

Background: Aberrant processing of the pro-IGF-II transcript produces pro- and big-IGF-II, which are secreted in a range of cancers.
Results: These induce potent receptor activation and cell proliferation and retard ternary complex formation with ALS and IGFBP-3 and -5.
Conclusion: They elicit unique biological responses that can be completely different from IGF-II.
Significance: Understanding the effects induced by these individual isoforms is crucial to elucidate their role in tumorigenesis.
Keywords: Cancer Biology, Cell Proliferation, Glycosylation, Insulin-like Growth Factor (IGF), Receptor Tyrosine Kinase, ALS, Bioavailable, IGF-IIR, IGFBP
Abstract
Insulin-like growth factor II (IGF-II) is a major embryonic growth factor belonging to the insulin-like growth factor family, which includes insulin and IGF-I. Its expression in humans is tightly controlled by maternal imprinting, a genetic restraint that is lost in many cancers, resulting in up-regulation of both mature IGF-II mRNA and protein expression. Additionally, increased expression of several longer isoforms of IGF-II, termed “pro” and “big” IGF-II, has been observed. To date, it is ambiguous as to what role these IGF-II isoforms have in initiating and sustaining tumorigenesis and whether they are bioavailable. We have expressed each individual IGF-II isoform in their proper O-glycosylated format and established that all bind to the IGF-I receptor and both insulin receptors A and B, resulting in their activation and subsequent stimulation of fibroblast proliferation. We also confirmed that all isoforms are able to be sequestered into binary complexes with several IGF-binding proteins (IGFBP-2, IGFBP-3, and IGFBP-5). In contrast to this, ternary complex formation with IGFBP-3 or IGFBP-5 and the auxillary protein, acid labile subunit, was severely diminished. Furthermore, big-IGF-II isoforms bound much more weakly to purified ectodomain of the natural IGF-II scavenging receptor, IGF-IIR. IGF-II isoforms thus possess unique biological properties that may enable them to escape normal sequestration avenues and remain bioavailable in vivo to sustain oncogenic signaling.
Introduction
Insulin-like growth factor II (IGF-II)2 is a small polypeptide ligand that is structurally and functionally related to IGF-I. Elegant genetic experiments have established a role for IGF-II as a major embryonic growth factor in mice, with IGF-I (together with growth hormone) being the dominant axis regulating postnatal somatic growth (1). However, IGF-II is the major circulating IGF in adult humans (2, 3), raising questions about its biological role after birth. IGF-II is able to elicit cellular signaling by binding and activating either the IGF-I receptor (IGF-IR), the two alternatively spliced isoforms of the structurally related insulin receptor (IR-A and IR-B) or, as highlighted in several studies, the IGF-IIR or cation-independent mannose-6-phosphate receptor (4, 5). The multiple receptors engaged by this ligand offer several redundant avenues to propagate its biological effects. Crucially, in many cancers including hepatocellular carcinoma, loss of maternal imprinting of the IGF-II allele contributes to tumorigenesis by activating biallelic expression of IGF-II and subsequent elevation of intratumoral levels of IGF-II mRNA and protein (6–8). Activation of IGF-IR, and possibly IR, signaling is then triggered, which aids the growth of the tumor (9).
IGF-II is translated as a 156-amino acid pro-ligand consisting of the N-terminal mature IGF-II sequence of 67 amino acids, together with a C-terminal 89 amino acid E-domain, which is glycosylated by up to four O-linked sugars (10). Sequential cleavage by pro-convertase enzymes at several intermediate consensus sites (amino acids 104, 87, and 67) yields the mature IGF-II ligand. The bioavailability of IGF-II in vivo is controlled by sequestration of the ligand into a binary complex with one of six IGF-binding proteins, IGFBP-1 to 6 (11, 12). Furthermore, a ternary complex can form by recruiting the auxillary protein, acid labile subunit (ALS), to binary complexes containing IGFBP-3 and IGFBP-5 (11, 12). The IGF·IGFBP-3·ALS ternary complex is thought to exist as the majority species in the bloodstream and is believed to impede paracellular transport of the ligand across vascular endothelial barriers (13).
Although some intermediate forms of IGF-II are present in normal adult serum, in cancer, processing of IGF-II is commonly incomplete, with a sharp increase in high molecular weight IGF-II isoforms detected in patient serum. These isoforms consist of a heterogenous mix of unprocessed pro-IGF-II (1–156), so-called “big” IGF-II (1–104), and big IGF-II (1–87) and are, in the majority, heterogeneously O-glycosylated. These isoforms have been directly linked to non-islet cell tumor hypoglycemia (NICTH), a pathological sequela of several cancers including hepatocellular carcinoma, gastric cancer, and colon cancer (14). Because serum isolated from NICTH patients is a rich source of heterogeneous IGF-II isoforms, the majority of studies have utilized pooled isolates from this origin or from conditioned cell culture media. However, it is the heterogeneous nature of IGF-II in these samples that makes it difficult to unequivocally establish the biological properties of the individual IGF-II isoforms.
For example, studies utilizing pooled isolates from bovine serum have indicated that the IGF-II isoforms can bind and activate IGF-IR (15). However, results showing that pooled IGF-II isoforms from NICTH serum or tumor tissue form binary complexes with IGFBP-1 to -6 (16) contrast with studies demonstrating pooled IGF-II isoforms from conditioned Ewing's sarcoma media not forming binary complexes with IGFBP-2 and IGFBP-3 (17). Similarly, evidence shows that glycosylated IGF-II isoforms from NICTH tumor tissue can escape ternary complex with IGFBP-3 and ALS by retarding ALS recruitment (16), but it is unknown what contribution the individual IGF-II isoforms make in this respect and what effect this has on bioavailability. Certainly, our studies in vivo have shown selective depletion of IGF-II isoforms, not mature IGF-II, from hepatocellular carcinoma tumors treated with DX-2647, an IGF-II neutralizing antibody (18). Because DX-2647 only binds free IGF-II, this implies that partially processed IGF-II isoforms are more bioavailable within the local tumor microenvironment, with implications for the role of these isoforms in tumor progression.
Previous attempts to answer questions related to the properties of individual IGF-II isoforms have used recombinant, unglycosylated proteins expressed in Escherichia coli. This has led to yet more discrepancies; for example, whether the induction of Akt signaling by pro-IGF-II 1–156 is less than (2) or greater than (19) that induced by mature IGF-II. What these studies fail to address is the fact that the majority of IGF-II isoforms are O-glycosylated and how this post-translational modification may impact on the biological activity. We directly address this issue by expressing the three major human O-glycosylated IGF-II isoforms associated with cancer: 1–156, 1–104, and 1–87, as recombinant proteins in human 293F cells. We then compared the performance of these homogenous preparations in a suite of biochemical and biological assays to mature IGF-II.
EXPERIMENTAL PROCEDURES
Cell Lines
The R−IGF1R, R−IR-A, and R−IR-B cell lines used in this study were cultured and maintained as described elsewhere (20). The suspension-adapted HEK293FreestyleTM (293F) cell line was obtained from Invitrogen and cultured in suspension using 293Freestyle medium (Invitrogen) in a shaking incubator at 100–140 rpm, 37 °C, 5% CO2, and 70% humidity. The cells were split every 2–3 days at a seeding density of 0.2–0.5 × 106 cells/ml.
