

Background: The anaerobic biosynthesis of cobalamin involves contraction of a cobalt-containing porphinoid.
Results: The contractase, CbiH60, contains a [4Fe-4S] center and catalyzes the conversion of cobalt factor III into cobalt-precorrin-4 in the presence of SAM and DTT.
Conclusion: Ring contraction requires cobalt-precorrin-3 as a substrate.
Significance: The high yield synthesis of cobalt-precorrin-4 will allow for further characterization of this pathway.
Keywords: Biosynthesis, Electron Paramagnetic Resonance (EPR), NMR, S-Adenosylmethionine (SAM), Site-directed Mutagenesis, CbiH60, Fe-S Cluster
Abstract
The anaerobic pathway for the biosynthesis of cobalamin (vitamin B12) has remained poorly characterized because of the sensitivity of the pathway intermediates to oxygen and the low activity of enzymes. One of the major bottlenecks in the anaerobic pathway is the ring contraction step, which has not been observed previously with a purified enzyme system. The Gram-positive aerobic bacterium Bacillus megaterium has a complete anaerobic pathway that contains an unusual ring contraction enzyme, CbiH60, that harbors a C-terminal extension with sequence similarity to the nitrite/sulfite reductase family. To improve solubility, the enzyme was homologously produced in the host B. megaterium DSM319. CbiH60 was characterized by electron paramagnetic resonance and shown to contain a [4Fe-4S] center. Assays with purified recombinant CbiH60 demonstrate that the enzyme converts both cobalt-precorrin-3 and cobalt factor III into the ring-contracted product cobalt-precorrin-4 in high yields, with the latter transformation dependent upon DTT and an intact Fe-S center. Furthermore, the ring contraction process was shown not to involve a change in the oxidation state of the central cobalt ion of the macrocycle.
Introduction
Cobalamin, the biological form of vitamin B12, the anti-pernicious anemia factor (1), is a modified tetrapyrrole that belongs to the same family of compounds as hemes, chlorophylls, heme d1, coenzyme F430, and siroheme (2). The biosynthesis of the coenzyme form of vitamin B12, adenosylcobalamin, requires approximately 30 enzymatic steps, making the pathway one of the largest found in nature. Like all other modified tetrapyrroles, cobalamin is derived from a common precursor, uroporphyrinogen III, which is synthesized from 5-aminolevulinic acid in three highly conserved steps (3). Cobalamin differs from other modified tetrapyrroles in that its macrocycle is reduced in size because of the loss of the methylene bridge between pyrrole rings A and D (4, 5). This ring contraction is necessary to ensure the tight coordination of the central cobalt ion, which is found at the epicenter of the corrin structure.
There are two genetically distinct routes for adenosylcobalamin synthesis that are referred to as the aerobic and anaerobic pathways (6). The earlier intermediates of the cobalamin pathway are given the term precorrin-n, where n refers to the number of methyl groups that have been added to the developing corrin frame. Along the early steps of the anaerobic pathway, however, some of the intermediates were found to be more oxidized and are termed factors (5, 7–9), where, for instance, factor II would represent the oxidized version of precorrin-2. Overall, the two pathways for cobalamin synthesis are quite similar because the sequences in which the modifications that take place during corrin-ring synthesis, such as the addition of the peripheral methyl groups and amidations, are identical (6). Thus, overall, many of the biosynthetic enzymes along the two pathways share a degree of similarity and are clearly orthologous.
The major differences between the pathways relate to the timing of cobalt insertion, where cobalt is added at an early stage in the anaerobic pathway, and the mechanism of ring contraction, where molecular oxygen is required in the aerobic pathway (4, 10). Indeed, in comparison with the aerobic pathway, much less is known about the anaerobic pathway for corrin synthesis (11). This report deals with the enzyme associated with anaerobic ring contraction (CbiH) and addresses questions about the oxidation state of both the substrate and the product.
For the anaerobic biosynthesis of cobalamin, uroporphyrinogen III is initially methylated at positions C2 and C7 to produce precorrin-2, which then undergoes a NAD+-dependent dehydrogenation to yield factor II (sirohydrochlorin) (12). This isobacteriochlorin acts as the substrate for cobalt insertion by the cobaltochelatase CbiX (or in some organisms CbiK) to give cobalt factor II (8, 13). Methylation of cobalt factor II at C20 by CbiL produces cobalt factor III and this appears to be the substrate for the ring contraction step (Fig. 1) (9, 14). Here, CbiH methylates at C17 and also mediates the formation of a δ-lactone on ring A, thereby promoting contraction of the macrocycle. The product of the reaction is thought to be cobalt factor IV. However, this reaction is also known to proceed with cobalt-precorrin 3 as a substrate, with cobalt-precorrin 4 as the product (5, 15). The low yield of the overall reaction, typically below 5%, has meant that the reaction has remained poorly characterized and has not been observed with a purified enzyme system. It has been suggested that the central cobalt ion of the intermediate may play a role in the catalytic mechanism underpinning ring contraction (5). The poor yield of either cobalt-precorrin-4 or cobalt factor IV has also meant that the remaining steps in the anaerobic synthesis of the corrin ring have not been fully elucidated.
FIGURE 1.

The anaerobic ring contraction step in relation to the structure of adenosylcobalamin. The ring contraction step is mediated by CbiH60 in B. megaterium. The pathway is thought to involve either the transformation of cobalt-precorrin-3 into cobalt-precorrin-4 or the oxidized versions of these intermediates (cobalt factor III and cobalt factor IV). The numbering system used for the corrin ring is shown with adenosylcobalamin.
