

Background: PC synthesis by the CDP-choline pathway is regulated by CCTα.
Results: Increased expression of nuclear CCTα in ras-transformed intestinal epithelial contributes to PC synthesis that is required for anchorage-independent growth.
Conclusion: An expanded pool of nuclear CCTα is involved in malignant transformation by ras.
Significance: CCTα is a potential target to restore anchorage-dependent growth sensitivity of cancer cells.
Keywords: Cancer, Epithelial Cell, Nuclear Membrane, Phosphatidylcholine, Ras, CTP:phosphocholine Cytidylyltransferase, Anoikis
Abstract
Cancer cells have enhanced lipogenic capacity characterized by increased synthesis of fatty acids and complex lipids, including phosphatidylcholine (PC). As the rate-limiting enzyme in the CDP-choline pathway for PC synthesis, CTP:phosphocholine cytidylyltransferase α (CCTα) is implicated in the provision of membranes and bioactive lipids necessary of cell proliferation. In this study, we assessed the role of CCTα in malignant intestinal epithelial cells transformed with activated H-ras (IEC-ras). Three IEC-ras clones had significant up-regulation CCTα expression, but PC synthesis and in vitro activity of CCTα were similar to control IEC. RNA interference of CCTα in adherent IEC-ras did not affect PC synthesis, confirming that the enzyme was relatively inactive. However, CCTα silencing in ras-transformed IEC reduced anchorage-independent growth, a criterion for malignant transformation, as well as tumorigenicity in mice. Relative to their adherent counterparts, detached IEC-ras had increased PC synthesis that was attenuated by inducible CCTα silencing. Detachment of IEC-ras was accompanied by increased CCTα phosphorylation and cytosolic enzyme activity. We conclude that the expanded pool of CCTα in IEC-ras is activated by detachment. This provides the increased PC biosynthetic capacity that contributes to malignant transformation of intestinal epithelial cells when detached from the extracellular matrix.
Introduction
Tumor cells have profound alterations in metabolic pathways that facilitate survival in hypoxic/anoxic environments and provide cellular biomass for accelerated growth (1). Channeling of the glycolytic product pyruvate into acetyl-CoA and increased expression of biosynthetic genes for fatty acids, cholesterol, and other lipids provide cancer cells with enhanced lipogenic capacity (2). Increased activity of ATP-citrate lyase (3, 4), acetyl-CoA carboxylase (5), fatty acid synthase (6, 7), and stearoyl-CoA desaturase (8), as a consequence of Akt/phosphatidylinositol-3 kinase stimulation of sterol regulatory element-binding protein-1 activity (9), contributes to a dependence on de novo fatty acid synthesis for proliferation and evasion of apoptosis. Because this is a relatively early event in malignant transformation that affects multiple lipid metabolic pathways, pharmacological targeting of the lipogenic capacity of tumors could be a viable therapeutic approach.
The majority of de novo synthesized fatty acids in transformed cells are channeled into glycerophospholipids (10, 11), the primary components of cellular membranes and precursors for signaling molecules that are essential for sustained cell proliferation. This raises the possibility that the sensitivity of tumor cells to inhibition of fatty acid synthesis could be due to reduced synthesis of complex lipids or products thereof. Phosphatidylcholine (PC),2 the major glycerophospholipid in eukaryotic cells, comprises 40–60% of membrane mass and is synthesized de novo by the CDP-choline (Kennedy) pathway (12). ras-transformed cells often display increased PC synthesis and catabolism (13, 14), and tumor tissue has significantly elevated levels of choline metabolites, indicative of increased choline uptake and/or synthesis and catabolism of PC (15, 16). Increased levels of phosphocholine (pCholine) in transformed cells and tumor tissue are linked to increased activity of the α-isoform of choline kinase (CK) (13, 14, 17, 18). ras-dependent activation of choline kinase α gene expression involves a phosphatidylinositol 3-kinase/RhoA pathway (19). In turn, elevated choline kinase activity is proposed to stimulate mitogen-activated protein kinase and Akt activity by production of a PC-derived lipid activator, possibly phosphatidic acid (20, 21). Pharmacological or RNA interference (RNAi) inhibition of CKα suppresses cell proliferation and anchorage-independent growth, suggesting an important role in carcinogenesis (22, 23).
Although elevated CK expression could increase flux through the CDP-choline pathway, it is generally not considered to be rate-limiting for PC synthesis because the pCholine pool in cells is large compared with other intermediates. In most cases, the rate-limiting step in the pathway is the conversion of pCholine to CDP-choline catalyzed by CTP:phosphocholine cytidylyltransferase (CCT), an amphitropic enzyme that is stimulated by reversible binding to membranes in response to increased fatty acids or type II lipids, such as diacylglycerol (24). The Pcyt1b gene encodes CCTβ2 and CCTβ3 isoforms in rodents as well as C-terminal truncated CCTβ1 in humans, all of which are cytoplasmic and have a restricted tissue distribution (25, 26). Pcyt1a encodes the ubiquitously expressed CCTα isoform, a nuclear enzyme that is essential for cell viability and embryonic development (25, 27, 28). CCTα regulates PC synthesis during cell division (29, 30) and programmed cell death (apoptosis) (27, 31–33), yet changes in enzyme activity and expression are not a consistent feature of oncogenic transformation. For instance, CCT activity in ras-transformed keratinocytes and fibroblasts is increased (34) or decreased (13, 14, 35), respectively. Bakovic et al. (36) showed that H-ras-transformed fibroblasts have increased CCTα mRNA (2–4-fold) and protein (1.8-fold) expression, yet enzyme activity was decreased by 40%. Surprisingly, pharmacological inhibition of the Ras/MAPK signaling pathway reduced CCTα mRNA and protein expression but had no effect on enzyme activity, suggesting that ras transformation expanded an inactive pool of enzyme.
Cancer cells, particularly those transformed with oncogenic ras, have elevated pCholine as a result of increased choline kinase expression and increased PC synthesis and turnover. However, this seems at odds with reports of reduced CCT expression and/or activity in the same or related studies. Given these inconsistencies, we investigated the role of CCTα in ras transformation of intestinal epithelial cells (IEC)-18. Normal epithelial cells grow in contact with the extracellular matrix (ECM), and detachment of such cells from the ECM triggers a form of apoptosis called anoikis (37). By contrast, malignant cells are typically anoikis-resistant and grow as three-dimensional masses in the absence of adhesion to the ECM, one of the most stringent criteria for malignant transformation (38). Oncogenic ras is a well known inhibitor of anoikis, and numerous studies indicate that ras-induced anoikis-resistance is required for the malignant transformation of IEC into tumors in vivo (39, 40). Here we provide evidence that oncogenic ras triggers CCTα up-regulation in non-malignant IEC-18 and that this is required for the ability of such cells to grow in the absence of adhesion to the ECM and form tumors in vivo. This dependence on CCTα expression was linked to increased PC synthesis in cells that were denied adhesion to the ECM. This represents a novel mechanism by which CCTα and the CDP-choline pathway contribute to the anoikis resistance of IEC-18 transformed with oncogenic ras.
EXPERIMENTAL PROCEDURES
Materials
AlexaFluor-conjugated donkey anti-goat antibodies, TRIzol, Lipofectamine 2000, and SYBR Green quantitative polymerase chain reaction (qPCR) master mix were purchased from Invitrogen. The CCTα antibody was generated in rabbits as described previously (41). The CCTβ2 antibody was generously provided by Dr. Suzanne Jackowski (St. Jude Children's Research Hospital, Memphis, TN). IRDye800- and IRDye680-conjugated secondary antibodies were purchased from LI-COR Biosciences (Lincoln, NE). A β-actin monoclonal antibody, and lentiviral pLKO.1 vectors encoding shRNAs directed against CCTα and CCTβ, as well as non-targeting (shNT) and shGFP controls, were purchased from Sigma-Aldrich. [3H]Choline and phospho[14C]choline was purchased from PerkinElmer Life Sciences. The lamin A/C antibody was from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). A CKα-specific antibody was from Proteintech Group (Chicago IL). Eight-week-old female athymic mice (BALB/c) were from Charles River Laboratories (Wilmington, MA) and housed in the Carleton Animal Care Facility (Dalhousie University) for 1 week prior to experiments.