Antibodies and Reagents
Europium-labeled IGF-II, human IGF-IR extracellular protein domains, and the human IR-A extracellular protein domains were all produced in-house as described (21, 22). Unglycosylated pro-IGF-II (1–156) and big-IGF-II (1–104), both produced in E. coli, were purchased from GroPep (Australia). Purified human IGFBP-2, IGFBP-3, and IGFBP-5 have been described elsewhere (16). The anti-IR mAb 83-7 and the anti-IGF-IR mAb 24-31 were produced in-house (23, 24). The anti-IGFBP-2 and anti-IGFBP-3 polyclonal antibodies were purchased from Santa Cruz Biotechnology. The anti-IGFBP-5 mAb was purchased from Abcam, and europium-labeled anti-phosphotyrosine monoclonal antibody (PY20) and europium assay enhancement solution were purchased from PerkinElmer Life Sciences.
Production and Purification of IGF-II Isoforms
The open reading frame for pro-IGF-II (1–156) containing three mutations, R104A, R87E, and R68E (10), and a C-terminal FLAG tag was synthesized by GeneArt and cloned into the pEE6.4 mammalian expression vector (Lonza Biologics). The resulting construct was used as template for PCR amplification of big-IGF-II (1–104) with a C-terminal FLAG tag using forward primer 5′-CCGCTCGAGATGGGCATCCCTATGGG-3′ and reverse primer 5′-ATAGTTTAGCGGCCGCTCACTTGTCGTCGTCGTCCTTGTAGTCGCGTCTCAGTCTCTGGG-3′ to incorporate a C-terminal FLAG tag. Big-IGF-II (1–87) was similarly amplified from the template using the 1–104 forward primer and reverse primer 5′-ATAGTTTAGCGGCCGCTCACTTGTCGTCGTCGTCCTTGTAGTCGCCCACGGGG- TATCTG-3′ to incorporate a C-terminal FLAG tag. Both big-IGF-II isoforms were then cloned into mammalian expression vectors. Suspension-adapted 293F cells were transfected with the plasmids using 2 mg of polyethylenimine, and expression was conducted for 7 days. Supernatants were then clarified by centrifugation and 0.2-μm filtration. Supernatants containing 1–156, 1–104, and 1–87 were purified by affinity chromatography on an anti-FLAG affinity column (Mini-Leak Low; Kem-En-Tec A/S, Copenhagen, Denmark) by passing the supernatants over two anti-FLAG columns linked in series (0.5 ml/min) at 4 °C overnight. The columns were washed with 20 column volumes of Tris-buffered saline containing 0.02% sodium azide (TBSA), pH 8.0. FLAG-tagged protein was eluted from the column using FLAG peptide (DYKDDDDK in TBSA, pH 8.0) at two different concentrations by recirculating 25 ml of a 0.25 mg/ml solution of the FLAG peptide for 30 min, followed by a second elution step using 50 ml of a 0.025 mg/ml solution of the FLAG peptide. Elutions 1 and 2 were pooled and concentrated using an Amicon Ultra-15 centrifugal concentrator with a 10-kDa molecular mass cutoff (Millipore) and further polished by size exclusion chromatography over a Superdex 75 column (Pharmacia; 10/30) in TBSA at 0.5 ml/min.
Production and Purification of the IGF-IIR Extracellular Domain 11–13 Fragment and Europium-labeled Human Acid Labile Subunit (Eu-ALS)
The pJB6 plasmid containing cDNA for extracellular domains 11–13 of IGF-IIR were kindly provided by Prof. E. Yvonne Jones (University of Oxford, Oxford, UK). The human ALS cDNA sequence was amplified by PCR to incorporate a C-terminal His tag using the pALTER/ALS plasmid as template. The product was subcloned into the pEE6.4 mammalian expression vector. Transfection of cultures of suspension-cultured 293F cells were performed as described above. The supernatants were then clarified by centrifugation and 0.2-μm filtration and then applied to a 1-ml HisTrap FF column equilibrated with HisA buffer (20 mm phosphate, 0.5 m NaCl, pH 7.4). The column was washed with HisA containing 20 mm imidazole, and then the protein was eluted with HisA containing 50 mm imidazole and then 300 mm imidazole. The unbound eluate was collected, applied to a second HisTrap column, and similarly eluted. Pooled eluates were then subject to size exclusion chromatography on a Superdex 200 column, where fractions within the uniform IGF-IIR extracellular domain 11–13 or ALS peak were pooled and concentrated. Approximately 100 μg of purified ALS was then labeled with Europium using a DELFIA®Eu-labeling kit at an Eu-N1-(p-isothiocyanatobenzyl)-diethylenetriamine-N1,N2,N3,N3-tetraacetic acid:ALS ratio of 15:1 according to the manufacturer's instructions.
Glycosylation Analysis by Mass Spectrometry and N-terminal Sequencing
Purified ligands were subjected to tryptic digest overnight. Peptide fragments were then separated by HPLC using a Vydac C8 column (2.1 mm × 15 cm) and a gradient of 5–70% Buffer B (0.1% TFA in 70% acetonitrile) in 50 ml at a flow rate of 0.2 ml/min at room temperature. Glycosylated fragments were identified on visual examination of the chromatograph for peak heterogeneity. Select peaks were then subject to Edman degradation and N-terminal sequencing. To analyze the approximate identity of the glycans attached to peptide fragments, the individual peak fractions were analyzed by MALDI-TOF mass spectrometry as previously described (25). The approximate identity of each glycan present in each peptide fraction was then elucidated by subtracting the calculated mass of the peptide backbone from the observed mass of the peak in the MALDI MS profile and matching the molecular weight to that of a known glycan.
Europium-based Phosphotyrosine Detection Assays
Glycosylated IGF-II isoforms and mature IGF-II were tested for their ability to elicit activation of the IGF-IR, IR-A, and IR-B using the Eu-phosphotyrosine assay described elsewhere (26).
Europium-based IGF-IR and IR Competition Binding Assays
Glycosylated IGF-II isoforms and mature IGF-II were tested for their ability to bind the IGF-IR, IR-A, and IGF-IIR using Eu-IGF-II competition assays described elsewhere (26).
Europium-based IGFBP Binary Complex Competition Assays
Unlabeled IGF-II or glycosylated IGF-II isoforms were serially diluted 6-fold in PBS containing 0.25% human serum albumin and 2 μm diethylenetriaminepentaacetic acid at pH 6.5. Next, 10 ng of IGFBP-2, -3, and 5 and 100,000 counts of Eu-IGF-II were added to each dilution and incubated overnight at 4 °C. White-walled ELISA plates were coated with anti-IGFBP antibodies at 4 °C overnight in bicarbonate buffer, pH 9.6. The next day, the plates were washed four times in TBS containing 0.1% Tween 20 (TBST) and then blocked in 0.5% ovalbumin in TBS for 90 min at room temperature. After another wash, the complexes were added to relevant wells in triplicate and incubated for 2 h at room temperature. The plates were washed again before Europium enhancement solution was added for 20 min at room temperature. The time-resolved fluorescence per well was then read on a Wallac 1420 Multilabel Counter.
Europium-based IGFBP Ternary Complex Formation Assays
White-walled ELISA plates were coated with anti-IGFBP-3 or anti-IGFBP-5 antibody at 4 °C overnight in bicarbonate buffer, pH 9.6. The next day, glycosylated and unglycosylated IGF-II isoforms were prepared in PBS containing 0.25% human serum albumin and 2 μm diethylenetriaminepentaacetic acid at pH 6.5. Afterward, 40,000 counts/reaction of Eu-ALS were added followed by 10 ng/reaction of IGFBP-3 or IGFBP-5. The entire mixture was incubated for 2 h at room temperature. The plates were washed in TBST and blocked in 0.5% ovalbumin in TBS. At the end of ternary complex formation, the plates were washed, and complexes were added to relevant wells in triplicate or quadruplicate and incubated for 2 h at room temperature. The plates were washed again before Europium enhancement solution was added for 20 min at room temperature. The time-resolved fluorescence/well was then read on a Wallac 1420 Multilabel Counter.
