

Background: Phosphorylation of nuclear factor-κB (NF-κB) subunits is critical for NF-κB activity.
Results: Mutation of phospho-acceptor sites within the p65 Rel homology domain influences NF-κB activity in a gene-dependent manner by altering p65 and RNA polymerase II promoter recruitment.
Conclusion: Differential p65 phosphorylation serves as a code to target NF-κB transcriptional activity to distinct gene subsets.
Significance: Our data provide insight into how NF-κB transcriptional specificity is achieved.
Keywords: Chromatin, Chromatin Immunoprecipitation (ChiP), Gene Expression, NF-κB Transcription Factor, Phosphorylation, RNA Polymerase II, Transcription, Transcription Promoter
Abstract
Phosphorylation of NF-κB plays an important role in modulating transcriptional activity of NF-κB independently of inhibitor of κB (IκB) proteins. For the p65 subunit, multiple phosphorylation sites have been mapped in and adjacent to both the N-terminal Rel homology domain and the C-terminal transactivation domain. Their impact on NF-κB-dependent transcription, however, has never been assessed at a broader level. In this study, we evaluate the importance of differential p65 phosphorylation on four serine acceptor sites in the Rel homology domain for the expression of an array of NF-κB-dependent genes in endothelial cells. We find that inhibition of p65 phosphorylation on these serine residues targets NF-κB activity to distinctive gene subsets in a κB enhancer element-specific context. We show that the phosphorylation-dependent alterations in gene and protein expression are reflective of the amount of p65 and phosphorylated RNA polymerase II (p-RNAP II) bound to respective gene promoter regions. Depending on the gene subset, impaired gene expression was either a result of decreased p65 promoter recruitment or of a failure of bound p65 to recruit p-RNAP II. In conclusion, our findings demonstrate that site-specific p65 phosphorylation targets NF-κB activity to particular gene subsets on a global level by influencing p65 and p-RNAP II promoter recruitment.
Introduction
Gene expression regulation by NF-κB is fundamental for controlling many important cellular processes, including inflammatory and immune responses, cell proliferation, development, and apoptosis (1–3). In resting cells, NF-κB transactivators are sequestered in the cytosol by association with IκB proteins (4). Upon stimulation with pro-inflammatory inducers, IκB proteins are phosphorylated, ubiquitinated, and degraded (5–7). This event constitutes the major step for initiation of NF-κB-dependent transcription as it allows the transcription factor to enter the nucleus where it can bind to regulatory sequences in gene promoters/enhancers. However, it has been shown that this step alone is often not sufficient to initiate gene expression.
Inducible post-translational modification of NF-κB subunits especially by phosphorylation has proven to be equally important for NF-κB to efficiently induce transcription of target genes (8). For the p65 subunit, which constitutes the most potent transcriptional activator of the NF-κB protein family (9), 12 phospho-acceptor sites have been mapped. Five sites (Ser-205, Thr-254, Ser-276, Ser-281, and Ser-311) are located in or adjacent to the N-terminal RHD,2 and seven residues (Thr-435, Ser-468, Thr-505, Ser-529, Ser-535, Ser-536, and Ser-547) are contained in the C-terminal transactivation domain. Dependent on the stimulus and the modification site, phosphorylation of p65 regulates NF-κB transcriptional activity by different mechanisms. Ser-276 and Ser-311 phosphorylation in the RHD is enhancing p65 interaction with the transcriptional co-activator cAMP-response element-binding binding protein, thus activating NF-κB-dependent gene expression (10, 11). Phosphorylation in the transactivation domain at Thr-435, Thr-505, or Ser-547 can either activate or inhibit NF-κB-dependent transcription by impeding or promoting the interaction with histone deacetylase-1 (HDAC1) (12–14). Phosphorylation at Ser-529 and Ser-536 was shown to alter the association with basal components of the transcriptional machinery (15, 16), and Ser-205, Ser-276, Ser-281, and Ser-536 phosphorylation regulates p65 subcellular localization (17–19). Attachment of phospho-groups to Thr-254 and Ser-468 is determining the binding to the suppressor of cytokine signaling 1 (SOCS1) ubiquitin ligase complex thereby regulating p65 protein stability (20, 21).
Whereas degradation of IκB and subsequent nuclear translocation of NF-κB serves as a general step in NF-κB activation that affects expression of all dependent genes, there is strong evidence that the phosphorylation of p65 regulates the activity of NF-κB in a gene-specific context. Several studies have shown that reconstitution of p65-deficient mouse embryonic fibroblasts with p65 S205A, S276A, S281A, T435A, S468A, and S536A mutants leads to reduction of NF-κB transcriptional activity confined to certain genes (12, 20, 22–25). Also, loss of glycogen synthase kinase-3β (GSK-3β), which phosphorylates p65 at Ser-468 (26), has profound effects on the expression of interleukin-6 (IL-6) and chemokine (CC motif) ligand 2 (CCL2) genes, but it only minimally impacts IκBα (NFKBIA) and chemokine (CXC motif) ligand 2 (CXCL2) expression (27). Silencing of NF-κB-dependent transcription by nuclear IκB is dependent on p65 Ser-536 phosphorylation and targets TNF and IL-6 genes but not the interleukin-8 (IL-8) gene (28).
Because existing reports only include a limited amount of NF-κB responsive genes, it is still unclear, however, to what extent differential phosphorylation of NF-κB leads to changes in its transcriptional profile at a global level. In this study, we investigated the effects of impaired p65 serine phosphorylation in and adjacent to the RHD on the expression of an array of NF-κB-dependent genes.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
The mouse endothelial cell line bEND.3 was obtained from ATCC. Mouse embryonic fibroblasts (3T3) isolated from p65−/− mice were a kind gift of Dr. A. Beg (Moffitt Cancer Center, Tampa, FL) and were described previously (29). All cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Cellgro) supplemented with 10% fetal bovine serum (Atlanta Biologicals) and 100 units/ml penicillin G and 100 μg/ml streptomycin B (both Cellgro) in a humidified atmosphere containing 5% CO2. Tumor necrosis factor (TNF; Invitrogen) was used at a final concentration of 10 ng/ml. Stable shRNA cell pools and p65 transfectants were selected in the presence of 1.5 μg/ml puromycin (Sigma) and 250 μg/ml hygromycin B (InvivoGen), respectively.
