

Background: The VDAC receptor, a major mitochondrial protein, is also present in the plasma membrane of normal brain cells.
Results: VDAC binds t-PA and stimulates plasminogen activation in normal and injured brain cells.
Conclusion: Formation of the ternary VDAC·t-PA·plasminogen complex enhances both t-PA activity and plasminogen activation.
Significance: This ternary complex may play a significant role in normal and injured brain physiology.
Keywords: Amyloid, Fibrinolysis, Neurophysiology, Plasminogen Regulation, Tissue Plasminogen Activator (tPA)
Abstract
The voltage-dependent anion channel (VDAC), a major pore-forming protein in the outer membrane of mitochondria, is also found in the plasma membrane of a large number of cells where in addition to its role in regulating cellular ATP release and volume control it is important for maintaining redox homeostasis. Cell surface VDAC is a receptor for plasminogen kringle 5, which promotes partial closure of the channel. In this study, we demonstrate that VDAC binds tissue-type plasminogen activator (t-PA) on human neuroblastoma SK-N-SH cells. Binding of t-PA to VDAC induced a decrease in Km and an increase in the Vmax for activation of its substrate, plasminogen (Pg). This resulted in accelerated Pg activation when VDAC, t-PA, and Pg were bound together. VDAC is also a substrate for plasmin; hence, it mimics fibrin activity. Binding of t-PA to VDAC occurs between a t-PA fibronectin type I finger domain located between amino acids Ile5 and Asn37 and a VDAC region including amino acids 20GYGFG24. These VDAC residues correspond to a GXXXG repeat motif commonly found in amyloid β peptides that is necessary for aggregation when these peptides form fibrillar deposits on the cell surface. Furthermore, we also show that Pg kringle 5 is a substrate for the NADH-dependent reductase activity of VDAC. This ternary complex is an efficient proteolytic complex that may facilitate removal of amyloid β peptide deposits from the normal brain and cell debris from injured brain tissue.
Introduction
The voltage-dependent anion channel (VDAC)2 is a small, 30–35-kDa protein originally identified in the outer membrane of mitochondria where it functions as the major pore-forming protein (1). VDAC is also found in the plasma membrane of a large number of cells (2) where it plays several roles, including the regulation of cellular ATP release and volume control (3). Cell surface VDAC is an NADH-dependent reductase (4), and it may play a significant role in the maintenance of redox homeostasis in normal cells (2).
Studies in rats show that VDAC is densely localized in regions of the brain, including the caudate nucleus, hippocampus, hypothalamus, and cerebellum (5). VDAC is a receptor for plasminogen kringle 5 (K5), which can induce a decrease in intracellular pH and hyperpolarization of the mitochondrial membrane (6). The interaction of plasminogen (Pg) with VDAC also promotes partial closure of the channel, leading to inhibition of the transport of ATP and other metabolites across the mitochondrial membrane (7). Both Pg and tissue-type Pg activator (t-PA) are present in neurons and microglia, and both are constitutively active in the central nervous system (CNS) (8). In addition to its participation in fibrinolysis, t-PA is proposed to have roles in synaptic plasticity, long term potentiation, and neuronal migration (9). Although the urokinase-type Pg activator (u-PA) is expressed in the inflamed CNS of both rodents and humans (10), it is not generally found in the normal CNS (8). Therefore, it is still hypothesized that activation of Pg by t-PA is a key process that participates not only in normal remodeling but also in neurodegenerative disease processes in the CNS (9).
Pg and t-PA in the normal brain are concentrated in the hippocampus (11), a region that also contains high concentrations of VDAC (5). Because Pg/t-PA activities are actively involved in brain pathologies (12), we hypothesized a role for VDAC as a regulator of the functional interaction between these two proteins in the brain.
For these studies, we used the human neuroblastoma SK-N-SH cell line as an in vitro model, and this cell line expresses VDAC on the cell surface (13). Receptor binding assays demonstrated that both Pg and t-PA bind with high affinity to sites on these cells. Proteins known to act as t-PA receptors are the low density lipoprotein receptor-related protein receptor (LRP) (14), annexin II (15), and the mannose receptor (CD-206) (16); however, VDAC has not previously been identified as a t-PA-binding protein. In this report, we demonstrate that t-PA binds to human VDAC at a site near its NH2-terminal region. In addition to binding Pg, VDAC binds to and stimulates t-PA activity upon Pg activation. In vitro experiments demonstrated that after plasmin is formed VDAC becomes its substrate, thereby behaving in a manner analogous to fibrin. Furthermore, after Pg was activated by t-PA, VDAC reduced Pg K5 via its NADH-dependent oxidoreductase activity, which may ultimately be a necessary mechanism for inhibiting the K5 proapoptotic effects on the cell surface. We also investigated this phenomenon in injured brain cells and found that overexpression of VDAC enhanced t-PA-mediated Pg activation.
EXPERIMENTAL PROCEDURES
Materials
Porcine pancreatic elastase, gastric mucosa pepsin, trypsin inhibitor, potassium ferricyanide, β-NADH, glutathione (GSH), and iodoacetamide were purchased from Sigma. Na125I was obtained from PerkinElmer Life Sciences. The chromogenic substrates VLK-pNA (S-2251), IPR-pNA (S-2288), and EGR-pNA (S-2444) were purchased from Diapharma (West Chester, OH). 3-(N-Maleimidylpropionyl)biocytin (MPB) was purchased from Molecular Probes (Eugene, OR). Horseradish peroxidase-conjugated streptavidin was purchased from Calbiochem EMD Biosciences. The amyloid β peptide analogues Aβ 1–42, Aβ 1–28, Aβ 25–35, Aβ 29–40, and Aβ 31–42 were purchased from Bachem Americas, Inc. (Torrance, CA). The VDAC 10GKSARDVFTKGYGFGLIKLDL30 (Gly10–Leu30) and t-PA 509CQGDSGGPLVC519 peptides were obtained from Genemed Synthesis, Inc. (San Antonio, TX).
Proteins
Human Pg purified by affinity chromatography on l-lysine-Sepharose (17) was digested with elastase and fractionated by gel and affinity chromatography to obtain essentially pure mini-Pg (mPg), K1–3, K4, and K5 (18–21). A functionally active human microplasminogen (μPg) without kringle structures was produced by incubation of Pg with urokinase-free plasmin at an alkaline pH (22). Human t-PA was purchased from American Diagnostica Inc. (Stamford, CT). Human u-PA was purchased from Calbiochem. Human VDAC was produced in Escherichia coli and purified from clones obtained from Genecopeia (Germantown, MD) as described previously (23). Radioiodination of proteins was performed by the method of Markwell (24). The 125I label was incorporated at 2 × 107 cpm/nmol of protein, and the radioactivity was measured with a GE Healthcare-LKB Biotechnology 1272 γ-counter.
Antibodies
The goat polyclonal IgG against human Pg (H-14), mouse monoclonal IgG against human LRP1 (A2MRα-2), rabbit polyclonal IgG against human annexin II (H-50), rabbit polyclonal IgG against human CD-206 (H-300), goat polyclonal IgG against the NH2-terminal region of human VDAC (N-18), and goat polyclonal IgG against the COOH-terminal region of human VDAC (C-20) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The rabbit polyclonal IgG against cytochrome c oxidase IV (COX IV) was purchased from Cell Signaling Technology (Beverly, MA). The rabbit antibody against the 21-amino acid sequence Lys235–Lys255 of human VDAC was prepared and purified as described previously (6). Goat IgG was purchased from Sigma. Anti-rabbit IgG F(ab′)2-FITC was purchased from Jackson ImmunoResearch Laboratories (West Grove, PA), and anti-goat IgG F(ab′)2-phycoerythrin-Cy7 was purchased from Santa Cruz Biotechnology.
Cell Culture
Human neuroblastoma SK-N-SH cells were obtained from the American Type Culture Collection (Manassas, VA) and grown in minimum Eagle's medium containing 2 mm l-glutamine, 1.5 g/liter sodium bicarbonate, 0.1 mm non-essential amino acids, 1.0 mm sodium pyruvate, 10% fetal bovine serum (FBS), and 100 units/ml penicillin/streptomycin, all purchased from Invitrogen.
t-PA Binding Analysis
The cells were grown in 96-well strip culture plates until the monolayers were confluent. The cells were then rinsed in Hanks' balanced salt solution (HBSS). All binding assays were performed at 4 °C in serum-free RPMI 1640 medium containing 2% bovine serum albumin (BSA). Increasing concentrations of 125I-labeled t-PA were incubated with the cells for 60 min. Free and bound ligands were separated by aspirating the incubation mixture and washing the cell monolayers rapidly three times with RPMI 1640 medium containing 2% BSA. The cells were stripped from the plates, and the radioactivity was determined. The bound t-PA was calculated after the subtraction of nonspecific binding, which was measured in the presence of 50 μm unlabeled t-PA. The Kd and Bmax were determined after Scatchard plot analyses using the statistical program GraphPad Prism® 5 from GraphPad Software, Inc. (San Diego, CA).