Cell Proliferation Assay
Cellular proliferation assays were conducted essentially as previously described (27) utilizing the CellTiter Glo Luminescent Cell Viability Assay (Promega) according to the manufacturer's instructions.
RESULTS
Production of Glycosylated IGF-II Isoforms
All isoforms were successfully expressed and secreted into medium after transfection. Initial chromatogram profiles suggested that some of the secreted pro-IGF-II 1–156 had, despite the mutations made, still been cleaved into smaller isoforms (data not shown), suggesting a functional redundancy at the pro-convertase cleavage sites. Fractions corresponding to the expected molecular weights were pooled and concentrated for all isoforms, with yields ranging from 1.9 to 2.4 mg from 1 liter of crude supernatant. Coomassie-stained gels showed proteins in each isolated fraction were of correct size and, as evidenced by smearing of the sample, most likely O-glycosylated (gels for pro-IGF-II 1–156 fractions are shown in supplemental Fig. S1). This was confirmed by Edman degradation and N-terminal sequencing, which was conducted on glycosylated peptide fragments derived from tryptic digest of pure populations of each ligand. These peptide fragments were confirmed as originating from E-domain regions 66–83, 69–83, and 131–155 and revealed that the correct residues of Ser71, Ser72, Ser75, and Thr139 were O-glycosylated (data not shown). Mass spectrometry analyses of these same fragments show molecular weights consistent with an O-glycan backbone decorated with a variable number of sialic acid residues, consistent with previous reports (25).
Glycosylated IGF-II Isoforms Display a Similar Binding Affinity to Mature IGF-II for Both IGF-IR and IR
We initially investigated the ability of increasing concentrations of the glycosylated isoforms to competitively displace europium-labeled IGF-II for binding to purified human recombinant IGF-IR and IR-A ectodomain trapped on antibody-coated plates. Competitive binding curves for binding to IGF-IR (Fig. 1A) and IR-A (Fig. 1B), with the calculated IC50 values for each ligand on each receptor (Table 1), are shown. Binding of the isoforms to the IGF-IR was quite similar to IGF-II, with no major differences for 1–156 or 1–104 but a 10-fold reduction in binding affinity for 1–87. A similar binding profile was observed for IR-A, with comparison with IGF-II showing an approximate 20-fold decrease for 1–87 but similar binding affinities for the other ligands tested.
FIGURE 1.
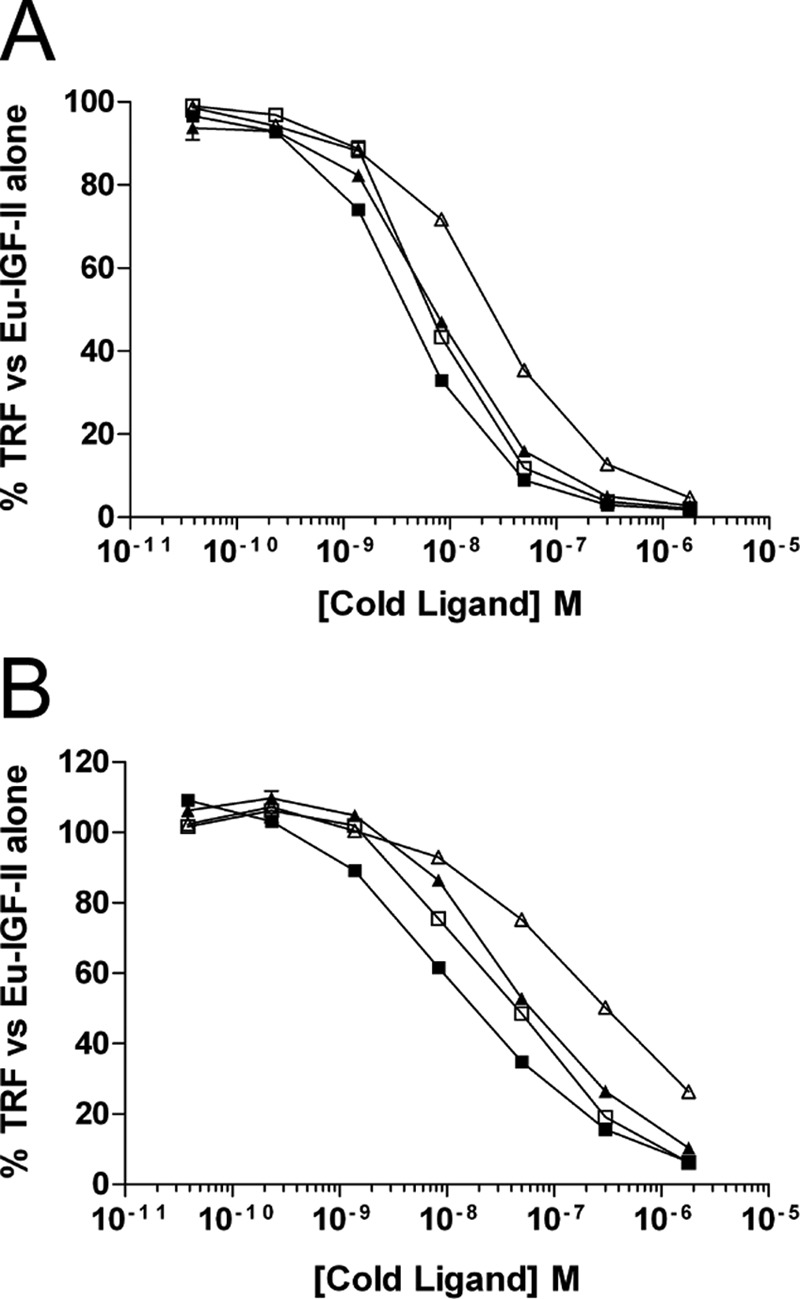
Competition binding curves of glycosylated IGF-II isoforms to purified extracellular domains of IGF-IR (A) or IR-A (B). Increasing amounts of each ligand were incubated in complex with 25 ng of purified receptor and 100,000 counts of Eu-IGF-II. Complexes were trapped on antibody-coated plates, and the time resolved fluorescence (TRF) was measured. The results are expressed as percentages of the TRF measured for Eu-IGF-II·receptor complex in the absence of competitor and are expressed as the means ± S.E. ■, IGF-II; □, glycosylated 1–156; ▴, glycosylated 1–104; ▵, glycosylated 1–87.
TABLE 1.
Inhibition of europium-labeled IGF-II binding to purified IGF-IR and IR-A ectodomains by mature IGF-II and glycosylated IGF-II isoforms
| Ligand | IGF-IR |
IR-A |
||
|---|---|---|---|---|
| IC50 | IC50 relative to IGF-II | IC50 | IC50 relative to IGF-II | |
| nm | % | nm | % | |
| Mature IGF-II | 4.4 ± 0.39 | 100 | 21.36 ± 5.58 | 100 |
| 1–87 | 45.6 ± 18.1 | 10 | 518.4 ± 307.8 | 4 |
| 1–104 | 8.23 ± 0.71 | 53 | 43 ± 2.9 | 50 |
| 1–156 | 6.67 ± 0.1 | 66 | 32 ± 1.6 | 67 |
Glycosylated IGF-II Isoforms Show Distinct Abilities to Activate IGF-IR, IR-A, and IR-B
The ability of each glycosylated isoform to activate IGF-IR, IR-A, and IR-B was tested using a panel of mouse fibroblast cell lines expressing the human receptors for IGF-IR, IR-A, and IR-B. Maximum receptor activation was determined by comparing all data to the average activation obtained using either 33 nm IGF-I, for IGF-IR, or 1 μm insulin, for IR-A and IR-B, because these doses have previously been reported to stimulate maximum activation of these receptors (26). The dose response curves for IGF-IR (Fig. 2A), IR-A (Fig. 2B), and IR-B (Fig. 2C) are shown, as well as the calculated EC50 values for each ligand on each receptor (Table 2).
