

Background: The binding of ligands to the orthosteric pocket induces ligand-specific conformational changes all through domains of G protein-coupled receptors.
Results: Loss of function and gain of function mutations in the transmembrane domain spontaneously induce changes similar to a ligand-specific conformation in the extracellular domain.
Conclusion: Coupling between domains determines the overall signaling state of GPCRs.
Significance: Targeting domain interfaces might be the future for GPCR-specific drug development.
Keywords: 7-Helix Receptor, Angiotensin II, G Protein-coupled Receptors (GPCR), Ligand-binding Protein, Site-directed Mutagenesis, AT1R, ECL2, GPCR, Conformational Change, Domain Coupling
Abstract
The topology of the second extracellular loop (ECL2) and its interaction with ligands is unique in each G protein-coupled receptor. When the orthosteric ligand pocket located in the transmembrane (TM) domain is occupied, ligand-specific conformational changes occur in the ECL2. In more than 90% of G protein-coupled receptors, ECL2 is tethered to the third TM helix via a disulfide bond. Therefore, understanding the extent to which the TM domain and ECL2 conformations are coupled is useful. To investigate this, we examined conformational changes in ECL2 of the angiotensin II type 1 receptor (AT1R) by introducing mutations in distant sites that alter the activation state equilibrium of the AT1R. Differential accessibility of reporter cysteines introduced at four conformation-sensitive sites in ECL2 of these mutants was measured. Binding of the agonist angiotensin II (AngII) and inverse agonist losartan in wild-type AT1R changed the accessibility of reporter cysteines, and the pattern was consistent with ligand-specific “lid” conformations of ECL2. Without agonist stimulation, the ECL2 in the gain of function mutant N111G assumed a lid conformation similar to AngII-bound wild-type AT1R. In the presence of inverse agonists, the conformation of ECL2 in the N111G mutant was similar to the inactive state of wild-type AT1R. In contrast, AngII did not induce a lid conformation in ECL2 in the loss of function D281A mutant, which is consistent with the reduced AngII binding affinity in this mutant. However, a lid conformation was induced by [Sar1,Gln2,Ile8] AngII, a specific analog that binds to the D281A mutant with better affinity than AngII. These results provide evidence for the emerging paradigm of domain coupling facilitated by long range interactions at distant sites on the same receptor.
Introduction
The superfamily of G protein-coupled receptors (GPCRs)2 includes the Glutamate, Rhodopsin, Adhesive, Frizzled and Secretin families, of which the Rhodopsin family with ∼750 GPCRs is the largest (1). GPCRs respond to a wide array of stimuli that include sensory signals, protein and peptide hormones, amino acids, bio-amines, lipids, nutrients, and metabolites. When agonists bind, the GPCRs switch to an activated conformation and recruit either G proteins or other proteins, which initiate intracellular signaling cascades. The extraordinary ability to elicit responses to diverse environmental cues makes the GPCR superfamily a fascinating target for drug development for a broad spectrum of diseases.
Structural features common to all GPCRs include a transmembrane domain (TMD) containing seven helices connected by three intracellular loops and three extracellular loops (ECL) (1, 2) (see Fig. 1). In the majority of GPCRs, the natural or orthosteric ligand-binding site resides in the core of the TMD. Crystal structures have revealed variability in the position of the orthosteric ligand-binding pocket in different GPCRs and conservation in the TMD core extending to the G protein-coupling domain (3). These structural findings broadly match those of earlier pharmacological, biochemical and biophysical studies on a variety of GPCRs, suggesting a general coupling mechanism for GPCRs activating different G protein isoforms. However, the mechanism of efficacies of different ligands for a given GPCR that can exhibit a wide range of activities remains unclear.
FIGURE 1.

Model of rat AT1R. A, secondary structure model of rat AT1R. ECL2 residues substituted with reporter cysteines are highlighted in dark blue. Cysteine residues involved in formation of disulfide bonds are shown in yellow. Nonessential cysteines are replaced with alanine as described previously (11). The gain of function substitution residue Asn111 on TMIII is highlighted in green. The loss of function substitution residue Asp281 on ECL3 is shown in a red circle. The interactions of Asn111 and Asp281 with AngII previously mapped by site-directed mutagenesis are shown with solid lines. The residues involved in both AngII and losartan binding are boxed. B, model of rat AT1R showing position of residues Asn111 and Asp281 relative to Cys180 located in the middle of ECL2 in the AT1R. The backbone Cα trace of AT1R model is shown in gray, and the ECL2 region is highlighted in magenta. The Cα-Cα distances of Asn111 (green) and Asp281 (red) from Cys180 (yellow) are shown as dashed lines. The distance between Cys180 and Asp281 in AngII- and losartan-bound states did not change substantially (not shown). The native residues replaced by Cys reporters are represented as blue sticks. PyMOL (version 0.99rc6) was used to visualize the protein structures and generate images.
The binding of a ligand induces ligand-specific conformational changes that have been detected through all domains in GPCRs that establish a new equilibrium of activity states (4). Activating ligands (agonists) stabilize a receptor conformation that enables coupling to G proteins, and inhibiting ligands (inverse agonists/antagonists) stabilize a conformation that inhibits signaling. The structural changes that occur in the TMD and the cytoplasmic domain indicate that activating ligands induce movement of TM helices. The TM helical motion seen in the crystal structures of the activated states of β2-adrenergic receptor (β2AR) and opsin is essential for activation of signaling (5). In contrast, very little information exists on the dynamics of the ligand pocket in the TMD and the extracellular domain (ECD), which consists of short interhelical ECL1, ECL2, and ECL3 and the N-terminal tail (see Fig. 1A). Mutations in the ECD result in misfolding and dysfunction of GPCRs (6). The greatest diversity in structure is seen in ECD of GPCRs (3); however, tethering of ECL2 to TM3 helix through a disulfide bond is conserved in ∼90% GPCRs. ECL2 is a part of the ligand-binding pocket in different GPCR structures and has an integral role in activation (3, 7). In rhodopsin, the ECL2 interacts extensively with the ligand. Upon activation of rhodopsin, ECL2 is displaced, which is tightly coupled to the reorientation of helices in the TMD (8, 9). Coupling between the ECD and the orthosteric ligand-binding pocket is also seen in the β2AR. For instance, drugs that bind within the TMD of β2AR affect intramolecular interactions between ECL2 and ECD (10). Therefore, whether mutations in the TMD that alter activation states of a GPCR affect the conformation of ECL2 is a significant and timely research question.
The present study explores the effect of mutations in the TM domain on basal and ligand-induced conformation of ECL2 by introducing gain and loss of function mutations in the hormone-binding pocket in the angiotensin II type 1 receptor (AT1R). The AT1R is a rhodopsin family GPCR with an essential role in regulating blood pressure. Several studies have elucidated the importance of the ECL2 in AT1R in ligand binding and receptor activation. The ECL2 has been shown to be cross-linked with ligands and is targeted by autoantibodies that activate AT1R in several pathologies, such as preeclampsia, malignant hypertension, and refractory vascular allograft rejection (6). In the ligand-free basal state, ECL2 of AT1R exists in an open conformation, and ECL2 conformation is modulated in a ligand-specific manner (11). AngII binding is shown to stabilize a less accessible conformation of ECL2, which is referred to as a “lid” conformation. The AngII-induced lid conformation differs from a lid conformation that is stabilized when the AT1R-selective inverse agonist losartan is bound. In this study, reporter cysteine accessibility mapping (RCAM) analysis demonstrated a lid conformation in the ligand-free state of the gain of function mutant (N111G in TMIII; see Fig. 1), which was not observed in the loss of function mutant (D281A in TMVII; see Fig. 1). Thus, ECL2 dynamics are affected by perturbations in distant regions of the receptor. In addition, binding of different ligands differentially modulates conformational change induced by the gain or loss of function mutations in the ECL2. Our studies elucidated the role of the ECL2 as a structural unit of GPCRs that is rearranged not only upon ligand binding, but also by perturbation in TMD. Moreover, a new insight into the dynamic behavior of GPCRs not addressable by the static, inactive state crystal structures is revealed.