Plasmid Constructs
Sequence and cloning of murine p65 and luciferase shRNAs in pSiren-RetroX (Clontech) were described elsewhere (30). The bicistronic retroviral vector pLXIH was obtained by exchanging the neomycin resistance cassette of pLXIN (Clontech) with the hygromycin resistance gene. Insertion of human p65 WT, S205A, S276A, S281A, and S311A into pLXIH was carried out by standard cloning procedures.
Gene Knockdown and Stable Transfectants
Endogenous p65 expression was suppressed in bEND.3 by retrovirally delivered shRNA. The resulting bEND.3 p65 knockdown cells as well as 3T3 p65−/− cells were retrovirally reconstituted with p65 WT or p65 phosphorylation-deficient mutants. Retroviral production, cellular infection with retrovirus-containing supernatants, and selection for positive cell pools was performed as described (22).
Immunoblotting and Immunofluorescence
For immunoblotting, cells were lysed in RIPA buffer (50 mm Tris-HCl, pH 8, 150 mm sodium chloride, 1 mm EDTA, 1% Igepal CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 1× protease inhibitors (Roche Applied Sciences)), and equal volumes were mixed with SDS sample buffer, boiled, and analyzed on 10% SDS-polyacrylamide gels. Proteins were transferred to PVDF membranes (Millipore), blocked with 5% milk in TBS, 0.1% Tween 20 (TBST), and incubated with polyclonal anti-p65 (C-20; Santa Cruz Biotechnology) or monoclonal anti-β-actin antibody (AC-74; Sigma). Membranes were washed in TBST and incubated with donkey anti-rabbit or goat anti-mouse secondary antibodies conjugated to horseradish peroxidase (Santa Cruz Biotechnology), and protein bands were visualized with Chemiluminescence Reagent (Santa Cruz Biotechnology) on a Kodak Image Station 2000R. For immunofluorescence, cells were stimulated with TNF for 0, 30, and 60 min and processed as described previously (19). The ratio of cytoplasmic to nuclear p65 localization was determined as described elsewhere (31).
Gene Expression Analysis
Transcriptional profiles of NF-κB-dependent genes were determined by quantitative real time PCR under untreated conditions and after 3 h of TNF stimulation. Genes were compiled after ranking induction levels in seven datasets derived from short term cytokine-induced endothelial cells deposited in the Gene Expression Omnibus database (www.ncbi.nlm.nih.gov). The following datasets were used: GSE973, GSE973, GSE2639, GSE2638, GSE5883, GSE8166, and GSE9647. The first 200 ranked genes were tested for inclusion in two curated gene lists for NF-κB-regulated genes (Dr. T. Gilmore laboratory, Boston University and Bonsai Bioinformatics Software Server, Lille University of Science and Technology, France). Only genes listed in at least one of the databases were considered to be NF-κB-dependent. Hence, mRNA expression levels of 70 potentially NF-κB-regulated genes and two housekeeping genes used for normalization were analyzed by quantitative RT-PCR using SYBR Green chemistry (Fermentas) on a Chromo4 continuous fluorescence monitoring thermocycler (MJ Research) as described previously (22). A complete list of tested genes as well as primer sequences can be found in the supplemental Table 1. Three independent experiments were performed. In the final analysis, only genes were included that were called in all TNF-induced datasets and showed at least 2-fold induction of mRNA levels in p65 WT-reconstituted cells over the untreated or TNF-stimulated empty vector control. A total of 37 genes fulfilled all criteria (see Fig. 2). In 3T3 cells, eight genes were tested for induction of mRNA expression (see supplemental Fig. 1).
FIGURE 2.

Comparison of expression profiles of 37 genes in p65 WT, S205A-, S276A-, S281A-, and S311A-expressing bEND.3 cells after 3 h of TNF stimulation. Stably transduced p65 WT and mutant bEND.3 cells were either left untreated or stimulated with 10 ng/ml TNF for 3 h; RNA was isolated, and the induction of NF-κB-dependent genes was tested by quantitative RT-PCR. Depending on the mRNA expression patterns obtained in p65 WT and mutant cells, the tested genes were subdivided into three groups as outlined in the main text. Arrows indicate the allowable range of variation for a given mRNA/p65 variant combination to assign the gene to the respective groups. Double arrowheads signal that the variation is permitted beyond the indicated levels. Asterisk, either p65 S276A- or S281A-derived mRNA expression levels remain below half of p65 WT mRNA levels. Data are represented as mean ± S.E. (n = 3). All datasets were calculated relative to the mean of unstimulated, pooled controls.
Fluorescence-activated Cell Sorting (FACS) Analysis
Cells were collected in 1× citric saline (135 mm potassium chloride, 15 mm sodium citrate), spun down, and incubated with Alexa Fluor 647 anti-mouse CD54 (ICAM-1), PerCP/Cy5.5 anti-mouse CD106 (VCAM-1), and FITC anti-mouse H-2Kd antibodies (all Biolegend) for 20 min on ice. Antibodies were titrated using mouse splenocytes or bone marrow cells to achieve optimal signal-to-noise ratio. Fluorochrome-matched isotype controls were used to validate detection specificity. Analysis was performed on a six-channel (Accuri C6, BD Biosciences) cytometer. Cells were gated according to their forward and side scatters to eliminate debris and dead cells. Gates were validated by TOPRO-3 and fluorescein-diacetate labeling to identify dead and live cells, respectively. Analysis of median fluorescent intensity was performed on 20,000 live cells.
