

Background: Phosphoregulation of focal adhesion (FA) proteins regulates FA dynamics underlying cell migration.
Results: Dynamic phosphoregulation of PEAK1 at Tyr-665 is essential for control of FA dynamics and cell migration.
Conclusion: Src family kinases (SFKs) and PEAK1 function cooperatively to control FA dynamics and cell migration.
Significance: PEAK1 is a novel SFK-dependent FA regulator and a promising anti-cancer drug target.
Keywords: Actin, Adhesion, Cell Invasion, Cell Migration, Cytoskeleton, Phosphorylation, Src, Focal Adhesions, PEAK1, Paxillin
Abstract
Pseudopodium-enriched atypical kinase 1 (PEAK1) is a recently described tyrosine kinase that associates with the actin cytoskeleton and focal adhesion (FA) in migrating cells. PEAK1 is known to promote cell migration, but the responsible mechanisms remain unclear. Here, we show that PEAK1 controls FA assembly and disassembly in a dynamic pathway controlled by PEAK1 phosphorylation at Tyr-665. Knockdown of endogenous PEAK1 inhibits random cell migration. In PEAK1-deficient cells, FA lifetimes are decreased, FA assembly times are shortened, and FA disassembly times are extended. Phosphorylation of Tyr-665 in PEAK1 is essential for normal PEAK1 localization and its function in the regulation of FAs; however, constitutive phosphorylation of PEAK1 Tyr-665 is also disruptive of its function, indicating a requirement for precise spatiotemporal regulation of PEAK1. Src family kinases are required for normal PEAK1 localization and function. Finally, we provide evidence that PEAK1 promotes cancer cell invasion through Matrigel by a mechanism that requires dynamic regulation of Tyr-665 phosphorylation.
Introduction
Cell migration is a tightly regulated process comprising a concerted sequence of steps that include leading edge protrusion, adhesion to the extracellular matrix (ECM),3 translocation of the cell body in the direction of migration, and finally, release of the ECM at the rear of the cell (1, 2). Each of these steps is dependent upon several critical signaling events, protein-protein interactions, and spatiotemporal regulation of integral components of the migration machinery. The process by which cells adhere to and then detach from the ECM is closely coupled to the ability of the cell to migrate effectively and therefore has been of great interest in the cell migration field. However, the pathways involved in the regulation of cell migration are still incompletely understood.
In the migrating cell, actin-rich protrusions extend from the cell body in multiple directions. These protrusions frequently ruffle backward toward the cell; however, in the direction of cell migration, protrusions are stabilized by the formation of integrin-dependent focal adhesions (FAs) (3, 4). FAs anchor the cell to the ECM and provide traction for cell migration (5). As the cell moves forward, FAs are modified and must eventually disassemble to facilitate continued cell migration (3).
Assembly, maturation, and disassembly of FAs are controlled by protein kinases and phosphatases that target FA components (6–9). Src family kinases (SFKs) play a critical role in the regulation of FA dynamics and cell migration (10, 11). SFKs that are recruited to FAs phosphorylate multiple FA-associated proteins, including focal adhesion kinase (FAK), paxillin, and p130CAS (12–14). These phosphorylation events lead to activation of the GTPase Rac1, which promotes lamellipodium protrusion, FA formation, and cell migration (15, 16).
Here, we describe the regulation of FA dynamics and cell motility by pseudopodium-enriched atypical kinase 1 (PEAK1, KIAA2002, Sgk269). We originally identified PEAK1 in a proteomics analysis of proteins enriched in the pseudopodia of migrating cells (17). PEAK1 promotes cancer metastasis and is expressed at high levels in human pancreatic and colon cancer (17, 18). Additionally, PEAK1 associates with the actin cytoskeleton and FAs in migrating cells and promotes the elongation of developing FAs (17). However, the mechanism by which PEAK1 promotes cell migration remains incompletely understood. PEAK1 is phosphorylated by SFKs downstream of the EGF receptor and integrins, suggesting that PEAK1 function may be regulated by SFK signaling (17, 19). In this study, we examined the role of PEAK1 in FA dynamics to better understand the function of PEAK1 in cell migration and cancer progression. We found that PEAK1 plays an instrumental role in regulating the formation, maturation, and disassembly of FAs, which are pivotal for cell migration. The proper function of PEAK1 is dependent on dynamic phosphorylation of Tyr-665. Blocking PEAK1 phosphorylation and mimicking constitutive phosphorylation at Tyr-665 both impair PEAK1 function.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
Fibronectin, vitronectin, PP2, and dimethyl sulfoxide were purchased from Sigma. Lipofectamine 2000 was purchased from Invitrogen. Dulbecco's modified Eagle's medium (DMEM) was purchased from Invitrogen. Matrigel and Transwell dishes were purchased from BD Biosciences. 35-mm glass-bottom dishes were purchased from MatTek Corp. (Ashland, MA).
Total ERK, PEAK1, and PEAK1 Tyr(P)-665 antibodies were purchased from Millipore. Src-specific (36D10) and α-tubulin (DM1A) antibodies were purchased from Sigma. GFP (ab290) antibody was purchased from Abcam (Cambridge, MA). Protein G-Sepharose beads, HRP-conjugated donkey anti-rabbit and sheep anti-mouse secondary antibodies were from GE Healthcare.
Cell Culture
Cells were cultured at 37 °C with 5% CO2 in DMEM supplemented with 10% FBS, 1% sodium pyruvate, 1% l-glutamine/gentamycin, and 1% penicillin/streptomycin. For transfection, 4 × 105 cells/well were plated in 6-well plates and incubated overnight at 37 °C. Cells were transfected in antibiotic-free, complete DMEM using Lipofectamine 2000 according to the manufacturer's instructions. HT1080 cells stably expressing shCntrl, shPEAK1 _2 (sh_2), or sh3′-UTR (sh_3) were generated through lentiviral transduction.