EXPERIMENTAL PROCEDURES
Model and Molecular Dynamics (MD) Simulations
The models of rat AT1R (Protein Data Bank code 2AC6), rat AT1R with AngII (Protein Data Bank code 2AH3), and rat AT1R with losartan were obtained as described previously (11). We used the software package NAMD and CHARMM 22 to perform MD simulations essentially as described earlier (11). Each of the three model structures was subjected to 1000 steps of energy minimization, and then the energy-minimized structure was heated from 0 to 310 °C in 31 °C intervals over the course of 20,000 steps. Simulations were carried out for 14 ns. The root mean square deviation of the backbone of AT1R with AngII or losartan was stabilized after 7 ns. MD simulations simulation of ECL2 residues (Ile165–Pro192) were carried out using the molecular modeling software VEGA ZZ 2.0.7 with selected loop constraints. The ECL2 simulation was carried out by imposing Cys101–Cys180 disulfide bond as a constraint in different states. The disulfide bond constraint was relaxed for 1 ns at the end of 14 ns of simulation. We used PyMOL (DeLano Scientific LLC) to visualize the output. Upon completion of MD simulation of ECL2 and the side chains of the residues, we created a library of ECL2 conformations consisting of 345 nonredundant frames. Frames 1–100 come from simulation of the empty state, frames 101–245 come from AngII-bound state, and frames 246–345 come from losartan-bound state. To select the frame that fit the experimental data for each state in each of the mutant background, this library was manually scanned, and the frames that best fit the data are displayed in each instance.
Cysteine Scanning Mutagenesis
The N-terminal HA-tagged synthetic rat AT1R gene, which is cloned in pMT3 vector, was used for construction of all mutants in this study as described previously (12, 13). CYS−AT1R construct and single-cysteine mutants were generated by PCR mutagenesis. N111G or D281A mutations were simultaneously introduced into these single-cysteine mutants by PCR mutagenesis as described previously (12, 13).
Cell Culture and Western Blotting
COS1 cells were grown and maintained in DMEM, supplemented with penicillin/streptomycin (100 units/ml) and 10% FBS. The cells were transfected using FuGENE 6 transfection reagent (Roche Applied Science) according to the manufacturer's instructions. 48 h post-transfection, the cells were harvested and lysed in mammalian protein extraction reagent (Pierce) containing protease (Sigma) and phosphatase inhibitors (Thermo Scientific). A total of 50 μg of cell lysate protein was run on 10% SDS-PAGE (Invitrogen) and blotted onto nitrocellulose membranes (Bio-Rad). Each membrane was probed with mouse anti-HA monoclonal antibody (Roche Applied Science) and HRP-conjugated anti-mouse IgG (GE Healthcare). The receptor expression was detected using ECL Plus Western blotting detection system (Amersham Biosciences).
Saturation Binding Assay
COS1 cells were harvested 72 h post-transfection and suspended in binding buffer (140 mm NaCl, 5.4 mm KCl, 1 mm EDTA, 0.006% BSA, 25 mm HEPES, pH 7.4). Assays were performed under equilibrium conditions, with [125I]-[Sar1,Ile8]AngII (Dr. Robert Speth, University of Mississippi) concentrations ranging between 0.125 and 12 nm (specific activity, 2176 Ci/mmol) for 1 h at room temperature as previously described (12, 13). Nonspecific binding was measured in the presence of 10−5 m [127I]-[Sar1Ile8]AngII (Bachem). 25 μl of binding buffer or [127I]-[Sar1Ile8]AngII was mixed with 50 μl of increasing concentrations of [125I]-[Sar1,Ile8] and 50 μl of cell suspension in total volume of 125 μl in duplicate in 96-well plates. The cells were harvested by filtering the binding mixture through Whatman GF/C glass fiber filters (102 × 256 mm), which were extensively washed with washing buffer (20 mm sodium phosphate, 100 mm NaCl, 10 mm MgCl2, 1 mm EGTA, pH 7.2). The bound ligand fraction was determined as the cpm remaining on the membrane. The binding kinetics were analyzed by nonlinear curve-fitting program LigandR, which yields the means ± S.D. for the Kd and Bmax values.
Calcium Mobilization Assay
Calcium mobilization experiments were performed using a FLIPR Calcium 5 assay kit (Molecular Devices) according to the manufacturer's instructions. The cells were plated into black Costar clear-bottomed 96-well plates (50,000 cells/well) 24 h post-transfection and incubated in DMEM supplemented with 10% FBS overnight and then replaced with serum-free culture medium for 4 h at 37 °C. 50 μl of calcium assay reagent was added to each well. The plates were incubated for 30 min at 37 °C and allowed to return to room temperature 30 min before reading. The cells were stimulated by adding eight different concentrations of AngII (ranging between 0 and 10 μm), and fluorescence was measured every 2 s for 2 min on a fluorometric imaging plate reader (FlexStation; Molecular Devices). The data were analyzed by averaging maximum-minimum relative fluorescence units generated during the calcium flux assay. The EC50 values were calculated by using sigmoidal dose-response curve fit in GraphPad PrismR.
MTSEA-Biotin Labeling
MTSEA-biotin labeling experiments were repeated at least three times as described previously (11). Briefly, 48 h post-transfection, the cells were washed twice with PBS, harvested using nonenzymatic cell dissociation solution (Sigma), and suspended in PBS. 500-μl aliquots of cells were incubated with 10 mm MTSEA-biotin (Toronto Research Chemicals) at 25 °C for 30 min. The cells were then resuspended in Nonidet P-40 lysis buffer (1% Nonidet P-40, 20 mm Tris, pH 7.4, 137 mm NaCl, 20% glycerol) with protease and phosphatase inhibitors. 750 μg of cell lysate protein was incubated overnight with 4 μl of mouse anti-HA monoclonal antibody in lysis buffer. The receptors were immunoprecipitated with protein G-agarose beads (Millipore). The receptor-bound beads were then washed four times with cold PBS, and the proteins were extracted by heating at 60 °C in NuPAGE LDS sample buffer for 10 min. The samples were blotted as described above and probed with streptavidin-HRP (Amersham Biosciences). To control for differences in receptor expression levels, the same blot was reprobed with anti-HA antibody as described above. MTSEA-biotin accessibility of each receptor was obtained by normalizing the biotinylation level of each receptor to its expression level. The same procedure was repeated following treatment of cells with ligands for 10 min at room temperature prior to harvesting. The results are expressed as the means ± S.E. and analyzed by unpaired t test for statistical significance to compare variables between mutants and CYS−AT1R using GraphPad Prism®. The significance is set at p < 0.05.
RESULTS
Modeling ECL2 in Relation to AT1R Residues Asn111 and Asp281
The AT1R model in Fig. 1 is validated previously for compliance with experimentally determined interactions of AngII and losartan, as well as previous RCAM data (11, 14). The Cα distances between Cys180 in the middle of ECL2 and Asn111 on TMIII and Asp281 on TMVII in the empty state of the receptor is shown. The distances between Cys180 and Asn111 in the AngII-bound and losartan-bound models were nearly the same. Similarly, the distance between Cys180 and Asp281 in the AngII-bound and losartan-bound models also did not change substantially. The mutation effects of Asn111 and Asp281 on pharmacology, signaling and in mimicking different activation states of the receptor is well documented (15–25). Functional interaction between receptor regions that are more than seven residues apart in the primary sequence and located at a distance greater than the hydrogen bonding distance (4 Å) in the tertiary structure is defined as long range interaction. The >10 Å distance observed in the model indicates that conformation of ECL2 is likely affected through multiple interactions with the neighboring domains of the receptor and not through a direct effect of mutation of Asn111 and Asp281. Thus, local effects of above mentioned mutations could have global influence on the dynamics of ECL2 in AT1R.