Chromatin Immunoprecipitation (ChIP)
ChIP analysis was carried out as described (30) with the following variations. bEND.3 cells required 16 cycles of 15-s sonication pulses at 20% amplitude to yield 0.3–1.5-kb DNA fragments. For immunoprecipitation, 2 μg of anti-p65 (C-20; Santa Cruz Biotechnology) and 0.8 μg of anti-p-RNAP II (CTD4H8; Upstate) antibodies were used. Monoclonal anti-p-RNAP II antibody was coupled to 2 μg of rabbit anti-mouse IgG (Jackson ImmunoResearch) before capturing with protein A-Sepharose. A list of primer sequences and targeting κB sites can be found in supplemental Table 2.
RESULTS
Inhibition of p65 Phosphorylation on Different Serines in the RHD Targets NF-κB-dependent Transcription to Distinctive Gene Subsets
The important conclusion that inhibition of p65 phosphorylation at serines 205, 276, 281, and 311 leads to impaired NF-κB activation after TNF stimulation is derived from the expression analysis of a limited number of genes (10, 19, 23). Here, we investigate the dependence of NF-κB-driven gene expression on these p65 phosphorylation sites on a broader level by constitutively knocking down endogenous WT p65 in the murine endothelial cell line bEND.3 and reconstituting the resulting cell pool with a control vector, human p65 WT or S205A, S276A, S281A, and S311A. p65 knockdown as well as equal protein expression of the introduced p65 forms was verified by Western blotting (Fig. 1A). Reconstituted p65 WT followed the characteristic wave of nuclear import and export after TNF stimulation (Fig. 1B), indicating its successful functional integration into the NF-κB pathway. p65 phospho-mutant nuclear translocation after 30 min of TNF stimulation was similar to p65 WT; however, as also seen in fibroblasts (19), basal nuclear p65 levels were higher in S205A-, S276A-, and S281A-expressing cells, and after 60 min of TNF stimulation, p65 S205A, S276A, and S281A were still nuclear, whereas p65 WT and the S311A mutant relocated to the cytoplasm (Fig. 1, B and C). To examine global NF-κB activity in these cells, RNA was prepared, and expression of 37 NF-κB-dependent genes, meeting our selection criteria (see “Experimental Procedures”), was analyzed in resting and 3-h TNF-treated cells. As a general finding, the transcriptional activity of p65 205, 276, and 281 serine to alanine mutants was impaired, whereas p65 S311A was inducing gene expression to equal levels as p65 WT on almost all genes tested (Fig. 2). Depending on whether their expression was or was not affected by p65 phosphorylation deficiency, we subdivided the tested genes into three regulatory groups. The first group includes 11 genes, which failed to be induced by all p65 Ser-205, Ser-276, and Ser-281 mutants. All but one (Cxcl5) of these genes were equally induced by p65 WT and S311A. Group II consists of 13 genes that were efficiently induced by p65 WT, S311A and S205A mutants. Thirteen group III genes were equally induced by all p65 variants. Supporting these results, we found that protein expression of selected NF-κB-dependent genes matched the mRNA levels in p65 WT and mutant cells (Fig. 3). p65 knockdown resulted in substantial but not complete repression of endogenous p65 protein (Fig. 1A). To minimize the probability that any of our observed effects were caused by residual endogenous p65 levels, we compared mRNA expression of representative genes for each regulatory group with those from p65−/− fibroblasts reconstituted with p65 WT and mutants. As shown in supplemental Fig. 1, expression profiles of all genes tested were similar in bEND.3 and 3T3 cells, except for minor variations for Cxcl10 and H2-k1, which could be cell type-specific.
FIGURE 1.

Generation and characterization of wild type and phosphorylation-deficient p65-expressing bEND.3 cells. A, Western blot analysis of p65 expression in bEND.3 cells transduced with luciferase (luc) control and p65-targeting shRNAs (+). c-Myc-tagged human p65 wild type (wt) and amino acids 205, 276, 281, 311 serine to alanine (SA) mutants were stably introduced into successful p65 knockdown cells. The fusion of p65 with N-terminal c-Myc leads to a shift in molecular weight of 10 amino acids reflected by slower migration of human c-Myc-p65 in SDS-PAGE. IB, immunoblot. B, subcellular localization of p65 variants in bEND.3 in the absence or presence of 10 ng/ml TNF for 30 and 60 min. DAPI staining is included as nuclear reference. Bar, 20 μm. C, semi-automated quantification of cytosolic and nuclear p65 immunofluorescence (n = 128–248 cells, derived from three experiments). Nuclear/cytosolic values of >1 indicate predominantly nuclear p65.
FIGURE 3.