Pg and t-PA Binding Rates to Apoptotic Cells
Confluent SK-N-SH cell monolayers were incubated with increasing concentrations of staurosporine (STS), a known inducer of apoptosis (25), in a cell culture chamber for 16 h at 37 °C. At the end of this incubation, the cells were rinsed in serum-free RPMI 1640 medium. Increasing concentrations of unlabeled Pg or t-PA at 4 °C in serum-free RPMI 1640 medium containing 2% BSA were incubated with cells for 60 min in separate culture plates. The free ligand was separated by aspirating the incubation mixture and washing the cell monolayers rapidly three times with RPMI 1640 medium containing 2% BSA. The bound t-PA was then assessed by measuring the rate of t-PA activity on the chromogenic substrate S-2288 (0.3 mm) in serum- and phenol red-free RPMI 1640 medium. The resulting t-PA hydrolysis of S-2288 was monitored by measuring the change in absorbance at a wavelength of 405 nm over time using a Molecular Devices Thermomax kinetic plate reader. In separate plates processed as described above, the bound Pg was monitored by measuring the resulting hydrolysis of S-2251 at a wavelength of 405 nm as described for t-PA. The cell-bound Pg and t-PA were calculated from calibration curves measuring the rate of activity of t-PA or plasmin (Pg + t-PA) versus concentration of the ligand.
Determination of Pg Activation Rate
Coupled assays were used to evaluate the initial rate of Glu-Pg activation by t-PA by monitoring the amidolytic activity of the generated plasmin (Pm) (26). Glu-Pg was incubated in 96-well microtiter plates at 37 °C in 20 mm HEPES, pH 7.4 in a total volume of 200 μl with the plasmin substrate S-2251 (0.3 mm). Pg activation was initiated by the addition of 0.55 nm t-PA. The resulting Pm hydrolysis of S-2251 was monitored as described above. The initial velocities (νi) were calculated from plots of A405 nm versus time2 using the equation νi = b(1 + Km/S0)/ϵke (27) where Km is the apparent Michaelis constant of S-2251 hydrolysis by Pm, ke is the empirically determined catalytic rate constant for Pm hydrolysis of S-2251 (3.2 × 104 m min−1(mol of Pm)−1), and ϵ is the molar extinction coefficient of p-nitroanilide at 405 nm (8800 m−1 cm−1) (28).
Heat-induced Aggregation of VDAC
VDAC solutions (0.5 mg/ml) in 20 mm HEPES, pH 7.4 were heated for 30 min in a thermostated bath at 40, 50, 60, 70, and 100 °C followed by cooling to room temperature. The temperatures and heating time were chosen based on the kinetics of the aggregation process that occurs in globular proteins during heat-induced denaturation (29).
Determination of Plasmin, t-PA, and u-PA Amidolytic Activities
Plasmin amidolytic activity was determined after incubation of Glu-Pg with u-PA (2 pm) in 20 mm HEPES, pH 7.4 in a total volume of 175 μl. The plasmin substrate VLK-pNA (0.3 mm; 25 μl) was added to the mixture, and substrate hydrolysis was monitored at 405 nm as described above. The t-PA amidolytic activity was measured by incubating t-PA (5.5 nm) with the chromogenic substrate S-2288 (0.3 mm) for 20 min at 37 °C in 20 mm HEPES, pH 7.4 in a total volume of 200 μl. The reaction was monitored at 405 nm. The u-PA amidolytic activity was determined by incubating u-PA (20 pm) with the chromogenic substrate S-2444 (0.3 mm), and substrate hydrolysis was monitored as described above.
Flow Cytometric Analyses of Living, Apoptotic, and Dead Cells
SK-N-SH cells seeded at 6 × 105 cells/well in 6-well culture plates were allowed to grow until they reached 80% confluence. The cells were then incubated with increasing concentrations of STS as described above. After this period, both STS-treated and non-treated cells were rinsed with PBS and harvested using a cell scraper. The cells were counted manually with a hemocytometer and resuspended in HBSS at a density of 1 × 106 cells/ml. After rinsing twice with 1 ml of 0.5% BSA in HBSS, the cells were incubated with anti-VDAC N-18 antibody (4 μg/106 cells) and anti-COX IV antibody (4 μg/106 cells) or an isotype control, goat IgG (4 μg/106 cells) or rabbit IgG (4 μg/106 cells), in 0.5% BSA in HBSS for 1 h at 4 °C. After two washes with 1 ml of 0.5% BSA in HBSS, secondary anti-goat IgG F(ab′)2-phycoerythrin-Cy7 and anti-rabbit IgG F(ab′)2-FITC were added to the cells at a concentration of 1 μg/106 cells in 0.5% BSA in HBSS and incubated for 30 min at 4 °C in the dark. The cells were then washed twice with 1 ml of HBSS and resuspended in 1 ml of 1× Annexin V Binding Buffer (BD Biosciences). Each sample (200 μl) was transferred to a 96-well plate, and 5 μl of 7-aminoactinomycin (BD Biosciences) and 5 μl of annexin V (BD Biosciences) were added to each well for staining according to the manufacturer's instructions to assess the number of dead and apoptotic cells, respectively. After a 10-min incubation at room temperature in the dark, we assessed antibody binding on a Guava EasyCyte Plus instrument (Millipore, Billerica, MA). The data analyses were performed using FlowJo version 7.5 software and GraphPad Prism 5.0.
Determination of VDAC NADH-ferricyanide Reductase Activity
The assays were performed at 37 °C in 1-ml reactions containing VDAC (100 nm) and β-NADH (25 μm) in 50 mm Tris-HCl, pH 8.0. The reaction was initiated by the addition of potassium ferricyanide (500 nm; control) or Pg fragments (200 nm). The reaction rates were measured at 340 nm following a decrease in absorbance for 10 min. The rate of enzymatic activity was calculated using an extinction coefficient of 6220 m−1 cm−1 for NADH at this wavelength.
Measurement of VDAC mRNA Levels by Reverse Transcription
Total RNA from untreated or STS-treated SK-N-SH cells was extracted by a single step method using the RNeasy® Mini kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. Total RNA was reverse transcribed with 1 μg of RNA in a 20-μl reaction mixture using Moloney murine leukemia virus reverse transcriptase (200 units) and oligo(dT) as the primer for 1 h at 42 °C. The resulting cDNA (5 μl) was used as a template, and a 237-bp segment of the VDAC1 cDNA was amplified using a 21-mer upstream primer (5′-GGCCTGACGTTTACAGAGAAA-3′) that was identical to the positions corresponding to amino acids Gly68–Lys74 and a 21-mer downstream primer (5′-CTCGTAACCTAGCACCAGAGC-3′) complementary to positions Ala141–Glu147 of the amino acids encoded in the VDAC1 mRNA. A 302-bp segment of the human β-actin cDNA, which was the constitutive internal control, was co-amplified using a set of PCR primers (30) provided in an R&D Systems kit (Minneapolis, MN). The amplification was performed in a Whatman Biometra T3 thermal cycler for 35 cycles (one cycle, 94 °C for 45 s and 72 °C for 45 s). The PCR products were analyzed on 1.2% (w/v) agarose-ethidium bromide gels. The gels were photographed, and the intensities of the VDAC1 and β-actin mRNA bands were evaluated and quantified as VDAC1/β-actin ratios.
Gel Electrophoresis and Immunoblotting
Recombinant VDAC was analyzed on a 4–20% polyacrylamide gel (1.2 mm thick; 14 × 10 cm) containing 0.1% SDS under reducing conditions. A discontinuous Laemmli buffer system was used (31). The gels were stained with 0.25% Coomassie Brilliant Blue R-250. The proteins were electroblotted onto nitrocellulose membranes (32). The molecular weights were determined using a set of dye-conjugated Mr markers (Fermentas Life Sciences, Glen Burnie, MD). The membranes were thoroughly rinsed with PBS and then incubated with 3% BSA in PBS for 1 h at room temperature to block the non-conjugated areas. For the detection of Pg forms, the membranes were incubated with a goat anti-human Pg IgG (Santa Cruz Biotechnology) followed by incubation with a rabbit anti-goat IgG conjugated to horseradish peroxidase and detected with a peroxidase substrate as described previously (33).