Construction of the ECL2-Cys Reporters in Gain of Function and Loss of Function CYS−AT1R Background
Previously, we engineered the MTSEA-biotin-insensitive N-terminal HA-tagged CYS−AT1R, which lacks six native nonessential Cys residues to prevent interference with the reporter Cys residues (Fig. 1). Essential Cys residues involved in disulfide bonds, Cys18–Cys274 and Cys101–Cys180, that stabilize the receptor were not mutated. The AT1R and CYS−AT1R have similar ligand binding and signal transduction characteristics (11). Therefore, CYS−AT1R is used as a surrogate template to introduce a reporter Cys in ECL2 and either N111G in TMIII or D281A in TMVII.
To scrutinize the conformation in different regions of ECL2, a reporter Cys was substituted at either 174, 176, 182, or 187 in ECL2 in the background of N111G-CYS−AT1R and D281A-CYS−AT1R. These reporter positions are highly sensitive to ligand-specific conformation changes in the ECL2 in the CYS−AT1R background (11). Expression and properties of 10 mutants created for this study and the corresponding single reporter controls in CYS−AT1R are listed in Table 1.
TABLE 1.
Characterization of ECL2 single cysteine mutants of N111G-CYS−AT1R and D281A-CYS−AT1R
The Kd value for CYS−AT1R was 5.10 nm, and the Bmax value was 7.46 pmol/mg in experiments (n = 3). Fold change in Kd and Bmax values for each mutant were derived from [125I]-[Sar1,Ile8]AngII saturation binding assay. Fold change in EC50 values were derived from [Sar1]AngII-induced calcium mobilization assay. All of the mutants in this table were N-terminally HA-tagged and at CYS−AT1R background.
| Mutant | Bmax | ΔKd | ΔEC50 | Ca2+ response |
|
|---|---|---|---|---|---|
| Basal | Maximum | ||||
| pmol/mg | |||||
| CYS−AT1R | 7.46 | 1.0 | 1.0 | 1.0 | 2.8 |
| N174C | 1.03 | 1.4 | 1.2 | 1.1 | 2.7 |
| N176C | 0.91 | 1.3 | 1.0 | 0.9 | 2.4 |
| F182C | 0.91 | 1.4 | 1.5 | 1.0 | 2.5 |
| R187C | 8.40 | 1.6 | 1.6 | 0.8 | 2.2 |
| N111G-CYS−AT1R | 2.76 | 0.2 | 1.2 | 1.3 | 3.2 |
| N111G-N174C | 2.78 | 0.7 | 1.1 | 1.3 | 2.8 |
| N111G-N176C | 2.30 | 0.2 | 1.4 | 1.2 | 2.8 |
| N111G-F182C | 2.39 | 0.5 | 1.3 | 1.3 | 2.7 |
| N111G-R187C | 2.79 | 0.5 | 1.1 | 1.2 | 2.8 |
| D281A-CYS−AT1R | 6.71 | 14.3 | 17.5 | 0.9 | 1.1 |
| D281A-N174C | 4.37 | 14.5 | 33.2 | 1.0 | 1.2 |
| D281A-N176C | 7.88 | 13.9 | 29.9 | 1.0 | 1.2 |
| D281A-F182C | 3.50 | 15.6 | 10.1 | 0.9 | 1.0 |
| D281A-R187C | 8.57 | 15.1 | 10.2 | 0.9 | 1.1 |
Effect of ECL2-Cys Reporter Substitutions on Gain of Function and Loss of Function Phenotypes
The expression of the mutants in transiently transfected COS1 cells determined by Western blotting is shown in Fig. 2. Each mutant yielded a ≈42-kDa glycosylated, functional form of the receptor protein in COS1 cells, suggesting that Cys substitutions did not significantly interfere with the overall folding and transport to cell surface. Variability caused by transient transfection was not significant. The expression level of mutant N111G-N176C and D281A-F182C was reduced but did not affect further studies. Cell surface receptor expression (Bmax) measured by saturation binding assay in intact cells is shown in Table 1. The cell surface receptor expression was sufficient to perform RCAM analysis for all mutants. All of the reporter Cys mutants in the N111G-CYS−AT1R background bound the AngII analog with higher affinity compared with the CYS−AT1R receptor. On the other hand, reporter Cys mutants in D281A-CYS−AT1R background exhibited lower affinity (14–16-fold) toward the AngII analog. AngII binding elicited Ca2+ mobilization response in all N111G mutants with similar EC50 values (1–1.5-fold). EC50 values in D281A reporter mutants (10–33-fold) were higher compared with CYS−AT1R. Because the expression levels of the D281A mutants are similar to the CYS−AT1R (Table 1), the differences in the cell surface expression of the receptors is not the basis for diminished signaling; instead, the decreased response indicates the loss of efficiency in ligand-mediated activation. Thus, introducing Cys reporters in ECL2 did not alter the gain of function and loss of function characteristics previously shown for the selected mutants (21, 22, 28–30).
FIGURE 2.
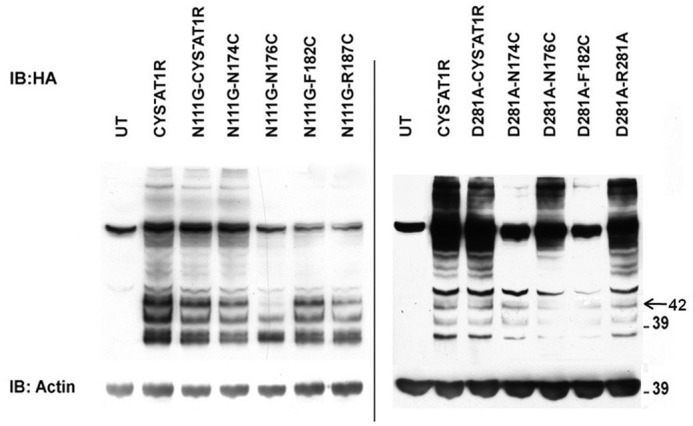
Expression of ECL2 single-cysteine mutants of N111G-CYS−AT1R and D281A-CYS−AT1R. Expression of HA-CYS−AT1R and HA-tagged single-cysteine mutants of N111G-CYS−AT1R and D281A-CYS−AT1R in transiently transfected COS1 cells was analyzed. Untransfected (UT) cells served as negative controls. Actin expression levels are shown as loading controls. IB, immunoblot.
MTSEA-Biotin Reaction with Reporter Cys
We used MTSEA-biotin, a thiol-labeling reagent that preferentially reacts with water-accessible cysteines and does not react with buried or disulfide bonded cysteines (26) (Fig. 3A). The reaction of MTSEA-biotin with the thiol group of an accessible cysteine can be quantified by streptavidin-HRP reaction, which is a measure of accessibility of a reporter cysteine introduced in the receptor (11). The MTSEA-biotin accessibility of single-cysteine mutants of ECL2 was compared in different ligand-bound states (Fig. 3B). We used N111G-CYS−AT1R, as the control, and the labeling of single-cysteine mutants was quantified in comparison with labeling of the control in the same experiment. Any significant measurement above the reactivity of control indicates accessibility of the particular reporter cysteine. The modification levels of single-cysteine mutants were determined by normalizing the streptavidin-HRP signal of immunoprecipitated receptors to the HA signal to minimize the influence of different cell surface expression levels on the level of MTSEA-biotin reactivity. The receptor topology was preserved during MTSEA-biotin reaction as closely as possible to the native state in the plasma membrane by using adherent cells rather than fractionated membranes. Immunoprecipitation of representative mutants differentially labeled with MTSEA-biotin in the presence and absence of ligands is shown in Fig. 3B.
