doi: 10.1007/s00418-004-0668-y. [DOI] [PubMed] [Google Scholar]
- Candi E, Oddi S, Paradisi A, Terrinoni A, Ranalli M, Teofoli P, Citro G, Scarpato S, Puddu P, Melino G. Expression of Transglutaminase 5 in Normal and Pathologic Human Epidermis. J Invest Dermatol. 2002;119:670–677. doi: 10.1046/j.1523-1747.2002.01853.x. [DOI] [PubMed] [Google Scholar]
- Candi E, Paradisi A, Terrinoni A, Pietroni V, Oddi S, Cadot B, Jogini V, Meiyappan M, Clardy J, Finazzi-Agro A, Melino G. Transglutaminase 5 is Regulated by Guanine-Adenine Nucleotides. Biochem J. 2004;381:313–319. doi: 10.1042/BJ20031474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K, Siegel M, Piper JL, Yuan L, Cho E, Strnad P, Omary B, Rich KM, Khosla C. Chemistry and Biology of Dihydroisoxazole Derivatives: Selective Inhibitos of Human Transglutaminase 2. Chem & Bio. 2005;12:469–475. doi: 10.1016/j.chembiol.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Dardik R, Leor J, Skutelsky E, Castel D, Holbova R, Schiby G, Shaish A, Dickneite G, Loscalzo J, Inbal A. Evaluation of the Pro-Angiogenic Effect of Factor XIII in Heterotopic Mouse Heart Allografts and FXIII-Deficient Mice. Thromb Haemost. 2006;95:546–550. doi: 10.1160/TH05-06-0409. [DOI] [PubMed] [Google Scholar]
- De Laurenzi V, Melino G. Gene Disruption of Tissue Transglutaminase. Mol Cell Biol. 2001;21:148–155. doi: 10.1128/MCB.21.1.148-155.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fesus L, Szondy Z. Transglutaminase 2 in the Balance of Cell Death and Survival. FEBS Lett. 2005;579:3297–3302. doi: 10.1016/j.febslet.2005.03.063. [DOI] [PubMed] [Google Scholar]
- Fisher M, Jones RA, Huang L, Haylor JL, El Nahas M, Griffin M, Johnson TS. Modulation of Tissue Transglutaminase in Tubular Epithelial Cells Alters Extracellular Matrix Levels: a Potential Mechanism of Tissue Scarring. Matrix Biol. 2009;28:20–31. doi: 10.1152/physrev.00044.2008. [DOI] [PubMed] [Google Scholar]
- Greenberg C, Birckbichler PJ, Rice RH. Transglutaminases: Multifunctional Cross-Linking Enzymes that Stabilize Tissues. J FASEB. 1991;5:3071–3077. doi: 10.1096/fasebj.5.15.1683845. [DOI] [PubMed] [Google Scholar]
- Grenard P, Bates MK, Aeschilmann D. Evolution of transglutaminase genes: identification of a transglutaminase gene cluster on human chromosome 15q15. J Biol Chem. 2001;276(35):33066–33078. doi: 10.1074/jbc.M10255320. [DOI] [PubMed] [Google Scholar]
- Griffin M, Casadio R, Bergamini CM. Transglutaminases: nature’s biological glues. Biochem J. 2002;368:377–396. doi: 10.1042/BJ20021234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjivassiliou M, Aeschlimann P, Strigun A, Sanders DS, Woodroofe N, Aeschlimann D. Autoantibodies in Gluten Ataxia Recognize a Novel Neuronal Transglutaminase. Ann Neurol. 2008;64(3):332–43. doi: 10.1002/ana.21450. [DOI] [PubMed] [Google Scholar]
- Hasegawa G, Suwa M, Ichikawa Y, Ohtsuka T, Kumagai S, Kikuchi M, Sato Y, Saito Y. A Novel Function of Tissue-Type Transglutaminase: Protein Disulphide Isomerase. Biochem J. 2003;373:793–803. doi: 10.1042/BJ20021084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiiragi T, Sasaki H, Nagafuchi A, Sabe H, Shen SC, Matsuki M, Yamanishi K, Tsukita S. Transglutaminase Type I and its Crosslinking Activity are Concentrated at Adherens Junctions in Simple Epithelial Cells. J Biol Chem. 1999;274:34148–34154. doi: 10.1074/jbc.274.48.34148. [DOI] [PubMed] [Google Scholar]
- Ho KC, Quarmby VE, French FS, Wilson EM. Molecular Cloning of Rat Prostate Transglutaminase Complementary DNA. The Major Androgen-Regulated Protein DP1 of Rat Dorsal Prostate and Coagulating Gland. J Biol Chem. 1992;267(18):12660–7. [PubMed] [Google Scholar]
- Hwang KC, Gray CD, Sweet WE, Moravec CS, Im MJ. Adrenergic Receptor Coupling with Gh in the Failing Human Heart. Circulation. 1996;94:718–726. doi: 10.1161/01.CIR.94.4.718. [DOI] [PubMed] [Google Scholar]
- Iismaa SE, Chung L, Wu MJ, Teller DC, Yee VC, Graham RM. The core domain of the tissue transglutaminase Gh hydrolyzes GTP and ATP. Biochemistry. 1997;36:11655–11664. doi: 10.1021/bi970545e. [DOI] [PubMed] [Google Scholar]
- Iismaa SE, Mearns BM, Lorand L, Graham RM. Transglutaminases and Disease: Lessons From Genetically Engineered Mouse Models and Inherited Disorders. Pysiol Rev. 2009;89:991–1023. doi: 10.1152/physrev.00044.2008. [DOI] [PubMed] [Google Scholar]