DNA Constructs and Stable Cell Lines
Constructs encoding mCherry-paxillin and mCherry-actin were kind gifts from Steve Hanks (Vanderbilt University, Nashville, TN). GFP (empty vector (EV)) and GFP-PEAK1 (WT PEAK1) were described previously (14), as were sh_2 and shCntrl (17). sh_3 (18) was purchased from Open Biosystems/The RNA Consortium (Thermo Scientific). HT1080 cells stably expressing shCntrl, sh_2, or sh_3 were generated by lentiviral transduction, and positively expressing cells were isolated with puromycin selection or FACS. Human c-Src-specific ON-TARGET plus siRNA SMART pool was from Thermo Scientific. PEAK1, with point mutations, was generated using the QuikChange® II XL Site-directed Mutagenesis kit, according to the manufacturer's instructions (Stratagene). Human PEAK1 cDNA in the GFP-TriEx4 vector was used as a template for mutagenesis. The following primers were used to generate PEAK1-Y665F_sense: 5′-ccacaagtgtaataagccatacttttgaagaaatagaaacagaaagcaaagtgcc-3′; PEAK1-Y665F_antisense: 5′-ggcactttgctttctgtttctatttcttcaaaagtatggcttattacacttgtgg-3′; PEAK1-Y665E_sense: 5′-ccacaagtgtaataagccatactgaggaagaaatagaaacagaaagcaaagtgcc-3′; and PEAK1-Y665E_antisense: 5′-gcactttgctttctgtttctatttcttcctcagtatggcttattacacttgtgg-3′.
Quantitative PCR, Immunoblotting, and Immunoprecipitation Assays
Total RNA from HT1080 shCntrl, sh_2, and sh_3 stable cell lines was extracted using the RNA EasyKit from Macherey-Nagel (Bethlehem, PA) and reverse-transcribed using an iScript cDNA Synthesis Kit (Bio-Rad). Quantitative PCRs were performed using TaqmanFast Universal PCR Mastermix 2×, Taqman PEAK1, and hypoxanthine phosphoribosyltransferase 1 (used as a control) primer probes, and the Step One Plus Real-Time PCR system instrument (Applied Biosystems).
Immunoblotting was performed using PVDF membranes and 8% SDS-polyacrylamide gels. Membranes were blocked in 1% BSA or 5% nonfat milk.
Cell extracts were harvested for immunoprecipitation assays using radioimmune precipitation assay (RIPA) buffer with protease and phosphatase inhibitors added. Protein concentrations were normalized to 1 mg/ml. Cell extracts were precleared with a 10% slurry of protein G beads in RIPA buffer. Precleared extracts were incubated with 1 μg/ml anti-GFP antibody (ab290) overnight at 4 °C, and proteins were pulled down with 10% protein G slurry in prepared RIPA at 4 °C for 1 h.
Microscopy
Total internal reflection fluorescence (TIRF), epifluorescence, and phase contrast imaging were performed using a Nikon Eclipse Ti inverted microscope running Nikon Elements software and equipped with an in vivoTM climate-controlled chamber and a Nikon electronic programmable stage and Perfect Focus. This microscope is also outfitted with 488- and 561-nm solid state laser lines and an Andor iXonEM+ camera for TIRF, and Plan Fluor 10× (N.A. 0.30), and APO TIRF 60× oil immersion (N.A. 1.45) objectives. Phase contrast and epifluorescence images were collected with a Hamamatsu Orca CCD camera.
Adhesion and Migration Assays
HT1080 cells co-expressing GFP-tagged PEAK1 (GFP-PEAK1, WT PEAK1), GFP-tagged PEAK1 Tyr-665 mutants (Y665E or Y665F), or GFP (EV) and mCherry-paxillin, or with siRNA, as indicated, were plated at low density on glass-bottom dishes coated with fibronectin for 1 h at 37 °C or at 4 °C overnight and allowed to attach in complete DMEM at 37 °C. The cultures were washed briefly with sterile PBS and incubated in serum-free medium along with drugs or vehicle, as indicated, for 1–2 h. Immediately before filming, the medium was replaced with phenol red-free complete DMEM containing 40 μm PP2 or vehicle (dimethyl sulfoxide), as indicated. Time-lapse TIRF images were collected in both the red and green channels using Nikon Elements software at 60× every 30 s for 1 h, and the images were analyzed using Metamorph software. FA lifetime was determined by calculating the time that passed between the first appearance of mCherry-paxillin in a developing adhesion and the last frame in which that same tagged molecule was visible. PEAK1 (WT or point mutants, as indicated) in FAs was similarly assessed. Lag time was calculated by subtracting the amount of time that elapsed between the first appearance of mCherry-paxillin and the first appearance of GFP-tagged PEAK1 or PEAK1 mutants in developing adhesions. Adhesion assembly and disassembly were analyzed as described previously (11, 20). Briefly, the background-subtracted fluorescence intensity of individual adhesions was tracked over time for at least 15 individual adhesions in a minimum of 5 cells/condition as they assembled or disassembled, and these values were used to generate a t½ value (half-life) for FA assembly or disassembly.
Random migration of 5 × 104 cells plated in fibronectin-coated 6-well plates was assessed using Metamorph software. Images were captured using a 10× phase contrast objective. Time-lapse images were collected every 5 min for at least 10 h for each condition. Transwell and Matrigel invasion assays were performed as described previously (21).
Statistical Analysis
All data represent means ± S.E. p values were determined by GraphPad, using Student's t test.
RESULTS
PEAK1 Regulates FA Dynamics
We demonstrated previously that PEAK1 gene silencing in MDA-MB-435 cells inhibits the ability of these cells to establish xenografts in mice (14). We also showed that PEAK1 overexpression promotes cell migration in vitro (17). In this study, first we examined random cell migration by video imaging HT1080 fibrosarcoma cells in which PEAK1 was silenced by transduction with lentivirus encoding either of two short hairpin RNAs (shRNAs). Fig. 1A shows that PEAK1 was silenced by 40–50% when we used shRNA targeting the coding region of PEAK1 (sh_2) or the 3′-UTR region of PEAK1 (sh_3). Although incomplete, PEAK1 gene silencing was associated with a significant decrease in random cell migration. Representative migration maps are shown in Fig. 1, B–D, and summarized results are presented in Fig. 1E.
FIGURE 1.