FIGURE 3.

MTSEA-biotin accessibility of representative mutants. A, structure of MTSEA-biotin. The region of the molecule that is modified with reporter Cys is shown in red. B, immunoprecipitated receptors were probed with anti-HA (left panel) or streptavidin-HRP (right panel) to estimate receptor pulldown and biotinylation levels, respectively. The same blot is used for probing with HA and streptavidin-HRP. The blots for representative mutants N111G-N174C and N111G-N176C are shown under three experimental conditions: in the absence of ligand (top panel), in the presence of 1 μm AngII (middle panel), and in the presence of 10 μm losartan (bottom panel). The 42-kDa monomeric receptor band was used for determination of MTSEA-biotin accessibility. The HA signal intensity and streptavidin-HRP signal intensity of each sample are compared with the N111G-CYS−AT1R in the same gel as indicated by the numbers below the bands. The corresponding plots show the MTSEA-biotin relative accessibility, which is the ratio of relative streptavidin-HRP signal to relative HA signal for a particular sample. Relative MTSEA-biotin accessibility of each mutant is compared with the N111G-CYS−AT1R in the same gel. The insets show schematic representations of reporter cysteines that point up when inaccessible (shown as SH in green) and point down when accessible, when reacted with MTSEA-biotin (shown as S-SR in red). IB, immunoblot.
Effect of N111G and D281A Mutations on Relative Accessibility of ECL2-Cys Reporters
The accessibility pattern of four Cys reporters in ECL2 shows distinctly different basal conformation of ECL2 in different genetic background. For example, in the empty state of the CYS−AT1R, a different degree of accessibility is seen for reporter Cys at positions 174, 176, and 187, but position 182 is inaccessible (Fig. 4A). This pattern is consistent with two accessible segments of ECL2 separated by the Cys106–Cys180 disulfide bond tethering ECL2 to TM3, which we reported in our previous study (11).
FIGURE 4.

MTSEA-biotin accessibility maps of ECL2 single-cysteine mutants in the empty states. The MTSEA-biotin relative accessibility of mutants are expressed as the means ± S.E., n = 3. The red line shown on the graph designates the significance cut off that determines the accessibility of mutants. Mutants with significantly higher accessibility compared with control are indicated with red asterisks. A–C, MTSEA-biotin accessibility maps of ECL2 mutants in HA-CYS−AT1R (A), N111G-CYS−AT1R (B), and D281A-CYS−AT1R (C) are shown in the absence of ligand. Note that the scales in A and B are different from that in C. D–F, the molecular dynamics of ECL2 frames that best fit accessibility data are shown in CYS−AT1R (D), N111G-CYS−AT1R (E), and D281A-CYS−AT1R (F). TM helices are shown in gray. The ECL2 backbone is shown in magenta. The side chains replaced by Cys reporter residues are shown in red when accessible and green when inaccessible. The disulfide-bonded cysteines Cys101 (TMIII) and Cys180 (ECL2) are shaded in yellow.
The accessibility of ECL2 Cys reporters in the empty state of N111G-CYS−AT1R is shown in Fig. 4B. The mean ± S.E. observed in three independent experiments indicates inaccessibility of reporter Cys at positions 174, 182, and 187. Significantly higher accessibility (means ± S.E.) was observed for reporter Cys at position 176 when compared with the open conformation of ECL2 observed in the empty state of CYS−AT1R. Thus, introduction of the gain of function N111G mutation reduced the accessibility of ECL2 on both sides of the tethered Cys106–Cys180 bond. This change in ECL2 conformation is suggestive of a lid-like conformation of ECL2, which appears to be induced in this mutant in the absence of any ligand.
Interestingly, in the empty state of D281A genetic background, the reporter Cys located at positions 174, 176, and 182 are accessible, but Cys at 187 is not (Fig. 4C). This pattern suggests that the 174–182 segment of ECL2, which includes the tethering disulfide bond, is open, whereas the region containing position 187 is buried. This mutation causes a loss of function phenotype; hence the observed change of ECL2 conformation is functionally significant.
Conformation of ECL2, which fit the reporter accessibility data in the empty states of CYS−AT1R, N111G-CYS−AT1R, and the D281A-CYS−AT1R, is shown in Fig. 4 (D–F). When the 345 frames in ECL2 conformation library are searched (see “Experimental Procedures”), frames 1–20 matched the open conformation for empty state of CYS−AT1R, out of which the best frame is shown in Fig. 4D, consistent with our previous study (11). However, the same analysis for the empty state of N111G-CYS−AT1R matched none of the 1–100 frames but matched frames 193–199, which correspond to the AngII-bound state of the CYS−AT1R. This MD simulation result means that the spontaneous ECL2 conformation in this mutant background appears to be similar to the ligand-induced lid conformation. Analysis of empty state of D281A-CYS−AT1R matched frames 97–100. Because none of the 101–345 frames matched the accessibility data of D281A mutant empty state, we speculate that this mutation stabilized an inactive empty state that may be only transiently present in the WT receptor.
Ligand Modulation of ECL2 Conformation in the N111G Mutant Background
The relationship between the binding of ligands in the orthosteric ligand pocket and the accessibility of the reporter Cys176 is shown in Fig. 5. In the AngII-bound state, both CYS−AT1R and N111G-CYS−AT1R produced a Ca2+ response (Fig. 5A). The dose-dependent change of accessibility of the reporter Cys176 (Fig. 5, B and C) indicated a similar trend in both CYS−AT1R and N111G-CYS−AT1R, but the variation is higher in the N111G-CYS−AT1R. The inverse agonist candesartan inhibited these receptors but with different efficacies (Fig. 5D). The reporter Cys176 accessibility decreased in both genetic backgrounds but was maximal at 10 μm candesartan in the N111G-CYS−AT1R (Fig. 5, E and F).
FIGURE 5.

Concentration-dependent change in MTSEA-biotin accessibility. A, AngII and induced calcium mobilization in CYS−AT1R and N111G-CYS−AT1R transfected COS1 cells is shown upon stimulation with 0–1 μm AngII. B and C, the accessibility of N176C is shown at different concentrations of AngII in CYS−AT1R (B) and N111G-CYS−AT1R (C). D, dose-response inhibition of CYS−AT1R and N111G-CYS−AT1R with 0–10 μm candesartan is shown. E and F, the accessibility of N176C is shown at different concentrations of candesartan in CYS−AT1R (E) and N111G-CYS−AT1R (F). The data indicate the higher variability in the accessibility measurements when the concentration of ligands is closer to ≈Kd in both CYS−AT1R and N111G-CYS−AT1R and indicate the rationale behind using saturation concentrations.