Reduction of Pg Forms by VDAC
Pg forms were incubated with VDAC for 2 h at 37 °C in PBS. The mixture was then incubated with MPB (100 μm) for 30 min at room temperature followed by quenching of the unreacted MPB with GSH (200 μm) for 10 min at room temperature. Unreacted GSH and other free sulfhydryls in the system were blocked with iodoacetamide (400 μm) for 10 min at room temperature. The proteins were then mixed with a non-reducing Laemmli buffer and separated by 10% SDS-PAGE. Then the proteins were transferred to nitrocellulose membranes, thoroughly rinsed with PBS, and incubated with 3% BSA in PBS for 1 h at room temperature to block non-conjugated areas. Finally, the membranes were incubated with streptavidin-peroxidase to detect the MPB-labeled proteins (34).
Statistics
GraphPad Prism version 5.0 software (GraphPad Software, Inc.) was used to determine the standard deviation of the experimental data. The significance of differences between the controls and different treatments was determined by a one-way analysis of variance and unpaired Student's t tests.
RESULTS
Binding of t-PA to Human Neuroblastoma SK-N-SH Cells
t-PA bound to SK-N-SH cells in a dose-dependent manner with high affinity. Analysis of the binding isotherm (Fig. 1A) identified a large number of binding sites for t-PA on the surface of these cells (Bmax = 9 × 105 ± 2 × 105 molecules/cell), and a Scatchard plot analysis (Fig. 1A, inset) indicated a high affinity (Kd = 34 ± 4 nm) of t-PA for these cells. We then studied the binding of t-PA (100 nm) to cells in the presence of antibodies to known t-PA receptors, LRP, annexin II, and CD-206 (14–16), and compared their effect with antibodies against VDAC NH2- or COOH-terminal regions. We also assessed the antagonistic effect on t-PA binding to SK-N-SH cells with a peptide including amino acid residues 10GKSARDVFTKGYGFGLIKLDL30 (Gly10–Leu30) of the VDAC NH2-terminal region and the amyloid β peptide analogues Aβ 1–42, Aβ 1–28, Aβ 25–35, Aβ 29–40, and Aβ 31–42, which are known to bind to t-PA (35). Furthermore, we also evaluated the effect of unlabeled t-PA and the 509CQGDSGGPLVC519 peptide, which contains Ser513 at the t-PA active site. The results (Fig. 1B) show almost no inhibition of t-PA binding to SK-N-SH cells in the presence of antibodies against LRP, annexin-II, and CD-206 or the anti-COOH-terminal region of VDAC, whereas t-PA binding to these cells was significantly inhibited by antibodies against the NH2-terminal region of VDAC. Only the Aβ 1–42, Aβ 25–35, Aβ 29–40, and Aβ 31–42 peptides significantly inhibited t-PA binding to the SK-N-SH cells, whereas the Aβ 1–28 peptide did not have a significant effect. Similarly, the VDAC NH2-terminal domain peptide (Gly10–Leu30) inhibited t-PA binding to these cells. Unlabeled t-PA significantly inhibited 125I-labeled t-PA binding to the cells, whereas the t-PA 509CQGDSGGPLVC519 peptide did not affect binding.
FIGURE 1.

Binding of t-PA to human neuroblastoma SK-N-SH cells and immobilized VDAC. Experiments were performed on cells grown in 96 strip-well tissue culture plates (2 × 104 cells/well) or in plates containing VDAC immobilized on 96 strip-well tissue culture plates. A, increasing concentrations of 125I-labeled t-PA were added to SK-N-SH cell monolayers. Inset, Scatchard plot analysis of the binding isotherm. B, inhibition of binding of 125I-labeled t-PA (100 nm) to SK-N-SH cell monolayers by a single concentration (500 nm) of different competitors identified under the horizontal axis. C, increasing concentrations of 125I-labeled t-PA were added to VDAC immobilized on 96 strip-well tissue culture plates. Inset, Coomassie Brilliant Blue staining of 10% SDS-PAGE (non-reducing conditions) of human recombinant VDAC. D, inhibition of binding of 125I-labeled t-PA (100 nm) to immobilized VDAC by a single concentration (500 nm) of different competitors identified under the horizontal axis. The data are the means ± S.D. (error bars) from experiments performed in triplicate (n = 6). *, p < 0.05 versus non-treated cells (NT). B/F, bound/free.
Binding of t-PA to Immobilized VDAC
125I-Labeled t-PA bound to immobilized VDAC in a dose-dependent manner (Fig. 1C) with an affinity within the same order of magnitude as that observed for SK-N-SH cells (43 ± 8 nm). As observed above, the binding of t-PA to VDAC was inhibited by an antibody against the NH2-terminal region of VDAC and the amyloid β peptide analogues Aβ 1–42, Aβ 25–35, Aβ 29–40, and Aβ 31–42 and the VDAC NH2-terminal domain peptide (Fig. 1D). The Aβ 1–28 peptide did not significantly inhibit binding. As expected, unlabeled t-PA significantly inhibited 125I-labeled t-PA binding, whereas the 509CQGDSGGPLVC519 peptide did not affect 125I-labeled t-PA binding to immobilized VDAC. These results confirm the observation described above, suggesting that t-PA binds to the VDAC receptor in SK-N-SH cells within its NH2-terminal region. These amyloid β peptide analogues contain four glycine residues between residues 29 and 35, which are known as the GXXXG repeat motif. These residues facilitate the conversion of an α-helical or random Aβ to a β-sheet and eventual fibril formation (36). These fibrils bind to t-PA and modify its kinetic properties (37). VDAC also contains a GXXXG repeat motif at amino acids 20GYGFG24. Our results suggest that this motif may be involved in the interaction between t-PA and VDAC.
Effect of VDAC on the Enzymatic Activity of Plasminogen Activators and Plasmin
The Pg activators t-PA and u-PA were incubated with VDAC. The amidolytic activity of t-PA or u-PA (10 nm) was determined with the chromogenic substrates S-2288 and S-2244 (6 mm), respectively. Increasing amounts of VDAC stimulated the amidolytic activity of t-PA (Fig. 2A), whereas the amidolytic activity of u-PA remained unchanged (Fig. 2B). Human Pg (0.1 μm) was incubated with u-PA (10 nm) for 60 min prior to the plasmin amidolytic activity assay with the chromogenic substrate S-2251 (6 mm). Increasing amounts of VDAC did not change the plasmin activity (Fig. 2C), suggesting that VDAC only affects t-PA activity. We also assessed the effect of VDAC on t-PA amidolytic activity in the presence of tranexamic acid, which inhibits kringle-dependent binding of t-PA to fibrin or cell receptors (38, 39). Increasing amounts of tranexamic acid (1–10 mm) did not change the stimulatory activity of VDAC (100 nm) on t-PA amidolytic activity (Fig. 2D), suggesting that interaction of t-PA with VDAC is kringle-independent.
FIGURE 2.

Effect of VDAC on t-PA, u-PA, and Pm amidolytic activities. The amidolytic activity of t-PA, u-PA, and Pm was determined with the chromogenic substrates S-2288, S-2244, and S-2251, respectively. Increasing amounts of VDAC were added to wells in 96-well culture plates containing a single concentration of each protease (10 nm t-PA or u-PA and 100 nm Pm). Changes in absorbance were recorded for 20 min at a wavelength of 405 nm. A, effect of VDAC on t-PA amidolytic activity. B, effect of VDAC on u-PA amidolytic activity. C, effect of VDAC on Pm amidolytic activity. D, effect of increasing concentrations of tranexamic acid (TXA) on t-PA (10 nm) amidolytic activity in the presence of VDAC (100 nm). The data are the means ± S.D. (error bars) from experiments performed in triplicate (n = 6).