Cell Culture
Rat IEC-18 and three independently derived clones of IEC-18 that are transformed with H-ras (referred to as ras3, ras4, and ras7) (39) were cultured at 37 °C in a humidified 5% CO2 atmosphere in α-minimal essential medium (MEM) supplemented with 5% fetal bovine serum (FBS), d-glucose (3.6 mg/ml), insulin (12.74 μg/ml), penicillin (600 μg/ml), streptomycin (100 μg/ml), and glutamine (2.92 mg/ml) (IEC-MEM). IEC-18 and ras-transformed cells were transfected with pEGFP or pCCTα-GFP (41) using Lipofectamine 2000 according to the manufacturer's instructions.
Lentivirus Production and Transduction
Lentivirus was produced in HEK293T cells (cultured in Dulbecco's minimal essential medium with 10% FBS) by polyethyleneimine-mediated transfection with plasmids encoding packaging factors (pCMVΔ8.2), vesicular stomatitis virus glycoprotein envelope protein (pCMV-VSVG), and shRNA (pLKO.1). pLKO.1 shRNAs encode the following targeting sequences: shCCTα1, CCTAAGGACATCTACAAGAA; shCCTα2, CCTGTGAGAGTTTATGCGGAT; shCCTβA, GCATGTTTGTTCCAACACAAA; shCCTβB, GCTACTTGTTGGTAGGAGTTT. Non-targeting shNT (CAACAAGATGAAGAGCACCAA) or shGFP (TACAACAGCCACAACGTCTAT) served as controls. After 48 h, virus-containing medium was harvested and filtered, and Polybrene (1 μg/ml) was added. The medium was applied to IEC and IEC-ras cells for 24 h, followed by selection for 48–72 h in IEC-MEM containing 1 or 2 μg/ml puromycin. The effectiveness of CCT silencing was determined by immunoblotting or qPCR.
Lentiviral shRNAs under the control of the doxycycline promoter (TRIPz-shCCTα1, TGACTTTTCCTCCACATC; TripZ shCCTα2, AAGTCAAATATTCCATCGG) were expressed in ras4 cells and selected with puromycin as described above. After culturing on agarose for 24 h, shRNAs were induced with or without 1 μg/ml doxycycline for 48 h prior to the start of experiments (see Fig. 9, C and D)
FIGURE 9.

Detached IEC-ras cells have sustained growth and PC synthesis. A, IEC, ras3, ras4, and ras7 cells were assayed for viability using a colonogenicity assay as described under “Experimental Procedures.” Results are the mean and S.D. of three experiments. B, IEC-18 (■), ras3 (□), ras4 (○), and ras7 (▵) cells were cultured on plastic dishes or dishes coated with 1% SeaPlaque agarose in MEM containing [3H]choline (2 μCi/ml) for 0–72 h. At the indicated times, cells were harvested, and [3H]choline incorporation into PC, pCholine, GPC, and CDP-choline was determined and quantified relative to cellular protein. The ratio of specific incorporation into PC and choline metabolites of detached versus adherent cells was determined as an estimate of relative biosynthetic rates. Results are the mean and S.D. (error bars) of five separate experiments. C, ras4 cells were transduced with TripZ lentiviral shCCTα3 or shCCTα4 and selected under adherent conditions. Cells were then cultured on agarose for 24 h, followed by induction of shRNAs with 1 μg/ml doxycycline for 48 h, at which time [3H]choline (1 μCi/ml) incorporation into PC and water-soluble metabolites was measured by a 3-h pulse. Results are expressed relative to cells that were not induced with doxycycline and are the mean and S.D. of four experiments. *, p < 0.05 compared with uninduced cells. D, lysates from ras4 cells cutured on agarose for 48 h in the absence or presence or doxycycline were immunoblotted for CCTα.
Immunofluorescence Microscopy
Cells cultured on glass coverslips were fixed with 4% (w/v) paraformaldehyde in phosphate-buffered saline (PBS) for 15 min and permeabilized with 0.5% (w/v) Triton X-100 for 10 min at 4 °C. Coverslips were incubated sequentially with a primary antibody in PBS plus 1% (w/v) BSA at 20 °C for 1 h followed by an AlexaFluor-conjugated secondary antibody for 1 h at 20 °C. Coverslips were mounted on glass slides with Mowiol® 4-88, and images were captured using a Zeiss LSM510 META confocal microscope equipped with a ×100 oil immersion (1.4 numerical aperture) objective. Microscope settings for imaging of lamin A/C in IEC-18 and IEC-ras cells were identical, but the gain was adjusted when imaging CCTα in IEC-ras cells due to the level of overexpression.
Immunoblotting
Total cell lysates were prepared in SDS-PAGE buffer (12.5% SDS, 30 mm Tris-HCl (pH 6.8), 12.5% glycerol, and 0.01% bromphenol blue), sonicated for 20 s, and heated to 90 °C for 5 min prior to SDS-8% PAGE and transfer to nitrocellulose. Proteins were visualized by immunoblotting with primary antibodies for 1–2 h at 20 °C followed by secondary antibodies conjugated to horseradish peroxidase (enhanced chemiluminescence detection) or IRDye800 and IRDye680 (detection using an Odyssey infrared imaging system). Depending on the method, protein expression was quantified relative to load controls (actin or oxysterol-binding protein) using densitometry and ImageJ software version 1.4 or Odyssey Application software version 3.0.
CCT Enzyme Assays
IEC-18, ras3, ras4, and ras7 cells were permeabilized on ice for 1 min with 10 mm Tris-HCl (pH 7.4), 0.25 m sucrose, 2.5 mm EDTA, 1 mm DTT, protease inhibitor mixture, and 300 mg/ml digitonin as described previously (33). Soluble and particulate membrane fractions were assayed in the presence or absence of PC/oleate vesicles by monitoring the conversion of phospho[14C]choline to CDP-[14C]choline (42).
mRNA Quantitation
mRNA extracted from cells using TRIzol was used for first strand cDNA synthesis using the ThermoScript RT-PCR system (Invitrogen). qPCR was performed on a Eppendorf Realplex Thermocycler using the ΔΔCt method and 3–4 serial dilutions of each template (43). Primer sets that amplify within the catalytic domain-encoded regions CCTα and CCTβ are as follows: CCTα forward, GCCCGGGCTCTGATGCAA; CCTα reverse, TGCTGCACCGCGTCGTAAC; CCTβ forward, GCAGATGTTCCAGGAGAGGAGTAG; CCTβ reverse, GAAGGGGGAGAGGTCTTGTTTG; GAPDH forward, TGATGACATCAAGGTGGTGAA; GAPDH reverse, TCCTTGGAGGCCATGTGGGCCAT.
Fluorescence-activated Cell Sorting (FACS)
Adherent or detached ras4 cells were harvested, washed in PBS, trypsinized to reduce aggregates, and fixed in 5 ml of ethanol. Prior to analysis, cells were resuspended in PBS containing 50 μg/ml propidium iodide. Samples were analyzed using a FACSCalibur system (BD Biosciences), and cell cycle profiles were analyzed using ModiFit LT 3.0 software.
[3H]Choline Incorporation into PC and Choline Metabolites
IEC-18 and IEC-ras cells growing in monolayers were incubated with choline-free IEC-MEM for 2 h prior to incubation with [3H]choline (1.5 μCi/ml) for 3 h. For cells cultured in suspension on agarose monolayers, [3H]choline (1.5 μCi/ml) was added directly to dishes without a medium change for the indicated times (see the legend to Fig. 9). Cells were harvested from dishes in 1 ml of methanol/water (5:4, v/v), an aliquot was removed for protein quantification, and 3 ml of CHCl3 was added. After phase separation by centrifugation, the aqueous fraction containing CDP-choline, glycerophosphocholine (GPC), and pCholine was resolved by thin-layer chromatography in water/ethanol/ammonium hydroxide, (50:95:6, v/v/v) and visualized by spraying with 1% (w/v) phosphomolybdate in chloroform/methanol (1:1, v/v) and 1% (w/v) stannous chloride in 3 n HCl. Choline-labeled metabolites were identified using standards, and radioactivity was measured by liquid scintillation counting. [3H]Choline incorporation into PC was determined by liquid scintillation counting of an aliquot of the CHCl3 phase (>98% of radioactivity was in the [3H]PC, as determined by thin layer chromatography).