In this paper we report on the characterization of CbiH60 from Bacillus megaterium, an organism that has been previously used as a commercial producer of cobalamin. CbiH enzymes are generally composed of ∼250 amino acid residues (∼30 kDa) and display sequence similarity to the class III methyltransferases found in both aerobic and anaerobic vitamin B12 pathways (6, 16). However, the CbiH60 from B. megaterium is a protein of 540 amino acids in length with a predicted molecular mass of 60 kDa. The N-terminal regions of CbiH60 (amino acids 1–247) houses the ring contraction activity, whereas the C-terminal region displays similarity to part of the nitrite and sulfite reductase family of proteins. The nitrite and sulfite reductase domain has the closest similarity to the nitrite reductase, NirB, from Paenibacillus spp. with 30% identity and 55% similarity. This region harbors four conserved cysteine residues, 402CXXC405 and 439CXXXC443. The function of the nitrite and sulfite reductase-like domain remains uncharacterized, although it has been considered nonessential for cobalamin biosynthesis (17) because deletion of this region does not appear to prevent cobalamin biosynthesis. Herein, we report an efficient method for the homologous production of large quantities of CbiH60 and show that the enzyme contains a [4Fe-4S] center. Moreover, we demonstrate that the enzyme requires reducing equivalents for the quantitative transformation of cobalt factor III into cobalt-precorrin-4.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
Most chemicals were purchased from Sigma. Other materials were provided by the following suppliers: restriction and modification enzymes purchased from Promega and New England Biolabs; molecular biology kits from Qiagen (QIAprep® Miniprep kit and QIAquick® gel extraction kit); chelating-Sepharose fast flow resin and PD10 columns from GE Healthcare; tryptone and yeast extract from Oxoid; primers from Fisher Scientific; and QuikChange II site-directed mutagenesis kit from Stratagene. Sequencing analysis was performed by GATC Biotech (Germany and UK).
Cloning of cbiH60
B. megaterium DSM509 cbiH60 was PCR-amplified from genomic DNA with the 5′ primer designed to incorporate a BglII restriction site (in bold type) (CATAGATCTGGAAAGGTAAACTGTTAGTTATTGG). The 3′ primer contained an EagI restriction site (in bold type) (CATCGGCCGATCTCCGCACGGAGCTG), with the stop codon removed to allow a fusion with a C-terminal His6 tag. The PCR fragment and pC-His1622 plasmid were digested with BglII and EagI, followed by purification, ligation, and transformation of Escherichia coli DH10B, with 100 μg/ml of ampicillin for selection. For plasmid list, please see Supplemental Table S1.
Mutagenesis of the B. megaterium CbiH60
The following point mutations were introduced into the pC-His1622-cbiH60 plasmid to allow conserved cysteine amino acids to be changed into alanine at positions C402A and C443A (Supplemental Table S2), following the instructions of the QuikChange II site-directed mutagenesis kit from Stratagene. All of the mutations were verified through DNA sequencing (GATC Biotech).
Overproduction and Purification of B. megaterium CbiH60 and C402A,C443A Mutants
After protoplast transformation (18) of B. megaterium DSM319, recombinant strains were grown in 1 liter of LB containing 10 μg ml−1 tetracycline at 30 °C. Upon reaching an A578 of 0.3, protein production was induced with 0.25% (w/v) xylose and left to grow overnight at 30 °C. The cells were collected by centrifugation (5000 rpm, 4 °C, 15 min) and resuspended in 15 ml of binding buffer (20 mm Tris-HCl, pH 8.0, 500 mm NaCl, 5 mm imidazole). The cells were lyzed by sonication, followed by centrifugation at 17,500 rpm, 4 °C, 20 min. Purification of CbiH60 and all subsequent incubations were carried out in an anaerobic glovebox (Belle Technology, Weymouth, UK) with <2 ppm oxygen. Recombinant CbiH60 was purified using metal affinity chromatography by washing with 20 mm Tris-HCl, pH 8.0, 500 mm NaCl containing increasing concentrations of imidazole (5, 30, 70, and 400 mm). The purified protein was buffer exchanged into 20 mm Tris-HCl, pH 8.0, and 400 mm NaCl using a PD-10 desalting column.
Preparation of Cobalt-Precorrin-3 and Cobalt Factor III
The plasmid pETcoco-2-cobA-hemB-hemC-hemD-sirC-cbiX-cbiL was provided by Dr. Stefanie Frank. This was modified from pETcoco-2-ABCDC, which was originally constructed by Dr. Evelyne Deery (9). In addition, it contains the cbiXS and cbiL from Methanobacter thermoautotrophicus, fused to N-terminal His6 tags. After production of the proteins in E. coli, the His6-tagged proteins were purified and transferred to the glovebox, and buffer was exchanged as described previously (9). These proteins were then incubated with 5-aminolevulinic acid (2 mg), SAM2 (10 mg), NAD+(2 mg), and cobalt (500 μg), at a temperature of 37 °C. To prevent protein precipitation, cobalt was gradually titrated as the reaction proceeded. For the synthesis of cobalt-precorrin-3, the same method was applied, but instead NAD+ was omitted. Because of instability, cobalt-precorrin-3 had to be freshly prepared and used within 24 h.
Mass Spectrometry
Tetrapyrroles were injected onto an Ace 5 AQ column (2.1 × 150 mm, 5 μm, Advanced Chromatography Technologies) that was attached to an Agilent 1100 series HPLC coupled to a micrOTOF-Q (Bruker) mass spectrometer and equipped with online diode array, run at a flow rate of 0.2 ml min−1. Tetrapyrroles were routinely separated with a linear gradient of acetonitrile and 0.1% (v/v) trifluoroacetic acid. Mass spectra were obtained using an Agilent 1100 liquid chromatography system connected to a Bruker micrOTOF II MS, using electrospray ionization in the positive mode.
EPR
Samples were prepared and then flash frozen in liquid nitrogen. EPR experiments were performed on a Bruker ELEXSYS E500 spectrometer operating at X-band, employing a Super High Q cylindrical cavity (Q factor = ∼16,000) equipped with an Oxford Instruments ESR900 liquid helium cryostat linked to an ITC503 temperature controller. The experimental parameters are given in the captions to the relevant figures.