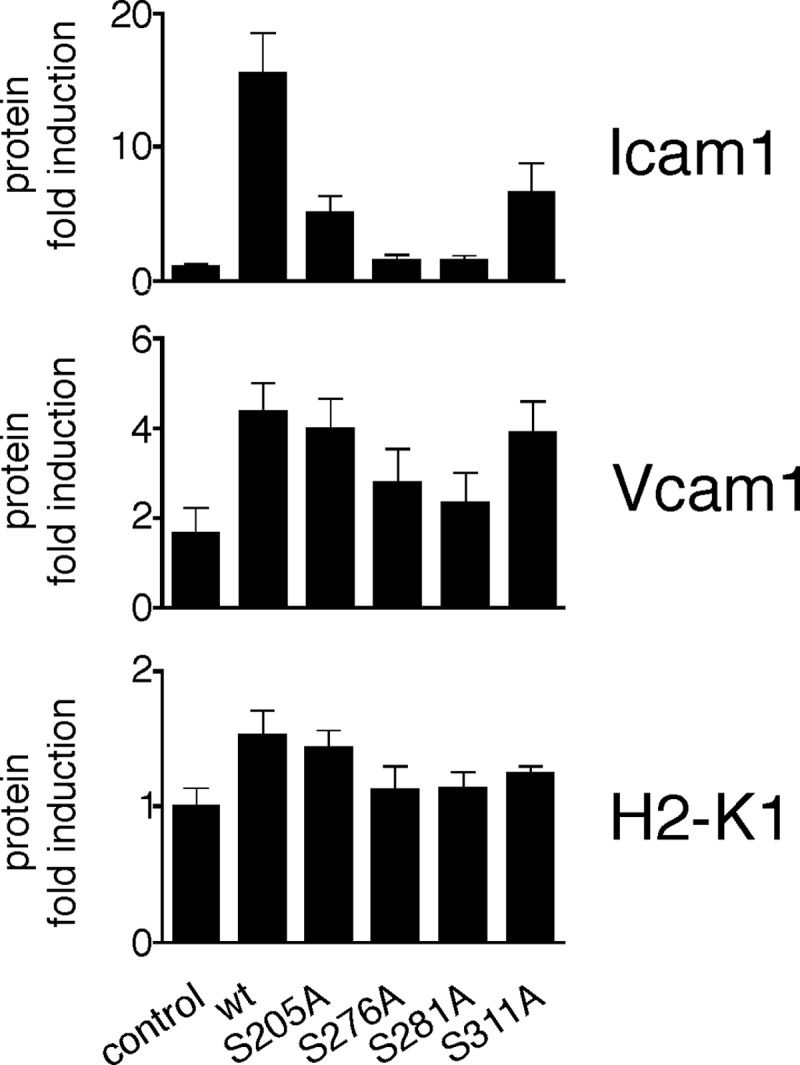
Protein expression levels of three selected genes as observed by FACS analysis after 3 h of TNF treatment. Untreated or 3-h TNF-treated retrovirally transduced p65 WT and mutant bEND.3 cells were processed for FACS analysis as described under “Experimental Procedures.” Data represent mean ± S.E. (n = 3), calculated relative to the mean of unstimulated, pooled controls.
Using isolated κB consensus sites to drive reporter gene expression, we have previously established that p65 phosphorylation-deficient forms are driving transcription in a cis-acting element-specific context (22). Here, we investigated whether the κB site preference of hypo-phosphorylated p65 mutants was also reflected in binding to endogenous gene promoters. Because the location and DNA sequence of functional κB sites has not been established for the majority of genes analyzed in this study, we restricted our analysis to selected genes of each induction group whose κB sites were functionally characterized and compared their annotated DNA sequences (Fig. 4).
FIGURE 4.

Relation of mRNA expression of two selected genes from each group to their promoter κB consensus sequences. The sequences of decameric κB sites for the tested genes (Ccl20 (38), Icam1 (39), Selp (40), Cxcl2 (41), Vcam1 (42), Cxcl10 (43), H2-k1 (44), and Nfkbie (45)) and their position relative to the translational start site are listed. Sequences for each group were subjected to a sequence conservation analysis using the tool WebLogo (46), where the relative height of letters corresponds to the relative frequency of bases at each position.
We found that the group affiliation of genes was largely dictated by the composition of the 5′ half-sites of the κB consensus sequence. The more guanines located in the N terminus of the κB site, the less the expression of the tested gene was susceptible to p65 Ser-to-Ala mutations. Whereas κB-binding sites in group I had preference for thymidine or maximal two guanines at the 5′ end, group II and III consensus sites were characterized by three or more guanine residues. The highest conservation was observed in group III with all but one κB site featuring four to five guanine residues at the 5′ end. In addition, all 5′ half-sites of group III genes were entirely composed of purines, a feature not as prevalent in κB sites of group I and II genes. The 3′ half-site showed higher homology spanning all groups with sites featuring two cytosines at the 3′ end preceded by two pyrimidines. In general, group I κB elements were found to be more diverse, whereas group II sites were similar, differing only in maximal two-base substitutions. Group III showed the highest homology with Cxcl10 site 2 and H2-k1, as well as H2-k1 and Nfkbie κB elements being identical except for a one-base shift.
In summary, we confirm that differential p65 phosphorylation directs NF-κB activity to particular subsets of genes. The transcriptional specificity of NF-κB p65 phospho-mutants is thereby, as suggested previously (22), dictated by the structure of the κB element in the promoter of respective genes.
Impaired p65 Phosphorylation Mutant Gene Expression Is Caused by Defective p65 and p-RNAP II Promoter Recruitment
Next, we investigated whether differential DNA-binding properties could account for differential transcriptional activities of p65 mutants by performing ChIP with a p65-specific antibody from resting, 0.5, and 3 h TNF-stimulated p65 knockdown, WT, or mutant-expressing bEND.3 cells. Maximal p65 binding was achieved after 0.5 h TNF stimulation for all p65 variants regardless of the expression profile of the corresponding gene (Fig. 5, left panel). p65 binding to gene promoters in group I was apparent for p65 WT but was markedly reduced for all p65 mutants (Fig. 5, left panel). This is in concordance with the observed gene expression patterns for p65 Ser-205, Ser-276, and Ser-281 mutants but is surprising for p65 S311A, which showed transcriptional activity comparable with the WT protein. Within group II, p65 S205A, S276A, and S311A were similarly recruited to the promoter as p65 WT, whereas p65 S281A DNA binding was impaired (Fig. 5, left panel). This suggests that reduced expression of group II genes in p65 S276A cells is not solely mediated by reduced p65 DNA binding, while the inability of the S281A mutant to be efficiently recruited to the promoter correlated well with its reduced transcriptional activity. In group III, promoter binding profiles of all p65 proteins were equal (Fig. 5, left panel), reflecting the observed mRNA expression patterns of genes belonging to this group.