Clonogenicity, Soft Agar, and Tumor Growth Assays
A clonogenicity assay was used to measure cell viability as a function of colony formation following anchorage-independent growth on SeaPlaque agarose-coated plates for up to 72 h (44). Briefly, IEC-18, ras3, ras4, and ras7 cells were trypsinized, and 500 cells were seeded onto 60-mm dishes coated with 2 ml of 1% (w/v) SeaPlaque agarose in MEM. To calculate survival, an equivalent number of cells were seeded onto dishes without agarose to determine maximum colony number under adherent growth conditions. Cells cultured on agarose for 24, 48, and 72 h were transferred to plastic dishes and cultured in IEC-MEM. After 3–4 days, colonies were visualized by staining with Crystal Violet, and survival was calculated relative to adherent controls.
Colony formation in soft agar was determined by spreading 500 IEC-18 or IEC-ras cells suspended in 0.3% (w/v) bacto-agar/IEC-MEM onto 60-mm dishes coated with 0.5% (w/v) agar/IEC-MEM. Colonies were counted after 3–4 days and expressed as a percentage relative to the number of colonies formed by an equivalent number of cells cultured on plastic dishes.
To assess whether CCTα expression was required for solid tumor growth, ras4 cells (2 × 105 cells) transduced with lentiviral shGFP, shCCTα1, or shCCTα2 were injected into the right flank of athymic mice. Tumor volume was monitored daily using a skin fold caliper and calculated using an ellipsoidal volume approximation (0.5 × (length × width2)) (45). All procedures were certified by the Dalhousie University Committee on Laboratory Animals (protocol number 11-022).
RESULTS
Phosphatidylcholine Metabolism in H-ras-transformed IEC
Epithelial cell-derived carcinomas account for >90% of all cancers, of which 25–50% are characterized by expression of oncogenic forms of ras (46). To assess whether PC metabolism is altered in ras-transformed epithelial cells, we utilized non-malignant spontaneously immortalized rat IEC-18 and three independently derived clones of this cell line that are transformed with an oncogenic form of H-ras (ras3, ras4, and ras7) (39). IEC-18 and its ras-transformed clones were cultured in monolayer and pulse-labeled with [3H]choline to measure incorporation into PC and other choline metabolites (Fig. 1). After 3 h, [3H]choline incorporation into the PC fraction of all three ras clones was not significantly different from IEC-18. ras3 and ras4 cells had significantly reduced [3H]choline incorporation into pCholine, indicative of CCT activation, and increased GPC due to degradation by phospholipase A activity. However, this shift in [3H]choline metabolites accounted for only 2–3% of total [3H]PC and was not evident in ras7 cells. Similar results were obtained when cells were labeled under steady state with [3H]choline for 24 h, and release of water-soluble choline metabolites into the medium was similar for all cell lines (results not shown). Thus, under adherent culture conditions, precursor-labeling experiments indicate that the CDP-choline pathway in IEC-18 is minimally affected by oncogenic ras.
FIGURE 1.
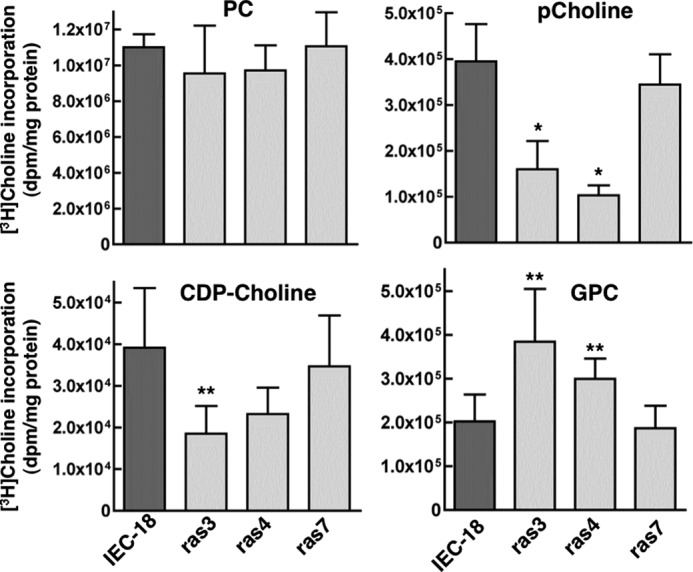
[3H]Choline incorporation into PC and choline metabolites of IEC-18 and IEC-ras cells. IEC-18, ras3, ras4, and ras7 cells were incubated with [3H]choline for 3 h, and incorporation into PC, pCholine, GPC, and CDP-choline was measured as described under “Experimental Procedures.” Results are the mean and S.D. (error bars) of three independent experiments. *, p < 0.01; **, p < 0.05 compared with IEC.
Although choline metabolism was not robustly affected in ras-transformed IEC-18, this may not reflect changes in CCTα expression because most cells contain a substantial pool of soluble inactive enzyme (47), and the membrane remodeling activity of CCTα does not require catalytic activity (48, 49). Indeed, the expression of CCTα mRNA in ras3 and ras7 cells was increased 8-fold compared with expression in IEC-18, whereas expression in ras4 cells was unaffected (Fig. 2A). CCTα protein expression was also increased in ras3 (20-fold), ras4 (5-fold), and ras7 cells (8-fold) relative to IEC-18 (Fig. 2B). qPCR analysis using a primer set that detects all CCTβ transcripts revealed a 5–7-fold increase in ras3 and ras7 cells but not in ras4 cells (Fig. 2A). CCTβ protein was detected by immunoblotting using an antibody specific for CCTβ2 that should also detect the CCTβ3 protein (Fig. 2B). IEC and ras cells expressed a single peptide at ∼40 kDa that was tentatively identified as CCTβ2 because there was no evidence of an additional lower molecular mass protein corresponding to N-terminal truncated CCTβ3 (50). CCTβ2 protein expression is significantly increased in ras7 cells (2.4-fold) but not in ras3 and ras4 cells, indicating a lack of correlation with mRNA levels.
FIGURE 2.
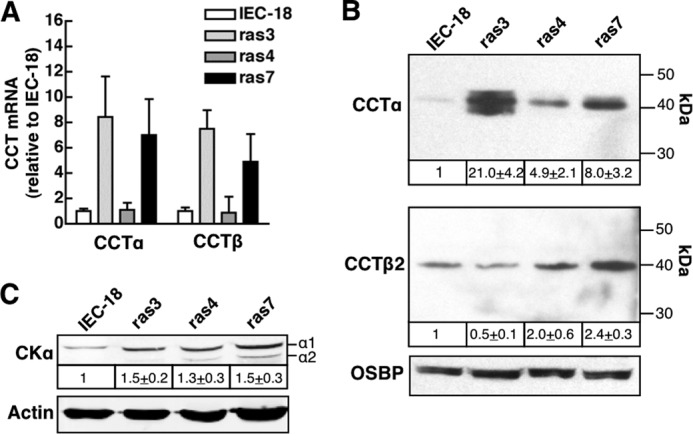
CCTα expression in adherent and detached IEC-18 and IEC-ras cells. A, CCTα and CCTβ mRNA were quantified by qPCR, normalized to GAPDH expression, and expressed relative to IEC-18. Results are the mean and S.D. (error bars) of 4–6 experiments. B, total cell lysates prepared from IEC-18 and IEC-ras cells cultured on plastic dishes were resolved by SDS-8% PAGE and immunoblotted for CCTα, CCTβ, and oxysterol-binding protein (OSBP), as described under “Experimental Procedures.” CCTα and CCTβ expression was normalized to the load control (oxysterol-binding protein) and expressed relative to IEC-18 (mean and S.D.). C, cell lysates prepared as described above were immunoblotted for the α1 and α2 isoforms of CK, and total CK was quantified relative to actin. Results are the mean and S.D. of four experiments.