NMR
The sample for NMR analysis of purified cyano-cobalt(III) factor IV was prepared as follows. An excess of the purified CbiH60 (100 μm) was incubated with 50 μm cobalt(II) factor III, 1 mm SAM, 10 mm DTT in Buffer L (20 mm HEPES, pH 7.5, 100 mm NaCl) in a total volume of 100 ml. After overnight incubation at 37 °C, the reaction was stopped by heating to 65 °C for 30 min to precipitate CbiH60, which was subsequently removed by centrifugation (4000 rpm, 10 min). The product, cobalt(II)-precorrin-4 was then bound to a DEAE column, washed, and eluted at 400 mm NaCl. The pH was adjusted to 4.0 with 1 m acetic acid before applying to a LiChroprep RP18 column equilibrated with water (adjusted to pH 4.0). The pigment was then briefly washed with water and 10% ethanol and eluted in 50% ethanol. At this point, cobalt(II)-precorrin-4 had oxidized into cobalt(II) factor IV. After lyophilization, ∼2.5 mg of pure cobalt(II) factor IV was dissolved in deuterium oxide (99.9% atom D) containing 25 mm KCN, to convert the intermediate into cyano-cobalt(III) factor IV. The sample was transferred into a Wilmad screw-cap 5 mm NMR tube. All NMR experiments were carried out using a 14.1 T Bruker Avance III spectrometer (600 MHz 1H resonance frequency) equipped with a QCI (+ 19F) cryoprobe). Resonance assignment was completed using a range of two-dimensional NMR experiments, including 13C-1H heteronuclear single quantum coherence (HSQC) and heteronuclear multiple bond correlation, as well as 1H-1H homonuclear correlation spectroscopy, rotating frame Overhauser effect (ROESY), and total correlation spectroscopy NMR experiments. All two-dimensional data sets were recorded with 2048 by 256 points in the direct and indirect dimensions, respectively. 13C-1H HSQC data sets were recorded using 1H and 13C spectral widths of 9615 and 24897 Hz, 13C-1H heteronuclear multiple bond correlation data sets were recorded using 1H and 13C spectral widths of 7211 and 33474 Hz. All 1H-1H correlation data sets were recorded with direct and indirect dimension spectral widths of 7211 and 7200 Hz, respectively.
RESULTS
CbiH60 Contains an [4Fe-4S] Center
B. megaterium DSM509 CbiH60 is insoluble when overexpressed in E. coli. To produce soluble CbiH60, homologous production in B. megaterium DSM319 was attempted. The B. megaterium DSM509 cbiH60 was cloned from pAR8766 (19) into pC-His1622 (20) to allow the recombinant protein to be produced with a C-terminal His6 tag and with 9 extra amino acid residues (MVQTSSKIW) incorporated into the N-terminal region of the protein. After transformation of B. megaterium DSM319, recombinant CbiH60 was homologously overproduced and purified by metal ion affinity chromatography, allowing the isolation of ∼20 mg liter−1 of CbiH60.
The purified CbiH60 migrates on SDS-PAGE (Fig. 2A) with an approximate molecular mass of 65 kDa, representing the 60 kDa of CbiH60 plus an extra 5 kDa added for the His6 tag and extra sequence at the N terminus. Under anaerobic conditions, CbiH60 purified with a yellow/brown color, with associated absorption peaks at 340 and 410 nm (Fig. 2B), and an A280/A410 ratio of ∼4:1. This finding is indicative of the presence of an iron-sulfur center (21). The CbiH60 enzyme has four conserved cysteines composed from two spatially close 402CXXC405 and 439CXXXC443 motifs found within the nitrite and sulfite reductase-like domain. If the protein was left exposed to air, the [4Fe-4S] absorbance (300–420 nm) decreased with time (3–4 h), with the A280 remaining constant (Fig. 2C). The iron content of the anaerobically purified protein was determined by a ferrozine-based colorimetric assay (22) and estimated to contain 4.8 ± 0.9 mol of iron/mol of CbiH60.
FIGURE 2.

Purified B. megaterium CbiH60 UV-visible and EPR spectra. A, SDS gel showing purified CbiH60 proteins used in this study. The molecular mass markers are shown under lane M; lane WT contains ∼3 μg of CbiH60; lane C402A contains ∼1.5 μg of C402A CbiH60; and lane C443A contains ∼1.5 μg of C402A CbiH60. B, UV-visible spectra of 50 μm CbiH60 as purified (solid line) and after reduction with excess dithionite (dotted line). C, UV-visible spectra of 50 μm CbiH60 exposed to air and scanned every 10 min. Arrows show a loss of absorbance between 300 and 450 nm, indicating the loss of the [4Fe-4S] cluster. D, EPR of 200 μm (12 mg ml−1) CbiH60 (buffer-exchanged in Buffer H), reduced with excess dithionite (10 mm) and frozen in liquid nitrogen. From the intensity of the signal, ∼20–25 μm (∼10%) of reduced [4Fe-4S]1+is present. The [4Fe-4S]1+ spectrum of CbiH60 was recorded at 12 K, employing a microwave frequency of 9.383984 GHz, microwave power of 1 milliwatt, a modulation frequency of 100 kHz, and a modulation amplitude of 5 G. A spectrum of CbiH60 was also recorded at 70 K, which resulted in a loss of the [4Fe-4S]1+ signal. This indicated t1hat the iron-sulfur center was of the [4Fe-4S] class.
To confirm the identity of the redox group, EPR spectroscopy was undertaken. The protein was first reduced with 10 mm dithionite (Eo′ =−0.66 V at pH 7), which leads to a bleaching of its color and a shift in the UV-visible spectrum with a broad decrease in the shoulder between 350 and 410 nm (Fig. 2B). The EPR spectrum at 15 K provided evidence of a reduced [4Fe-4S]1+ center exhibiting axial anisotropy with g values g‖ = 2.04, g⊥ = 1.93. Further evidence of a [4Fe-4S] center was obtained when it was observed that the spectrum was lost when the temperature was raised to 70 K (Fig. 2D). This is a distinguishing test to differentiate [4Fe-4S] and [2Fe-2S] centers (10). An attempt to measure the redox potential of the protein was also attempted, but unfortunately this could not be fully determined because of the instability of the protein during the titration.
The Role of CbiH60 and the [4Fe-4S] Center
The activity of CbiH60 was tested using either cobalt-precorrin-3 or cobalt factor III as the substrate. When CbiH60 was incubated with cobalt factor III and SAM, there was no change in either the visual or UV-visible spectral appearance. Significantly, when DTT was added to the incubation, a change in the appearance of the green cobalt factor III to a red color was observed. The UV-visible spectrum of cobalt factor III, which has a Soret peak at 392 nm, also changed, giving rise to a spectrum with a broad absorbance between 300 and 350 nm. Moreover, the loss of the absorbance peak at 595 nm, corresponding to the d bands, suggests that cobalt factor III had been transformed into a new intermediate (Fig. 3).