Endogenous PEAK1 gene silencing impairs cell migration and FA dynamics. A, PEAK1-specific shRNA significantly decreases endogenous PEAK1 mRNA levels in HT1080 cells, as determined by quantitative PCR. B, representative tracings every 5 min for 10 h of three shCntrl cells. C and D, sh_2 cells (C) or sh_3 cells (D) migrating on fibronectin. Distances are in micrometers. E, quantification of random migration represented in Fig. B–D. n = 18–20 cells/condition. F–H, inhibition of endogenous PEAK1 decreasing FA lifetimes (F) and t½ assembly (G) but increasing the t½ disassembly of mCherry-paxillin compared with shCntrl cells. n = 15–22 individual adhesions/analysis from ≥5 cells per condition. Data represent mean ± S.E. (error bars). *, p < 0.05; **, p < 0.005; ***, p < 0.0001.
Next, we tested whether PEAK1 regulates FA dynamics. Cells in which PEAK1 was silenced with sh_2 or sh_3 demonstrated a decrease in FA lifetime (Fig. 1F), accompanied by a decrease in assembly time (Fig. 1G) and an increase in disassembly time (Fig. 1H), as determined by monitoring mCherry-paxillin fluorescence by TIRF time-lapse microscopy. These results, which are summarized in supplemental Table S1, suggest that PEAK1 stabilizes FAs overall, while extending the time during which FAs enlarge (t½ assembly) but, at the same time, promoting more rapid FA disassembly.
Regulation of FA Dynamics by PEAK1 Is Controlled by PEAK1 Phosphorylation
We and others have shown that PEAK1 phosphorylation is significantly increased by Src in cancer cells (17, 19). In PEAK1, Tyr-665 is an SH2-binding site and predicted SFK target (17, 22, 23). As such, Tyr-665 is thought to be an important site for the control of PEAK1 function; therefore, we mutated Tyr-665 to further evaluate the function of PEAK1 in FA dynamics and cell migration. Fig. 2A shows that fusion proteins of GFP with wild-type (WT) PEAK1 and PEAK1 in which Tyr-665 is mutated to Glu (Y665E) or Phe (Y665F) were expressed at comparable levels in HT1080 cells; however, endogenous PEAK1 is expressed in relatively low levels in these cells compared with the exogenously expressed form of the protein. Because we previously reported that PEAK1 localizes with the actin cytoskeleton and with FAs (17), we tested whether mutation of Tyr-665 alters the subcellular localization of PEAK1. Time-lapse TIRF microscopy imaging of mCherry-actin and PEAK1 showed that Y665E mutation had no effect on PEAK1 localization with actin (Fig. 2B and supplemental Movies 1 and 2). By contrast, Y665E demonstrated a substantial decrease in co-localization with mCherry-paxillin in FAs (Fig. 2C).
FIGURE 2.

Phosphoregulation of Tyr-665 controls PEAK1 localization to FAs and FA dynamics. A, immunoblot (IB) showing PEAK1expression in HT1080 cells transiently expressing EV or GFP-tagged WT PEAK1, Y665E, or Y665F constructs. A short exposure time (top) reveals exogenous expression of WT or mutant PEAK1 constructs, but endogenous PEAK1 is not visible. A longer exposure (middle) was necessary to detect endogenous PEAK1. Tubulin (bottom) was used as a loading control. B, TIRF images of GFP-tagged WT PEAK1 or Y665E and mCherry-actin localization in HT1080 cells. C, TIRF images of GFP-tagged WT PEAK1 or Y665E and mCherry-paxillin localization in HT1080 cells. D, expression of WT PEAK1 but not Y665E increasing paxillin lifetime in FAs. n = 15–21 individual adhesions from ≥5 cells per condition. E, WT PEAK1 but not Y665E increases the t½ of paxillin assembly compared with EV controls. n = 15–19 individual adhesions from ≥5 cells per condition. F, WT PEAK1 expression decreasing the t½ of paxillin disassembly, but Y665E increasing the t½ of paxillin disassembly compared with EV control. n = 16–19 individual adhesions from mtequ]5 cells per condition. Data represent mean ± S.E. (error bars). *, p < 0.05; **, p < 0.005; ***, p < 0.001. F, HT1080 cells were co-transfected with GFP-tagged WT PEAK1, Y665E, or Y665F and mCherry-actin, then plated on fibronectin-coated glass-bottom dishes. TIRF images are shown. Scale bars, 10 μm in full images and 5 μm in enlargements, respectively.
Fig. 2D and supplemental Table S1 show that expression of WT PEAK1 (supplemental Movie 4) significantly increased FA lifetime; however, Y665E (supplemental Movie 5) failed to lengthen the FA lifetime. Similarly, Y665E failed to reproduce the increase in t½ FA assembly (Fig. 2E) and the decrease in t½ disassembly (Fig. 2F) that were observed in cells transfected with WT PEAK1. Mutation of Tyr to Glu typically produces a model of constitutive phosphorylation (24–27). Our results with Y665E suggest that constitutive phosphorylation of PEAK1 at Tyr-665 is inactivating.
Because Y665E failed to regulate FA dynamics, we hypothesized that Y665E would also fail to promote cell migration. To test this hypothesis, first we examined random cell migration. As shown in the representative cell migration maps in Fig. 3A and in the results summary (Fig. 3B), PEAK1 overexpression increased random cell migration (compared with EV) whereas Y665E actually decreased random cell migration. In Transwell migration assays, once again WT PEAK1 promoted cell migration, and Y665E failed to reproduce the effects of the wild-type protein (Fig. 3C). Thus, the ability of PEAK1 to regulate FA dynamics correlates with its ability to regulate cell migration.
FIGURE 3.

The phosphorylation state of Tyr-665 regulates PEAK1-mediated migration. A, representative tracings of three randomly migrating HT1080 cells per condition (EV, WT PEAK1, and Y665E) every 10 min for 10 h. Distance is in micrometers. B, quantification of data represented in A. n = 15–16. C, WT PEAK1 expression increases migration in a Transwell compared with EV control cells. Y665E expression abrogates this effect. n = 15–20. Data represent mean ± S.E. (error bars). ***, p < 0.001.
To block Tyr-665 phosphorylation, we mutated this amino acid to Phe (Y665F). Y665F demonstrated unchanged localization with mCherry-actin, compared with WT PEAK1 (Fig. 4A and supplemental Movies 1 and 3). Y665F also demonstrated unchanged co-localization with mCherry-paxillin, compared with WT PEAK1 (Fig. 4B and supplemental Movies 4 and 6). However, like the phosphomimetic form of the kinase, Y665F was ineffective in its ability to regulate FA dynamics. Y665F significantly decreased FA lifetime, compared with those measured in cells transfected with EV (Fig. 4C). Y665F failed to lengthen FA assembly time similarly to WT PEAK1 (Fig. 4D). However, Y665F did shorten the disassembly time (Fig. 4E).