CKα expression and activity is increased in ras-transformed cells (16, 19), suggesting that discrepant choline metabolite profiles in ras3, -4, and -7 cells could be due to altered expression of this enzyme. However, immunoblotting for CKα revealed a 30–50% increase in protein expression for the α1 and α2 isoforms in all three ras cells compared with IEC-18 (Fig. 2C).
CCTα is localized in the nucleoplasm of most cultured cells but translocates to the nuclear envelope and nucleoplasmic reticulum upon fatty acid activation (41, 48, 51). Similarly, CCTα was detected by indirect immunofluorescence throughout the interior of uniform, elliptically shaped IEC-18 nuclei with no evidence of co-localization at the nuclear envelope with lamin A/C (Fig. 3). In contrast, the nuclei of IEC-ras cells were irregularly shaped with numerous folds, indentations, and protrusions that stained for lamin A/C (Fig. 3). Localization of CCTα in IEC-ras cells was heterogeneous both within individual cells and between cell lines; there was evidence of nucleoplasmic CCTα as well as co-localization with lamin A/C at the nuclear envelope and in nuclear folds and protrusions, particularly in ras4 and ras7 cells.
FIGURE 3.

CCTα is localized to the nucleoplasm and nuclear envelope in IEC-ras cells. IEC-18, ras3, ras4, and ras7 cells cultured on glass coverslips were immunostained with the following primary and secondary antibodies: rabbit anti-CCTα and AlexaFluor488-conjugated goat anti-rabbit; goat anti-lamin A/C and AlexaFluor594-conjugated donkey anti-goat. Images are 1-μm optical sections through the mid-nuclei captured using a Zeiss LSM520 Meta confocal microscope.
Increased CCTα protein expression and partial localization on the nuclear envelope in IEC-ras cells suggests that enzyme activity is elevated. On the other hand, PC synthesis and choline metabolites in IEC-ras cells were not significantly affected (Fig. 1). To resolve this, we assayed CCT activity in the soluble (cytosolic) and particulate (membrane) fractions of IEC-18, ras3, ras4, and ras7 cells using the digitonin permeabilization method (33) (Fig. 4A). Despite increased CCTα expression in both the soluble and membrane fractions of IEC-ras cells (Fig. 4B), CCT activity in the absence of oleate was no different from that in IEC-18 controls. With the exception of ras4 cytosolic activity, enzyme activity in the soluble and membrane fractions from all four cell lines was not activated by PC/oleate vesicles. Control experiments confirmed that the activity of recombinant CCTα was increased 4–5-fold by PC/oleate vesicle under the same conditions (results not shown). Thus, the increased nuclear pool of CCTα in IEC-ras cells appears to be catalytically inactive.
FIGURE 4.
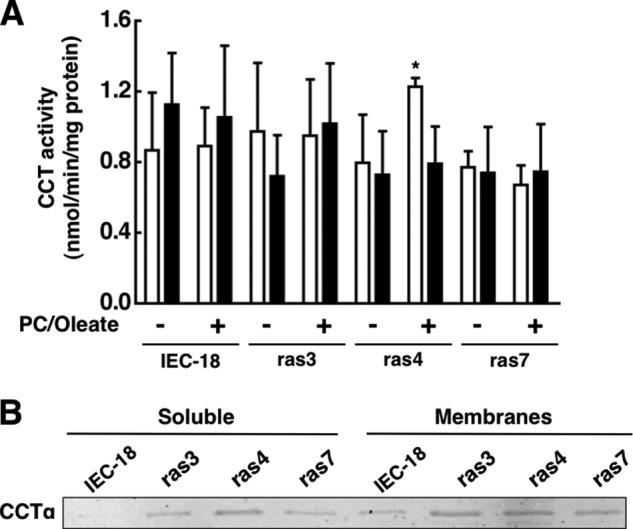
CCTα activity in cytosol and membranes of IEC-18 and IEC-ras cells. A, CCT activity was measured in the soluble (open bars) and membrane particulate fractions (black bars) from IEC-18 and IEC-ras in the presence or absence of PC/oleate vesicles as described under “Experimental Procedures.” Results are the mean and S.D. (error bars) of three experiments. B, the soluble and membrane particulate fraction (25 μg) of IEC-18 and IEC-ras cells were separated by SDS-8% PAGE and immunoblotted for CCTα. *, p < 0.05 compared with IEC-18 activity assayed under the same conditions.
CCTα Is Necessary for the Anchorage-independent Growth of IEC-ras Cells
The increased pool of CCTα in monolayer cultures of IEC-ras cells did not contribute to PC synthesis but could be involved in anchorage-independent growth and anoikis. IEC-ras cells are resistant to anoikis and grow as anchorage-independent, three-dimensional multicellular spheroids due to activation and suppression of anti- and pro-apoptotic pathways, respectively (40, 52, 53). As a first step in assessing whether increased expression of CCTα contributes to anchorage-independent growth of IEC-ras cells, we identified two lentiviral shRNAs that provided optimal silencing of gene expression. As an example, shCCTα1 transduction into IEC-18 and ras cells reduced CCTα expression by 80–90%, which in the case of ras4 and ras7 cells was similar to the level of expression in IEC-18 transduced with non-targeting (shNT) lentivirus (Fig. 5A). Silencing in ras4 cells using shCCTα1 or shCCTα2 (two separate shRNAs complementary to different regions of the CCTα mRNA) reduced expression of CCTα to the level of shNT-transduced IEC-18 but did not affect CCTβ2 expression (Fig. 5B) (data not shown). Based on these experiments, ras4 cells were used in subsequent experiments. As well, CCTβ mRNA and protein expression in ras4 cells were similar to IEC-18, thus precluding potential involvement of this CCT isoform (Fig. 2, A and B).
FIGURE 5.
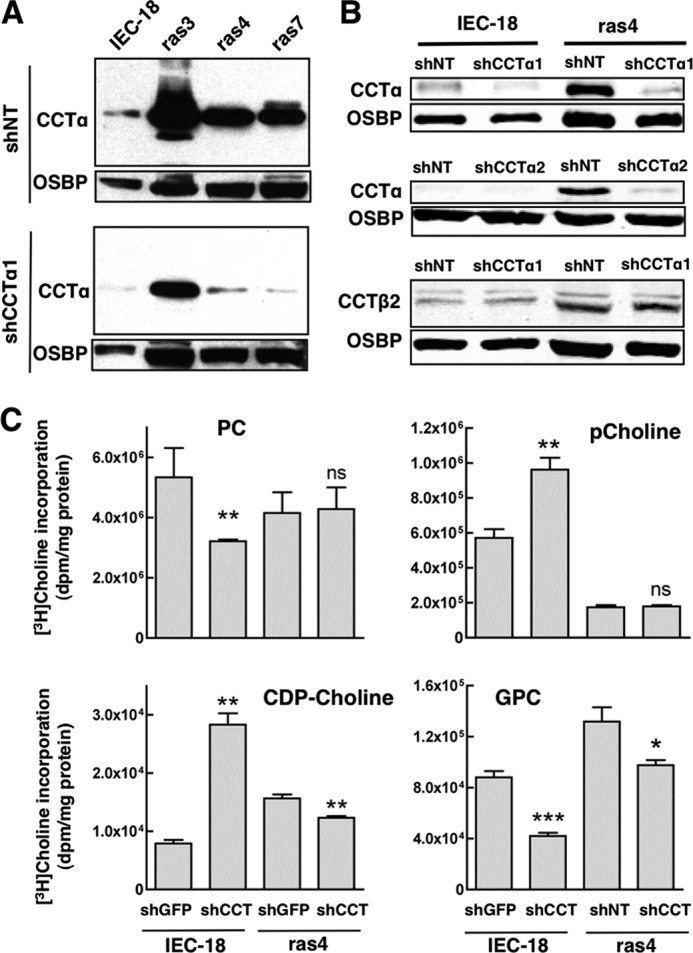
Effect of CCTα silencing on PC metabolism in IEC-18 and IEC-ras cells. A, lentivirus encoding an shRNA against CCTα (shCCTα1) or a non-targeting shRNA control (shNT) were expressed in IEC-18 and IEC-ras cells and selected with puromycin for 24 h. Total cell lysates were resolved by SDS-8% PAGE and immunoblotted for CCTα and oxysterol-binding protein (OSBP). B, knockdown of CCTα by lentiviral shCCTα1 and shCCTα2 was determined in IEC-18 and ras4 cells. CCTβ2 expression was determined in shCCTα1-transduced cells. C, IEC-18 and ras4 cells transduced with shGFP or shCCTα1 were pulse-labeled with [3H]choline (2 μCi/ml) for 3 h. Cells were harvested, and [3H]choline incorporation into PC, pCholine, CDP-choline, and GPC was measured as described under “Experimental Procedures.” Results are the mean and S.D. (error bars) of three independent experiments. *, p < 0.1; **, p < 0.05; ***, p > 0.01; ns, not significant compared with shGFP controls.