FIGURE 2.
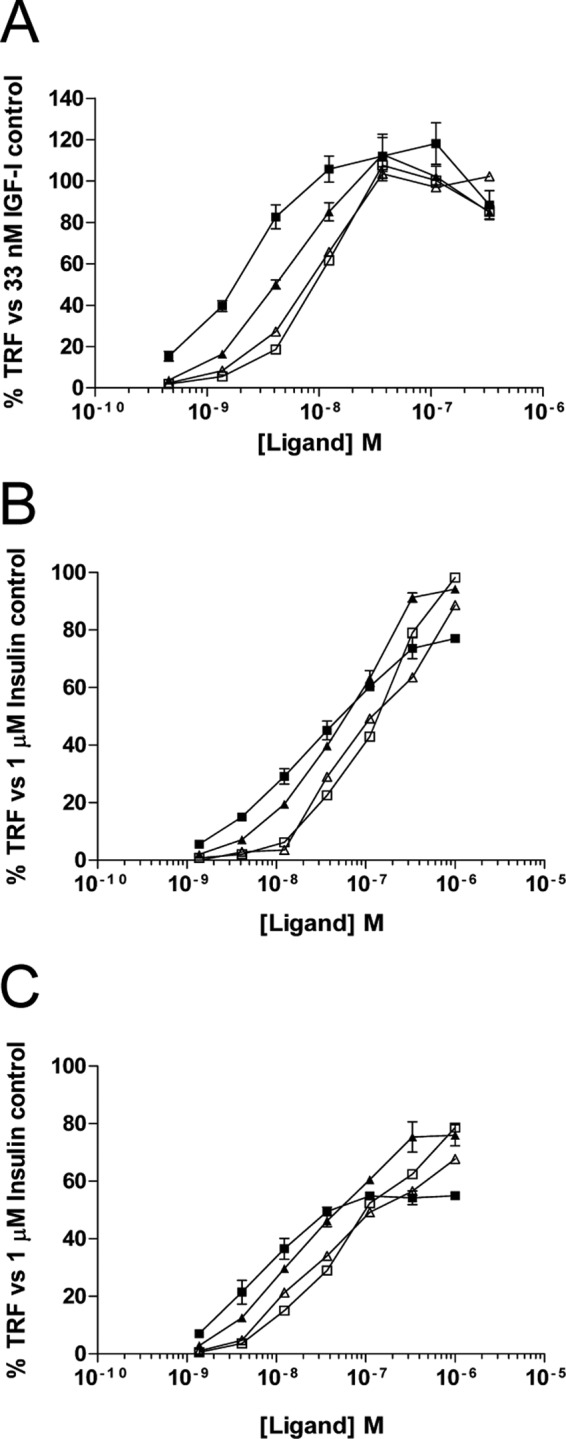
Activation of the IGF-IR and insulin receptors by mature IGF-II and glycosylated IGF-II isoforms. Mouse fibroblasts expressing either IGF-IR, IR-A, or IR-B were serum-starved overnight before treatment with increasing concentrations of each ligand. The cells were then lysed in ice-cold buffer containing phosphatase inhibitor and bound to plates coated with anti-IGF-IR or anti-IR antibody. Phosphorylated receptor was detected using a Europium-labeled pan-phosphotyrosine antibody and TRF measured. A, IGF-IR activation by IGF-II and its isoforms. The graph shows the mean percentage TRF as compared with a 33 nm IGF-I stimulated receptor ± S.E. B, IR-A activation by IGF-II and its isoforms. The graph shows the mean percentage TRF as compared with a 1 μm insulin stimulated receptor ± S.E. C, IR-B activation by IGF-II and its isoforms. The graph shows the mean percentage TRF as compared with a 1 μm insulin-stimulated receptor ± S.E. ■, IGF-II; □, glycosylated 1–156; ▴, glycosylated 1–104; ▵, glycosylated 1–87.
TABLE 2.
Stimulation of tyrosine phosphorylation of the IGF-IR, IR-A, and IR-B by mature IGF-II and glycosylated IGF-II isoforms
| Ligand | IGF-IR |
IR-A |
IR-B |
|||
|---|---|---|---|---|---|---|
| EC50 | EC50 relative to IGF-II | IC50 | EC50 relative to IGF-II | EC50 | EC50 relative to IGF-II | |
| nm | % | nm | % | nm | % | |
| Mature IGF-II | 2.37 ± 0.45 | 100 | 18.75 ± 3.91 | 100 | 6.39 ± 0.19 | 100 |
| 1–87 | 9.02 ± 1.5 | 26 | 128.4 ± 24.6 | 15 | 32.92 ± 0.67 | 19 |
| 1–104 | 4.76 ± 0.4 | 50 | 44.86 ± 7.9 | 42 | 17.63 ± 3.43 | 36 |
| 1–156 | 14.68 ± 2 | 16 | 244.7 ± 103.7 | 8 | 62.84 ± 10.87 | 10 |
For each receptor, differences were observed between the binding IC50 and the activation EC50 for 1–87 and 1–156, relative to IGF-II. Isoform 1–104 generally matched the relative affinities compared with IGF-II for both binding and activation of IGF-IR and IR-A. In contrast, whereas 1–87 possessed a 10- and 20-fold weaker IC50 for binding to IGF-IR and IR-A (Fig. 1 and Table 1), respectively, its activation EC50 relative to IGF-II was only 4- and 6-fold lower for IGF-IR and IR-A (Fig. 2 and Table 2), respectively. This demonstrates that, despite binding the receptors most weakly, 1–87 was able to induce a potent activation response. Conversely, whereas 1–156 showed strong binding to IGF-IR and IR-A with a 1.3-fold weaker IC50 relative to IGF-II, its activation kinetics were quite poor, with a 6- and 12-fold lower EC50 for IGF-IR and IR-A, respectively.
All ligands reached a saturating dose for IGF-IR activation (Fig. 2A). However, not all isoforms induced saturation of IR-A and IR-B activation, whereas IGF-II did so at an approximate dose of 333 nm for IR-A and 111 nm for IR-B (Fig. 2, B and C). At 1 μm, each isoform had stimulated IR-A to 90–100% maximum phosphorylation and IR-B to ∼80% maximum phosphorylation. Interestingly, despite a lower overall degree of receptor activation for IR-B, the EC50 values measured for this receptor in comparison with IR-A were approximately 3–4-fold lower for each ligand used (Table 2).
Binary Complex Formation Is Not Impeded by Glycosylated IGF-II Isoforms, but Recruitment of ALS to Ternary Complex Is Severely Hindered
To see whether the glycosylated isoforms differed in their ability to be sequestered into binary complexes with selected IGFBPs, increasing concentrations of each ligand were tested for their ability to displace europium-labeled IGF-II for binding to IGFBP-2 (Fig. 3A), IGFBP-3 (Fig. 3B), and IGFBP-5 (Fig. 3C). Complexes were preformed in tubes and then trapped onto plates coated with the relevant anti-IGFBP antibody. There was very little variance in the ability of each glycosylated isoform to displace Eu-IGF-II tracer, with 2-fold differences being the maximum observed (Table 3).