FIGURE 3.
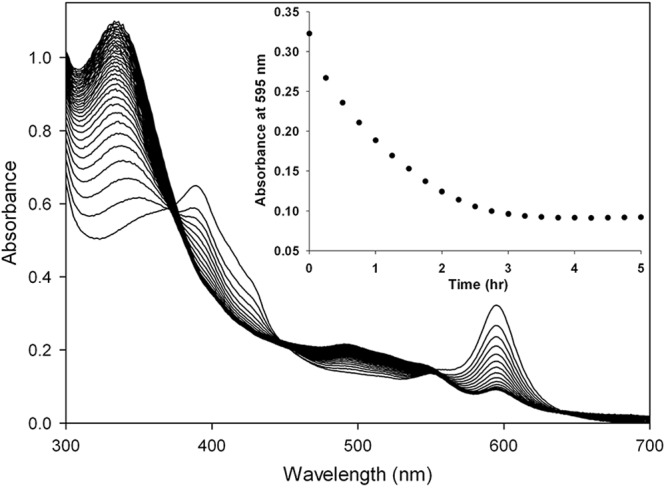
Conversion of cobalt(II) factor III to cobalt(II)-precorrin-4 by UV-visible spectroscopy. The reaction was performed at 37 °C, with 5 μm cob(II)alt factor and 20 μm CbiH60 in the presence of 1 mm SAM and 10 mm DTT. Excess CbiH60 was added to ensure complete turnover. UV-visible scans were measured every 15 min. The inset shows the loss of absorbance at 595 nm (d bands) over time.
To determine the identity of this new species, LC-MS was employed. A minor peak (17 min) of cobalt-precorrin-4 (950 m/z) and a major peak (18 min) of cobalt factor IV (948 m/z) were detected (supplemental Fig. S2), but only in the reaction containing CbiH60, cobalt factor III, SAM, and DTT. In the equivalent reaction that lacked DTT, only cobalt factor III was observed, suggesting that the presence of an artificial electron source (DTT) was essential for turnover. Interestingly, if CbiH60 was instead incubated with cobalt-precorrin-3 and SAM, the reaction did not require the presence of DTT, because both the products cobalt-precorrin-4 and cobalt factor IV were formed, although at slightly lower levels because of the spontaneous oxidation of cobalt-precorrin-3 to cobalt factor III during the reaction. It is important to highlight this latter detail because both cobalt-precorrin-3 and cobalt-precorrin-4 are unstable and quickly (hours) oxidize into cobalt factor III and cobalt factor IV, respectively, despite protection under anaerobic conditions (O2 < 2 ppm). This instability is also observed with precorrin-2 that oxidizes more slowly (days) into factor II. These oxidation events can be prevented by the presence of the reducing agent DTT. Interestingly, in an enzyme-free system, DTT was observed to reduce cobalt factor IV into cobalt-precorrin-4 but could not reduce cobalt factor III into cobalt-precorrin-3.
The Role of the 4Fe-4S Center in the Activity of CbiH60
To study the role of the [4Fe-4S] center, two separate point mutations were made in cbiH60 to generate cysteine to alanine replacements at positions C402A and C443A. These were chosen so as to prevent the [4Fe-4S] center from forming. After purification of the C402A and C443A variants of CbiH60, both mutant enzymes were found to have decreased solubility in comparison with the wild-type CbiH60. However, enough soluble protein could be recovered to test for enzymatic activity. The C402A and C443A mutants both lacked any visible coloration and had decreased absorbance between 350 and 420 nm, when compared with wild-type CbiH60 (data not shown), suggesting that the [4Fe-4S] cluster had not formed.
After overnight incubation of the wild-type enzyme and the cysteine variants with the substrates cobalt-precorrin-3, cobalt factor III, and SAM in the presence and absence of DTT, the purified intermediates of the incubations were analyzed by LC-MS. With cobalt-precorrin-3 as the substrate, both cobalt factor IV and cobalt-precorrin-4 were formed at similar levels by the C402A or C443A variants in comparison with the wild-type CbiH60. However, with cobalt factor III as the substrate, only low levels of (<5%) of cobalt factor IV could be detected with the C402A and C443A variants, when compared with the wild-type CbiH60. Thus, the cysteine mutations give rise to proteins that are able to utilize cobalt-precorrin-3 as substrate but are unable to mediate the transformation of cobalt factor III. A summary of the reactions is given in Table 1.
TABLE 1.
Relative activities of the CbiH60 wild type and C402A and C443A mutants
Cobalt-precorrin-3 or cobalt-factor III was used as the substrate (5 μm) and incubated with 10 μm of CbiH60, 500 μm SAM, and 1 mm DTT in a 2.5-ml reaction. The reactions were left overnight at 37 °C, stopped with 1% (v/v) acetic acid, and purified by reverse phase chromatography. Bound tetrapyrroles were eluted in 2 ml of ethanol, freeze-dried, and resuspended in 200 μl of 1% (v/v) acetic acid. 50 μl was analyzed by LC-MS, with cobalt-precorrin-4 (CP4) and cobalt-factor IV (CFIV) eluting at 17 and 18 min, respectively. The products are represented as high (++) or low (+), relative to the CbiH60 wild-type reaction. Low product formation indicates a turnover of less than 10%, relative to the CbiH60 wild-type reaction.
| Substrate | CbiH60 | CP4 | CFIV |
|---|---|---|---|
| Cobalt-precorrin-3 | − | − | − |
| Wild-type | + | ++ | |
| C402A | + | ++ | |
| C443A | + | ++ | |
| Cobalt-factor III | − | − | − |
| Wild-type | + | ++ | |
| C402A | − | + | |
| C443A | − | + |
EPR of the CbiH60 Catalyzed Ring Contraction Step
To investigate the ring contraction reaction further, EPR was utilized to probe for changes in the environment of the paramagnetic cobalt(II) ion and the [4Fe-4S] center in CbiH60 during the reaction. The samples were prepared with an excess of CbiH60 (150 μm) to cobalt factor III (∼100 μm). Reactions were made with combinations of SAM (5 mm) and DTT (10 mm). The reactions were started at 37 °C, and the samples were removed at time 0 and after overnight incubation (24 h). Under these conditions the reaction did not go to completion. Only the reaction containing cobalt factor III, CbiH60, SAM, and DTT showed a change in the UV-visible spectrum and color, turning brown rather than red, indicating a mixture of both cobalt factor III and the product cobalt-precorrin-4. It was estimated that only ∼20–25% turnover had been achieved (data not shown).