We further tested whether CCTα knockdown affected [3H]choline incorporation into PC and metabolites of the CDP-choline pathway. Following lentiviral transduction of shCCTα1, adherent IEC-18 and ras4 cells were pulse-labeled with [3H]choline, and incorporation into PC and water-soluble metabolites was measured (Fig. 5C). Consistent with its rate-limiting role in the pathway, CCTα silencing in IEC-18 resulted in a significant 40% reduction in PC synthesis and accumulation of pCholine compared with shNT controls. Reduced PC synthesis was also partially balanced by reduced degradation to [3H]GPC. In contrast, [3H]choline incorporation into PC and pCholine were unaffected in shCCTα1-transduced ras4 cells. There was a slight reduction in [3H]GPC, but this accounted for only 4% of [3H]choline incorporation into PC. This further suggests that the large reservoir of nuclear enzyme in adherent IEC-ras cells has minimal impact on PC synthesis.
To determine if CCTα overexpression was involved in anchorage-independent growth, ras4 cells transduced with control and shCCT lentivirus were subjected to clonogenicity and soft agar growth assays (Fig. 6, A and B). When suspended in agar for 5 days, IEC-18 produced <1% of the colonies formed by adherent controls (results not shown). In contrast, ras4 cells expressing control shNT formed colonies in soft agar (deprived of adhesion to the ECM) to a similar extent as adherent counterparts. However, this growth was reduced by 50% when CCTα expression was targeted with either shCCTα1 or shCCTα2 (Fig. 6A). Using a clonogenicity assay that measures that ability of detached cells to form colonies when provided with a suitable substrate on which to grow, IEC-18 had a progressive loss of viability when cultured in suspension for up to 72 h that was not affected by reducing CCTα expression (Fig. 6B). Similar to the soft agar growth assay, the >90% viability of detached ras4 cells was suppressed to 20–40% when CCTα expression was reduced with shCCTα1.
FIGURE 6.
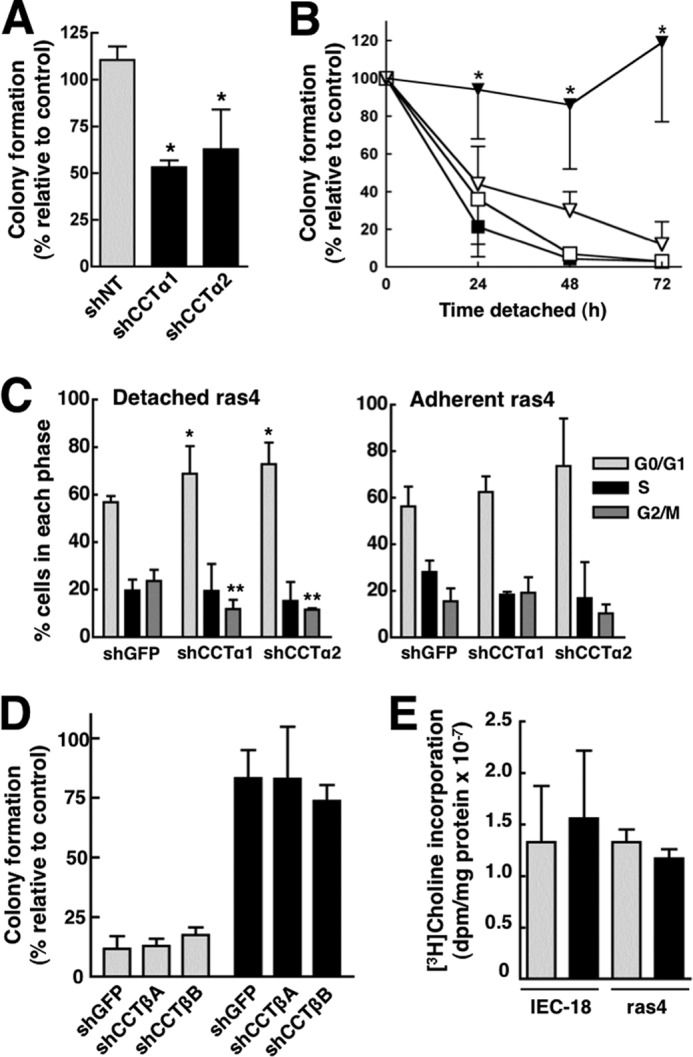
Knockdown of CCTα, but not CCTβ, sensitizes ras4 cells to anoikis. A, lentiviral shCCTα1, shCCTα2, or a non-targeting control (shNT) were transduced into ras4 cells and selected with puromycin for 48 h. Cells (500) were suspended in 0.3% agar, and colonies were quantified after 5 days in culture. Results are the mean and S.D. of three experiments. *, p < 0.05 compared with shNT ras4 cells. B, IEC-18 (squares) and ras4 cells (triangles) transduced with shNT (■ and ▾) or shCCTα1 (□ and ▿) were cultured on SeaPlaque agarose for the indicated times and transferred to plastic dishes, and colonies were quantified after 3–4 days as described under “Experimental Procedures.” Results are the mean and S.D. (error bars) of 5–6 experiments. *, p < 0.05 compared with shNT ras4 cells. C, the cell cycle distribution of shGFP-transduced (control), shCCTα1-transduced, or shCCTα2-transduced ras4 cells was determined under adherent and 24-h detached conditions as described under “Experimental Procedures.” Results are the mean and S.D. of three experiments. *, p < 0.05; **, p < 0.005, compared with shGFP control. D, IEC-18 (gray bars) and ras4 cells (black bars) were transduced with lentiviral shGFP, shCCTβ1, or shCCTβ2 and cultured on SeaPlaque agarose for 48 h prior to seeding on plastic dishes for 3–4 days as described in B. Results are the mean and S.D. of 3–6 experiments. E, [3H]choline incorporation into PC was measured in adherent IEC-18 and ras4 cells transduced with shGFP (gray bars) or shCCTβ2 (black bars) as described in the legend to Fig. 6. Results are the mean and S.D. of three experiments.
To determine how CCTα silencing prevented detachment-independent growth of ras4 cells, we initially tested whether apoptosis was increased. However, CCTα silencing did not increase apoptosis in ras4 cells as measured by several different methods (results not shown). In contrast, FACS analysis of detached ras4 cells transduced with two different CCTα shRNAs showed a significant 10–15% increase in G0/G1 and a reduction in the G2/M component compared with control (Fig. 6C). This partial G0/G1 block due to CCTα silencing was not observed in adherent ras4 cells (Fig. 6C).
Finally, we tested whether reduction of CCTβ expression in IEC-18 and ras4 cells with two different shRNAs affected cell viability as measured by a clonogenicity assay (Fig. 6D). Both CCTβ shRNAs reduced mRNA expression by >85% in IEC-18 and ras4 cells as quantified by qPCR (results not shown). Relative to the control shRNA, reduction of CCTβ expression did not affect the viability and anchorage-independent growth of 48-h detached IEC-18 and ras4 cells. CCTβ silencing in IEC-18 or ras4 cells also did not affect [3H]choline incorporation into PC (Fig. 6E) or other choline metabolites (results not shown).