FIGURE 3.
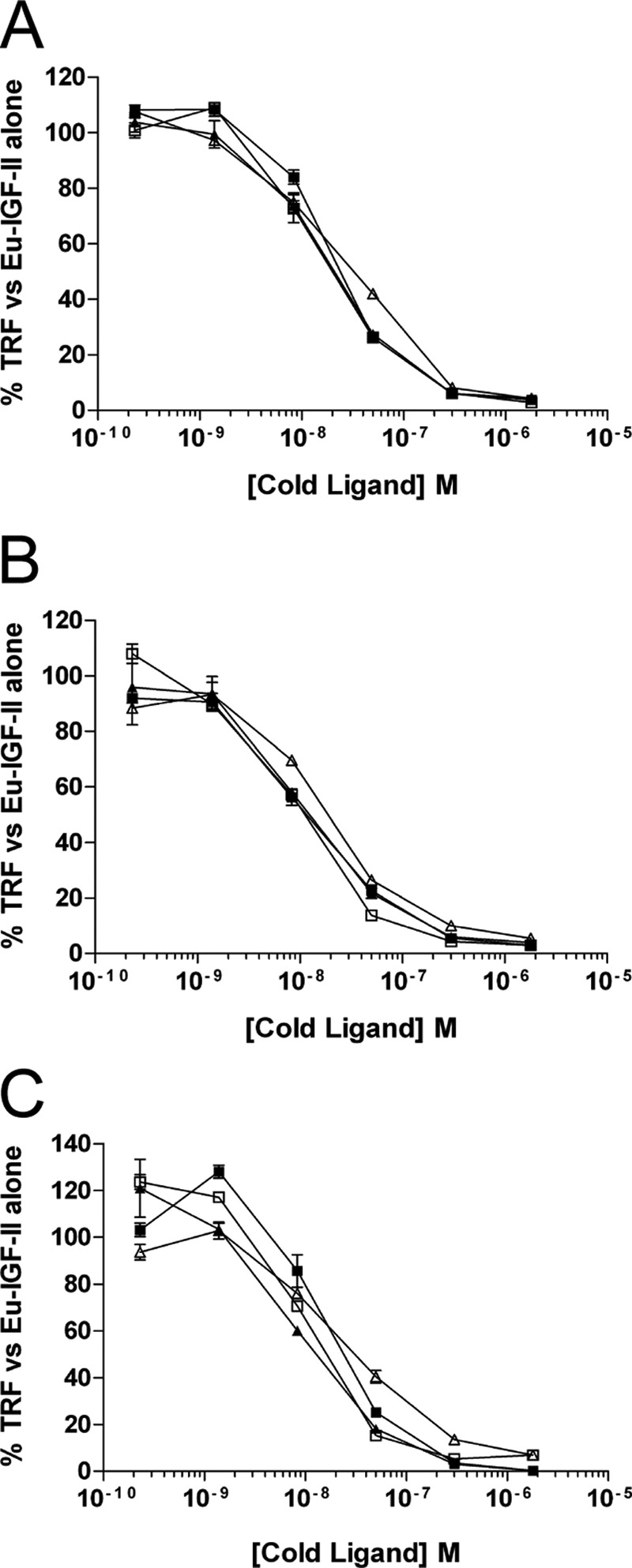
Competition binding curves of glycosylated IGF-II isoforms to purified IGFBP-2 (A), IGFBP-3 (B), and IGFBP-5 (C). Increasing amounts of each ligand were incubated in complex with 10 ng of purified IGFBP and 100,000 counts of Eu-IGF-II. The complexes were trapped on antibody-coated plates, and TRF was measured. The results are expressed as percentages of the TRF measured for Eu-IGF-II·IGFBP complex in the absence of competitor and are expressed as the means ± S.E. ■, IGF-II; □), glycosylated 1–156; ▴, glycosylated 1–104; ▵, glycosylated 1–87.
TABLE 3.
Inhibition of europium-labeled IGF-II binding to IGFBP-2, IGFBP-3, and IGFBP-5 by mature IGF-II and glycosylated IGF-II isoforms (binary complex formation)
| Ligand | IGFBP-2 |
IGFBP-3 |
IGFBP-5 |
|||
|---|---|---|---|---|---|---|
| IC50 | IC50 relative to IGF-II | IC50 | IC50 relative to IGF-II | IC50 | IC50 relative to IGF-II | |
| nm | % | nm | % | nm | % | |
| Mature IGF-II | 17.59 ± 2.29 | 100 | 16.73 ± 4.64 | 100 | 12.88 ± 2.13 | 100 |
| 1–87 | 28.63 ± 4.22 | 61 | 15.34 ± 4.39 | 109 | 22.37 ± 5.3 | 58 |
| 1–104 | 26.91 ± 7.77 | 65 | 25.09 ± 13.5 | 67 | 12.88 ± 2.94 | 100 |
| 1–156 | 36.76 ± 11.42 | 48 | 10.96 ± 2.85 | 153 | 14.85 ± 5.27 | 87 |
When the glycosylated isoforms were tested at a fixed ligand concentration (10 ng) for ternary complex formation with Eu-ALS and IGFBP-3 (Fig. 4A) or IGFBP-5 (Fig. 4C), all showed significant impairment of ternary complex formation (p < 0.0001). Specifically, glycosylated 1–87 displayed the most potent inhibition of Eu-ALS incorporation (∼90% reduction) followed by 1–104 (∼80% reduction) and 1–156 (∼65% reduction), compared with IGF-II. The unglycosylated 1–104 and 1–156 showed no significant difference to the IGF-II control in the presence of IGFBP-3 but were significantly lower than IGF-II with IGFBP-5 (p < 0.0001). A more in-depth analysis into ternary complexes in the presence of increasing amounts of glycosylated and unglycosylated 1–156 and IGFBP-3 (Fig. 4B) showed that, whereas mature IGF-II·IGFBP complex efficiently bound the Eu-ALS, binary complexes formed with glycosylated 1–156 showed an impaired ability to recruit Eu-ALS (p < 0.001). Lower amounts of unglycosylated 1–156 produced a profile of a rapid reduction in the level of Eu-ALS recruitment to ternary complex. Surprisingly, over several increasing amounts of unglycosylated 1–156, Eu-ALS binding continued to rise and did not reach saturation until 40 ng of ligand was used (data not shown). This was well above the saturating ligand amounts of 5 ng for IGF-II and 10 ng for glycosylated 1–156. This indicates that unglycosylated 1–156 may facilitate a greater amount of ternary complex formation at higher ligand concentrations, in contrast to previous reports showing pools of unglycosylated high molecular weight IGF-II isoforms forming identical ternary complexes to mature IGF-II (28). Overall, all glycosylated isoforms were able to inhibit ternary complex formation with both IGFBP-3 and IGFBP-5, with some inhibition of IGFBP-5 ternary complex formation associated with unglycosylated 1–104 and 1–156.
FIGURE 4.