Fig. 6A shows activation of N111G-CYS−AT1R by AngII and [Sar1,Ile4,Ile8]AngII. Although both the peptides produced higher calcium responses in the N111G-CYS−AT1R, the increase over basal is small. In contrast, AngII produced a full response in the CYS−AT1R, and [Sar1,Ile4,Ile8]AngII did not produce a response. This finding is similar to the observation reported for WT and N111G-AT1R (20). Consistent with the small change in calcium response, treatment of N111G-CYS−AT1R with AngII (Fig. 6B) and [Sar1,Ile4,Ile8]AngII (Fig. 6C) produced only small changes in the overall accessibility of ECL2 reporters. The accessibility changes seen for Cys176 with both ligands indicate higher variability, which is consistent with the instability of conformation, which has been suggested for constitutively activated GPCRs in general. The accessibility data for the AngII-bound state of N111G-CYS−AT1R best fit with MD simulation frames 189–200, which are frames from the AngII-bound state. Treatment with 1 μm losartan, a weak inverse agonist, did not change the ECL2 conformation, and treatment with 1 μm candesartan, a strong inverse agonist, decreased accessibility of Cys176 (Fig. 6, D and E). At higher concentrations, both losartan and candesartan (10 μm) completely reduced the accessibility of ECL2, which is indicative of the inactive receptor state. Inhibition of basal activity of N111G-CYS−AT1R by losartan and candesartan confirmed suppression of the constitutive activity when used at high concentrations (Fig. 6E). This is consistent with our previous experimental observation that N111G receptor has a lower affinity for antagonists, and losartan cannot fully suppress basal activation of the receptor function imparted by the N111G mutation (19, 20). The candesartan-bound state matched with frames 309–312, which are frames from the losartan-bound state (Fig. 6, F and G).
FIGURE 6.

MTSEA-biotin accessibility maps of ECL2 single-cysteine mutants in the N111G-AT1R in the presence of ligands. A, AngII and [Sar1,Ile4,Ile8]AngII induced calcium mobilization in CYS−AT1R and N111G-CYS−AT1R. CYS−AT1R and N111G-CYS−AT1R transfected COS1 cells are stimulated with 0–1 μm AngII or [Sar1,Ile4,Ile8]AngII. B and C, MTSEA-biotin accessibility maps of ECL2 mutants in N111G-CYS−AT1R in the presence of AngII (B) and [Sar1,Ile4,Ile8]AngII (C) are shown. B–E, the gray shading shows the accessibility pattern of ECL2 mutants in the absence of ligand. D, molecular dynamics simulation frames of ECL2 that fit accessibility data in the presence of AngII (orange spheres) are shown. The peptide and ECL2 backbones are shown in gray. The ECL2 backbone that corresponds to the best of all frames is shown in magenta. The side chains replaced by Cys reporter residues are shown in red when accessible and green when inaccessible. E, dose response inhibition of N111G-CYS−AT1R with 0–10 μm losartan or candesartan is shown. F and G, MTSEA-biotin accessibility maps of ECL2 mutants in N111G-CYS−AT1R in the presence of losartan (F) and candesartan (G) are shown. H, molecular dynamic simulation frames of ECL2 in the presence of losartan (cyan spheres) are shown.
Ligand Modulation of ECL2 Conformation in the D281A Mutant Background
The accessible conformation of ECL2 observed in the empty state of D281A-CYS−AT1R did not change when treated with 1 μm AngII (Fig. 7A). AngII binding to D281A-CYS−AT1R is impaired as indicated by the high Kd and EC50 values observed (Table 1). Feng and co-workers (19) reported that binding affinity for the D281A mutant is restored when the positively charged ARG2 of AngII is substituted with a neutral residue, Gln2. Therefore, we tested the effect of the analog [Sar1,Gln2,Ile8]AngII on accessible ECL2 conformation. Binding of [Sar1,Gln2,Ile8]AngII to D281A-CYS−AT1R actually induced accessibility changes in ECL2, rendering all reporter cysteines inaccessible (Fig. 7B). Thus, rescuing peptide ligand binding to the D281A mutant using an AngII analog alters the conformation of ECL2. Treatment with 1 μm losartan decreased the accessibility of position 182 but did not suppress accessibility at position 174 and 176 (Fig. 7C). Thus, losartan binding altered the ECL2 conformation as anticipated. The accessibility data for the AngII-bound state of D281A-CYS−AT1R best fit with MD simulation frames 229–231, and that for the [Sar1,Gln2,Ile8]AngII-bound state best fit with frames 228–234, all of which are frames from the AngII-bound state (Fig. 7, D and E). The losartan-bound state matched with frames 271–278, which come from the losartan-bound state (Fig. 7E).
FIGURE 7.

A–C, MTSEA-biotin accessibility maps of ECL2 single-cysteine mutants in the D281A-CYS−AT1R in the presence of AngII (A), [Sar1,Gln2,Ile8]AngII (B), and losartan (C) are shown. D–F, molecular dynamics simulations of ECL2 in the presence of AngII (D, orange spheres), [Sar1,Gln2,Ile8]AngII (E), and losartan (F, cyan spheres) are shown. The peptide backbone is shown in gray. The ECL2 backbone that corresponds to the best of all frames is shown in magenta. The side chains replaced by Cys reporter residues are shown in red when accessible and green when inaccessible.
DISCUSSION
The most important observation in this study is the discovery of a lid conformation of ECL2 in the gain of function AT1R mutant in absence of any ligand. In contrast, the loss of function D281A mutant displayed an open conformation in the empty state that failed to produce a lid conformation in response to AngII binding. Previously, we demonstrated ligand-specific modulation of ECL2 conformation in AT1R. In the empty state of AT1R, 10 of 22 ECL2 residues examined were accessible, which is defined as an open conformation. In the ligand-bound state, only four residues are accessible, defined as a lid conformation in which the specific accessible residues differ when agonist and antagonist bind to AT1R (11). ECL2 is a critical structural element ideally positioned to interact with the bound ligands, as well as the TM helices and other extracellular segments of the receptor. Logical questions that followed our previous work on the AT1R are: (i) whether the ECL2-lid conformation is present in constitutively activated and inhibited states of the AT1R, (ii) whether the mutations located in distant sites on TM helices modulate ECL2 conformation, and (iii) whether the mutation-induced ECL2 conformation is further modulated by ligands. To answer these questions, we utilized the well characterized gain of function (N111G) and loss of function (D281A) mutants of AT1R. The mutation-induced conformational changes were analyzed by RCAM analysis of four Cys reporters introduced in ECL2. In our previous study RCAM was shown to depict specific conformational changes in ECL2 upon binding of the ligand in the orthosteric site of cell surface AT1R in the intact cells under conditions used for signal transduction studies (27–31). The RCAM analysis in this study indicated the presence of a distinct ECL2 lid conformation in the empty state of the gain of function AT1R mutant but not in the loss of function receptor mutant. The mutation-induced conformation of ECL2 was reversible by specific ligands that selectively alter the activity of gain of function and loss of function mutant receptors as shown in Fig. 6 for N111G and in Fig. 7 for D281A. Two mutations examined are located outside the ECL2, thus providing new insights into long range interactions between domains of AT1R.
Of the four Cys reporters used, Cys174 and Cys176 monitor the accessible ECL2 segment on the N-terminal side of the Cys101-Cys180 disulfide bond tethering ECL2 to TMIII. Cys182 is located in a region of low MTSEA-biotin reactivity, and Cys187 is in the accessible ECL2 segment on the C-terminal side of the Cys101-Cys180 disulfide bond. The accessibility pattern of these reporters in the empty state of CYS−AT1R (surrogate for the basal state wild-type AT1R) matched the results reported in our previous work (11). The accessibility map of the same reporters in mutant backgrounds differed from ECL2 conformational states of the CYS−AT1R. Observed accessibility patterns in the empty states of N111G-CYS−AT1R and D281A-CYS−AT1R indicate mutation-induced conformational change in ECL2. To avoid experimental confounds, the MTSEA-biotin reaction was carried out on adherent cells expressing these receptors rather than fractionated membranes. Furthermore, the accessibility of each Cys reporter was compared in empty and ligand-bound states concurrently for each mutant and the CYS−AT1R control. The data suggested that accessibility of each Cys reporter is modulated to different extents in empty state and agonist/antagonist-bound states of the mutants. The reversible effect of each mutation on function and ligand-specific accessibility pattern of Cys reporters is a hallmark of functional relevance of altered topology of ECL2 in these mutants.