FIGURE 5.

NF-κB p65 and p-RNAP II promoter binding. ChIP analysis was carried out by RT-PCR in p65 WT and mutant-expressing bEND.3 cells left untreated or treated with 10 ng/ml TNF for 0.5 and 3 h. Chromatin was isolated, and DNA was immunoprecipitated with p65 and p-RNAP II antibodies. Data are represented as mean ± S.E. (n = 3).
Because mRNA expression patterns of some genes could not be explained by p65 DNA binding properties alone, we next examined the levels of promoter-bound p-RNAP II, thought to be indicative for transcriptionally active genes (Fig. 5, right panel) (32). Similarly to p65, p-RNAP II reached maximum binding at 0.5 h after TNF addition on most promoters, but recruitment was sustained at Vcam1, Cxcl10, H2-k1, and Nfkbie promoters. We found that the p65 Ser-311 mutant was more efficiently recruiting p-RNAP II to promoters of genes in group I than the other mutants, which could account for the high mRNA induction levels despite lower p65 binding. Other than that, p-RNAP II binding profiles matched the ones for p65 in groups I and III; however, in group II this was not the case. Binding of p-RNAP II to group II promoters was especially impaired with p65 Ser-276 mutants despite the presence of p65, providing a possible explanation for the deficit in transcription of group II genes in S276A cells. In summary, we find that inhibition of p65 phosphorylation influences NF-κB-dependent gene expression by altering p65 and in some cases also p-RNAP II promoter recruitment.
p65 Mutant-dependent Impairment of Groups I and II Gene Expression Is Caused by Different p65 and p-RNAP II Promoter Binding Dynamics
We calculated the ratio of promoter bound p-RNAP II to bound p65 WT and mutants for each regulatory group. Thereby we found that in groups I and III relative p-RNAP II to p65 ratios were equal for all p65 proteins (Fig. 6), indicating that p65 completely dictates the binding of p-RNAP II to cognate DNA sequences in these groups. Therefore, the reduced expression of genes in group I is a result of low p65 DNA binding capability of Ser to Ala mutants, which subsequently leads to less recruitment of p-RNAP II. Similarly, all p65 mutants are tethered to group III promoters as efficiently as WT p65 leading to efficient p-RNAP II recruitment and gene transcription. In contrast, in group II, ratios of bound p-RNAP II compared with p65 were different for p65 WT and mutants (Fig. 6). In more detail, p-RNAP II to p65 binding ratios were highest for p65 WT, followed by p65 S311A, S205A, S276A, and S281A. This suggests that whereas p65 mutants are able to bind to gene promoters of this group, they fail to efficiently recruit p-RNAP II. As reflected by the higher p-RNAP II to p65 ratios, more p-RNAP II is required to initiate transcription in this group in comparison to the others. In conclusion, p65 mutant transcriptional impairment is dependent on the regulatory group either caused by deficient p65 promoter binding alone or by a combination of reduced p65 and p-RNAP II DNA binding.
FIGURE 6.

p-RNAP II promoter binding expressed as ratio to p65 promoter binding. ChIP data obtained from Ccl20, Icam1, Selp, Cxcl2, Vcam1, Cxcl10, H2-k1, and Nfkbie promoter sites were used to determine the ratio of promoter-bound p-RNAP II to p65 in the three regulatory groups. Higher y values signify relatively more bound p-RNAP II than p65.
DISCUSSION
Inducible phosphorylation of the NF-κB p65 subunit plays an important role in shaping the potential of nuclear NF-κB to drive transcription. However, it has not been clearly determined what gene subsets are regulated by different phosphorylation events. Thus, in this study we examined the consequences of impaired p65 phosphorylation at previously identified phosphorylated serines 205, 276, 281, and 311 on the expression of a larger group of NF-κB-dependent genes.
We found that inhibition of p65 phosphorylation at Ser-205, Ser-276, and Ser-281 severely impaired the transcription of many tested genes after TNF stimulation (Fig. 2), establishing the importance of these functional sites for enabling pro-inflammatory responses. Our finding is in line with earlier observations where fibroblasts isolated from p65 S276A knock-in mice displayed a significant reduction of NF-κB-dependent transcription after TNF addition (23). At the same time, we showed previously that mutation of p65 at Ser-205, Ser-276, and Ser-281 phospho-acceptor sites impairs gene expression after LPS/IFNγ stimulation (22). In contrast to the clearly reduced transcriptional activity of p65 mutants carrying alanine substitutions at positions 205, 276, and 281, inhibition of p65 phosphorylation at position 311 only led to a minor reduction of mRNA levels of some NF-κB target genes. In support of our data, Ser-311 phosphorylation was not essential for p65 transcriptional activity in an NF-κB-dependent reporter assay (22), and so far decreased mRNA expression as a result of p65 S311A mutation was only reported for the Il-6 gene (10). Taken together with data from this study, where solely Cxcl5 mRNA expression was decreased by more than 50% in p65 S311A cells, it seems possible that Ser-311 phosphorylation is only needed for transcription of a minority of NF-κB-dependent genes.
NF-κB transcriptional activity is regulated at multiple levels, and phosphorylation of the NF-κB dimer may influence every stage of the activation cascade. First, stimulus-induced degradation of IκB proteins leads to nuclear import of active NF-κB dimers. In Drosophila, it has been shown that this step is supported by phosphorylation of the p65-homolog Dorsal involving Ser-317 (corresponding to mammalian p65 Ser-281) (18); however, we could not detect such a function in mammalian cells. Despite the tendencies of cells expressing S205A, S276A, and S281A to have higher basal nuclear p65 levels, nuclear translocation after 30 min of TNF exposure was not different from p65 WT and S311A-expressing cell pools. Whereas p65 WT and S311A followed the classical wave of nuclear import and export after TNF stimulation, p65 S205A, S276A, and S281A were retained in the nucleus (Fig. 1, B and C). This defect was attributed earlier to lower IκBα levels in these cells due to insufficient Nfkbia synthesis (19). In contrast to Nfkbia, we show here that Nfkbie expression is not dependent on p65 phosphorylation status, providing a possible explanation why TNF inducibility is maintained in phosphoserine mutant cells despite the lack of IκBα. The nuclear persistence of p65 mutants did not result in transcriptional activation of the majority of target genes (Fig. 2), further confirming that NF-κB nuclear localization alone is insufficient to initiate the transcriptional program.