Cobalt possesses 27 electrons and a nucleus with nuclear spin I = . The electronic configuration of cobalt is [Ar]3d74s2; thus, the cobalt(II) ion has configuration [Ar]3d7, with the odd number of electrons rendering the ion paramagnetic; cobalt(III) and cobalt(I) ions are consequently diamagnetic. The seven 3d electrons can be arranged within the five d orbitals to give either one unpaired electron, the low spin S = ½ state, or three unpaired electrons, the high spin S = state. In cobalt-centered tetrapyrroles, the solvent provides a fifth, axial ligand described as a base on form. The cobalt(II) ion is in the low spin state with the single unpaired electron in either the dz2 or dyz orbital. The EPR spectra of complexes bearing the unpaired electron in dz2 are characterized by an axial line shape with g⊥ of 2.20–2.30 and g‖ = ∼2.00. The I = nuclear spin splits both g‖ and g⊥ into eight lines via the hyperfine effect, but the small perpendicular splitting, A⊥, is unresolved, whereas the larger parallel splitting, A‖, of ∼100–150 G gives rise to eight resolved, or partially resolved, lines. The g factors and A values are very sensitive to changes in metal coordination geometry. For the control incubations, free cobalt(II) factor III is in a low spin base-on form. This leads to a g‖ value of 2.28 with typical hyperfine splitting, as observed previously (9). The addition of SAM leads to changes in the hyperfine splitting of A⊥, but not A‖ (Fig. 4A). A similar but spectrally different effect is also observed with the addition of DTT to cobalt(II) factor III (Fig. 4A). Changes in A⊥ suggest that both SAM and DTT can interact with cobalt(II) factor III, although this interaction is not through axial ligation of the cobalt(II) ion (Fig. 4A) because g⊥ = 2.002 and A‖ = 113 G in all spectra. For CbiH60 control incubations, only a small amount of reduced [4Fe-4S]1+ cluster was observed and remains unchanged by the addition of either SAM or DTT (supplemental Fig. S4). For the reaction containing cobalt(II) factor III, CbiH60 plus SAM and/or DTT, slow changes in both the g‖ and g⊥ tensors and their associated hyperfine splitting occurs over 24 h (Fig. 4, B and C). Focusing on the parallel effects, these effects comprise a slow change from the aforementioned cobalt(II) factor III values to g⊥ = 1.990 and A‖ = 120 G. After 24 h all of the reactions apparently contain mixtures of cobalt(II) factor III and this new species (Fig. 4D).
FIGURE 4.
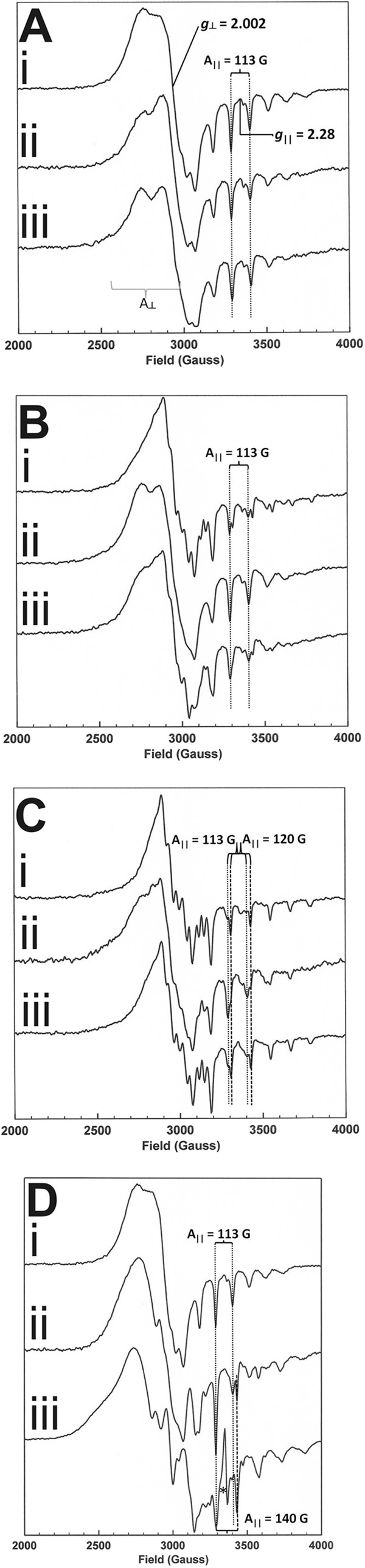
EPR of the anaerobic ring contraction reaction. Concentrations of 150 μm CbiH60, 100 μm cobalt(II) factor III (CFIII), 1 mm SAM, and 10 mm DTT were used in all of the following reactions and incubated at 37 °C. A, spectrum i, CFIII; spectrum ii, CFIII + SAM; spectrum iii, CFIII + DTT. B, reaction of CbiH60 with CFIII recorded immediately after mixing, together with SAM (spectrum i), DTT (spectrum ii), or SAM + DTT (spectrum iii). C, reaction of CbiH60 with CFIII recorded after 24 h of incubation, together with SAM (spectrum i), DTT (spectrum ii), or SAM + DTT (spectrum iii). D, EPR spectra of purified intermediates. Spectrum i, CFIII; spectrum ii, anaerobically purified cobalt(II)-precorrin-4; spectrum iii, aerobically purified cobalt(II) factor IV. The asterisk indicates the presence of a small amount tetrapyrrole radical signal in spectrum iii) (estimated to be less than 1% of total tetrapyrrole).