The N111G and D281A mutations effectively induced different activity states in the CYS−AT1R surrogate as originally shown in the AT1R. Mimicry of activated state AT1R conformation in the N111G mutant is very well established. Asn111 is a key residue responsible for coupling ligand binding to conformational changes leading to AT1R activation. The N111G mutation in AT1R increases affinity and efficacy (3–10-fold) for agonists, AngII, and AngII analogs and lowers the affinity (3–315-fold) for losartan and other inverse agonists (20). The agonist bias of N111G-AT1R conformation is also reflected in agonist-independent IP3 production, ≈50% more basal activity compared with wild-type AT1R and full activation by AngII analogs that can only partially activate the wild-type AT1R. The basal activity of N111G-AT1R can be inhibited by AT1R-selective blockers, albeit at 3–315-fold higher concentrations compared with wild-type AT1R (16, 19, 20, 22, 23). The conformation of the TMD of the N111G-AT1R has been shown to be altered to the activated state (12, 13, 32–36). The RCAM results in Fig. 6 showed that ECL2 in N111G-CYS−AT1R spontaneously mimics a lid conformation in the absence of agonist, which is similar to AngII-induced folding of ECL2 observed in the CYS−AT1R. Further change in reporter accessibility upon binding AngII and [Sar1,Ile4,Ile8]AngII in N111G-CYS−AT1R supports the concept that the N111G mutant is sensitive to both these ligands (17). A different degree of ECL2 conformational change in response to losartan and candesartan binding was also anticipated. Being a stronger inverse agonist, candesartan was more effective in reducing the overall accessibility of ECL2 than losartan when used at a 1 μm concentration. This observation reflects the well known ability of constitutively active GPCRs to discriminate between different inverse agonists, which stabilize the inactive conformation of the receptor. It is known that binding of inverse agonists induces the most pronounced stabilizing effect on the extracellular region of the receptor. The agonist binding promotes a conformational change around the intracellular domain while having little impact on other structural elements of the receptor (37).
The AT1R conformation that disfavors high affinity AngII binding and exhibits a poor agonist efficacy state is produced by mutation of Asp281 located in TMVII. The loss of function effect of D281A mutation is reported independently by different laboratories. The Asp281 side chain is the primary docking point for the positively charged Arg2 side chain of AngII. This interaction is essential for high affinity AngII binding and receptor activation (15, 18, 21, 24). The binding affinity of losartan and other nonpeptide antagonists is not significantly altered in the D281A mutant. The inactive state of the receptor produced by D281A mutation is further validated by analysis of ECL2 conformation in this study. RCAM results demonstrated open ECL2 conformation in the empty state for the D281A mutant. Altered accessibility of Cys174 and Cys182 reporters upon losartan binding resembles the losartan-induced conformation of ECL2 in the CYS−AT1R. Thus, the D281A mutant is normal in the empty state and in response to losartan. However, the defect in the D281A mutant is specific toward AngII; the ECL2 conformation did not change to a lid conformation upon exposing the D281A mutant to AngII. This was anticipated because when Asp281 is substituted with a neutral hydrophobic Ala side chain, the affinity of AngII is reduced (Table 1), but the affinity is regained by replacing the Arg2 side chain in AngII with a neutral side chain, such as Gln2 (15). This expectation was indeed borne out when the D281A mutant AT1R was compensated by Gln2 substituted analog of AngII in the RCAM experiment. The reporter accessibility pattern of ECL2 in the mutant was similar to a lid conformation. This observation suggests that the AngII-specific defect of the D281A mutant is overcome by the mutant-specific AngII analog [Sar1Gln2Ile8]AngII.
The study of conformational dynamics of the extracellular regions of a GPCR is relevant to better understanding of the mechanism of efficacies of different ligands for a given GPCR that can exhibit a wide range of activities. Analytical approaches capable of probing dynamic conformational states of GPCRs elucidate the plasticity of the receptor that is critical for function but not revealed by crystal structures. Techniques such as hydrogen/deuterium exchange coupled with mass spectrometry, nuclear magnetic resonance spectroscopy, site-directed labeling with spin probes, or fluorescent probes have been used to probe ligand-induced conformational changes in ECD in rhodopsin (38) and β2AR (10). Conformational changes in ECD have been analyzed using other sensitive but indirect methods in other GPCRs that are not amenable to purification (6, 7, 11, 38, 39). The RCAM data in this study provide new insights regarding two different modes of regulating ECL2 conformation. First, the effects of TMD mutation suggest cooperativity between TMD and ECL2. N111G mutation in TMIII spontaneously induces lid, an active state conformation of the ECL2, and D281A mutation in TMVII has the opposite effect. These mutation effects must occur via long range interaction with ECL2. The N111G mutation is ∼13.4 Å from the disulfide bond that links TMIII to ECL2, and the D281A mutation is ∼13.8 Å from the conserved disulfide bond in ECL2. A local structural perturbation caused by each mutation could be transmitted to distant domains, leading to global consequences.
Empirical support for long range communication between domains of GPCRs is very common. The extensively studied retinitis pigmentosa mutations of human rhodopsin suggest that the folding of the receptor polypeptide generates cooperativity between different domains. Retinitis pigmentosa mutations located in the TM and cytoplasmic domains of rhodopsin are associated with disruption of the unique disulfide bond of rhodopsin in the ECD (i.e., intradiscal domain) and loss of retinal binding in the TM domain (40, 41). Evidence suggests that the intradiscal domain is structurally coupled to the TM domain, because mutations in the intradiscal domain can severely affect the ability of rhodopsin to bind 11-cis-retinal in the TM domain. In rhodopsin without any mutations, light activation, which takes place in TM domain, has been shown to displace ECL2 (9). A similar long range conformational effect is seen in both rhodopsin and β2AR through NMR studies. Drugs that bind within the TMD of β2AR stabilize distinct conformations of the ECD (10). Rotation and/or tilting of the TM helices and orthosteric binding site changes associated with a constitutively active β2AR mutant in which mutation is in the cytoplasmic domain are documented (42). In a recent crystallographic study, a cytoplasmic domain directed nanobody binding induced subtle changes in binding pocket at >35 Å distance, and these changes led to active state orientation of critical serine residues that favor agonist binding (43). Induced conformational changes in the ECL2 have been documented in several GPCRs, including the D2 dopamine receptor and C5A complement receptor (44, 45) upon ligand occupancy of the orthosteric pocket in the TM domain. Long range conformational changes associated with ligand binding have been shown in ECL1 of glucagon receptor (46); the N terminus, ECL1, and extracellular ends of all TM helices of Saccharomyces cerevisiae G protein-coupled receptor Ste2p are known (47–49). These observations suggest that a mutation in a particular domain of the receptor causes a local structural perturbation that is eventually transmitted to other domains, leading to a global consequence in terms of affinity toward ligands and G protein. The conformational change in the extracellular domain thus seems a core function of ligand binding and receptor activation in GPCRs. This mechanism is presumably shared by the entire rhodopsin family of receptors, despite their selectivity for a diverse group of ligands. Understanding the intrinsic coupling between different domains of a GPCR will help explain the flexibility essential for its biological function and transduction of signal across the membrane. Domain coupling has been shown to be critical for the biological activity of many different protein families with signaling and structural roles, as well as in enzymes (50, 51).