Effect of VDAC on t-PA-mediated Plasminogen Activation
Initial velocities of Pg activation by t-PA were obtained under steady-state conditions. The velocity curves in the absence or presence of VDAC (100 nm) show a dose-dependent increase in the activation velocity (Fig. 3A). The rates of plasmin formation were plotted against the initial Pg concentration, and the data were extrapolated to the Michaelis-Menten equation by nonlinear regression using GraphPad Prism version 5 to determine the kcat and Km (Table 1). The kinetic constants obtained for t-PA are consistent with previously reported values (27). The stimulatory effect of VDAC was caused primarily by a decrease in Km and an increase in Vmax. The kcat/Km for the activation reaction calculated from these data increased from 0.84 to 5.27 μm−1 s−1, resulting in an overall 6.27-fold increase in activation efficiency. We also evaluated whether increasing concentrations of the anti-VDAC N-18 IgG (Fig. 3B) and the anti-VDAC 235–255 IgG (Fig. 3C) could inhibit the activation of Pg (200 nm) by t-PA (10 nm) in the presence of VDAC (100 nm). We observed a decreasing rate of Pg activation as the concentration of both antibodies increased, confirming that the formation of a ternary complex among t-PA, Pg, and VDAC is necessary to activate this mechanism. Because protein aggregation has been reported as a major cofactor for t-PA-mediated plasmin formation on injured brain cells (40), we also assessed whether aggregated VDAC (produced by heat denaturation) could act as a cofactor for t-PA-mediated plasmin formation. Although exposure of VDAC to heat induced a modest decrease in the rate of plasmin formation by t-PA (Fig. 3D), the protein exposed to 100 °C continued to stimulate Pg activation (2-fold), supporting the role of aggregated VDAC as a major cofactor in this mechanism.
FIGURE 3.

Kinetics of Pg activation by t-PA in the presence of native VDAC, native VDAC in the presence of anti-VDAC antibodies, or heat-denatured VDAC. Coupled assays were used to evaluate the initial rate of Glu-Pg activation by t-PA by monitoring the amidolytic activity of the generated Pm. A, Glu-Pg was preincubated in 96-well microtiter plates for 15 min at 37 °C in 20 mm HEPES, pH 7.4 in a total volume of 200 μl in the absence (●) or presence (▴) of VDAC (100 nm) followed by the addition of the plasmin substrate VLK-pNA (0.3 mm) and t-PA (0.55 nm). B, a single concentration of Glu-Pg (200 nm) was incubated as above with t-PA (0.55 nm) in the presence of increasing concentrations of the anti-VDAC N-18 IgG. C, a single concentration of Glu-Pg (200 nm) was incubated as above with t-PA (0.55 nm) in the presence of increasing concentrations of anti-VDAC 235–255 IgG. D, a single concentration of Glu-Pg (200 nm) was incubated as above with t-PA (0.55 nm) in the presence of a single concentration of VDAC (100 nm) heated to increasing temperatures. The data are the means ± S.D. (error bars) from experiments performed in triplicate (n = 6).
TABLE 1.
Kinetic parameters of Glu-Pg activation by t-PA in the presence of VDAC
| Protein | Vmax | Km | kcat | kcat/Km | kcat/Km -fold increase |
|---|---|---|---|---|---|
| mol Pm/min × 10−13 | μm | s−1 | μm−1 s−1 | ||
| None | 54.2 | 0.19 | 0.16 | 0.84 | |
| VDAC | 111.1 | 0.11 | 0.58 | 5.27 | 6.27 |
The Role of VDAC in Injured Cells
The function of VDAC in apoptotic or dead cells was further investigated in SK-N-SH neuroblastoma cells exposed to increasing concentrations of STS. Cells cultured in 96-well plates were incubated for 16 h in the presence of 100, 200, and 500 nm STS. Binding of t-PA and Pg was then assessed. The results show that binding of t-PA (Fig. 4A) and Pg (Fig. 4B) increased as the concentration of STS increased. Using flow cytometric techniques, we evaluated the levels of VDAC in untreated live and dead cells and found that the dead cells had a higher percentage of VDAC-positive cells (Fig. 4C). We then found that a larger population of both live and apoptotic cells treated with STS showed expression of VDAC compared with the untreated cell populations (Fig. 4D). The results are summarized in Table 2. Analyses of the mRNA levels by PCR amplification showed a decrease of VDAC1 mRNA expression in cells treated with STS compared with untreated SK-N-SH cells (Fig. 5A). Because a common process in apoptotic cells is the migration of mitochondria toward the surface (41) and VDAC is a major protein in the outer membrane of mitochondria (1), we investigated whether its expression on the cell surface was correlated with the expression of another major mitochondrial membrane enzyme, COX IV (42), on SK-N-SH cells treated with STS. We found that increases in VDAC (Fig. 5B) and COX IV (Fig. 5C) are correlated (Fig. 5D) in cells treated with STS. These experiments suggest that the increase of VDAC expression at the surface of injured cells is the result of mitochondrial migration toward the surface.
FIGURE 4.

Effect of STS on t-PA or Pg binding and VDAC expression in SK-N-SH cells. Experiments were performed on cells grown in 96-well tissue culture plates (2 × 104 cells/well). A, increasing concentrations of t-PA were added to SK-N-SH cell monolayers in the absence or presence of three different STS concentrations (100, 200, and 500 nm). B, increasing concentrations of Pg were added to SK-N-SH cell monolayers in the absence or presence of three different STS concentrations (100, 200, and 500 nm). C, analysis of VDAC expression on live and dead untreated SK-N-SH neuroblastoma cells by flow cytometric analysis. D, flow cytometric analyses of VDAC expression on SK-N-SH neuroblastoma cells incubated with two STS concentrations (200 and 500 nm) for 16 h. The data are the mean ± S.D. (error bars). ***, p < 0.001 compared with the untreated control. *, p < 0.05; **, p < 0.01.
TABLE 2.
Effect of staurosporine on VDAC expression by SK-N-SH neuroblastoma cells
The data are the mean ± S.D.
| Treatment | Cell population |
|
|---|---|---|
| VDAC-positive live cells | VDAC-positive apoptotic cells | |
| % | ||
| 0.2 μm staurosporine | 36.2 ± 3.8a | 27.4 ± 4.3a |
| 0.5 μm staurosporine | 31.1 ± 4.6a | 33.1 ± 5.1a |
| Untreated | 17.0 ± 3.8 | 15.6 ± 3.6 |
a p < 0.001 compared with the untreated control.
FIGURE 5.

Effect of STS on VDAC and COX IV expression at the surface of SK-N-SH cells. A, cell monolayers in 6-well culture plates (2.5 × 106 cells/well) were incubated in culture medium containing increasing concentrations of STS (50, 100, 200, and 500 nm) at 37 °C overnight. Isolation of the total RNA and measurements of VDAC1 mRNA by reverse transcription-PCR were performed as described under “Experimental Procedures.” Ethidium bromide-stained gels were photographed and analyzed by densitometric scanning. VDAC1 mRNA levels were expressed as relative VDAC1 mRNA/β-actin mRNA ratios. The values represent the means ± S.D. (error bars) of three separate experiments. B, flow cytometric analysis of VDAC expression on live, dead, and apoptotic SK-N-SH neuroblastoma cells incubated with two STS concentrations (200 and 500 nm) for 16 h. C, flow cytometric analysis of COX IV expression on live, dead, and apoptotic SK-N-SH neuroblastoma cells incubated with two STS concentrations (200 and 500 nm) for 16 h. D, flow cytometric analysis of co-expression of VDAC and COX IV on live, dead, and apoptotic SK-N-SH neuroblastoma cells incubated with two STS concentrations (200 and 500 nm) for 16 h. The data are the mean ± S.D. (error bars). ***, p < 0.001 compared with the untreated control.
We also evaluated the in vitro effect of VDAC on t-PA-mediated activation of Pg structural forms, such as mPg or μPg. The results demonstrate that VDAC also stimulated t-PA-mediated activation of these Pg structural forms (Fig. 6A). A similar action was observed with the Gly10–Leu30 peptide of the VDAC NH2-terminal domain (Fig. 6B), confirming that this VDAC region is involved in the binding and stimulation of t-PA activity. VDAC binds Pg or mPg via K5 (6); however, t-PA-mediated activation of μPg, which lacks K5, was also stimulated in addition to t-PA-mediated activation of Pg or mPg. Furthermore, the VDAC Gly10–Leu30 peptide, which does not bind to either Pg form, also stimulated t-PA activity. It is clear from this experiment that μPg in the solution phase (Fig. 6, A and B, lanes 5 and 6, respectively) is as good a substrate for the VDAC·t-PA complex or the Gly10–Leu30 peptide·t-PA complex as Pg or mPg (Fig. 6, A and B, lanes 2 and 4, respectively). Therefore, although VDAC bound to both t-PA and Pg, only the catalytic efficiency of t-PA was enhanced as a consequence of this interaction as shown above in Table 1.
FIGURE 6.