The new species dominates in the presence of SAM alone, being some 72% of the final mixture. The level of conversion is slightly reduced in the presence of DTT and SAM (63%), whereas cobalt(II) factor III is clearly still the major species in the presence of DTT alone with the new species, accounting for only 10% of the mixture. This indicates a possible change in the axial ligation state of the cobalt ion and could be interpreted as ternary complex formation, with binding of the cobalt ion to an amino acid ligand provided by CbiH60. This has previously been observed with CbiL and cobalt(II) factor II in the presence of SAM (9). The EPR spectra do not indicate a correlation between the extent of this process and changes in the oxidation state of the cobalt ion. Only the cobalt(II) state is visible to EPR spectroscopy; therefore either oxidation or reduction of the cobalt ion renders it undetectable by EPR. However, the experiments employing CbiH60, cobalt(II) factor III, and DTT, which showed the smallest yield of the new species after 24 h, see above, showed the greatest loss of cobalt(II), 52%, whereas the reactions where DTT was replaced with SAM showed the highest yield of the new species and yet the smallest loss of cobalt(II), 12%.
Isolation and Characterization of Cobalt(II)-Precorrin-4
Pure cobalt-precorrin-4 produced from the reaction containing CbiH60, cobalt(II) factor III, SAM, and DTT is light red in color with a Soret peak at ∼320 nm (Supplemental Fig. S1). After aerobic exposure the intermediate rapidly converts into cobalt factor IV, a dark red intermediate. This process also occurs under anaerobic conditions, taking a few hours. Concentrated samples of both reduced (as purified) and oxidized (exposed to air) forms of the intermediate were analyzed by EPR (Fig. 4D). The anaerobically purified sample gave rise to a pair of overlapped low spin cobalt(II) spectra, one having g⊥ and A‖ values of 2.002 and 113 G, respectively, which is similar to those of cobalt(II) factor III, although the intensity pattern of the lines might indicate that it is not factor III. The other contributor to the overlap is a spectrum with g⊥ = 1.993 and A‖ = 144 G, which we tentatively attribute to cobalt(II) factor IV because it is the only species found in the air-oxidized sample. Both spectra suggest that ∼100% of the cobalt is in the cobalt(II) oxidation state.
To confirm the structure of the intermediate cobalt(II) factor IV, it was purified to homogeneity in its free acid form and converted into the green-yellow cyano-cobalt(III) factor IV derivative to allow for NMR analysis. Data sets of two-dimensional HSQC (supplemental Fig. S3), heteronuclear multiple bond correlation, total correlation spectroscopy, homonuclear correlation spectroscopy, and ROESY were collected measuring resonance of naturally abundant 13C and 1H. The uniqueness of cobalt(II) factor IV is the presence of the δ-lactone ring. This was shown to be present as a major and a minor tautomeric form, both derived from the C-20 methyl group with NOE connections to the ring D acetate (C18a), observed in the ROESY spectrum. In addition, methyl groups were found to be at positions C2, C7, and C17, whereas the C20 methyl group was present in the δ-lactone ring (for carbon assignments, ROESY connections, and 1H 13C HSQC data; Fig. 5). This is consistent with the enzyme CbiH60 catalyzing both the C17 methylation and the ring contraction event. The carbon assignments (supplemental Table S3) are in close agreement with the previously published spectra of cobalt factor IV heptamethyl ester (5). Together with the EPR analysis, these data clearly demonstrate that CbiH60 is able to convert cobalt(II) factor III into cobalt(II)-precorrin-4 in the presence of SAM and DTT.
FIGURE 5.

NMR analysis of cyano-cobalt(III) factor IV. A, structure of cyano-cobalt(III) factor IV with the CHX atom definitions labeled in red and used for the assignment of the NMR chemical shifts (supplemental Table S3). B, the structure of cyano-cobalt(III) factor IV with the NOE contacts observed in the ROESY spectrum shown with blue arrows. C and D, two regions of the 13C 1H HSQC spectrum of the HSQC spectrum of cyano-cobalt(III) factor IV with major resonances labeled. C, methyl groups; D, methylene bridges.
DISCUSSION
The activity of CbiH has previously been observed using E. coli lysates with the enzyme overproduced and combined in a one-pot incubation with precorrin-3, cobalt, and SAM as substrates (5). However, we have found this approach difficult to repeat and with only limited turnover (<5%) to cobalt-precorrin-4 (23). To undertake a more detailed analysis of this transformation, we chose to characterize the CbiH60 enzyme from B. megaterium.
Initial recombinant production in E. coli found that CbiH60 is insoluble. Previous research in our group had demonstrated the successful application of using B. megaterium to produce and purify the cobaltochelatase, CbiXL (24). With CbiH60, we repeated this strategy, while also utilizing B. megaterium plasmids that had been recently developed for affinity purification (20). Using this approach, we found that CbiH60 could be obtained in soluble yields of 20 mg liter−1. Noticeably, the purified enzyme was found to be yellow/brown in color, which EPR analysis revealed as a reducible [4Fe-4S] center. Iron-sulfur centers have sprung up in a variety of pathway enzymes related to tetrapyrrole biosynthesis, some with no obvious function. For example, such redox centers have been observed in the class II chelatase enzymes, CbiXL, in B. megaterium (24), SirB in Arabidopsis thaliana (25), and the human ferrochelatase (26). Following the basic characterization of CbiH60, we wanted to investigate its activity and substrate specificity for the anaerobic ring contraction reaction, while also scrutinizing the role of the [4Fe-4S] center.
The anaerobic pathway seems to proceed through the oxidized factor intermediates, which is favored for the ease of cobalt insertion (8, 9, 11, 24, 27). Indeed purified CbiH60 showed it could transform cobalt factor III into cobalt-precorrin-4, with a dependence on SAM as a methyl donor and an artificial electron source (DTT). In contrast, the conversion of cobalt-precorrin-3 into cobalt-precorrin-4 only requires SAM. To determine the requirement for an electron source to activate the enzyme with cobalt factor III as the substrate, it seemed likely that the iron-sulfur center was involved. This hypothesis was tested by making cysteine to alanine replacements at C402A and C443A to disrupt the [4Fe-4S] cluster. As a result of this, activity was decreased by more than 95% in both mutants when compared with wild-type CbiH60. Interestingly, with cobalt-precorrin-3 as the starting substrate, the activity of wild-type CbiH60 and the C402A and C443A variants are comparable, signifying that the [4Fe-4S] cluster is important for activity only with cobalt factor III as the substrate. From these data, we suggest that the [4Fe-4S] cluster is involved in the reduction of cobalt factor III into cobalt-precorrin-3, prior to its transformation into cobalt-precorrin-4. This is likely to be a gated mechanism involving SAM because the reduction of cobalt factor III into cobalt-precorrin-3 only takes place in the presence of SAM.