Second, the ligand-specific modulation of ECL2 conformation in both mutants illustrates further regulation of coupled domains by ligands. AngII makes contact with multiple residues on the TMD, ECL3, and the N terminus to facilitate conformational changes in ECL2 (i.e., a distinctly ligand-facilitated step in receptor function). We speculate that this initial ligand contact, resulting in reduced entropy of ECL2, is the driving force for coupled transition of conformation in the TMD. Classical drug efficacy theories have postulated that ligand-regulated isomerization of protein conformation underlies activation and inhibition of GPCR function (4). Based on the observation that ligands bind within TMD in well known GPCRs, different mechanistic models have been proposed to describe how positional conserved ligand-binding residues in TMD modulate GPCR function. Recent crystallographic studies, however, revealed variability in the position of ligand-binding pocket of GPCRs. Still unresolved is how conformation of TMD changes when it lacks the inherent ability to bind the physiological ligand. As is the case in some GPCRs, the activating ligands do not reach the TM domain. Other ECD motions, either subtle or large scale, could be involved in coupling ligand binding to TMD conformational changes in such GPCRs. The existence of both aspects of the mechanism in GPCRs leads to a more diverse and complex process of ligand sensing and may explain the variability of the efficacy observed under different experimental conditions. The emerging role of ECD in AT1R and other GPCRs make the components of this domain an attractive target for rational drug design and improvement of new classes of ligands including allosteric modulators and biased ligands of GPCRs.
Acknowledgments
We thank J. Kemp and Z. P. Jara for critical reading of the manuscript and members of the S. V. Naga Prasad Lab for fruitful discussions and advice.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 HL57470 (to S. K.). This work was also supported by National Research Service Award HL007914 (to H. U.).
- GPCR
- G protein-coupled receptor
- ECL
- extracellular loop
- RCAM
- reporter cysteine accessibility mapping
- AngII
- angiotensin II
- AT1R
- angiotensin II type 1 receptor
- MTSEA-biotin
- N-biotinylaminoethyl methanethiosulfonate
- TM
- transmembrane
- TMD
- TM domain
- β2AR
- β2-adrenergic receptor
- ECD
- extracellular domain
- MD
- molecular dynamics.
REFERENCES
- 1. Lagerström M. C., Schiöth H. B. (2008) Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 7, 339–357 [DOI] [PubMed] [Google Scholar]
- 2. Rosenbaum D. M., Rasmussen S. G., Kobilka B. K. (2009) The structure and function of G-protein-coupled receptors. Nature 459, 356–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Katritch V., Cherezov V., Stevens R. C. (2012) Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol. Sci. 33, 17–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vaidehi N., Kenakin T. (2010) The role of conformational ensembles of seven transmembrane receptors in functional selectivity. Curr. Opin. Pharmacol. 10, 775–781 [DOI] [PubMed] [Google Scholar]
- 5. Altenbach C., Kusnetzow A. K., Ernst O. P., Hofmann K. P., Hubbell W. L. (2008) High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc. Natl. Acad. Sci. U.S.A. 105, 7439–7444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Unal H., Jagannathan R., Karnik S. (2012) Mechanism of GPCR-directed Autoantibody in Diseases. Adv. Exp. Med. Biol. 749, 187–199 [DOI] [PubMed] [Google Scholar]
- 7. Unal H., Karnik S. (2012) Domain coupling in GPCRs. The engine for induced conformational changes. Trends Pharmacol. Sci. 33, 79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yan E. C., Kazmi M. A., Ganim Z., Hou J. M., Pan D., Chang B. S., Sakmar T. P., Mathies R. A. (2003) Retinal counterion switch in the photoactivation of the G protein-coupled receptor rhodopsin. Proc. Natl. Acad. Sci. U.S.A. 100, 9262–9267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ahuja S., Hornak V., Yan E. C., Syrett N., Goncalves J. A., Hirshfeld A., Ziliox M., Sakmar T. P., Sheves M., Reeves P. J., Smith S. O., Eilers M. (2009) Helix movement is coupled to displacement of the second extracellular loop in rhodopsin activation. Nat. Struct. Mol. Biol. 16, 168–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bokoch M. P., Zou Y., Rasmussen S. G., Liu C. W., Nygaard R., Rosenbaum D. M., Fung J. J., Choi H. J., Thian F. S., Kobilka T. S., Puglisi J. D., Weis W. I., Pardo L., Prosser R. S., Mueller L., Kobilka B. K. (2010) Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature 463, 108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Unal H., Jagannathan R., Bhat M. B., Karnik S. S. (2010) Ligand-specific conformation of extracellular loop-2 in the angiotensin II type 1 receptor. J. Biol. Chem. 285, 16341–16350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miura S., Karnik S. S. (2002) Constitutive activation of angiotensin II type 1 receptor alters the orientation of transmembrane Helix-2. J. Biol. Chem. 277, 24299–24305 [DOI] [PubMed] [Google Scholar]
- 13. Miura S., Zhang J., Boros J., Karnik S. S. (2003) TM2-TM7 interaction in coupling movement of transmembrane helices to activation of the angiotensin II type-1 receptor. J. Biol. Chem. 278, 3720–3725 [DOI] [PubMed] [Google Scholar]
- 14. Baleanu-Gogonea C., Karnik S. (2006) Model of the whole rat AT1 receptor and the ligand-binding site. J. Mol. Model. 12, 325–337 [DOI] [PubMed] [Google Scholar]
- 15. Feng Y. H., Noda K., Saad Y., Liu X. P., Husain A., Karnik S. S. (1995) The docking of Arg2 of angiotensin II with Asp281 of AT1 receptor is essential for full agonism. J. Biol. Chem. 270, 12846–12850 [DOI] [PubMed] [Google Scholar]
- 16. Groblewski T., Maigret B., Larguier R., Lombard C., Bonnafous J. C., Marie J. (1997) Mutation of Asn111 in the third transmembrane domain of the AT1A angiotensin II receptor induces its constitutive activation. J. Biol. Chem. 272, 1822–1826 [DOI] [PubMed] [Google Scholar]
- 17. Miura S., Feng Y. H., Husain A., Karnik S. S. (1999) Role of aromaticity of agonist switches of angiotensin II in the activation of the AT1 receptor. J. Biol. Chem. 274, 7103–7110 [DOI] [PubMed] [Google Scholar]
- 18. Hjorth S. A., Schambye H. T., Greenlee W. J., Schwartz T. W. (1994) Identification of peptide binding residues in the extracellular domains of the AT1 receptor. J. Biol. Chem. 269, 30953–30959 [PubMed] [Google Scholar]
- 19. Noda K., Feng Y. H., Liu X. P., Saad Y., Husain A., Karnik S. S. (1996) The active state of the AT1 angiotensin receptor is generated by angiotensin II induction. Biochemistry 35, 16435–16442 [DOI] [PubMed] [Google Scholar]
- 20. Feng Y. H., Miura S., Husain A., Karnik S. S. (1998) Mechanism of constitutive activation of the AT1 receptor. Influence of the size of the agonist switch binding residue Asn111. Biochemistry 37, 15791–15798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Costa-Neto C. M., Miyakawa A. A., Oliveira L., Hjorth S. A., Schwartz T. W., Paiva A. C. (2000) Mutational analysis of the interaction of the N- and C-terminal ends of angiotensin II with the rat AT(1A) receptor. Br. J. Pharmacol. 130, 1263–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hunyady L., Vauquelin G., Vanderheyden P. (2003) Agonist induction and conformational selection during activation of a G-protein-coupled receptor. Trends Pharmacol. Sci. 24, 81–86 [DOI] [PubMed] [Google Scholar]