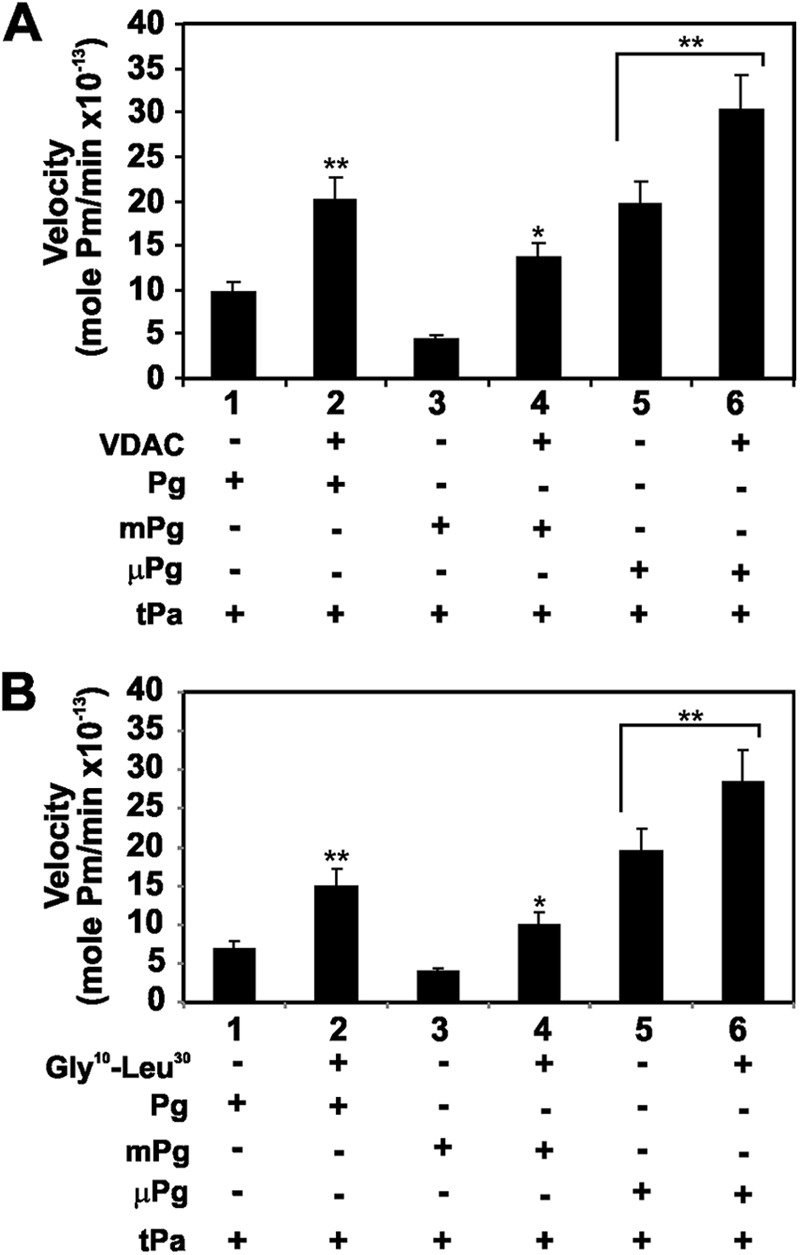
Effect of VDAC and the VDAC N-terminal domain Gly10—Leu30 peptide on the activation of Pg and its truncated forms by t-PA. Pg and its truncated forms at a single concentration (100 nm) were activated by t-PA (10 nm) in the presence of a single concentration of VDAC (100 nm). The amidolytic activity of the corresponding Pm forms was monitored with the chromogenic Pm substrate S-2251. A, t-PA-mediated activation of Pg, mPg, and μPg in the presence (+) or absence (−) of VDAC. B, t-PA-mediated activation of Pg, mPg, or μPg in the presence (+) or absence (−) of the VDAC N-terminal domain Gly10–Leu30 peptide. The data are the means ± S.D. (error bars). **, p < 0.001 and *, p < 0.05 from experiments (n = 6) performed in the presence of VDAC or Gly10–Leu30 peptide and compared with t-PA-mediated activation of the Pg forms in the absence of these proteins.
Control of Pg Activity by VDAC
Pg and Pm bind to VDAC via its K5 domains (6), and VDAC is a receptor that functions as a ferricyanide reductase on the cell surface, an activity that is maintained by the solubilized VDAC protein (43). We hypothesized that K5 could also serve as a substrate for this VDAC reductase activity. We found that the recombinant human VDAC is active as an NADH-ferricyanide reductase and as a K5-dependent reductase for all the Pg forms except μPg, which lacks this structural domain (Fig. 7A). The NADH-K5-dependent reductase activity was also tested in Pg fragments (200 nm) incubated with t-PA (5 nm) and VDAC (100 nm) for 2 h and then labeled with MBP. The fragments were resolved by 10% SDS-PAGE, transferred to a nitrocellulose membrane, and blotted with streptavidin-peroxidase to detect the MPB label. Only the Pg fragments containing the K5 domain incorporated MBP after reduction with VDAC (Fig. 7B). The results of this experiment also suggest that the NADH-dependent reductase activity is specific for Pg because no incorporation of MBP was observed in the t-PA size range (70 kDa; lanes 8 and 9). Because it was reported previously that plasmin is a key protease in the degradation of injured neuronal cells (40) and that this activity is stimulated by a cofactor that in turn is degraded by plasmin, we investigated whether plasmin degrades VDAC. The results demonstrate that VDAC in addition to its capacity to stimulate Pg activation may also be a substrate for plasmin. Proteolytic degradation was observed only when VDAC was incubated with Pg and t-PA and was absent when VDAC was incubated with t-PA alone, Pg alone, or Pg and t-PA in the presence of the plasmin inhibitor aprotinin (Fig. 7C). Although the samples were separated under reducing conditions, the appearance of higher molecular weight protein bands in the mixture containing plasmin-digested VDAC could be the result of nonspecific peptide aggregation (Fig. 7C, lane 3).
FIGURE 7.

NADH-dependent oxidoreductase activity of VDAC on Pg forms. The assays were performed at 37 °C in 1-jml reactions containing VDAC (100 nm) and β-NADH (25 μm) in 50 mm Tris-HCl, pH 8.0. The reaction was initiated by the addition of potassium ferricyanide (500 nm) (positive control) or Pg fragments (200 nm). The rates were measured as described under “Experimental Procedures.” A, effect of Pg forms on the NADH-dependent oxidoreductase activity of VDAC. The data are the means ± S.D. (error bars). B, reduction of Pg forms by VDAC. The assays were performed as described under “Experimental Procedures.” The three Pg forms were incubated with t-PA in the presence or absence of VDAC and then incubated with MPB (100 μm) for 30 min at room temperature followed by quenching of the unreacted MPB with GSH (200 μm) for 10 min at room temperature. Unreacted GSH and other free sulfhydryls in the system were blocked with iodoacetamide (400 μm) for 10 min at room temperature. The proteins were separated by 10% SDS-PAGE under non-reducing conditions, transferred to nitrocellulose membranes, and incubated with streptavidin-peroxidase to detect the MPB-labeled proteins. Lanes 1–3, blots of activated Pg, mPg, and μPg incubated with a goat anti-human Pg IgG followed by incubation with an alkaline phosphatase-conjugated secondary antibody and developed using 5-bromo-4-chloroindolyl-3-phosphate nitroblue tetrazolium. Lanes 4–6, blots of Pg, mPg, and μPg activated with t-PA in the absence of VDAC followed by incubation with MPB and separation by electrophoresis. The proteins were transferred to nitrocellulose and incubated with streptavidin-peroxidase. Lanes 7–9, blots of Pg, mPg, and μPg activated with t-PA in the presence of VDAC followed by incubation with MPB, separation by electrophoresis, transfer to nitrocellulose, and reaction with streptavidin-peroxidase. C, proteolytic degradation of VDAC by t-PA-mediated Pg activation. VDAC (10 μg) was incubated with Pg alone (0.5 μg), Pg (0.5 μg) and t-PA (0.1 μg), or Pg (0.5 μg) with t-PA (0.1 μg) and aprotinin (1 μg) for 2 h at 37 °C. The proteins were separated by 4–20% SDS-PAGE under reducing conditions and stained as described under “Experimental Procedures.”
DISCUSSION
The Pm proteolytic cascade is primarily involved with fibrin proteolysis and cell migration. This process begins with the localized synthesis and secretion of t-PA and u-PA, activating Pg to yield the active serine protease Pm, which then proteolyzes its target proteins (44). The role of the Pm system in the brain has been intensely scrutinized mainly because Pg and t-PA are synthesized in neurons and microglia (11) and because Pm generation has been implicated in neuronal loss (45, 46). Furthermore, the expression of Pg in the brain is critical for resistance to neurodegenerative disease and vice versa (47). On the cell surface, Pg binds to a large and heterogeneous number of receptors (48, 49). A common consequence of the association of Pg with cell surface receptors is the change in Km between Pg and its activators that promotes Pm formation even in the absence of fibrin, facilitates specific signaling pathways (49), and protects Pm from inhibition by the primary Pm inhibitor α2-antiplasmin (50), thereby facilitating its proteolytic activity. Thus, the finding that VDAC, a known Pg receptor (6), also functions as a t-PA-binding protein prompted us to investigate the physiology of the interaction between these three proteins.