Analysis of ternary complex formation and binding to IGF-IIR extracellular fragment. In all tests, complex was trapped on ELISA plates by either an IGFBP antibody, for IGFBP tests, or anti-His tag antibody for IGF-IIR. The TRF values was then recorded and analyzed. gly denotes O-glycosylated ligand, and ungly denotes E. coli-produced unglycosylated ligand. A, IGFBP-3 ternary complex formation using 10 ng of ligand, 10 ng of IGFBP, and 40,000 counts of Eu-ALS. The graph shows the percentage TRF measured for Eu-ALS recruitment to complexes involving each ligand as compared with the mature IGF-II control ± S.E. B, titration of mature IGF-II, glycosylated 1–156, or unglycosylated 1–156 in complex with 10 ng of IGFBP-3 and 40,000 counts of Eu-ALS. The results are expressed as percentages of input Eu-ALS TRF bound to the complex compared with an Eu-ALS·IGFBP-3 control and are expressed as the means ± S.E. ▴, IGF-II; ●, glycosylated 1–156; ○, unglycosylated 1–156. C, IGFBP-5 ternary complex formation using 10 ng of ligand, 10 ng of IGFBP, and 40,000 counts of Eu-ALS. The graph shows the percentage of TRF measured for Eu-ALS recruitment to complexes involving each ligand as compared with the mature IGF-II control ± S.E. D, competition binding curves of glycosylated IGF-II isoforms to the purified IGF-IIR extracellular domain 11–13 fragment. The results are expressed as a percentage of the TRF measured for Eu-IGF-II·receptor complex in the absence of competitor and are expressed as the means ± S.E. ■, IGF-II; □, glycosylated 1–156; ▴, glycosylated 1–104; ▵, glycosylated 1–87.
Glycosylated IGF-II Isoforms Display Individual IGF-IIR Binding Characteristics
Another pathway of sequestration of IGF-II in vivo is internalization and degradation after binding to its cognate receptor, the IGF-IIR. To test the ability of the glycosylated isoforms to bind this receptor, we conducted Eu-IGF-II competition assays against the purified IGF-IIR extracellular domain 11–13 fragment, which contains all sequences necessary and sufficient to bind IGF-II. Complexes were formed and bound on anti-His tag antibody-coated plates, and the results were compared with profiles obtained for mature IGF-II (Fig. 4D). In juxtaposition with previous receptors, 1–104 bound IGF-IIR with similar affinity to mature IGF-II. However, startlingly different profiles were observed for 1–156 and 1–87. Isoform 1–156 bound IGF-IIR with far greater magnitude than mature IGF-II, whereas isoform 1–87 bound IGF-IIR very poorly compared with mature IGF-II. These results indicate that the size and glycosylation status of the E-domain is crucial for decreasing the affinity of individual isoform species for IGF-IIR and that glycosylated 1–87 is very effective at evading binding to IGF-IIR.
All Glycosylated Isoforms of IGF-II Stimulate Proliferation Utilizing IGF-IR, IR-A, and IR-B
It is currently unclear as to whether IGF-II isoforms are able to stimulate proliferative responses using IGF-IR, IR-A, and IR-B. To address this, we tested concentrations of ligand, determined from Fig. 2, that stimulate either 10–40 or 60–90% of maximal receptor phosphorylation for each receptor. This was equal to 1 and 10 nm, respectively, for IGF-IR, or 10 and 100 nm, respectively, for IR-A and IR-B. Proliferative responses mediated via IGF-IR (Fig. 5, A and B), IR-A (Fig. 5, C and D), and IR-B (Fig. 5, E and F) were determined after 72 h and compared with vehicle controls. Although the more significant (p < 0.001) robust proliferative responses were observed for the IGF-IR and IR-A, utilization of the IR-B by all ligands also led to significant increases in cell proliferation over vehicle controls (p < 0.05). Because this response was observed at both concentrations of ligand used, our data indicated that only minimal activation of all three receptors was required to induce proliferation of cells.
FIGURE 5.

Proliferation induced by stimulation of individual receptors by mature IGF-II or its glycosylated isoforms. Mouse fibroblasts expressing IGF-IR, IR-A, or IR-B were serum-starved and treated with concentrations of each ligand, which would induce minimal and maximal activation of each receptor as determined in Fig. 2. After 72 h of treatment, cellular ATP levels were measured. Each graph shows the percentage proliferation as compared with vehicle controls ± S.E. A, IGF-IR at 1 nm ligand. B, IGF-IR at 10 nm ligand. C, IR-A at 10 nm ligand. D, IR-A at 100 nm ligand. E, IR-B at 10 nm ligand. F, IR-B at 100 nm ligand.
DISCUSSION
Using homogeneous preparations of recombinant, O-glycosylated IGF-II isoforms corresponding to those high molecular weight IGF-II isoforms associated with a number of cancers such as hepatocellular carcinoma, gastric carcinoma, and colon carcinoma (14), we have identified characteristics to suggest that increased bioavailability of one or more isoforms may provide a selective growth advantage to tumors.
Our studies have clarified a number of aspects of basic IGF-II isoform biology that were previously ambiguous. We have now confirmed that all IGF-II isoforms can bind and activate not only IGF-IR and IR-A but also IR-B. In particular, we have observed that the isoforms are able to activate the IR just as well as mature IGF-II at higher concentrations, and some appear to avoid saturation of the receptor, unlike mature IGF-II. Minimal activation of all three receptors was found to be sufficient to induce a significant increase in mouse fibroblast proliferation, with additional improvements in proliferation observed for IGF-IR and IR-A when the concentration of the IGF-II isoforms was increased. This is in contrast to the observations of Marks et al. (2), who used unglycosylated IGF-II isoforms that resulted in a modest 1.4-fold increase in cell number over vehicle controls and no additional concentration-specific improvement in cell proliferation when compared mature IGF-II.
The findings presented here have also confirmed that IGFBP-2, IGFBP-3, and IGFBP-5 bind all IGF-II isoforms similarly to mature IGF-II, in agreement with Bond et al. (16) but not with Elmlinger et al. (17), who reported that pooled isolates of IGF-II isoforms from Ewing's sarcoma cell conditioned media appeared to retard binary complex formation with IGFBP-2 and IGFBP-3. Our approach has also confirmed early findings that IGFBP-3-based ternary complex formation by glycosylated IGF-II isoforms is severely compromised (16) and has refined these findings to show that big-IGF-II 1–87 and big-IGF-II 1–104 are the most potent at preventing its assembly. However, we also found IGFBP-5-based ternary complex formation to be compromised in the presence of glycosylated IGF-II isoforms, in contrast to what had been reported by the same group (16). Curiously, we also observed that unglycosylated IGF-II isoform 1–156 facilitated greater IGFBP-3-based ternary complex formation but retarded ALS association with IGFBP-5. This is important because it implies that there are major conformational differences in the quaternary structures formed when the ternary complexes of IGFBP-3 and IGFBP-5 are formed with ALS in the presence of IGF-II isoforms. We believe the exclusion of ALS from binary complex may be caused by an ionic association of negatively charged sialic acid residues on O-linked sugars within the E-domain interacting with the positively charged basic amino acids present in the ALS-binding motif on both IGFBP-3 (29) and IGFBP-5 (30). This association would most likely block ALS binding to the domain and repulse it from the binary complex. Further experiments involving neuraminidase treatment of the glycosylated IGF-II isoforms to remove the positively charged sialic acid residues will be required to prove this theory.