FIGURE 4.

PEAK1 Tyr-665 phosphorylation is required to regulate PEAK1 localization and FA dynamics in HT1080 cells. A, TIRF images of GFP-tagged WT PEAK1 or Y665F and mCherry-actin localization in HT1080 cells. B, TIRF images of GFP-tagged WT PEAK1 or Y665F and mCherry-paxillin localization in HT1080 cells. Scale bars, 10 μm in full images and 5 μm in enlargements, respectively. C, expression of WT PEAK1 increasing paxillin lifetime in FAs, but Y665F decreasing it compared with EV. D, WT PEAK1 but not Y665F increasing the t½ of paxillin assembly compared with EV controls. n = 15–18 individual adhesions from ≥5 cells per condition. E, WT PEAK1 and Y665F expression both decreasing the t½ of paxillin disassembly compared with EV control. n = 16–19 individual adhesions from ≥5 cells per condition. Data represent mean ± S.E. (error bars). Control data representing EV and WT PEAK1 in D–F are the same as those represented in Fig. 2. *, p < 0.05; **, p < 0.005; ***, p < 0.0001.
In addition to our analysis of PEAK1-mediated FA dynamics using paxillin as an FA marker, we also analyzed WT and mutant PEAK1 dynamics in these cells. These data, summarized in supplemental Table S1, indicate that WT or mutant PEAK1 dynamics, including total lifetime, t½ of assembly, and t½ of disassembly, closely mirror those of paxillin under identical conditions. However, WT PEAK1 entered adhesions, on average, >1 min after adhesions were detectable by paxillin fluorescence. By contrast, Y665F entered FAs almost simultaneously with the initial detection of paxillin fluorescence (supplemental Table S1). These results indicate that modifying Tyr-665 so that it cannot be phosphorylated or so that it mimics a constitutively phosphorylated state alters the activity of PEAK1 in the regulation of FA dynamics.
Because Y665F expression affected PEAK1-mediated FA dynamics, we next sought to examine the effects of blocking Tyr-665 phosphorylation on PEAK1-mediated cell migration. Representative cell migration maps and a summary of results comparing random migration of WT PEAK1 and Y665F are shown in Fig. 5, A and B, respectively. Like the phosphomimetic mutant, Y665F failed to promote random cell migration. These results were confirmed using the Transwell model system, in which we also found that Y665F failed to promote cell migration like WT PEAK1 (Fig. 5C).
FIGURE 5.

PEAK1 Tyr-665 phosphoregulation is required to regulate cell migration. A, representative tracings of three randomly migrating cells per condition (EV, WT PEAK1, and Y665F) every 10 min for 10 h. Distance is in micrometers. B, quantification of data represented in A. Control data representing EV and WT PEAK1 in B and C are the same as those represented in Fig. 3. n = 15–16 cells per condition. Data represent mean ± S.E. (error bars). ***, p < 0.0001.
Invasion through Matrigel in vitro depends not only on the ability of the cell to migrate but also on its ability to remodel extracellular matrix (6). Because Matrigel invasion is frequently studied as a model of in vivo cancer cell invasion, we applied this model to study our mutated forms of PEAK1. Fig. 6 shows that PEAK1 overexpression in HT1080 cells increased invasion through Matrigel by >3-fold. By contrast, Y665E and Y665F failed to promote invasion. These results are consistent with our cell migration data and suggest that the effects of PEAK1 on FA maturation may regulate not only cell migration but also invasion.
FIGURE 6.
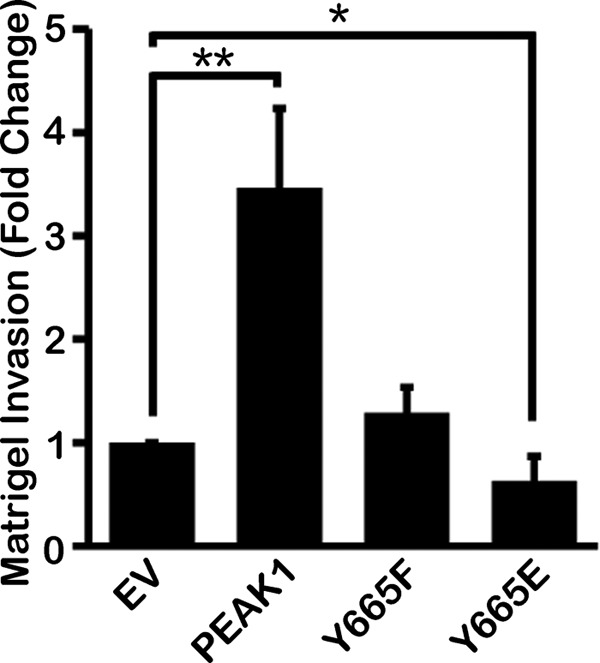
Phosphoregulation of Tyr-665 controls PEAK1-mediated invasion. WT PEAK1 promotes Matrigel invasion by HT1080 cells compared with EV controls, but Y665F fails to promote invasion and Y665E inhibits invasion to below basal levels. n = 36 fields quantified per condition. Data represent mean ± S.E. (error bars). *, p < 0.05; **, p < 0.005.