The activity of t-PA is strongly enhanced by fibrin, and t-PA is therefore predominantly responsible for plasmin-catalyzed fibrin dissolution in the maintenance of vascular hemostasis (51). In addition to fibrin, t-PA is activated by numerous unrelated proteins, including denatured forms of albumin, fibrinogen, and endostatin (35), all of which contain cross-β structure motifs that are typical of misfolded proteins with amyloid-like properties (52). These observations help explain the mechanism by which t-PA binds such diverse ligands. It is known that amyloid β peptides stimulate t-PA-mediated Pg activation (37) but have no effect on u-PA-mediated Pg activation (54). Our in vitro experiments suggest that the cofactor activity of VDAC is specific for t-PA and does not affect preformed plasmin, which also binds to VDAC (6). This specificity was further confirmed because VDAC also stimulated t-PA-mediated μPg activation. This Pg form lacks the K5 necessary to bind to VDAC, and it is present only in the solution phase. Although we have not tested the capacity of μPg to bind to VDAC, both K5 and μPg possess a benzamidine-binding site (19); therefore, we hypothesize that μPg binds to VDAC. In the cellular environment, this mechanism may act as an alternative to activation for μPg bound to glucose-regulated protein 78 (GRP78) (55). VDAC and GRP78 are co-localized at the cell surface (55). K5 binds GRP78 with an affinity 5-fold higher than that of μPg (55); however, K5 and μPg bind to different sites on GRP78 (55). From previous studies, we know that μPg binds to human umbilical vein endothelial cells and prostate cancer 1-LN cells (6, 55). Although we have not assessed μPg binding to neuroblastoma SK-N-SH cells or immobilized VDAC, we hypothesize that the reduction of K5 by VDAC on the surface may confer μPg a kinetic advantage over K5 for binding to VDAC on the cell surface.
The activation of Pg by t-PA produces plasmin, which specifically cleaves Aβ 1–40 in the NH2-terminal region between Arg5 and His6. The Aβ 6–40 peptide adopts a strong β-sheet secondary structure, which acts as a potent stimulator of t-PA activity in vitro (56). In situ generated amyloid β peptides have the potential to bind to VDAC, which by opening the channel may induce apoptosis if left unchecked (57). Maintaining VDAC in an open state requires the binding of aggregated amyloid β peptide; however, Pm prevents this aggregation (58) and in turn further stimulates t-PA activity (56). The effect of the amyloid β peptides on t-PA was only partially antagonized by tranexamic acid or fibrin monomers, suggesting that they bind via lysine-dependent or lysine-independent sites on t-PA (35). The lysine-independent amyloid β peptide-binding site is a fibronectin type I finger domain located in the NH2-terminal region that binds to the aggregated amyloid β peptide (34). Our results suggest that this site may be involved in the binding of t-PA to VDAC. As discussed above, the 25GSNKG29 repeat motif is present in all amyloid β peptides and significantly inhibits the binding of t-PA to SK-N-SH cells or VDAC. A similar motif, 20GYGFG24, is also present in the VDAC NH2-terminal domain and appears to mediate binding of VDAC to t-PA. Our data suggest that VDAC provides a binding site for t-PA via a mechanism that is independent of cross-β-sheet formation, similar to the binding of the Aβ 6–40 peptide (56). A further confirmation of the lack of cross-β-sheet formation between VDAC and t-PA was inferred from the inability of the t-PA peptide 509CQGDSGGPLVC519 containing the GDSGG motif to act as a competitor of t-PA binding to SK-N-SH cells or immobilized VDAC.
VDAC may act as a non-fibrin cofactor that stimulates t-PA-mediated Pg activation and as suggested by our data may act in either the normal native state or the aggregated state. It is known that protein aggregates formed in injured brain cells act as cofactors for t-PA-mediated Pg activation in a large number of nonviable brain cells, and this is independent of species, cell lineage, or type of injury (40). We found that a normal culture of SK-N-SH neuroblastoma cells contains numerous dead cells expressing VDAC in a greater ratio than VDAC expressed by living cells. Similarly, the induction of apoptosis in SK-N-SH cells by STS increased the expression of VDAC compared with a population of untreated cells. This overexpression of VDAC was accompanied by an increase in t-PA and Pg binding. It is known that t-PA-mediated plasmin generation coincides with rapid necrosis and formation of aggregates in injured neurons (40). During this stage, there is a significant reduction in cell size, which depends on plasmin-mediated proteolytic degradation (39). Our data suggest that during injury VDAC overexpressed at the cell surface may originate from the migration of mitochondria toward the surface, and we know from a previous study that mitochondrial VDAC binds Pg K5 (6). Furthermore, our data also suggest that VDAC in addition to facilitating t-PA-mediated plasmin generation may also serve as a plasmin substrate in a manner similar to fibrin (51).
In normal cells, VDAC interacts with Pg K5 and induces hyperpolarization of the mitochondrial membrane (6), which results in partial closure of the channel, leading to inhibition of ATP transport and other metabolites across the mitochondrial membrane (7). These non-fibrinolytic roles of Pg make it an important player in the CNS physiology of both healthy and diseased states (47). Pg K5 is a highly specific antiangiogenic molecule (59) that affects endothelial cell adhesion, proliferation, and migration (60). Thus, after Pg is converted to Pm on the cell surface, K5 is rapidly inactivated by disulfide reductases, such as phosphoglycerate kinase (61) and annexin II (62), and proteolysis of Pm occurs. Because annexin II also serves as a receptor of t-PA and Pg (49) and is able to reduce K5, a similar function for the VDAC in complex with t-PA and Pg is not an uncommon mechanism for the binding and release of these proteins from the cell surface. Because K5 binding to VDAC induces apoptosis (55), this mechanism would increase the protection of the cell from harm by this molecule at the cell surface level.
Based on the experimental evidence from our laboratory and others, we constructed a working model that illustrates the interactions and possible functions of VDAC, t-PA, and Pg on the cell surface (Fig. 8). Pg binds to a VDAC domain including amino acids Gly244–Leu257 via its K5 (6). t-PA binds via its fibronectin type I finger domain, which is located between amino acids Ile5 and Asn37, to a domain in VDAC including amino acids 20GYGFG24. Binding of t-PA to VDAC is facilitated by exposure of its NH2-terminal region, which in the plasma membrane is facing toward the surface of the cell (63). Pg binding to VDAC induces closure of the channel, whereas t-PA may be involved in the opening of the channel. Both functions may lead to apoptosis; however, once converted to Pm, the K5 region may be reduced by the NADH-dependent activity of VDAC, and Pm would be rapidly released from the surface of the channel. At the NH2 terminus, t-PA can be displaced from VDAC by amyloid β peptides on the surface. These amyloid β peptides have the potential to aggregate and bind to VDAC, leading to apoptosis (64). However, Pm prevents aggregation of these amyloid β peptides via cleavage of Aβ 1–40 between Arg5 and His6, a site in the RHDS sequence of Aβ known to promote cell adhesion (53), and maintains the capacity of the Aβ peptide to stimulate Pg activation or displace t-PA from VDAC. We hypothesize that such a mechanism may facilitate the rapid removal of Aβ peptides from the cell surface in a normal brain, thereby preventing apoptosis. However, in injured brain tissue, the fibrin-mimicking activity of aggregated VDAC may potentiate t-PA-mediated plasmin formation, thereby enhancing cell proteolytic degradation, which facilitates the removal of cellular debris.
FIGURE 8.

Model illustrating the interactions of VDAC with t-PA and Pg on the cell surface. Pg binds VDAC at a site including the amino acid sequence Gly244–Leu257. Binding of t-PA at a site including the amino acid sequence Gly20–Gly24 promotes activation of Pg by t-PA. The Pm generated may proteolyze the Aβ 1–42 peptide, which in turn may displace t-PA from the VDAC receptor by competing for binding to VDAC. The Pm-truncated Aβ 1–42 peptide cannot aggregate and is rapidly cleared from the receptor surface. Additionally, Pm can be released from the receptor through reduction of its K5 via the VDAC NADH-dependent oxidoreductase activity.