Second, dimer composition can dictate the preference of a homo- or heterodimer to bind certain cis-elements (33). Although not addressed in this study, we have previously shown that the composition of NF-κB complexes was not influenced by the absence of RHD phospho-acceptor sites when p65 was expressed in fibroblasts (22). It is therefore unlikely that differences in NF-κB dimer formation could account for the observed changes in gene transcription.
Third, NF-κB DNA binding itself and the associated recruitment of components of the basal transcriptional machinery as well as of co-activators to gene promoters are prerequisites for efficient target gene expression. As suggested by earlier studies, we found that irrespective of the location of the phospho-site, the effects of phosphorylation deficiency on NF-κB-controlled transcription are gene-dependent. This implies that p65 serine phosphorylation in the RHD exerts its effect on gene regulation downstream of IκBα degradation and nuclear import, potentially by directly interfering with promoter-associated transcriptional events. This is also supported by the fact that LPS/IFNγ and TNF stimulation result in the same regulatory pattern of gene expression (22, 23). Indeed, we found that the altered transcriptional potential of p65 phospho-mutants is a consequence of impaired p65 and p-RNAP II promoter binding to specific gene promoter regions, dictated by the nature of the κB enhancer element (Figs. 4 and 5). It has been shown earlier that phosphorylation of p65 is favorable for its DNA binding, as treatment with phosphatases strongly reduced TNF-induced NF-κB DNA binding activity, whereas addition of protein kinase A (PKA) enhanced its promoter association (34). In contrast, inhibition of p65 phosphorylation by dominant negative protein kinase C resulted in loss of NF-κB transcriptional activity without interfering with its DNA binding (35). Conflicting observations have also been reported for specific phospho-acceptor sites. Although inhibition of PKA-induced recombinant p65 phosphorylation at Ser-276 decreased NF-κB DNA binding affinity in vitro (11), kinase inhibition affecting Ser-276 phosphorylation did not alter NF-κB DNA binding as tested by EMSA in another study (36). Comparably, p65 isolated from TNF- or LPS-stimulated S276A knock-in cells had a similar DNA binding profile as p65 WT (23). While p65 S205A and S311A exhibited unchanged DNA binding capacity when compared with p65 WT, blockage of p65 Ser-281 phosphorylation did have a negative impact on NF-κB DNA complex formation in nuclear extracts obtained from fibroblasts (10, 22). Therefore, it seems dependent on the phospho-acceptor site and the gene studied whether p65 phosphorylation contributes to its DNA binding. Indeed, dependent on the gene induction group, we found differences in the p65 mutant DNA binding activity. Whereas p65 mutants were unable to bind κB consensus sites of group I genes, they were readily tethered to promoters of group II genes (Fig. 5), indicating that the transcriptional deficit in group II was caused by an inability of bound p65 to efficiently recruit phosphorylated RNAP II, rather than by p65 mutant DNA-binding deficiency. Also, we show that the expression of approximately one-third of all genes tested is unaffected by differential p65 phosphorylation (Fig. 2, group III), again suggesting that phosphorylation of p65 is not necessary for NF-κB DNA binding to all gene promoters.
The molecular mechanism for the promoter-specific NF-κB activity of differentially phosphorylated p65 remains elusive. Recently, we found that p65 S205A, S276A, and S281A phospho-mutants are, in contrast to p65 WT and S311A, highly ubiquitinated and that nuclear localization of these mutants is a prerequisite for ubiquitination (31). Importantly, ubiquitin permanently attached to the N-terminal end of p65 down-regulated NF-κB transcriptional activity in a gene-specific fashion similar to p65 hypo-phosphorylation (31). Although the indication is only correlative, this may suggest that ubiquitination is involved in the processes leading to p65 phospho-mutant transcriptional deficiency. We have shown earlier that the gene-specific effects of p65 mutants also apply to linear isolated κB sequences (22), which do not resemble the complex nature of endogenous gene promoters; hence, it is unlikely that histone-related promoter remodeling or interaction with other transcription factor-binding sites is involved. Indeed, from our data shown here, it seems certain that p65 hypo-phosphorylation results in NF-κB transcriptional impairment by altering DNA binding or p-RNAP II recruitment depending on the DNA sequence of the binding site. The findings are supportive of the important role of the κB DNA consensus sequence not only in recruiting NF-κB but also in the build up of a productive transcriptional initiation complex as shown previously (37).
Taken together, our study underlines the importance of site-specific p65 phosphorylation in the RHD for regulation of pro-inflammatory gene expression. Unlike its association with IκB proteins, which equally affects the expression of all NF-κB-dependent genes, differential p65 phosphorylation exerts its effects in a gene-specific manner, by having profound effects on the ability of NF-κB to bind to some κB enhancer elements but not to others.
Acknowledgment
We thank Dr. Amer A. Beg for providing us with p65−/− 3T3 fibroblasts.
This work was supported, in whole or in part, by National Institutes of Health Grant HL077308 (to J. A.). This work was also supported by American Heart Association Scientist Development Grant 10SDG2600298 (to K. H.).

This article contains supplemental Fig. 1 and Tables 1 and 2.
- RHD
- Rel homology domain
- p-RNAP II
- phosphorylated RNA polymerase II.