PEAK1 Functions with SFKs to Regulate Cell Migration
We hypothesized that SFKs work in concert with protein phosphatases to mediate the dynamic phosphorylation of PEAK1 at Tyr-665, which appears to be necessary for cell migration and invasion. Fig. 7A shows the results of an immunoblot in which we validated an anti-PEAK1 Tyr(P)-665 specific antibody that recognizes WT PEAK1 but not the Y665F mutant. To test our hypothesis that SFKs mediate PEAK1 Tyr-665 phosphorylation, we used SYF−/− MEFs, which are mouse embryonic fibroblasts that lack the SFKs: Src, Yes, and Fyn. Fig. 7B shows the results of an immunoprecipitation experiment in which GFP-tagged PEAK1 was transiently expressed in WT or SYF−/− MEFs, immunoprecipitated, and immunoblotted for PEAK1 Tyr(P)-665. These results show that, in the presence of SFKs, Tyr(P)-665 is increased substantially, compared with the negligible level observed in SYF−/− MEFs that lack SFKs. To further test the dependence of PEAK1 on SFK-mediated regulation, we transfected HT1080 cells with an siRNA pool that targeted human c-Src specifically. Fig. 7C shows an immunoblotting experiment in which we used an antibody that specifically recognizes c-Src. In cells that overexpress WT PEAK1 and in control cells, the siRNA decreased c-Src. Fig. 7D shows the results of Transwell migration experiments using HT1080 cells under these conditions. In cells that were transfected with control, nontargeting siRNA, PEAK1 promoted cell migration, as was anticipated. However, in cells in which c-Src was silenced, the promigratory activity of PEAK1 was neutralized. These results suggest that c-Src plays an instrumental role in regulating PEAK1 and its ability to control cell migration.
FIGURE 7.

SFKs promote Tyr-665 phosphorylation and are required for PEAK1-mediated migration. A, anti-PEAK1 Tyr(P)-665 phosphospecific antibody specifically recognizes WT PEAK1, but not Y665F in HEK293 cells transiently expressing GFP-tagged WT PEAK1 or GFP-tagged Y665F. Following Tyr(P)-665 analysis, this immunoblot (IB) was reprobed for GFP as a loading control. B, WT PEAK1 is robustly phosphorylated at Tyr-665 in WT MEF cells, but this is abrogated in SYF−/− MEFs, which lack the SFKs Src, Yes, and Fyn. Total cell extracts were probed for GFP as a loading control. IP, immunoprecipitation. C, transient siSrc expression decreased endogenous Src expression in HT1080 cells compared with nonspecific siCntrl in cells co-expressing EV or WT PEAK1. ERK is a loading control. D, WT PEAK1 expression increased Transwell migration, but is dependent on Src expression. n = 24–36 fields quantified per condition. Data represent mean ± S.E. (error bars). *, p < 0.05; ***, p < 0.001.
In HT1080 cells that were treated with the c-Src inhibitor, PP2, and in cells that were transfected with c-Src-specific siRNA, co-localization of GFP-PEAK1 with mCherry-paxillin was inhibited (Fig. 8A and B and supplemental Movies 7 and 8). In addition, the effects of PEAK1 overexpression on FA lifetime (Fig. 8C), assembly time (Fig. 8D), and disassembly time (Fig. 8E) were neutralized in cells transfected with c-Src-specific siRNA. c-Src gene silencing independently regulated FA turnover parameters, such as the disassembly time, as anticipated (11). In cells in which c-Src was silenced, PEAK1 overexpression decreased FA lifetime, as opposed to increasing lifetime. However, the pattern of PEAK1 effects on FA dynamics, resulting in increased cell migration, was clearly disrupted in c-Src gene-silenced cells, explaining the results presented in Fig. 7.
FIGURE 8.

Src activity is required for PEAK1 localization to FAs and PEAK1-mediated adhesion effects in HT1080 cells. A, WT PEAK1 fails to localize to nascent adhesions at the leading edge of HT1080 cells in which Src activity is inhibited by PP2, compared with those treated with dimethyl sulfoxide (DMSO) only. B, WT PEAK1 localizes to FAs at the leading edge of HT1080 cells co-expressing siCntrl, but fails to localize in those co-expressing siSrc. TIRF images are shown. Scale bars, 10 μm in full images and 5 μm in enlargements, respectively. C, in siCntrl-expressing cells, co-expression of WT PEAK1 increases paxillin lifetime in FAs compared with siCntrl cells co-expressing EV. In siSrc-expressing cells, co-expression of WT PEAK1 decreases paxillin lifetime in FAs compared with siSrc + EV co-expression. D, WT PEAK1 fails to affect adhesion assembly when co-expressed with siSrc. E, WT PEAK1 expression has no effect on FA disassembly in siSrc-expressing cells. Data represent mean ± S.E. (error bars). *, p < 0.05; **, p < 0.005; ***, p < 0.001.
DISCUSSION
Previous work has shown that PEAK1 regulates cell migration and cancer metastasis (17, 18, 28); however, the underlying mechanisms are currently not known. Here, we report that PEAK1 regulates cell migration by modulating FA dynamics. We found that PEAK1 expression increases cell migration and is dependent upon phosphoregulation of PEAK1 at Tyr-665 and Src activity. This is consistent with our analysis of PEAK1-mediated FA dynamics that indicate PEAK1 promotes prolonged FA assembly coupled with rapid disassembly. Inhibiting (Y665F) or mimicking (Y665E) Tyr-665 phosphorylation disrupted cell migration and perturbed the underlying FA dynamics, yielding decreased FA lifetimes and FA assembly; Y665E was unable to facilitate rapid disassembly. In addition, we showed that regulation of Tyr-665 is necessary for PEAK1-mediated Matrigel invasion. Src knockdown alone impaired migration and impaired FA disassembly, as is consistent with previous reports (11). In cells depleted of Src, PEAK1 expression was unable to promote migration beyond that seen in controls, and PEAK1-mediated FA dynamics were significantly hindered; both PEAK1-mediated FA assembly and disassembly were altered in the absence of Src. This is supported by reports that PEAK1 phosphorylation is increased by SFKs in cancer cell lines (19, 29). Src is predicted by mass spectrometric profiling to regulate Tyr-665 phosphorylation (17, 22, 23), and our data support the hypothesis that Src regulates PEAK1 through modulation of Tyr-665 to control FA dynamics and therefore, cell migration.
Lamellipodia are stabilized by the formation of new FAs at the leading edge. Currently, ∼180 identified proteins comprise the adhesome including paxillin, Src, FAK, and α-actinin (30, 31). Paxillin enters FAs early in assembly and is well established as an FA marker (11, 20, 32, 33). As assembly progresses, scaffolds such as α-actinin incorporate, marking FA maturation (11). FA dynamics are controlled in part by post-translational modification and conformational changes (34, 35). For example, Src-mediated phosphorylation triggers paxillin-promoted disassembly (32, 36, 37), which allows release of the ECM as new FAs form at the leading edge (11). We found that PEAK1 enters FAs after both paxillin (supplemental Table S1) and α-actinin (data not shown) and persists in adhesions after paxillin (supplemental Movie 4) is gone, suggesting that PEAK1 functions more in the later stages of FA development and turnover than in FA initiation (11, 38). These findings are consistent with PEAK1 increasing FA length (17) and promoting longer FA lifetimes (Figs. 1F and 2D and supplemental Table S1), yielding increased cell migration.