One consequence of the enhanced bioavailability of glycosylated IGF-II isoforms secreted by tumors in vivo is the potential to sustain prolonged oncogenic signaling, which may aid the growth of tumors. From clinical specimens, extraordinarily high concentrations of IGF-II have been measured from tumor fluid extracted from hemangiopericytoma patients, which ranges from 234–297 nm, 90–100% of which are in a glycosylated high molecular weight format (31, 32). Further analyses of these fluid samples showed a complete lack of ternary complex formation and a potent activation of the IR. Indeed, purified pools of these IGF-II isoforms were able to induce IR phosphorylation to a greater extent than mature IGF-II at high concentrations. Importantly, all of these clinical findings are mirrored by the data that we have obtained using homogenous preparations of each isoform in this study. The majority species found in most resected IGF-II-expressing tumors consists of the O-glycosylated big-IGF-II phenotype (14, 33, 34). These isoforms, we have now demonstrated, are impaired in their ability to mediate ternary complex formation and sequestration by the natural scavenging receptor, IGF-IIR (Fig. 4). We envisage a scenario that, once secreted from the tumor, the intrinsic properties of glyco-isoforms of IGF-II, as well as their residence within binary complexes, may create several advantages. First, it would certainly enable more effective paracellular penetration of vascular barriers to diffuse more completely throughout the tumor. In the local tumor microenvironment, the release of ligand from IGFBP is believed to occur by means of protease cleavage of IGFBP decreasing the affinity of the IGFBP for ligand and resulting in release (11). Here, tumors are known to secrete a very high local concentration of metalloproteinases, which efficiently cleave IGFBP (35). Theoretically, through a combination of increased tissue penetration and exploitation of protease secretion by tumor that may enable their release, binary complexes formed by glycosylated IGF-II isoforms may be a way of concentrating bioavailable and bioactive ligand at the site of the tumor. These IGF-II isoforms, in particular the big-IGF-II variants, would then be free to chronically activate IGF-IR, IR-A, and IR-B (Fig. 3) to initiate and sustain proliferation (Fig. 5) and to propagate this oncogenic axis.
We have also shown for the first time that a variety of IGF-II isoforms elicit potent activation of the IR-B, similar to that obtained with insulin (Fig. 3), which may contribute to the phenomenon of NICTH associated with tumors secreting high amounts of these isoforms into the serum. It is suspected that NICTH is caused by chronic stimulation of IR-B, either in peripheral muscles and organs or the tumor itself, resulting in a rapid efflux of glucose from the blood into tissues (36). Elevated secretion of big IGF-II isoforms that exhibit enhanced bioavailability by tumors may be a contributing factor to NICTH by direct stimulation of the IR. Hypoglycemia is usually ablated by surgical resection of the tumor followed by slow restoration of blood glucose to normal levels. Where surgical resection is not an option, the use of the IGF-II neutralizing antibody DX-2647 (18), which has very high affinity for IGF-II isoforms, is a viable therapeutic option to deploy to prevent this serious side effect in NICTH patients.
Acknowledgments
We thank Dr. George Lovrecz and Tram Pham (Fermentation Facility, Commonwealth Scientific and Industrial Research Organisation Division of Materials Science and Engineering, Parkville, Australia) for conducting all transfections. We thank Prof. E. Yvonne Jones (University of Oxford, Oxford, UK) for the generous gift of mammalian expression plasmid containing the cDNA encoding the IGF-IIR extracellular domain 11–13 fragment. We also thank Dr. Maria Soos (University of Cambridge, UK) for the hybridomas producing monoclonal antibodies 83-7 and 24-31.

This article contains supplemental Fig. S1.
- IGF
- insulin-like growth factor
- IGFBP
- IGF-binding protein
- ALS
- acid labile subunit
- IGF-IR
- IGF-I receptor
- IR
- insulin receptor
- NICTH
- non-islet cell tumor hypoglycemia
- Eu-ALS
- europium-labeled human ALS
- TRF
- time resolved fluorescence.
REFERENCES
- 1. DeChiara T. M., Robertson E. J., Efstratiadis A. (1991) Parental imprinting of the mouse insulin-like growth factor II gene. Cell 64, 849–859 [DOI] [PubMed] [Google Scholar]
- 2. Marks A. G., Carroll J. M., Purnell J. Q., Roberts C. T., Jr. (2011) Plasma distribution and signaling activities of IGF-II precursors. Endocrinology 152, 922–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Qiu Q., Yan X., Bell M., Di J., Tsang B. K., Gruslin A. (2010) Mature IGF-II prevents the formation of “big” IGF-II/IGFBP-2 complex in the human circulation. Growth Horm. IGF Res. 20, 110–117 [DOI] [PubMed] [Google Scholar]
- 4. El-Shewy H. M., Abdel-Samie S. A., Al Qalam A. M., Lee M. H., Kitatani K., Anelli V., Jaffa A. A., Obeid L. M., Luttrell L. M. (2011) Phospholipase C and protein kinase C-β2 mediate insulin-like growth factor II-dependent sphingosine kinase 1 activation. Mol. Endocrinol. 25, 2144–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brown J., Delaine C., Zaccheo O. J., Siebold C., Gilbert R. J., van Boxel G., Denley A., Wallace J. C., Hassan A. B., Forbes B. E., Jones E. Y. (2008) Structure and functional analysis of the IGF-II/IGF2R interaction. EMBO J. 27, 265–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Breuhahn K., Schirmacher P. (2008) Reactivation of the insulin-like growth factor-II signaling pathway in human hepatocellular carcinoma. World J. Gastroenterol 14, 1690–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chao W., D'Amore P. A. (2008) IGF2. Epigenetic regulation and role in development and disease. Cytokine Growth Factor Rev. 19, 111–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rainier S., Johnson L. A., Dobry C. J., Ping A. J., Grundy P. E., Feinberg A. P. (1993) Relaxation of imprinted genes in human cancer. Nature 362, 747–749 [DOI] [PubMed] [Google Scholar]
- 9. Gallagher E. J., LeRoith D. (2011) Minireview. IGF, insulin, and cancer. Endocrinology 152, 2546–2551 [DOI] [PubMed] [Google Scholar]
- 10. Duguay S. J., Jin Y., Stein J., Duguay A. N., Gardner P., Steiner D. F. (1998) Post-translational processing of the insulin-like growth factor-2 precursor. Analysis of O-glycosylation and endoproteolysis. J. Biol. Chem. 273, 18443–18451 [DOI] [PubMed] [Google Scholar]
- 11. Firth S. M., Baxter R. C. (2002) Cellular actions of the insulin-like growth factor binding proteins. Endocr. Rev. 23, 824–854 [DOI] [PubMed] [Google Scholar]
- 12. Holly J., Perks C. (2006) The role of insulin-like growth factor binding proteins. Neuroendocrinology 83, 154–160 [DOI] [PubMed] [Google Scholar]
- 13. Zapf J., Futo E., Peter M., Froesch E. R. (1992) Can “big” insulin-like growth factor II in serum of tumor patients account for the development of extrapancreatic tumor hypoglycemia? J. Clin. Invest. 90, 2574–2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fukuda I., Hizuka N., Ishikawa Y., Yasumoto K., Murakami Y., Sata A., Morita J., Kurimoto M., Okubo Y., Takano K. (2006) Clinical features of insulin-like growth factor-II producing non-islet-cell tumor hypoglycemia. Growth Horm. IGF Res. 16, 211–216 [DOI] [PubMed] [Google Scholar]