Because increased CCTα expression was correlated with the ability of ras4 cells to grow anchorage-independently, we tested whether CCTα overexpression and/or activation with oleate had a similar effect in IEC-18 (Fig. 7). Overexpression of GFP-tagged CCTα (8–10-fold) and/or activation with oleate did not improve the detachment-independent growth of IEC-18 as measured by the clonogenicity assay, indicating that CCTα is not sufficient to promote anoikis-resistance in the absence of ras transformation.
FIGURE 7.

Activation and/or overexpression of CCTα does not grant anchorage-independent growth to IEC-18. After transfection with pCCTα-GFP or empty vector for 24 h, IEC-18 (500 cells) were cultured on 60-mm dishes coated with SeaPlaque agarose in media supplemented without or with 300 μm oleate complexed to BSA for 72 h. Cells were then seeded on plastic dishes, and colony formation was determined. CCTα-GFP overexpression relative to endogenous CCTα was determined after SDS-8% PAGE and immunoblotting using a CCTα antibody. Results are the means and S.D. (error bars) of three separate experiments.
Capacity for anchorage-independent growth is critical for the ability of malignant cells to form primary tumors. We thus examined the effect of reducing CCTα expression on the tumorigenicity of ras4 cells in mice. To this end, ras4 cells expressing control shGFP, shCCTα1, or shCCTα2 were injected subcutaneously into the flanks of immunocompromised BALB/c mice, and tumor growth was measured (Fig. 8). Although ras4 cells transduced with shCCTα1 and shCCTα2 produced tumors similar in size to controls at day 1–7, expression of shCCTα1 caused a significant ∼50% reduction in tumor volume at days 8–12. ras4 cells transduced with shCCTα2 produced tumors with a significant 20% in reduction volume at day 12. The results of this xenograft experiment indicate that CCTα silencing reduces the tumorigenic potential of ras4 cells.
FIGURE 8.
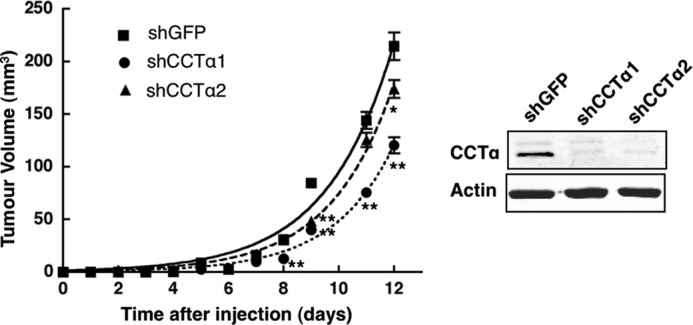
Tumor growth of ras4 cells is reduced by CCTα silencing. ras4 cells (2 × 105) expressing shCCTα1, shCCTα2, or shGFP were resuspended in sterile PBS and injected subcutaneously into the right flank of athymic mice. Tumor growth was measured daily using a skin fold caliper. Results are the mean and S.D. (error bars) of three separate experiments involving 8–12 mice. *, p < 0.05; **, p < 0.005 compared with shGFP. The inset shows a representative immunoblot of CCTα expression in ras4 cells prior to injection into mice.
CCTα-dependent PC Synthesis Facilitates Anchorage-independent Growth of ras Cells
We next used [3H]choline labeling to compare PC metabolism between adherent and anchorage-deprived IEC-18 and ras cells, as well as the influence of CCTα silencing on PC synthesis in detached ras cells (Fig. 9). IEC cells showed a progressive loss of viability after growth in suspension culture using a clonogenicity assay, whereas ras3, ras4, and ras7 cells displayed robust anchorage-independent growth after 72 h (Fig. 9A). These cells cultured in suspension on agarose-coated dishes were labeled with [3H]choline for 24, 48, or 72 h, and incorporation into PC and choline metabolites was compared with that of similarly treated adherent cells (Fig. 9B). Consistent with reduced IEC-18 viability, [3H]choline incorporation into PC and choline metabolites was almost completely inhibited by 72 h (Fig. 9B). IEC-ras cells had an initial decline in [3H]PC synthesis at 24 h that increased to <1.5-fold above adherent controls by 72 h. [3H]pCholine levels also declined at 24 h and remained lower than levels of their adherent counterparts, consistent with increased conversion to PC. With the exception of ras3, isotope incorporation into CDP-choline and GPC was similar in adherent and detached ras cells.
To test whether the detachment-dependent increase in PC synthesis in ras cells was dependent on CCTα, the enzyme was silenced using two inducible lentiviral shRNAs. For these experiments, ras4 cells transduced with doxycycline-inducible lentiviral shRNAs were cultured on agarose for 24 h, incubated with or without doxycycline for 48 h, and pulse-labeled with [3H]choline for 3 h. Relative to non-induced controls, [3H]choline incorporation into PC was significantly reduced by both CCTα-specific shRNAs (Fig. 9C). [3H]GPC was reduced with one shRNA, but all other metabolites were unaffected. Immunoblotting revealed that CCTα expression was suppressed by >70% in doxycycline-treated cells compared with uninduced controls (Fig. 9D). These results suggest that ras-induced proliferation of detached IEC is linked to increased CCTα expression and PC synthesis.
To determine how detachment from the ECM affects CCT expression, total cell extracts of IEC-18 and ras cells cultured under detached conditions for up to 72 h were immunoblotted for CCTα and β2 (Fig. 10A). CCTα and CCTβ2 in adherent IEC-18 and ras cells resolved as a single major species, suggesting that the proteins are hypophosphorylated (Fig. 2A). When IEC were denied anchorage by culturing on agarose, there was an immediate shift to a higher mass CCTα and CCTβ2 species. As cell viability and protein expression declined, the relative expression of these two species oscillated (Fig. 10A). In anoikis-resistant ras cells, a single species of CCTα and CCTβ2 was evident immediately after detachment (0 h), but at 6–24 h, a higher mass species appeared, and it become prominent by 72 h. Alkaline phosphatase treatment of ras cell lysates confirmed that the increase in molecular mass is due to hyperphosphorylation (results not shown). This shift in apparent molecular mass of CCTα on SDS-PAGE is due to phosphorylation of the C-terminal domain, which negatively regulates membrane association and activity (54). Thus, expression of CCTα and CCTβ2 is sustained in detached ras-transformed cells but eventually shifts to a hyperphosphorylated species.
FIGURE 10.
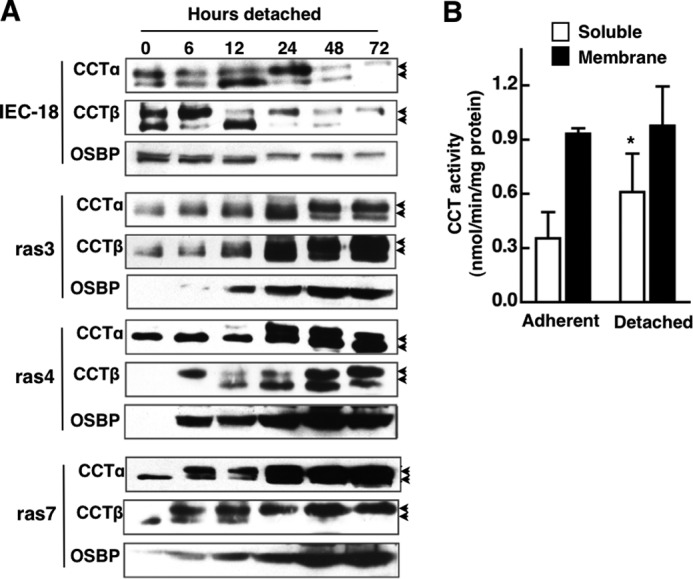
Effect of anchorage-independent growth on CCTα expression and activity. A, CCTα and CCTβ expression in cell lysates from IEC-18 and IEC-ras cells cultured on agarose for 0–72 h was analyzed by immunoblotting. Because of the high level of CCTα overexpression in IEC-ras cells, 4 times more IEC lysate was loaded, and exposure times were adjusted to improve resolution and comparison. The arrows denote the upper hyperphosphorylated and lower hypophosphorylated forms of CCTα. B, CCT activity in the soluble and membrane (particulate fractions) of adherent and 72-h detached ras4 cells. Results are the mean and S.D. (error bars) of three separate experiments. *, p < 0.05 compared with activity in the soluble fraction from adherent cells.