REFERENCES
- 1. Barkett M., Gilmore T. D. (1999) Control of apoptosis by Rel/NF-κB transcription factors. Oncogene 18, 6910–6924 [DOI] [PubMed] [Google Scholar]
- 2. Hayden M. S., West A. P., Ghosh S. (2006) NF-κB and the immune response. Oncogene 25, 6758–6780 [DOI] [PubMed] [Google Scholar]
- 3. Karin M., Lin A. (2002) NF-κB at the crossroads of life and death. Nat. Immunol. 3, 221–227 [DOI] [PubMed] [Google Scholar]
- 4. Baeuerle P. A., Baltimore D. (1988) IκB. A specific inhibitor of the NF-κB transcription factor. Science 242, 540–546 [DOI] [PubMed] [Google Scholar]
- 5. Henkel T., Machleidt T., Alkalay I., Krönke M., Ben-Neriah Y., Baeuerle P. A. (1993) Rapid proteolysis of IκB-α is necessary for activation of transcription factor NF-κB. Nature 365, 182–185 [DOI] [PubMed] [Google Scholar]
- 6. Spencer E., Jiang J., Chen Z. J. (1999) Signal-induced ubiquitination of IκBα by the F-box protein Slimb/β-TrCP. Genes Dev. 13, 284–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Traenckner E. B., Pahl H. L., Henkel T., Schmidt K. N., Wilk S., Baeuerle P. A. (1995) Phosphorylation of human IκB-α on serines 32 and 36 controls IκB-α proteolysis and NF-κB activation in response to diverse stimuli. EMBO J. 14, 2876–2883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang B., Yang X. D., Lamb A., Chen L. F. (2010) Post-translational modifications of NF-κB: another layer of regulation for NF-κB signaling pathway. Cell. Signal. 22, 1282–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schmitz M. L., Baeuerle P. A. (1991) The p65 subunit is responsible for the strong transcription activating potential of NF-κB. EMBO J. 10, 3805–3817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Duran A., Diaz-Meco M. T., Moscat J. (2003) Essential role of RelA Ser-311 phosphorylation by ζPKC in NF-κB transcriptional activation. EMBO J. 22, 3910–3918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhong H., Voll R. E., Ghosh S. (1998) Phosphorylation of NF-κB p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell 1, 661–671 [DOI] [PubMed] [Google Scholar]
- 12. O'Shea J. M., Perkins N. D. (2010) Thr-435 phosphorylation regulates RelA (p65) NF-κB subunit transactivation. Biochem. J. 426, 345–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rocha S., Garrett M. D., Campbell K. J., Schumm K., Perkins N. D. (2005) Regulation of NF-κB and p53 through activation of ATR and Chk1 by the ARF tumour suppressor. EMBO J. 24, 1157–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sabatel H., Di Valentin E., Gloire G., Dequiedt F., Piette J., Habraken Y. (2012) Phosphorylation of p65(RelA) on Ser(547) by ATM represses NF-κB-dependent transcription of specific genes after genotoxic stress. PLoS ONE 7, e38246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Buss H., Dörrie A., Schmitz M. L., Hoffmann E., Resch K., Kracht M. (2004) Constitutive and interleukin-1-inducible phosphorylation of p65 NF-κB at serine 536 is mediated by multiple protein kinases including IκB kinase (IKK)-α, IKKβ, IKKϵ, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J. Biol. Chem. 279, 55633–55643 [DOI] [PubMed] [Google Scholar]
- 16. Wang D., Westerheide S. D., Hanson J. L., Baldwin A. S., Jr. (2000) Tumor necrosis factor α-induced phosphorylation of RelA/p65 on Ser-529 is controlled by casein kinase II. J. Biol. Chem. 275, 32592–32597 [DOI] [PubMed] [Google Scholar]
- 17. Bohuslav J., Chen L. F., Kwon H., Mu Y., Greene W. C. (2004) p53 induces NF-κB activation by an IκB kinase-independent mechanism involving phosphorylation of p65 by ribosomal S6 kinase 1. J. Biol. Chem. 279, 26115–26125 [DOI] [PubMed] [Google Scholar]
- 18. Drier E. A., Huang L. H., Steward R. (1999) Nuclear import of the Drosophila Rel protein Dorsal is regulated by phosphorylation. Genes Dev. 13, 556–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hochrainer K., Racchumi G., Anrather J. (2007) Hypo-phosphorylation leads to nuclear retention of NF-κB p65 due to impaired IκBα gene synthesis. FEBS Lett. 581, 5493–5499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Geng H., Wittwer T., Dittrich-Breiholz O., Kracht M., Schmitz M. L. (2009) Phosphorylation of NF-κB p65 at Ser-468 controls its COMMD1-dependent ubiquitination and target gene-specific proteasomal elimination. EMBO Rep. 10, 381–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ryo A., Suizu F., Yoshida Y., Perrem K., Liou Y. C., Wulf G., Rottapel R., Yamaoka S., Lu K. P. (2003) Regulation of NF-κB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol. Cell 12, 1413–1426 [DOI] [PubMed] [Google Scholar]
- 22. Anrather J., Racchumi G., Iadecola C. (2005) cis-Acting, element-specific transcriptional activity of differentially phosphorylated nuclear factor-κB. J. Biol. Chem. 280, 244–252 [DOI] [PubMed] [Google Scholar]
- 23. Dong J., Jimi E., Zhong H., Hayden M. S., Ghosh S. (2008) Repression of gene expression by unphosphorylated NF-κB p65 through epigenetic mechanisms. Genes Dev. 22, 1159–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moreno R., Sobotzik J. M., Schultz C., Schmitz M. L. (2010) Specification of the NF-κB transcriptional response by p65 phosphorylation and TNF-induced nuclear translocation of IKKϵ. Nucleic Acids Res. 38, 6029–6044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nowak D. E., Tian B., Jamaluddin M., Boldogh I., Vergara L. A., Choudhary S., Brasier A. R. (2008) RelA Ser-276 phosphorylation is required for activation of a subset of NF-κB-dependent genes by recruiting cyclin-dependent kinase 9/cyclin T1 complexes. Mol. Cell. Biol. 28, 3623–3638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Buss H., Dörrie A., Schmitz M. L., Frank R., Livingstone M., Resch K., Kracht M. (2004) Phosphorylation of serine 468 by GSK-3β negatively regulates basal p65 NF-κB activity. J. Biol. Chem. 279, 49571–49574 [DOI] [PubMed] [Google Scholar]