The requirement for DTT and that CbiH60 seems to function as a single turnover enzyme suggests that a biological redox partner is required. It is intriguing that another redox protein CbiW, a putative thioredoxin, is found as the first gene encoded in the cobI operon in B. megaterium and is followed by cbiH60 and cbiXL (cbiW-cbiH60-cbiXL) (19). Significantly the cobaltochelatase, CbiXL, also contains a [4Fe-4S] center with a midpoint redox potential of E0 ≈ −170 ± 4 mV (24). Unfortunately, so far it has not been possible to characterize CbiW because of its insolubility when overproduced in either E. coli or B. megaterium. It is tantalizing to suggest that this protein could act as electron donor to both CbiH60 and CbiXL, assisting in the reduction of cobalt factor II and III to the corresponding cobalt-precorrins.
Probing the ring contraction reaction by EPR to monitor the paramagnetic cobalt(II) ion has also given some interesting insights. Early theories for anaerobic ring contraction had suggested the involvement of the cobalt ion in a redox-based mechanism (5, 28, 29). If this were correct, it would result in major changes in the EPR spectrum of a number of the intermediates, with either a loss of signal (cobalt reduction-oxidation) or a redefined spectrum resulting from changes in the environment of the cobalt ion. However, despite a low turnover (∼25%) for the reaction, the metal ion was found to remain as a low spin cobalt(II) species throughout the reaction, with neither a loss of signal from any reduction-oxidation of the cobalt ion nor any changes in the g‖ or g⊥ features to indicate formation of a tri-covalent Co(II) species. This result eliminates a cobalt redox change during ring contraction. We can confirm that the cobalt ion forms an axial bond with an unidentified ligand from the enzyme. Because ligand binding to the cobalt is incomplete after initial mixing, the tetrapyrrole might initially bind in the active site through weak interactions with the propionate and acetate side chains. Then to secure the tetrapyrrole into the correct orientation, the cobalt ion could act as anchor and bind to an unknown amino acid within the active site. We suggest that the cobalt ion is merely a passenger during ring contraction but can offer a means of substrate recognition in the enzyme, a common theme observed with the preceding reaction, catalyzed by CbiL (9).
The results herein have demonstrated that CbiH60 is able to catalyze the transformation of both cobalt factor III and cobalt-precorrin-3 into cobalt-precorrin-4. There are still important questions relating to how this takes place because the transformation involves the formation of a lactone ring and methylation at C17. Although the two events are not linked mechanistically, the fact that we do not observe lactone formation without methylation suggests that either SAM gates lactone formation or that lactone formation only occurs after methylation. Further investigations are required to distinguish between these possibilities.
Acknowledgments
We thank Dr. Kirsty McClean and Prof. Andy Munro for help with redox potentiometry experiments, Dr. Michelle Rowe for NMR technical assistance, Dr. Steffi Frank for the plasmid pETcoco-2-ABCDCXL, and Dr. Amanda Brindley for helpful discussions.
This work was supported by a Biotechnology and Biological Sciences Research Council studentship and Grants BB/I012079 and BB/E024203.

This article contains supplemental Tables S1–S3 and Figs. S1–S4.
- SAM
- S-adenosyl-l-methionine
- CFIII
- cobalt factor III
- EPR
- electron paramagnetic resonance
- HSQC
- heteronuclear single quantum coherence
- ROESY
- rotating frame Overhauser effect spectroscopy.
REFERENCES
- 1. Minot G. R., Murphy W. P. (1926) Treatment of pernicious anemia by a special diet. JAMA 87, 470. [PMC free article] [PubMed] [Google Scholar]
- 2. Warren M. J., Scott A. I. (1990) Tetrapyrrole assembly and modification into the ligands of biologically functional cofactors. Trends Biochem. Sci. 15, 486–491 [DOI] [PubMed] [Google Scholar]
- 3. Heinemann I. U., Jahn M., Jahn D. (2008) The biochemistry of heme biosynthesis. Arch. Biochem. Biophys. 474, 238–251 [DOI] [PubMed] [Google Scholar]
- 4. Scott A. I., Roessner C. A., Stolowich N. J., Spencer J. B., Min C., Ozaki S. I. (1993) Biosynthesis of vitamin B12. Discovery of the enzymes for oxidative ring contraction and insertion of the fourth methyl group. FEBS Lett. 331, 105–108 [DOI] [PubMed] [Google Scholar]
- 5. Scott A. I., Stolowich N. J., Wang J., Gawatz O., Fridrich E., Müller G. (1996) Biosynthesis of vitamin B12. Factor IV, a new intermediate in the anaerobic pathway. Proc. Natl. Acad. Sci. U.S.A. 93, 14316–14319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Warren M. J., Raux E., Schubert H. L., Escalante-Semerena J. C. (2002) The biosynthesis of adenosylcobalamin (vitamin B12). Nat. Prod Rep. 19, 390–412 [DOI] [PubMed] [Google Scholar]
- 7. Nussbaumer C., Imfeld M., Wörner G., Müller G., Arigoni D. (1981) Biosynthesis of vitamin B12. Mode of incorporation of factor III into cobyrinic acid. Proc. Natl. Acad. Sci. U.S.A. 78, 9–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Raux E., Leech H. K., Beck R., Schubert H. L., Santander P. J., Roessner C. A., Scott A. I., Martens J. H., Jahn D., Thermes C., Rambach A., Warren M. J. (2003) Identification and functional analysis of enzymes required for precorrin-2 dehydrogenation and metal ion insertion in the biosynthesis of sirohaem and cobalamin in Bacillus megaterium. Biochem. J. 370, 505–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Frank S., Deery E., Brindley A. A., Leech H. K., Lawrence A., Heathcote P., Schubert H. L., Brocklehurst K., Rigby S. E., Warren M. J., Pickersgill R. W. (2007) Elucidation of substrate specificity in the cobalamin (vitamin B12) biosynthetic methyltransferases. Structure and function of the C20 methyltransferase (CbiL) from Methanothermobacter thermautotrophicus. J. Biol. Chem. 282, 23957–23969 [DOI] [PubMed] [Google Scholar]