Footnotes
- VDAC
- voltage-dependent anion channel
- t-PA
- tissue-type plasminogen activator
- Pg
- plasminogen
- K
- kringle
- u-PA
- urokinase-type Pg activator
- LRP
- low density lipoprotein receptor-related protein receptor
- MPB
- 3-(N-maleimidylpropionyl)biocytin
- mPg
- mini-Pg
- μPg
- microplasminogen
- COX IV
- cytochrome c oxidase IV
- HBSS
- Hanks' balanced salt solution
- STS
- staurosporine
- Pm
- plasmin
- pNA
- p-nitroanilide
- Aβ
- amyloid β
- GRP78
- glucose-regulated protein 78.
REFERENCES
- 1. Colombini M. (1979) A candidate for the permeability pathway of the outer mitochondrial membrane. Nature 279, 643–645 [DOI] [PubMed] [Google Scholar]
- 2. De Pinto V., Messina A., Lane D. J, Lawen A. (2010) Voltage-dependent anion-selective channel (VDAC) in the plasma membrane. FEBS Lett. 584, 1793–1799 [DOI] [PubMed] [Google Scholar]
- 3. Okada S. F., O'Neal W. K., Huang P., Nicholas R. A., Ostrowski L. E., Craigen W. J., Lazarowski E. R., Boucher R. C. (2004) Voltage-dependent anion channel-1 (VDAC-1) contributes to ATP release and cell volume regulation in murine cells. J. Gen. Physiol. 124, 513–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baker M. A., Ly J. D., Lawen A. (2004) Characterization of VDAC1 as a plasma membrane NADH-oxidoreductase. BioFactors 21, 215–221 [DOI] [PubMed] [Google Scholar]
- 5. McEnery M. W., Dawson T. M., Verma A., Gurley D., Colombini M., Snyder S. H. (1993) Mitochondrial voltage-dependent anion channel. Immunochemical and immunohistochemical characterization in rat brain. J. Biol. Chem. 268, 23289–23296 [PubMed] [Google Scholar]
- 6. Gonzalez-Gronow M., Kalfa T., Johnson C. E., Gawdi G., Pizzo S. V. (2003) The voltage-dependent anion channel is a receptor for plasminogen kringle 5 on human endothelial cells. J. Biol. Chem. 278, 27312–27318 [DOI] [PubMed] [Google Scholar]
- 7. Banerjee J., Ghosh S. (2004) Interaction of mitochondrial voltage-dependent anion channel from rat brain with plasminogen protein leads to partial closure of the channel. Biochim. Biophys. Acta 1663, 6–8 [DOI] [PubMed] [Google Scholar]
- 8. Sappino A. P., Madani R., Huarte J., Belin D., Kiss J. Z., Wohlwend A., Vassalli J. D. (1993) Extracellular proteolysis in the adult murine brain. J. Clin. Investig. 92, 679–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Melchor J. P., Strickland S. (2005) Tissue plasminogen activator in central nervous system physiology and pathology. Thromb. Haemost. 93, 655–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cunningham O., Campion S., Perry V. H., Murray C., Sidenius N., Docagne F., Cunningham C. (2009) Microglia and the urokinase plasminogen activator receptor/uPA system in innate brain inflammation. Glia 57, 1802–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tsirka S. E., Rogove A. D., Bugge T. H., Degen J. L., Strickland S. (1997) An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J. Neurosci. 17, 543–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pawlak R., Rao B. S., Melchor J. P., Chattarji S., McEwen B., Strickland S. (2005) Tissue plasminogen activator and plasminogen mediate stress-induced decline of neuronal and cognitive functions in the mouse hippocampus. Proc. Natl. Acad. Sci. U.S.A. 102, 18201–18206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gonzalez-Gronow M., Cuchacovich M., Francos R., Cuchacovich S., del Pilar Fernandez M., Blanco A., Bowers E. V., Kaczowka S., Pizzo S. V. (2010) Antibodies against the voltage-dependent anion channel (VDAC) and its protective ligand hexokinase-I in children with autism. J. Neuroimmunol. 227, 153–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bu G., Williams S., Strickland D. K., Schwartz A. L. (1992) Low density lipoprotein receptor-related protein/α2-macroglobulin receptor is an hepatic receptor for tissue-type plasminogen activator. Proc. Natl. Acad. Sci. U.S.A. 89, 7427–7431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hajjar K. A., Jacovina A. T., Chacko J. (1994) An endothelial cell receptor for plasminogen/tissue plasminogen activator. I. Identity with annexin II. J. Biol. Chem. 269, 21191–21197 [PubMed] [Google Scholar]
- 16. Barrett-Bergshoeff M., Noorman F., Bos R., Rijken D. C. (1997) Monoclonal antibodies against the human mannose receptor that inhibit the binding of tissue-type plasminogen activator. Thromb. Haemost. 77, 718–724 [PubMed] [Google Scholar]
- 17. Deutsch D. G., Mertz E. T. (1970) Plasminogen: purification from human plasma by affinity chromatography. Science 170, 1095–1096 [DOI] [PubMed] [Google Scholar]
- 18. Sottrup-Jensen L., Zajdel M., Claeys H., Petersen T. E., Magnusson S. (1975) Amino-acid sequence of activation cleavage site in plasminogen: homology with “pro” part of prothrombin. Proc. Natl. Acad. Sci. U.S.A. 72, 2577–2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Váradi A., Patthy L. (1981) Kringle 5 of human plasminogen carries a benzamidine-binding site. Biochem. Biophys. Res. Commun. 103, 97–102 [DOI] [PubMed] [Google Scholar]
- 20. Cao Y., Chen A., An S. S., Ji R. W., Davidson D., Llinás M. (1997) Kringle 5 of plasminogen is a novel inhibitor of endothelial cell growth. J. Biol. Chem. 272, 22924–22928 [DOI] [PubMed] [Google Scholar]
- 21. Thewes T., Ramesh V., Simplaceanu E. L., Llinás M. (1987) Isolation, purification and 1H-NMR characterization of a kringle 5 domain fragment from human plasminogen. Biochim. Biophys. Acta 912, 254–269 [DOI] [PubMed] [Google Scholar]
- 22. Shi G. Y., Wu H. L. (1988) Isolation and characterization of microplasminogen. A low molecular weight form of plasminogen. J. Biol. Chem. 263, 17071–17075 [PubMed] [Google Scholar]
- 23. Shi Y., Jiang C., Chen Q., Tang H. (2003) One-step on-column affinity refolding purification and functional analysis of recombinant human VDAC1. Biochem. Biophys. Res. Commun. 303, 475–482 [DOI] [PubMed] [Google Scholar]
- 24. Markwell M. A. (1982) A new solid-state reagent to iodinate proteins. I. Conditions for the efficient labeling of antiserum. Anal. Biochem. 125, 427–432 [DOI] [PubMed] [Google Scholar]
- 25. Chae H. J., Kang J. S., Byun J. O., Han K. S., Kim D. U., Oh S. M., Kim H. M., Chae S. W., Kim H. R. (2000) Molecular mechanism of staurosporine-induced apoptosis in osteoblasts. Pharmacol. Res. 42, 373–381 [DOI] [PubMed] [Google Scholar]
- 26. Wohl R. C., Summaria L., Robbins K. C. (1980) Kinetics of activation of human plasminogen by different activator species at pH 7.4 and 37 °C. J. Biol. Chem. 255, 2005–2013 [PubMed] [Google Scholar]
- 27. Stack S., Gonzalez-Gronow M., Pizzo S. V. (1990) Regulation of plasminogen activation by components of the extracellular matrix. Biochemistry 29, 4966–4970 [DOI] [PubMed] [Google Scholar]
- 28. Erlanger B. F., Kokowsky N., Cohen W. (1961) The preparation and properties of two new chromogenic substrates of trypsin. Arch. Biochem. Biophys. 95, 271–278 [DOI] [PubMed] [Google Scholar]
- 29. Gimel J. C., Durand D., Nicolai T. (1994) Structure and distribution of aggregates formed after heat-induced denaturation of globular proteins. Macromolecules 27, 583–589 [Google Scholar]
- 30. Øbstevø R., Haug K. B., Lande K., Kierulf P. (2003) PCR-based calibration curves for studies of quantitative gene expression in human monocytes: development and evaluation. Clin. Chem. 49, 425–432 [DOI] [PubMed] [Google Scholar]