- 27. Steinbrecher K. A., Wilson W., 3rd, Cogswell P. C., Baldwin A. S. (2005) Glycogen synthase kinase 3β functions to specify gene-specific, NF-κB-dependent transcription. Mol. Cell. Biol. 25, 8444–8455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ghosh C. C., Ramaswami S., Juvekar A., Vu H. Y., Galdieri L., Davidson D., Vancurova I. (2010) Gene-specific repression of proinflammatory cytokines in stimulated human macrophages by nuclear IκBα. J. Immunol. 185, 3685–3693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Beg A. A., Sha W. C., Bronson R. T., Ghosh S., Baltimore D. (1995) Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature 376, 167–170 [DOI] [PubMed] [Google Scholar]
- 30. Anrather J., Racchumi G., Iadecola C. (2006) NF-κB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J. Biol. Chem. 281, 5657–5667 [DOI] [PubMed] [Google Scholar]
- 31. Hochrainer K., Racchumi G., Zhang S., Iadecola C., Anrather J. (2012) Monoubiquitination of nuclear RelA negatively regulates NF-κB activity independent of proteasomal degradation. Cell. Mol. Life Sci. 69, 2057–2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. O'Brien T., Hardin S., Greenleaf A., Lis J. T. (1994) Phosphorylation of RNA polymerase II C-terminal domain and transcriptional elongation. Nature 370, 75–77 [DOI] [PubMed] [Google Scholar]
- 33. Kunsch C., Ruben S. M., Rosen C. A. (1992) Selection of optimal κB/Rel DNA-binding motifs. Interaction of both subunits of NF-κB with DNA is required for transcriptional activation. Mol. Cell. Biol. 12, 4412–4421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Naumann M., Scheidereit C. (1994) Activation of NF-κB in vivo is regulated by multiple phosphorylations. EMBO J. 13, 4597–4607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Anrather J., Csizmadia V., Soares M. P., Winkler H. (1999) Regulation of NF-κB RelA phosphorylation and transcriptional activity by p21(ras) and protein kinase Cζ in primary endothelial cells. J. Biol. Chem. 274, 13594–13603 [DOI] [PubMed] [Google Scholar]
- 36. Vermeulen L., De Wilde G., Van Damme P., Vanden Berghe W., Haegeman G. (2003) Transcriptional activation of the NF-κB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1). EMBO J. 22, 1313–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Leung T. H., Hoffmann A., Baltimore D. (2004) One nucleotide in a κB site can determine cofactor specificity for NF-κB dimers. Cell 118, 453–464 [DOI] [PubMed] [Google Scholar]
- 38. Harant H., Eldershaw S. A., Lindley I. J. (2001) Human macrophage inflammatory protein-3α/CCL20/LARC/Exodus/SCYA20 is transcriptionally up-regulated by tumor necrosis factor-α via a nonstandard NF-κB site. FEBS Lett. 509, 439–445 [DOI] [PubMed] [Google Scholar]
- 39. Tanaka Y., Hayashi M., Takagi S., Yoshie O. (1996) Differential transactivation of the intercellular adhesion molecule 1 gene promoter by Tax1 and Tax2 of human T-cell leukemia viruses. J. Virol. 70, 8508–8517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pan J., Xia L., Yao L., McEver R. P. (1998) Tumor necrosis factor-α- or lipopolysaccharide-induced expression of the murine P-selectin gene in endothelial cells involves novel κB sites and a variant activating transcription factor/cAMP response element. J. Biol. Chem. 273, 10068–10077 [DOI] [PubMed] [Google Scholar]
- 41. Widmer U., Manogue K. R., Cerami A., Sherry B. (1993) Genomic cloning and promoter analysis of macrophage inflammatory protein (MIP)-2, MIP-1α, and MIP-1β, members of the chemokine superfamily of proinflammatory cytokines. J. Immunol. 150, 4996–5012 [PubMed] [Google Scholar]
- 42. Iademarco M. F., McQuillan J. J., Rosen G. D., Dean D. C. (1992) Characterization of the promoter for vascular cell adhesion molecule-1 (VCAM-1). J. Biol. Chem. 267, 16323–16329 [PubMed] [Google Scholar]
- 43. Ohmori Y., Hamilton T. A. (1993) Cooperative interaction between interferon (IFN) stimulus response element and κB sequence motifs controls IFNγ- and lipopolysaccharide-stimulated transcription from the murine IP-10 promoter. J. Biol. Chem. 268, 6677–6688 [PubMed] [Google Scholar]
- 44. Baldwin A. S., Jr., Sharp P. A. (1988) Two transcription factors, NF-κB and H2TF1, interact with a single regulatory sequence in the class I major histocompatibility complex promoter. Proc. Natl. Acad. Sci. U.S.A. 85, 723–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tian B., Nowak D. E., Jamaluddin M., Wang S., Brasier A. R. (2005) Identification of direct genomic targets downstream of the nuclear factor-κB transcription factor mediating tumor necrosis factor signaling. J. Biol. Chem. 280, 17435–17448 [DOI] [PubMed] [Google Scholar]
- 46. Crooks G. E., Hon G., Chandonia J. M., Brenner S. E. (2004) WebLogo. A sequence logo generator. Genome Res. 14, 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]