- 15. Valenzano K. J., Heath-Monnig E., Tollefsen S. E., Lake M., Lobel P. (1997) Biophysical and biological properties of naturally occurring high molecular weight insulin-like growth factor II variants. J. Biol. Chem. 272, 4804–4813 [DOI] [PubMed] [Google Scholar]
- 16. Bond J. J., Meka S., Baxter R. C. (2000) Binding characteristics of pro-insulin-like growth factor-II from cancer patients. Binary and ternary complex formation with IGF binding proteins-1 to -6. J. Endocrinol. 165, 253–260 [DOI] [PubMed] [Google Scholar]
- 17. Elmlinger M. W., Rauschnabel U., Koscielniak E., Weber K., Ranke M. B. (1999) Secretion of noncomplexed 'Big' (10–18 kD) forms of insulin-like growth factor-II by 12 soft tissue sarcoma cell lines. Horm. Res. 52, 178–185 [DOI] [PubMed] [Google Scholar]
- 18. Dransfield D. T., Cohen E. H., Chang Q., Sparrow L. G., Bentley J. D., Dolezal O., Xiao X., Peat T. S., Newman J., Pilling P. A., Phan T., Priebe I., Brierley G. V., Kastrapeli N., Kopacz K., Martik D., Wassaf D., Rank D., Conley G., Huang Y., Adams T. E., Cosgrove L. (2010) A human monoclonal antibody against insulin-like growth factor-II blocks the growth of human hepatocellular carcinoma cell lines in vitro and in vivo. Mol. Cancer Ther. 9, 1809–1819 [DOI] [PubMed] [Google Scholar]
- 19. Singh S. K., Moretta D., Almaguel F., De León M., De León D. D. (2008) Precursor IGF-II (proIGF-II) and mature IGF-II (mIGF-II) induce Bcl-2 and Bcl-X L expression through different signaling pathways in breast cancer cells. Growth Factors 26, 92–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Denley A., Brierley G. V., Carroll J. M., Lindenberg A., Booker G. W., Cosgrove L. J., Wallace J. C., Forbes B. E., Roberts C. T., Jr. (2006) Differential activation of insulin receptor isoforms by insulin-like growth factors is determined by the C domain. Endocrinology 147, 1029–1036 [DOI] [PubMed] [Google Scholar]
- 21. Cosgrove L., Lovrecz G. O., Verkuylen A., Cavaleri L., Black L. A., Bentley J. D., Howlett G. J., Gray P. P., Ward C. W., McKern N. M. (1995) Purification and properties of insulin receptor ectodomain from large-scale mammalian cell culture. Protein Expr. Purif. 6, 789–798 [DOI] [PubMed] [Google Scholar]
- 22. McKern N. M., Lou M., Frenkel M. J., Verkuylen A., Bentley J. D., Lovrecz G. O., Ivancic N., Elleman T. C., Garrett T. P., Cosgrove L. J., Ward C. W. (1997) Crystallization of the first three domains of the human insulin-like growth factor-1 receptor. Protein Sci. 6, 2663–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Soos M. A., O'Brien R. M., Brindle N. P., Stigter J. M., Okamoto A. K., Whittaker J., Siddle K. (1989) Monoclonal antibodies to the insulin receptor mimic metabolic effects of insulin but do not stimulate receptor autophosphorylation in transfected NIH 3T3 fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 86, 5217–5221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Soos M. A., Field C. E., Lammers R., Ullrich A., Zhang B., Roth R. A., Andersen A. S., Kjeldsen T., Siddle K. (1992) A panel of monoclonal antibodies for the type I insulin-like growth factor receptor. Epitope mapping, effects on ligand binding, and biological activity. J. Biol. Chem. 267, 12955–12963 [PubMed] [Google Scholar]
- 25. Jespersen S., Koedam J. A., Hoogerbrugge C. M., Tjaden U. R., van der Greef J., Van den Brande J. L. (1996) Characterization of O-glycosylated precursors of insulin-like growth factor II by matrix-assisted laser desorption/ionization mass spectrometry. J. Mass Spectrom. 31, 893–900 [DOI] [PubMed] [Google Scholar]
- 26. Denley A., Bonython E. R., Booker G. W., Cosgrove L. J., Forbes B. E., Ward C. W., Wallace J. C. (2004) Structural determinants for high-affinity binding of insulin-like growth factor II to insulin receptor (IR)-A, the exon 11 minus isoform of the IR. Mol. Endocrinol. 18, 2502–2512 [DOI] [PubMed] [Google Scholar]
- 27. Brierley G. V., Macaulay S. L., Forbes B. E., Wallace J. C., Cosgrove L. J., Macaulay V. M. (2010) Silencing of the insulin receptor isoform A favors formation of type 1 insulin-like growth factor receptor (IGF-IR) homodimers and enhances ligand-induced IGF-IR activation and viability of human colon carcinoma cells. Endocrinology 151, 1418–1427 [DOI] [PubMed] [Google Scholar]
- 28. Daughaday W. H., Trivedi B., Baxter R. C. (1993) Serum “big insulin-like growth factor II” from patients with tumor hypoglycemia lacks normal E-domain O-linked glycosylation, a possible determinant of normal propeptide processing. Proc. Natl. Acad. Sci. U.S.A. 90, 5823–5827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Firth S. M., Ganeshprasad U., Baxter R. C. (1998) Structural determinants of ligand and cell surface binding of insulin-like growth factor-binding protein-3. J. Biol. Chem. 273, 2631–2638 [DOI] [PubMed] [Google Scholar]
- 30. Firth S. M., Clemmons D. R., Baxter R. C. (2001) Mutagenesis of basic amino acids in the carboxyl-terminal region of insulin-like growth factor binding protein-5 affects acid-labile subunit binding. Endocrinology 142, 2147. [DOI] [PubMed] [Google Scholar]
- 31. Hoekman K., van Doorn J., Gloudemans T., Maassen J. A., Schuller A. G., Pinedo H. M. (1999) Hypoglycaemia associated with the production of insulin-like growth factor II and insulin-like growth factor binding protein 6 by a haemangiopericytoma. Clin. Endocrinol. (Oxf.) 51, 247–253 [DOI] [PubMed] [Google Scholar]
- 32. van Doorn J., Hoogerbrugge C. M., Koster J. G., Bloemen R. J., Hoekman K., Mudde A. H., van Buul-Offers S. C. (2002) Antibodies directed against the E region of pro-insulin-like growth factor-II used to evaluate non-islet cell tumor-induced hypoglycemia. Clin. Chem. 48, 1739–1750 [PubMed] [Google Scholar]
- 33. Hizuka N., Fukuda I., Takano K., Asakawa-Yasumoto K., Okubo Y., Demura H. (1998) Serum high molecular weight form of insulin-like growth factor II from patients with non-islet cell tumor hypoglycemia is O-glycosylated. J. Clin. Endocrinol. Metab. 83, 2875–2877 [DOI] [PubMed] [Google Scholar]
- 34. Ishikura K., Takamura T., Takeshita Y., Nakagawa A., Imaizumi N., Misu H., Taji K., Kasahara K., Oshinoya Y., Suzuki S., Ooi A., Kaneko S. (2010) Cushing's syndrome and big IGF-II associated hypoglycaemia in a patient with adrenocortical carcinoma. BMJ Case Rep. 2010, bcr07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nakamura M., Miyamoto S., Maeda H., Ishii G., Hasebe T., Chiba T., Asaka M., Ochiai A. (2005) Matrix metalloproteinase-7 degrades all insulin-like growth factor binding proteins and facilitates insulin-like growth factor bioavailability. Biochem. Biophys. Res. Commun. 333, 1011–1016 [DOI] [PubMed] [Google Scholar]
- 36. Eastman R. C., Carson R. E., Orloff D. G., Cochran C. S., Perdue J. F., Rechler M. M., Lanau F., Roberts C. T., Jr., Shapiro J., Roth J. (1992) Glucose utilization in a patient with hepatoma and hypoglycemia. Assessment by a positron emission tomography. J. Clin. Invest. 89, 1958–1963 [DOI] [PMC free article] [PubMed] [Google Scholar]