- 31. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 32. Towbin H., Staehelin T., Gordon J. (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U.S.A. 76, 4350–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gonzalez-Gronow M., Gawdi G., Pizzo S. V. (2002) Tissue factor is the receptor for plasminogen type 1 on 1-LN human prostate cancer cells. Blood 99, 4562–4567 [DOI] [PubMed] [Google Scholar]
- 34. Stathakis P., Fitzgerald M., Matthias L. J., Chesterman C. N., Hogg P. J. (1997) Generation of angiostatin by reduction and proteolysis of plasmin. Catalysis by a plasmin reductase secreted by cultured cells. J. Biol. Chem. 272, 20641–20645 [DOI] [PubMed] [Google Scholar]
- 35. Maas C., Schiks B., Strangi R. D., Hackeng T. M., Bouma B. N., Gebbink M. F., Bouma B. (2008) Identification of fibronectin type I domains as amyloid-binding modules on tissue-type plasminogen activator and three homologs. Amyloid 15, 166–180 [DOI] [PubMed] [Google Scholar]
- 36. Liu W., Crocker E., Zhang W., Elliott J. I., Luy B., Li H., Aimoto S., Smith S. O. (2005) Structural role of glycine in amyloid fibrils formed from transmembrane α-helices. Biochemistry 44, 3591–3597 [DOI] [PubMed] [Google Scholar]
- 37. Kingston I. B., Castro M. J., Anderson S. (1995) In vitro stimulation of tissue-type plasminogen activator by Alzheimer amyloid β-peptide analogues. Nat. Med. 1, 138–142 [DOI] [PubMed] [Google Scholar]
- 38. Hoylaerts M., Lijnen H. R., Collen D. (1981) Studies on the mechanism of the antifibrinolytic action of tranexamic acid. Biochim. Biophys. Acta 673, 75–85 [PubMed] [Google Scholar]
- 39. Bizik J., Stephens R. W., Grofova M., Vaheri A. (1993) Binding of tissue-type plasminogen activator to human melanoma cells. J. Cell. Biochem. 51, 326–335 [DOI] [PubMed] [Google Scholar]
- 40. Samson A. L., Borg R. J., Niego B., Wong C. H., Crack P. J., Yongqing T., Medcalf R. L. (2009) A nonfibrin macromolecular cofactor for tPA-mediated plasmin generation following cellular injury. Blood 114, 1937–1946 [DOI] [PubMed] [Google Scholar]
- 41. Elmore S. (2007) Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ostermeier C., Iwata S., Michel H. (1996) Cytochrome c oxidase. Curr. Opin. Struct. Biol. 6, 460–466 [DOI] [PubMed] [Google Scholar]
- 43. Baker M. A., Lane D. J., Ly J. D., De Pinto V., Lawen A. (2004) VDAC1 is a transplasma membrane NADH-ferricyanide reductase. J. Biol. Chem. 279, 4811–4819 [DOI] [PubMed] [Google Scholar]
- 44. Reuning U., Magdolen V., Wilhelm O., Fischer K., Lutz V., Graeff H., Schmitt M. (1998) Multifunctional potential of the plasminogen activation system in tumor invasion and metastasis (review). Int. J. Oncol. 13, 893–906 [DOI] [PubMed] [Google Scholar]
- 45. Chen Z. L., Strickland S. (1997) Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell 91, 917–925 [DOI] [PubMed] [Google Scholar]
- 46. Wang Y. F., Tsirka S. E., Strickland S., Stieg P. E., Soriano S. G., Lipton S. A. (1998) Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat. Med. 4, 228–231 [DOI] [PubMed] [Google Scholar]
- 47. Tsirka S. E., Bugge T. H., Degen J. L., Strickland S. (1997) Neuronal death in the central nervous system demonstrates a non-fibrin substrate for plasmin. Proc. Natl. Acad. Sci. U.S.A. 94, 9779–9781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gonzalez-Gronow M., Kaczowka S., Gawdi G., Pizzo S. V. (2008) Dipeptidyl peptidase IV (DPP IV/CD26) is a cell-surface plasminogen receptor. Front. Biosci. 13, 1610–1618 [DOI] [PubMed] [Google Scholar]
- 49. Das R., Pluskota E., Plow E. F. (2010) Plasminogen and its receptors as regulators of cardiovascular inflammatory responses. Trends Cardiovasc. Med. 20, 120–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Miles L. A., Hawley S. B., Baik N., Andronicos N. M., Castellino F. J., Parmer R. J. (2005) Plasminogen receptors: the sine qua non of cell surface plasminogen activation. Front. Biosci. 10, 1754–1762 [DOI] [PubMed] [Google Scholar]
- 51. Hoylaerts M., Rijken D. C., Lijnen H. R., Collen D. (1982) Kinetics of the activation of plasminogen by human tissue plasminogen activator. Role of fibrin. J. Biol. Chem. 257, 2912–2919 [PubMed] [Google Scholar]
- 52. Kranenburg O., Bouma B., Kroon-Batenburg L. M., Reijerkerk A., Wu Y. P., Voest E. E., Gebbink M. F. (2002) Tissue-type plasminogen activator is a multiligand cross-β structure receptor. Curr. Biol. 12, 1833–1939 [DOI] [PubMed] [Google Scholar]
- 53. Ghiso J., Rostagno A., Gardella J. E., Liem L., Gorevic P. D., Frangione B. (1992) A 109-amino-acid C-terminal fragment of Alzheimer's-disease amyloid precursor protein contains a sequence, -RHDS-, that promotes cell adhesion. Biochem. J. 288, 1053–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wnendt S., Wetzels I., Günzler W. A. (1997) Amyloid β peptides stimulate tissue-type plasminogen activator but not recombinant prourokinase. Thromb Res. 85, 217–224 [DOI] [PubMed] [Google Scholar]
- 55. Gonzalez-Gronow M., Kaczowka S. J., Payne S., Wang F., Gawdi G., Pizzo S. V. (2007) Plasminogen structural domains exhibit different functions when associated with cell surface GRP78 or the voltage-dependent anion channel. J. Biol. Chem. 282, 32811–32820 [DOI] [PubMed] [Google Scholar]
- 56. Van Nostrand W. E., Porter M. (1999) Plasmin cleavage of the amyloid β-protein: alteration of secondary structure and stimulation of tissue plasminogen activator activity. Biochemistry 38, 11570–11576 [DOI] [PubMed] [Google Scholar]
- 57. Marin R., Ramírez C. M., González M., González-Muñoz E., Zorzano A., Camps M., Alonso R., Díaz M. (2007) Voltage-dependent anion channel (VDAC) participates in amyloid β-induced toxicity and interacts with plasma membrane estrogen receptor α in septal and hippocampal neurons. Mol. Membr. Biol. 24, 148–160 [DOI] [PubMed] [Google Scholar]
- 58. Tucker H. M., Kihiko M., Caldwell J. N., Wright S., Kawarabayashi T., Price D., Walker D., Scheff S., McGillis J. P., Rydel R. E., Estus S. (2000) The plasmin system is induced by and degrades amyloid-β aggregates. J. Neurosci. 20, 3937–3946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ji W. R., Barrientos L. G., Llinás M., Gray H., Villarreal X., DeFord M. E., Castellino F. J., Kramer R. A., Trail P. A. (1998) Selective inhibition by kringle 5 of human plasminogen on endothelial cell migration, an important process in angiogenesis. Biochem. Biophys. Res. Commun. 247, 414–419 [DOI] [PubMed] [Google Scholar]
- 60. Park K., Chen Y., Hu Y., Mayo A. S., Kompella U. B., Longeras R., Ma J. X. (2009) Nanoparticle-mediated expression of an angiogenic inhibitor ameliorates ischemia-induced retinal neovascularization and diabetes-induced retinal vascular leakage. Diabetes 58, 1902–1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lay A. J., Jiang X. M., Kisker O., Flynn E., Underwood A., Condron R., Hogg P. J. (2000) Phosphoglycerate kinase acts in tumour angiogenesis as a disulphide reductase. Nature 408, 869–873 [DOI] [PubMed] [Google Scholar]
- 62. Kwon M., Caplan J. F., Filipenko N. R., Choi K. S., Fitzpatrick S. L., Zhang L., Waisman D. M. (2002) Identification of annexin II heterotetramer as a plasmin reductase. J. Biol. Chem. 277, 10903–10911 [DOI] [PubMed] [Google Scholar]
- 63. Thinnes F. P., Reymann S. (1997) New findings concerning vertebrate porin. Naturwissenschaften 84, 480–498 [DOI] [PubMed] [Google Scholar]
- 64. Thinnes F. P. (2011) Apoptogenic interactions of plasmalemmal type-1 VDAC and Aβ peptides via GxxxG motifs induce Alzheimer's disease—a basic model of apoptosis? Wien. Med. Wochenschr. 161, 274–276 [DOI] [PubMed] [Google Scholar]