CCTα activity was assayed in the soluble and particulate fractions (membrane) of 48-h detached and adherent ras4 cells (Fig. 10B). There was a significant 80% increase in soluble CCT activity from detached ras4 cells compared with adherent cells, but membrane-associated activities were comparable. In these experiments, enzyme activity in soluble and membrane fractions was not affected by PC/oleate vesicles (results not shown).
DISCUSSION
Increased fatty acid synthase expression and channeling of glycolytic products into fatty acid synthesis are a hallmark of transformed cells and are frequently associated with more aggressive tumors (55). Increased lipogenesis could provide an energy supply and storage format that enhances cell proliferation. Alternatively, fatty acids could be utilized for membrane biogenesis in rapidly dividing cells. For example, the CDP-choline pathway is exquisitely sensitive to fatty acid activation by virtue of the membrane-sensing properties of CCTα (47), and PC synthesis and choline metabolites are elevated in various cancer cells (15). A comparison between IEC-18 and three ras-transformed clones identified an unusual dichotomy within the CDP-choline pathway, a dramatic elevation of nuclear CCTα expression that that was inactive in adherent cells but contributed to anchorage-independent growth of IEC-ras cells by a mechanism that was dependent on PC biosynthesis.
In adherent IEC-18 and IEC-ras cells, [3H]choline incorporation into PC was similar, as was the total amount of radiolabeled choline incorporated into all metabolites. Radiolabeled water-soluble metabolites were variably altered between the three ras-transformed clones. Despite having apparently normal rates of PC biosynthesis, CCTα mRNA and protein were increased by 5–20-fold in all IEC-ras clones. However, increased CCTα expression in IEC-ras cells was not correlated with the levels of [3H]choline metabolites. For instance, CCTα expression was increased 8-fold in ras7 cells, but choline metabolites and PC were unchanged compared with IEC-18, whereas ras4 cells had less CCTα overexpression but significant changes in pCholine and GPC. The reason for this apparent discrepancy between CCT expression versus CDP-choline pathway activity was revealed when CCT was assayed in vitro; enzyme activity in the cytosolic and membrane fractions of IEC-ras cells was similar to control IEC-18, indicating that the large pool of enzyme was inactive and insensitive to activation by oleate. This was unexpected because CCTα in IEC-ras was partially localized with lamin A/C at the nuclear envelope and fractionated with membranes after digitonin permeabilization, a situation that should be correlated with increased activity (47). Our results agree with a previous study that showed modest increases in CCTα mRNA and protein but inhibition of CCT activity in H-ras-transformed murine fibroblasts (36). The conclusion that IEC-ras cells overexpress a biosynthetically inactive pool of CCTα is supported by the lack of effect of lentiviral shRNA silencing of CCTα on PC synthesis or the level of [3H]choline metabolites in ras4 cells cultured in monolayer.
Unlike CCTα, increased CCTβ expression was not a consistent feature of ras-transformed IEC-18. In addition, there was discordance between total mRNA and protein expression of the full-length CCTβ2 isoform, suggesting additional levels of post-transcriptional regulation or expression of an additional isoform that is not detected by the CCTβ2 antibody. This last explanation seems unlikely because the antibody should detect both rodent CCTβ2 and CCTβ3 species (26). CCTβ also did not appear to have a major role in PC synthesis relative to CCTα; CCTβ silencing in IEC-18 cells had no effect on PC synthesis (Fig. 6E), whereas reducing CCTα expression caused a 40% reduction (Fig. 5C). Although the ras4 cells chosen for anchorage-independent growth experiments did not have significantly elevated CCTβ expression, further reduction by RNAi did not affect ras4 cell viability or PC synthesis (Fig. 6, D and E). Collectively, this suggests that CCTβ expression is not increased by H-ras transformation and does not contribute to anoikis resistance, and it is subordinate to CCTα in terms of providing PC biosynthetic capacity.
A major finding of this study is that ras-induced CCTα up-regulation in IEC-18 is required to promote anchorage-independent growth and malignant transformation. Choline-labeling experiments showed that detached ras cells have increased PC synthesis and reduced [3H]pCholine levels indicative of CCTα activation (Fig. 9B). Unlike adherent ras4 cells, inducible RNAi silencing of CCTα after detachment from the ECM significantly attenuated PC synthesis, suggesting that ras-induced CCTα is activated by these growth conditions. The partial G1 arrest caused by CCTα silencing in detached ras4 cells is also consistent with inhibition of PC synthesis. CCTα is activated in early G1 by phospholipid degradation products and contributes to net PC synthesis during S phase (25, 29, 56). Cessation of PC synthesis in CHO cells expressing temperature-sensitive CCTα resulted in G1 arrest prior to induction of apoptosis (27). In the case of ras4 cells cultured on agarose, reducing CCTα expression by 70% caused partial inhibition of PC synthesis (Fig. 9C) and cell cycle arrest without induction of apoptosis (Fig. 6C) (results not shown).
Despite the apparent increase in PC synthesis in ras cells deprived of attachment to the ECM, CCTα and CCTβ underwent a time-dependent increase in phosphorylation, which is usually associated with the cytosolic inactive form of the enzymes. It is feasible that detachment triggers an essential phosphorylation event required for enzyme activation. However, phosphorylation of CCTα occurred 6–24 h after cells were cultured on agarose, suggesting that it is secondary to other factors that affect enzyme activity (e.g. release of lipid activators upon disengagement from the ECM). Despite the apparent increase in CCTα-dependent PC synthesis, there was only a modest increase in cytosolic CCT activity in detached ras4 cells compared with adherent cells. CCT activity in ras cells did not correlate well with protein expression and metabolic labeling; nor was it activated by exogenous oleate, suggesting a novel mode of enzyme regulation. The factors regulating CCTα in ras-transformed cells will require an in depth analysis of covalent modifications, nuclear localization, and lipid activation in both adherent and detached cell populations.
CCTα has been implicated in rat hepatoma proliferation (57) and several human cancers (58). Our results indicate that ras induction of CCTα promotes carcinogenesis by increasing the PC biosynthetic capacity of cells after detachment from the ECM. In this context, CCTα could promote metastasis by increasing the survival and anoikis resistance of cancer cells detached from the ECM.
Acknowledgment
Robert Zwicker provided excellent technical assistance with cell culture.
This work was supported by Canadian Institutes of Health Research Grants MOP62916 (to N. D. R.) and MOP88914 (to K. V. R.) and a Nova Scotia Health Research Foundation grant (to N. D. R.).
- PC
- phosphatidylcholine
- pCholine
- phosphocholine
- CCT
- CTP:phosphcholine cytidylyltransferase
- CK
- choline kinase
- ECM
- extracellular matrix
- GPC
- glycerophosphocholine
- IEC
- intestinal epithelial cell(s)
- qPCR
- quantitative polymerase chain reaction
- MEM
- minimal essential medium
- shNT
- non-targeting control shRNA.