- 23. Nikiforovich G. V., Mihalik B., Catt K. J., Marshall G. R. (2005) Molecular mechanisms of constitutive activity. Mutations at position 111 of the angiotensin AT1 receptor. J. Pept. Res. 66, 236–248 [DOI] [PubMed] [Google Scholar]
- 24. Lee C., Hwang S. A., Jang S. H., Chung H. S., Bhat M. B., Karnik S. S. (2007) Manifold active-state conformations in GPCRs. Agonist-activated constitutively active mutant AT1 receptor preferentially couples to Gq compared to the wild-type AT1 receptor. FEBS Lett. 581, 2517–2522 [DOI] [PubMed] [Google Scholar]
- 25. Clément M., Cabana J., Holleran B. J., Leduc R., Guillemette G., Lavigne P., Escher E. (2009) Activation induces structural changes in the liganded angiotensin II type 1 receptor. J. Biol. Chem. 284, 26603–26612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Akabas M. H., Stauffer D. A., Xu M., Karlin A. (1992) Acetylcholine receptor channel structure probed in cysteine-substitution mutants. Science 258, 307–310 [DOI] [PubMed] [Google Scholar]
- 27. Rakesh K., Yoo B., Kim I. M., Salazar N., Kim K. S., Rockman H. A. (2010) β-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci. Signal. 3, ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li M., Liu K., Michalicek J., Angus J. A., Hunt J. E., Dell'Italia L. J., Feneley M. P., Graham R. M., Husain A. (2004) Involvement of chymase-mediated angiotensin II generation in blood pressure regulation. J. Clin. Invest. 114, 112–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cotton M., Boulay P. L., Houndolo T., Vitale N., Pitcher J. A., Claing A. (2007) Endogenous ARF6 interacts with Rac1 upon angiotensin II stimulation to regulate membrane ruffling and cell migration. Mol. Biol. Cell 18, 501–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Escano C. S., Jr., Keever L. B., Gutweiler A. A., Andresen B. T. (2008) Angiotensin II activates extracellular signal-kinase independently of receptor tyrosine kinases in smooth muscle cells. Implications for blood pressure regulation. J. Pharmacol. Exp. Ther. 324, 34–42 [DOI] [PubMed] [Google Scholar]
- 31. Bonde M. M., Yao R., Ma J. N., Madabushi S., Haunsø S., Burstein E. S., Whistler J. L., Sheikh S. P., Lichtarge O., Hansen J. L. (2010) An angiotensin II type 1 receptor activation switch patch revealed through evolutionary traceanalysis. Biochem. Pharmacol. 80, 86–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boucard A. A., Roy M., Beaulieu M. E., Lavigne P., Escher E., Guillemette G., Leduc R. (2003) Constitutive activation of the angiotensin II type 1 receptor alters the spatial proximity of transmembrane 7 to the ligand-binding pocket. J. Biol. Chem. 278, 36628–36636 [DOI] [PubMed] [Google Scholar]
- 33. Martin S. S., Boucard A. A., Clément M., Escher E., Leduc R., Guillemette G. (2004) Analysis of the third transmembrane domain of the human type 1 angiotensin II receptor by cysteine scanning mutagenesis. J. Biol. Chem. 279, 51415–51423 [DOI] [PubMed] [Google Scholar]
- 34. Martin S. S., Holleran B. J., Escher E., Guillemette G., Leduc R. (2007) Activation of the angiotensin II type 1 receptor leads to movement of the sixth transmembrane domain. Analysis by the substituted cysteine accessibility method. Mol. Pharmacol. 72, 182–190 [DOI] [PubMed] [Google Scholar]
- 35. Domazet I., Martin S. S., Holleran B. J., Morin M. E., Lacasse P., Lavigne P., Escher E., Leduc R., Guillemette G. (2009) The fifth transmembrane domain of angiotensin II Type 1 receptor participates in the formation of the ligand-binding pocket and undergoes a counterclockwise rotation upon receptor activation. J. Biol. Chem. 284, 31953–31961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yan L., Holleran B. J., Lavigne P., Escher E., Guillemette G., Leduc R. (2010) Analysis of transmembrane domains 1 and 4 of the human angiotensin II AT1 receptor by cysteine-scanning mutagenesis. J. Biol. Chem. 285, 2284–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. West G. M., Chien E. Y., Katritch V., Gatchalian J., Chalmers M. J., Stevens R. C., Griffin P. R. (2011) Ligand-dependent perturbation of the conformational ensemble for the GPCR β2 adrenergic receptor revealed by HDX. Structure 19, 1424–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ahuja S., Smith S. O. (2009) Multiple switches in G protein-coupled receptor activation. Trends Pharmacol. Sci. 30, 494–502 [DOI] [PubMed] [Google Scholar]
- 39. Rana S., Baranski T. J. (2010) Third extracellular loop (EC3)-N terminus interaction is important for seven-transmembrane domain receptor function. Implications for an activation microswitch region. J. Biol. Chem. 285, 31472–31483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hwa J., Klein-Seetharaman J., Khorana H. G. (2001) Structure and function in rhodopsin. Mass spectrometric identification of the abnormal intradiscal disulfide bond in misfolded retinitis pigmentosa mutants. Proc. Natl. Acad. Sci. U.S.A. 98, 4872–4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stojanovic A., Hwang I., Khorana H. G., Hwa J. (2003) Retinitis pigmentosa rhodopsin mutations L125R and A164V perturb critical interhelical interactions. New insights through compensatory mutations and crystal structure analysis. J. Biol. Chem. 278, 39020–39028 [DOI] [PubMed] [Google Scholar]
- 42. Javitch J. A., Fu D., Liapakis G., Chen J. (1997) Constitutive activation of the β2 adrenergic receptor alters the orientation of its sixth membrane-spanning segment. J. Biol. Chem. 272, 18546–18549 [DOI] [PubMed] [Google Scholar]
- 43. Rasmussen S. G., Choi H. J., Fung J. J., Pardon E., Casarosa P., Chae P. S., Devree B. T., Rosenbaum D. M., Thian F. S., Kobilka T. S., Schnapp A., Konetzki I., Sunahara R. K., Gellman S. H., Pautsch A., Steyaert J., Weis W. I., Kobilka B. K. (2011) Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 469, 175–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shi L., Javitch J. A. (2004) The second extracellular loop of the dopamine D2 receptor lines the binding-site crevice. Proc. Natl. Acad. Sci. U.S.A. 101, 440–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Klco J. M., Wiegand C. B., Narzinski K., Baranski T. J. (2005) Essential role for the second extracellular loop in C5a receptor activation. Nat. Struct. Mol. Biol. 12, 320–326 [DOI] [PubMed] [Google Scholar]
- 46. Roberts D. J., Vertongen P., Waelbroeck M. (2011) Analysis of the glucagon receptor first extracellular loop by the substituted cysteine accessibility method. Peptides 32, 1593–1599 [DOI] [PubMed] [Google Scholar]
- 47. Lin J. C., Duell K., Konopka J. B. (2004) A microdomain formed by the extracellular ends of the transmembrane domains promotes activation of the G protein-coupled alpha-factor receptor. Mol. Cell Biol. 24, 2041–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Uddin M. S., Kim H., Deyo A., Naider F., Becker J. M. (2012) Identification of residues involved in homodimer formation located within a β-strand region of the N-terminus of a yeast G protein-coupled receptor. J. Recept. Signal Transduct. Res. 32, 65–75 [DOI] [PubMed] [Google Scholar]
- 49. Hauser M., Kauffman S., Lee B. K., Naider F., Becker J. M. (2007) The first extracellular loop of the Saccharomyces cerevisiae G protein-coupled receptor Ste2p undergoes a conformational change upon ligand binding. J. Biol. Chem. 282, 10387–10397 [DOI] [PubMed] [Google Scholar]
- 50. Wells J. A. (1990) Additivity of mutational effects in proteins. Biochemistry 29, 8509–8517 [DOI] [PubMed] [Google Scholar]
- 51. Post C. B., Ray W. J., Jr. (1995) Reexamination of induced fit as a determinant of substrate specificity in enzymatic reactions. Biochemistry 34, 15881–15885 [DOI] [PubMed] [Google Scholar]