Although the exact mechanism by which PEAK1 regulates FA maturation and dynamics is unknown, our combined mutational and functional FA dynamics, migration, and invasion studies clearly identify Tyr-665 as a critical amino acid that regulates the timing of entry and function of PEAK1 at FAs. We have provided evidence that these events are likely regulated by the phosphorylation state of Tyr-665. Phosphomimetic Tyr-665 disrupts FA disassembly to the same extent as shRNA-mediated knockdown of PEAK1 (Figs. 1H and 2F and supplemental Table S1). Our data indicate that phosphorylation of this site inhibits PEAK1-mediated FA dynamics and PEAK1 localization. We showed that the dephosphorylated mutant, Y665F, impaired FA dynamics and failed to promote cell migration (Figs. 4, C–E, and 5, A–C) and invasion (Fig. 6). Like WT PEAK1, Y665F promotes rapid FA disassembly. Unlike WT PEAK1, Y665F enters FAs simultaneously with paxillin (supplemental Table S1 and Movies 4 and 6) and fails to stabilize growing FAs; together, yielding shorter FA lifetimes and impaired migration. However, Y665F is able to localize robustly to FAs, whereas the phosphomimetic Y665E localizes poorly to FAs (Figs. 2C and 4B and supplemental Movies 4 and 5). Further, disassembly in Y665E-expressing cells is dramatically impaired compared with cells expressing WT PEAK1 or Y665F. From these studies, we conclude that dephosphorylation of Tyr-665 facilitates the translocation of PEAK1 to FAs whereas phosphorylation of this site impairs it. Together, these data suggest that tightly regulated cycles of PEAK1-Tyr-665 phosphorylation and dephosphorylation are necessary for proper FA turnover, cell migration, and invasion.
The tyrosine kinase(s) directly responsible for Tyr-665 phosphorylation has not been positively identified; however, Src is a likely candidate. In fact, informatics indicate Tyr-665 is a highly conserved Src consensus phosphorylation site within a Src SH2 binding motif (TTSVISHTYEEIETESK) (17, 22, 23), and SFKs increase PEAK1 phosphorylation in a number of cancer cell lines (19, 29). Consistent with these findings, we have shown that SFK expression dramatically increases Tyr-665 phosphorylation in MEF cells (Fig. 7B). Also, our data show that Src tightly regulates the spatiotemporal dynamics of PEAK1. Inhibition of Src clearly disrupted PEAK1 localization in migrating cells (Fig. 8, A and B, and supplemental Movies 7 and 8) and substantially muted the effect of PEAK1 on FA dynamics (Fig. 8,C–E). FA lifetimes were cut short, entry into new FAs was delayed, developing FAs were not stabilized, and FA disassembly was disrupted, despite WT PEAK1 overexpression. These data show that Src is required for PEAK1 function within FAs. Src regulates FAs, controlling their assembly, maturation, and disassembly (14) by affecting the spatiotemporal localization and activity of effectors like paxillin. Consistent with our findings, the inhibition or absence of SFKs is associated with hampered cell migration and specifically, with defects in FA disassembly and rear detachment (11). Notably, like Src, PEAK1 is especially important for promoting FA disassembly. We found that PEAK1 expression was unable to rescue cell migration defects in Src-depleted cells (Fig. 7D), and similarly, PEAK1 was unable to promote FA assembly and rapid disassembly in the absence of Src (Fig. 8, D and E, and supplemental Table S1). As with Src or PEAK1 inhibition, perturbation of Tyr-665 phosphorylation impedes cell migration, and dephosphorylation (Y665F) affects FA assembly to the same extent as Src or PEAK1 gene silencing (supplemental Table S1). Phosphomimetic Y665E prevents normal FA disassembly, similar to the slow disassembly seen with the inhibition of PEAK1 or Src. Although one may expect the phosphomimetic mutant to be constitutively active, we have shown that it is not. This suggests that regulation of Tyr-665 must be tightly controlled to promote PEAK1 function and supports our findings indicating the phosphorylation state of Tyr-665 plays an important role in PEAK1 localization to FAs. Together, these data support the hypothesis that Src controls PEAK1-mediated cell migration and invasion through FA modulation by dynamic phosphoregulation of Tyr-665.
This discovery that PEAK1 and Src work cooperatively to promote cell migration and invasion has important implications for cancer metastasis. The central role of Src in pancreatic ductal adenocarcinoma metastasis and progression is well documented (39). PEAK1 is also overexpressed in a variety of cancers, including pancreatic ductal adenocarcinoma, in which it associates with an Src-ErbB2 complex to promote cancer progression and metastasis (18). ErbB2-mediated Src activity increases cancer cell migration and invasion (40), and our previous study showed that PEAK1 mediates the formation of an active Src-ErbB2 complex that drives anchorage-independent growth and carcinogenesis (18). PEAK1-Src-ErbB2 function in a feed-forward loop; however, the mechanism through which this kinase complex promotes migration remains unknown. Downstream of ErbB2, Src-mediated FAK regulation controls FA formation and promotes invasion, and FAK co-localizes with ErbB2 in FAs at the leading edge (40, 41). PEAK1 may promote a Src/ErbB2 interaction necessary for downstream signaling to FAK in FAs; however, further studies are needed to examine this possibility. Our findings are consistent with the idea that PEAK1 and Src work closely together to modulate cell migration and cancer progression by modulating FA dynamics. Disrupting PEAK1-Src activities could be a fruitful avenue for therapeutic cancer treatment and could lead to new avenues for the detection and diagnosis of metastatic disease.
Acknowledgments
We thank the members of the Klemke and Gonias laboratories, Erin Zardouzian for excellent technical support, and Donna Webb for reading the manuscript. mCherry-paxillin and mCherry-actin constructs were kind gifts from Steve Hanks (Vanderbilt University, Nashville, TN) that were generated by Dominique Donato and Larissa Ryzhova.