- Johnson KA, Polewski M, erkeltaub RA. Transglutaminase 2 is Central to Induction of the Arterial Calcification Program by Smooth Muscle Cells. Circ Res. 2008;102:529–537. doi: 10.1161/CIRCRESAHA.107.154260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramoto N, Takizawa T, Matsuki M, Morioka H, Robinson JM, Yamanishi K. Development of Ichthyosiform Skin Compensates for Defective Permeability Barrier Function in Mice Lacking Transglutaminase 1. J Clin Invest. 2002;109:243–250. doi: 10.1172/JCI0213563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol. 2003;4:140–156. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- Mishra S, Murphy LJ. Tissue transglutaminase has intrinsic kinase activity: identification of transglutaminase 2 as an insulin-like growth factor-binding protein-3 kinase. J Biol Chem. 2004;279(23):23863–23868. doi: 10.1074/jbc.M311919200. [DOI] [PubMed] [Google Scholar]
- Nadalutti C, Viiri KM, Kaukinen K, Maki M, Lindfors K. Extracellular Transglutamianse 2 has a Role in Cell Adhesion, Whereas Intracellular Transglutaminase is Involved in Regulation of Endothelial Cell Proliferation and Apoptosis. Cell Prolif. 2011;44(1):49–58. doi: 10.1111/j.1365-2184.2010.00716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda N, Iismaa SE, Owens WA, Husain A, Mackay F, Graham RM. Targeted inactivation of Gh/tissue transglutaminase II. J Biol Chem. 2001;276:20673–20678. doi: 10.1074/jbc.M010846200. [DOI] [PubMed] [Google Scholar]
- Nurminskaya M, Kaartinen MT. Transglutaminases in Mineralized Tissues. Front Biosci. 2006;11:1591–1606. doi: 10.2741/1907. [DOI] [PubMed] [Google Scholar]
- Nurminskaya M, Linsenmayer TF. Identification and Characterization of Up-Regulated Genes During Chondrocyte Hypertrophy. Dev Dynam. 1996;206:260–271. doi: 10.1002/(SICI)1097-0177(199607)206:3<260::AID-AJA4>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Nurminskaya M, Magee C, Nurminsky D, Linsenmayer TF. Plasma Transglutaminase in Hypertophic Chondrocytes: Expression and Cell-Specific Intracellular Activation Produce Cell Death and Externalization. J Cell Biol. 1998;142:1135–1144. doi: 10.1083/jcb.142.4.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurminsky D, Shanmugasundaram S, Deasey S, Michaud C, Allen S, Hendig D, Dastjerdi A, Francis-West P, Nurminskaya M. Transglutaminase 2 regulates early chondrogenesis and glycosaminoglycan synthesis. Mech Dev. 2011;128:234–245. doi: 10.1016/j.mod.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistea A, Bakker ENTP, Spaan JAE, Hardeman MR, van Rooijen N, VanBavel E. Small Artery Remodeling and Erythrocyte Deformability in L-NAME-Induced Hypertension: Role of Transglutaminases. J Vasc Res. 2008;45:10–18. doi: 10.1159/000109073. [DOI] [PubMed] [Google Scholar]
- Small K, Feng JF, Lorenz J, Donnelly ET, Yu A, Im MJ, Dorn GW, 2nd, Liggett SB. Cardiac Specific Overexpression of Transglutaminase II (Gh) Results in a Unique Hypertrophy Phenotype Independent of Phospholipase C Activation. J Biol Chem. 1999;274:21292–21296. doi: 10.1074/jbc.274.30.21291. [DOI] [PubMed] [Google Scholar]
- Spina AM, Esposito C, Pagano M, Chiosi E, Mariniello L, Cozzolino A, Porta R, Illiano G. GTPase and Transglutaminase are Associated in the Secretion of the Rat Anterior Prostate. Biochem Biophys Res Commun. 1999;260(2):351–356. doi: 10.1006/bbrc.1999.0914. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Yokosaki Y, Higashikawa F, Saito Y, Eboshida A, Ochi M. The Integrin α5β1 Regulates Chondrocyte Hypertrophic Differentiation Induced by GTP-bound Transglutaminase 2. Matrix Biol. 2007;26:409–418. doi: 10.1016/j.matbio.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Thomazy V, Fesus L. Differential Expression of Tissue Transglutaminase in Human Cells. An Immunohistochemical Study. Cell Tissue Res. 1989;255:215–224. doi: 10.1007/BF00229084. [DOI] [PubMed] [Google Scholar]
- Trigwell SM, Lynch PT, Griffin M, Hargreaves AJ, Bonner PLR. An Improved Colorimetric Assay for the Measurement of Transglutaminase (Type II)-ε-(γ-Glutamyl) Lysine Cross-Linking Activity. Anal Biochem. 2004;330(1):164–166. doi: 10.1016/j.ab.2004.03.068. [DOI] [PubMed] [Google Scholar]
- Xu L, Begum S, Hearn JD, Hynes RO. GPCR56, an Atypical G Protein-Coupled Receptor, Binds Tissue Transglutaminase, TG2, Inhibits Melanoma Tumor Growth and Metastasis. Proc Natl Acad Sci. 2006;103:9023–9028. doi: 10.1073/pnas.0602681103. [DOI] [PMC free article] [PubMed] [Google Scholar]