- 10. Schroeder S., Lawrence A. D., Biedendieck R., Rose R. S., Deery E., Graham R. M., McLean K. J., Munro A. W., Rigby S. E., Warren M. J. (2009) Demonstration that CobG, the monooxygenase associated with the ring contraction process of the aerobic cobalamin (vitamin B12) biosynthetic pathway, contains an Fe-S center and a mononuclear non-heme iron center. J. Biol. Chem. 284, 4796–4805 [DOI] [PubMed] [Google Scholar]
- 11. Frank S., Brindley A. A., Deery E., Heathcote P., Lawrence A. D., Leech H. K., Pickersgill R. W., Warren M. J. (2005) Anaerobic synthesis of vitamin B12. Characterization of the early steps in the pathway. Biochem. Soc. Trans. 33, 811–814 [DOI] [PubMed] [Google Scholar]
- 12. Schubert H. L., Rose R. S., Leech H. K., Brindley A. A., Hill C. P., Rigby S. E., Warren M. J. (2008) Structure and function of SirC from Bacillus megaterium. A metal-binding precorrin-2 dehydrogenase. Biochem. J. 415, 257–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Romão C. V., Ladakis D., Lobo S. A., Carrondo M. A., Brindley A. A., Deery E., Matias P. M., Pickersgill R. W., Saraiva L. M., Warren M. J. (2011) Evolution in a family of chelatases facilitated by the introduction of active site asymmetry and protein oligomerization. Proc. Natl. Acad. Sci. U.S.A. 108, 97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Spencer P., Stolowich N. J., Sumner L. W., Scott A. I. (1998) Definition of the redox states of cobalt-precorrinoids. Investigation of the substrate and redox specificity of CbiL from Salmonella typhimurium. Biochemistry 37, 14917–14927 [DOI] [PubMed] [Google Scholar]
- 15. Santander P. J., Roessner C. A., Stolowich N. J., Holderman M. T., Scott A. I. (1997) How corrinoids are synthesized without oxygen. Nature's first pathway to vitamin B12. Chem. Biol. 4, 659–666 [DOI] [PubMed] [Google Scholar]
- 16. Kozbial P. Z., Mushegian A. R. (2005) Natural history of S-adenosylmethionine-binding proteins. BMC Struct. Biol. 5, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Raux E., Lanois A., Rambach A., Warren M. J., Thermes C. (1998) Cobalamin (vitamin B12) biosynthesis. Functional characterization of the Bacillus megaterium cbi genes required to convert uroporphyrinogen III into cobyrinic acid a,c-diamide. Biochem. J. 335, 167–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barg H., Malten M., Jahn M., Jahn D. (2005) in Microbial Processes and Products (Barredo J. L., ed), 1st Ed., pp. 165–184, Humana Press, Totowa, NJ [Google Scholar]
- 19. Raux E., Lanois A., Warren M. J., Rambach A., Thermes C. (1998) Cobalamin (vitamin B12) biosynthesis. Identification and characterization of a Bacillus megaterium cobI operon. Biochem. J. 335, 159–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Biedendieck R., Yang Y., Deckwer W. D., Malten M., Jahn D. (2007) Plasmid system for the intracellular production and purification of affinity-tagged proteins in Bacillus megaterium. Biotechnol. Bioeng 96, 525–537 [DOI] [PubMed] [Google Scholar]
- 21. Ollagnier-De Choudens S., Sanakis Y., Hewitson K. S., Roach P., Baldwin J. E., Münck E., Fontecave M. (2000) Iron-sulfur center of biotin synthase and lipoate synthase. Biochemistry 39, 4165–4173 [DOI] [PubMed] [Google Scholar]
- 22. Riemer J., Hoepken H. H., Czerwinska H., Robinson S. R., Dringen R. (2004) Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Anal. Biochem. 331, 370–375 [DOI] [PubMed] [Google Scholar]
- 23. Frank S. (2007) Biosynthesis of cobalamin (Vitamin B12). Characterisation of the anaerobic pathway in Methanobacter thermoautotrophicus. Ph.D. Thesis, Queen Mary University of London [Google Scholar]
- 24. Leech H. K., Raux E., McLean K. J., Munro A. W., Robinson N. J., Borrelly G. P., Malten M., Jahn D., Rigby S. E., Heathcote P., Warren M. J. (2003) Characterization of the cobaltochelatase CbiXL. Evidence for a 4Fe-4S center housed within an MXCXXC motif. J. Biol. Chem. 278, 41900–41907 [DOI] [PubMed] [Google Scholar]
- 25. Saha K., Webb M. E., Rigby S. E., Leech H. K., Warren M. J., Smith A. G. (2012) Characterisation of the evolutionary conserved iron-sulphur cluster of sirohydrochlorin ferrochelatase from Arabidopsis thaliana. Biochem. J. 444, 227–237 [DOI] [PubMed] [Google Scholar]
- 26. Dailey H. A., Finnegan M. G., Johnson M. K. (1994) Human ferrochelatase is an iron-sulfur protein. Biochemistry 33, 403–407 [DOI] [PubMed] [Google Scholar]
- 27. Brindley A. A., Raux E., Leech H. K., Schubert H. L., Warren M. J. (2003) A story of chelatase evolution. Identification and characterization of a small 13–15-kDa “ancestral” cobaltochelatase (CbiXS) in the archaea. J. Biol. Chem. 278, 22388–22395 [DOI] [PubMed] [Google Scholar]
- 28. Roessner C. A., Santander P. J., Scott A. I. (2001) Multiple biosynthetic pathways for vitamin B12. Variations on a central theme. Vitam Horm. 61, 267–297 [DOI] [PubMed] [Google Scholar]
- 29. Roessner C. A., Scott A. I. (2006) Fine-tuning our knowledge of the anaerobic route to cobalamin (vitamin B12). J. Bacteriol. 188, 7331–7334 [DOI] [PMC free article] [PubMed] [Google Scholar]