This work was supported, in whole or in part, by National Institutes of Health Grants CA097022 (to R. L. K.) and HL060551 (to S. L. G.).

This article contains supplemental Table S1 and Movies 1–8.
- ECM
- extracellular matrix
- EV
- empty vector
- FA
- focal adhesion
- FAK
- focal adhesion kinase
- GFP-PEAK1
- GFP-tagged wild-type PEAK1
- MEF
- mouse embryonic fibroblast
- PP2
- (4-amino)-5-(4-chlorophenyl)-7-(dimethylethyl)pyrozolo[3,4-d]pyrimidine
- PEAK1
- pseudopodium-enriched atypical kinase 1
- sh_2
- PEAK1-specific shRNA _2
- sh_3
- PEAK1-specific shRNA targeting the 3′-untranslated region
- shCntrl
- nonspecific control shRNA
- siCntrl
- nonspecific control siRNA
- siSrc
- Src-specific siRNA
- SFK
- Src family kinase
- TIRF
- total internal reflection fluorescence
- Tyr(P)-665
- phosphorylated tyrosine 665.
REFERENCES
- 1. Lauffenburger D. A., Horwitz A. F. (1996) Cell migration: a physically integrated molecular process. Cell 84, 359–369 [DOI] [PubMed] [Google Scholar]
- 2. Ridley A. J., Schwartz M. A., Burridge K., Firtel R. A., Ginsberg M. H., Borisy G., Parsons J. T., Horwitz A. R. (2003) Cell migration: integrating signals from front to back. Science 302, 1704–1709 [DOI] [PubMed] [Google Scholar]
- 3. Burridge K., Sastry S. K., Sallee J. L. (2006) Regulation of cell adhesion by protein-tyrosine phosphatases. I. Cell-matrix adhesion. J. Biol. Chem. 281, 15593–15596 [DOI] [PubMed] [Google Scholar]
- 4. Gardel M. L., Schneider I. C., Aratyn-Schaus Y., Waterman C. M. (2010) Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol. 26, 315–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bach C. T., Schevzov G., Bryce N. S., Gunning P. W., O'Neill G. M. (2010) Tropomyosin isoform modulation of focal adhesion structure and cell migration. Cell Adhesion Migration 4, 226–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Larsen M., Artym V. V., Green J. A., Yamada K. M. (2006) The matrix reorganized: extracellular matrix remodeling and integrin signaling. Curr. Opin. Cell Biol. 18, 463–471 [DOI] [PubMed] [Google Scholar]
- 7. Goetz J. G. (2009) Bidirectional control of the inner dynamics of focal adhesions promotes cell migration. Cell Adhesion Migration 3, 185–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beningo K. A., Dembo M., Kaverina I., Small J. V., Wang Y.-L. (2001) Nascent focal adhesions are responsible for the generation of strong propulsive forces in migrating fibroblasts. J. Cell Biol. 153, 881–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shattil S. J. (2005) Integrins and Src: dynamic duo of adhesion signaling. Trends Cell Biol. 15, 399–403 [DOI] [PubMed] [Google Scholar]
- 10. Bromann P. A., Korkaya H., Courtneidge S. A. (2004) The interplay between Src family kinases and receptor tyrosine kinases. Oncogene 23, 7957–7968 [DOI] [PubMed] [Google Scholar]
- 11. Webb D. J., Donais K., Whitmore L. A., Thomas S. M., Turner C. E., Parsons J. T., Horwitz A. F. (2004) FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 6, 154–161 [DOI] [PubMed] [Google Scholar]
- 12. Choi C. K., Zareno J., Digman M. A., Gratton E., Horwitz A. R. (2011) Cross-correlated fluctuation analysis reveals phosphorylation-regulated paxillin-FAK complexes in nascent adhesions. Biophys. J. 100, 583–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lock J. G., Wehrle-Haller B., Strömblad S. (2008) Cell-matrix adhesion complexes: master control machinery of cell migration. Semin. Cancer Biol. 18, 65–76 [DOI] [PubMed] [Google Scholar]
- 14. Carragher N. O., Frame M. C. (2004) Focal adhesion and actin dynamics: a place where kinases and proteases meet to promote invasion. Trends Cell Biol. 14, 241–249 [DOI] [PubMed] [Google Scholar]
- 15. Vallés A. M., Beuvin M., Boyer B. (2004) Activation of Rac1 by paxillin-Crk-DOCK180 signaling complex is antagonized by Rap1 in migrating NBT-II cells. J. Biol. Chem. 279, 44490–44496 [DOI] [PubMed] [Google Scholar]
- 16. Holcomb M., Rufini A., Barilà D., Klemke R. L. (2006) Deregulation of proteasome function induces Abl-mediated cell death by uncoupling p130CAS and c-CrkII. J. Biol. Chem. 281, 2430–2440 [DOI] [PubMed] [Google Scholar]
- 17. Wang Y., Kelber J. A., Tran Cao H. S., Cantin G. T., Lin R., Wang W., Kaushal S., Bristow J. M., Edgington T. S., Hoffman R. M., Bouvet M., Yates J. R., 3rd, Klemke R. L. (2010) Pseudopodium-enriched atypical kinase 1 regulates the cytoskeleton and cancer progression [corrected]. Proc. Natl. Acad. Sci. U.S.A. 107, 10920–10925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kelber J. A., Reno T., Kaushal S., Metildi C., Wright T., Stoletov K., Weems J. M., Park F. D., Mose E., Wang Y., Hoffman R. M., Lowy A. M., Bouvet M., Klemke R. L. (2012) KRas induces a Src/PEAK1/ErbB2 kinase amplification loop that drives metastatic growth and therapy resistance in pancreatic cancer. Cancer Res. 72, 2554–2564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Leroy C., Fialin C., Sirvent A., Simon V., Urbach S., Poncet J., Robert B., Jouin P., Roche S. (2009) Quantitative phosphoproteomics reveals a cluster of tyrosine kinases that mediates SRC invasive activity in advanced colon carcinoma cells. Cancer Res. 69, 2279–2286 [DOI] [PubMed] [Google Scholar]
- 20. Bristow J. M., Sellers M. H., Majumdar D., Anderson B., Hu L., Webb D. J. (2009) The Rho-family GEF Asef2 activates Rac to modulate adhesion and actin dynamics and thereby regulate cell migration. J. Cell Sci. 122, 4535–4546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Webb D. J., Nguyen D. H., Gonias S. L. (2000) Extracellular signal-regulated kinase functions in the urokinase receptor-dependent pathway by which neutralization of low density lipoprotein receptor-related protein promotes fibrosarcoma cell migration and Matrigel invasion. J. Cell Sci. 113, 123–134 [DOI] [PubMed] [Google Scholar]
- 22. Moritz A., Li Y., Guo A., Villén J., Wang Y., MacNeill J., Kornhauser J., Sprott K., Zhou J., Possemato A., Ren J. M., Hornbeck P., Cantley L. C., Gygi S. P., Rush J., Comb M. J. (2010) Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci. Signal. 3, ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rikova K., Guo A., Zeng Q., Possemato A., Yu J., Haack H., Nardone J., Lee K., Reeves C., Li Y., Hu Y., Tan Z., Stokes M., Sullivan L., Mitchell J., Wetzel R., Macneill J., Ren J. M., Yuan J., Bakalarski C. E., Villen J., Kornhauser J. M., Smith B., Li D., Zhou X., Gygi S. P., Gu T.-L., Polakiewicz R. D., Rush J., Comb M. J. (2007) Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 131, 1190–1203 [DOI] [PubMed] [Google Scholar]
- 24. Kassenbrock C. K., Anderson S. M. (2004) Regulation of ubiquitin protein ligase activity in c-Cbl by phosphorylation-induced conformational change and constitutive activation by tyrosine to glutamate point mutations. J. Biol. Chem. 279, 28017–28027 [DOI] [PubMed] [Google Scholar]
- 25. Wu Y., Spencer S. D., Lasky L. A. (1998) Tyrosine phosphorylation regulates the SH3-mediated binding of the Wiskott-Aldrich syndrome protein to PSTPIP, a cytoskeletal-associated protein. J. Biol. Chem. 273, 5765–5770 [DOI] [PubMed] [Google Scholar]
- 26. McEwen D. P., Meadows L. S., Chen C., Thyagarajan V., Isom L. L. (2004) Sodium channel β1 subunit-mediated modulation of Nav1.2 currents and cell surface density is dependent on interactions with contactin and ankyrin. J. Biol. Chem. 279, 16044–16049 [DOI] [PubMed] [Google Scholar]
- 27. Reszka A. A., Hayashi Y., Horwitz A. F. (1992) Identification of amino acid sequences in the integrin β1 cytoplasmic domain implicated in cytoskeletal association. J. Cell Biol. 117, 1321–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Simpson K. J., Selfors L. M., Bui J., Reynolds A., Leake D., Khvorova A., Brugge J. S. (2008) Identification of genes that regulate epithelial cell migration using an siRNA screening approach. Nat. Cell Biol. 10, 1027–1038 [DOI] [PubMed] [Google Scholar]
- 29. Hochgräfe F., Zhang L., O'Toole S. A., Browne B. C., Pinese M., Porta Cubas A., Lehrbach G. M., Croucher D. R., Rickwood D., Boulghourjian A., Shearer R., Nair R., Swarbrick A., Faratian D., Mullen P., Harrison D. J., Biankin A. V., Sutherland R. L., Raftery M. J., Daly R. J. (2010) Tyrosine phosphorylation profiling reveals the signaling network characteristics of basal breast cancer cells. Cancer Res. 70, 9391–9401 [DOI] [PubMed] [Google Scholar]
- 30. Zaidel-Bar R., Itzkovitz S., Ma'ayan A., Iyengar R., Geiger B. (2007) Functional atlas of the integrin adhesome. Nat. Cell Biol. 9, 858–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zaidel-Bar R., Geiger B. (2010) The switchable integrin adhesome. J. Cell Sci. 123, 1385–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nayal A., Webb D. J., Brown C. M., Schaefer E. M., Vicente-Manzanares M., Horwitz A. R. (2006) Paxillin phosphorylation at Ser273 localizes a GIT1-PIX-PAK complex and regulates adhesion and protrusion dynamics. J. Cell Biol. 173, 587–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zaidel-Bar R., Ballestrem C., Kam Z., Geiger B. (2003) Early molecular events in the assembly of matrix adhesions at the leading edge of migrating cells. J. Cell Sci. 116, 4605–4613 [DOI] [PubMed] [Google Scholar]
- 34. Geiger B., Bershadsky A., Pankov R., Yamada K. M. (2001) Transmembrane cross-talk between the extracellular matrix–cytoskeleton cross-talk. Nat. Rev. Mol. Cell Biol. 2, 793–805 [DOI] [PubMed] [Google Scholar]
- 35. Geiger B., Bershadsky A. (2002) Exploring the neighborhood: adhesion-coupled cell mechanosensors. Cell 110, 139–142 [DOI] [PubMed] [Google Scholar]
- 36. Zaidel-Bar R., Milo R., Kam Z., Geiger B. (2007) A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J. Cell Sci. 120, 137–148 [DOI] [PubMed] [Google Scholar]
- 37. Broussard J. A., Webb D. J., Kaverina I. (2008) Asymmetric focal adhesion disassembly in motile cells. Curr. Opin. Cell Biol. 20, 85–90 [DOI] [PubMed] [Google Scholar]
- 38. Donato D. M., Ryzhova L. M., Meenderink L. M., Kaverina I., Hanks S. K. (2010) Dynamics and mechanism of p130Cas localization to focal adhesions. J. Biol. Chem. 285, 20769–20779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nagaraj N. S., Washington M. K., Merchant N. B. (2011) Combined blockade of Src kinase and epidermal growth factor receptor with gemcitabine overcomes STAT3-mediated resistance of inhibition of pancreatic tumor growth. Clin. Cancer Res. 17, 483–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Feigin M. E., Muthuswamy S. K. (2009) ErbB receptors and cell polarity: new pathways and paradigms for understanding cell migration and invasion. Exp. Cell Res. 315, 707–716 [DOI] [PubMed] [Google Scholar]
- 41. Xu Y., Benlimame N., Su J., He Q., Alaoui-Jamali M. A. (2009) Regulation of focal adhesion turnover by ErbB signalling in invasive breast cancer cells. Br. J. Cancer 100, 633–643 [DOI] [PMC free article] [PubMed] [Google Scholar]