REFERENCES
- 1. Hsu P. P., Sabatini D. M. (2008) Cancer cell metabolism. Warburg and beyond. Cell 134, 703–707 [DOI] [PubMed] [Google Scholar]
- 2. Menendez J. A., Lupu R. (2007) Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 7, 763–777 [DOI] [PubMed] [Google Scholar]
- 3. Bauer D. E., Hatzivassiliou G., Zhao F., Andreadis C., Thompson C. B. (2005) ATP citrate lyase is an important component of cell growth and transformation. Oncogene 24, 6314–6322 [DOI] [PubMed] [Google Scholar]
- 4. Hatzivassiliou G., Zhao F., Bauer D. E., Andreadis C., Shaw A. N., Dhanak D., Hingorani S. R., Tuveson D. A., Thompson C. B. (2005) ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 8, 311–321 [DOI] [PubMed] [Google Scholar]
- 5. Brusselmans K., De Schrijver E., Verhoeven G., Swinnen J. V. (2005) RNA interference-mediated silencing of the acetyl-CoA-carboxylase-α gene induces growth inhibition and apoptosis of prostate cancer cells. Cancer Res. 65, 6719–6725 [DOI] [PubMed] [Google Scholar]
- 6. Menendez J. A., Vellon L., Mehmi I., Oza B. P., Ropero S., Colomer R., Lupu R. (2004) Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene overexpression in cancer cells. Proc. Natl. Acad. Sci. U.S.A. 101, 10715–10720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pizer E. S., Lax S. F., Kuhajda F. P., Pasternack G. R., Kurman R. J. (1998) Fatty acid synthase expression in endometrial carcinoma. Correlation with cell proliferation and hormone receptors. Cancer 83, 528–537 [PubMed] [Google Scholar]
- 8. Scaglia N., Chisholm J. W., Igal R. A. (2009) Inhibition of stearoylCoA desaturase-1 inactivates acetyl-CoA carboxylase and impairs proliferation in cancer cells. Role of AMPK. PLoS One 4, e6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Porstmann T., Griffiths B., Chung Y. L., Delpuech O., Griffiths J. R., Downward J., Schulze A. (2005) PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene 24, 6465–6481 [DOI] [PubMed] [Google Scholar]
- 10. Pizer E. S., Wood F. D., Pasternack G. R., Kuhajda F. P. (1996) Fatty acid synthase (FAS). A target for cytotoxic antimetabolites in HL60 promyelocytic leukemia cells. Cancer Res. 56, 745–751 [PubMed] [Google Scholar]
- 11. Swinnen J. V., Van Veldhoven P. P., Timmermans L., De Schrijver E., Brusselmans K., Vanderhoydonc F., Van de Sande T., Heemers H., Heyns W., Verhoeven G. (2003) Fatty acid synthase drives the synthesis of phospholipids partitioning into detergent-resistant membrane microdomains. Biochem. Biophys. Res. Commun. 302, 898–903 [DOI] [PubMed] [Google Scholar]
- 12. Li Z., Vance D. E. (2008) Phosphatidylcholine and choline homeostasis. J. Lipid Res. 49, 1187–1194 [DOI] [PubMed] [Google Scholar]
- 13. Ratnam S., Kent C. (1995) Early increase in choline kinase activity upon induction of the H-ras oncogene in mouse fibroblast cell lines. Arch. Biochem. Biophys. 323, 313–322 [DOI] [PubMed] [Google Scholar]
- 14. Teegarden D., Taparowsky E. J., Kent C. (1990) Altered phosphatidylcholine metabolism in C3H10T1/2 cells transfected with the Harvey-ras oncogene. J. Biol. Chem. 265, 6042–6047 [PubMed] [Google Scholar]
- 15. Glunde K., Serkova N. J. (2006) Therapeutic targets and biomarkers identified in cancer choline phospholipid metabolism. Pharmacogenomics 7, 1109–1123 [DOI] [PubMed] [Google Scholar]
- 16. Aboagye E. O., Bhujwalla Z. M. (1999) Malignant transformation alters membrane choline phospholipid metabolism of human mammary epithelial cells. Cancer Res. 59, 80–84 [PubMed] [Google Scholar]
- 17. Ramírez de Molina A., Gutiérrez R., Ramos M. A., Silva J. M., Silva J., Bonilla F., Sánchez J. J., Lacal J. C. (2002) Increased choline kinase activity in human breast carcinomas. Clinical evidence for a potential novel antitumor strategy. Oncogene 21, 4317–4322 [DOI] [PubMed] [Google Scholar]
- 18. Gallego-Ortega D., Ramirez de Molina A., Ramos M. A., Valdes-Mora F., Barderas M. G., Sarmentero-Estrada J., Lacal J. C. (2009) Differential role of human choline kinase α and β enzymes in lipid metabolism. Implications in cancer onset and treatment. PLoS One 4, e7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ramírez de Molina A., Gallego-Ortega D., Sarmentero J., Bañez-Coronel M., Martín-Cantalejo Y., Lacal J. C. (2005) Choline kinase is a novel oncogene that potentiates RhoA-induced carcinogenesis. Cancer Res. 65, 5647–5653 [DOI] [PubMed] [Google Scholar]
- 20. Yalcin A., Clem B., Makoni S., Clem A., Nelson K., Thornburg J., Siow D., Lane A. N., Brock S. E., Goswami U., Eaton J. W., Telang S., Chesney J. (2010) Selective inhibition of choline kinase simultaneously attenuates MAPK and PI3K/AKT signaling. Oncogene 29, 139–149 [DOI] [PubMed] [Google Scholar]
- 21. Chua B. T., Gallego-Ortega D., Ramirez de Molina A., Ullrich A., Lacal J. C., Downward J. (2009) Regulation of Akt(Ser473) phosphorylation by choline kinase in breast carcinoma cells. Mol. Cancer 8, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rodríguez-González A., Ramírez de Molina A., Fernández F., Ramos M. A., del Carmen Núñez M., Campos J., Lacal J. C. (2003) Inhibition of choline kinase as a specific cytotoxic strategy in oncogene-transformed cells. Oncogene 22, 8803–8812 [DOI] [PubMed] [Google Scholar]
- 23. Glunde K., Raman V., Mori N., Bhujwalla Z. M. (2005) RNA interference-mediated choline kinase suppression in breast cancer cells induces differentiation and reduces proliferation. Cancer Res. 65, 11034–11043 [DOI] [PubMed] [Google Scholar]
- 24. Cornell R. B., Taneva S. G. (2006) Amphipathic helices as mediators of the membrane interaction of amphitropic proteins, and as modulators of bilayer physical properties. Curr. Protein Pept. Sci. 7, 539–552 [DOI] [PubMed] [Google Scholar]
- 25. Jackowski S., Fagone P. (2005) CTP:phosphocholine cytidylyltransferase. Paving the way from gene to membrane. J. Biol. Chem. 280, 853–856 [DOI] [PubMed] [Google Scholar]
- 26. Jackowski S., Rehg J. E., Zhang Y. M., Wang J., Miller K., Jackson P., Karim M. A. (2004) Disruption of CCTβ2 expression leads to gonadal dysfunction. Mol. Cell Biol. 24, 4720–4733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cui Z., Houweling M., Chen M. H., Record M., Chap H., Vance D. E., Tercé F. (1996) A genetic defect in phosphatidylcholine biosynthesis triggers apoptosis in Chinese hamster ovary cells. J. Biol. Chem. 271, 14668–14671 [DOI] [PubMed] [Google Scholar]
- 28. Wang L., Magdaleno S., Tabas I., Jackowski S. (2005) Early embryonic lethality in mice with targeted deletion of the CTP:phosphocholine cytidylyltransferase α gene (Pcyt1a). Mol. Cell Biol. 25, 3357–3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jackowski S. (1994) Coordination of membrane phospholipid synthesis with the cell cycle. J. Biol. Chem. 269, 3858–3867 [PubMed] [Google Scholar]
- 30. Northwood I. C., Tong A. H., Crawford B., Drobnies A. E., Cornell R. B. (1999) Shuttling of CTP:phosphocholine cytidylyltransferase between the nucleus and endoplasmic reticulum accompanies the wave of phosphatidylcholine synthesis during the G0 → G1 transition. J. Biol. Chem. 274, 26240–26248 [DOI] [PubMed] [Google Scholar]
- 31. Anthony M. L., Zhao M., Brindle K. M. (1999) Inhibition of phosphatidylcholine biosynthesis following induction of apoptosis in HL-60 cells. J. Biol. Chem. 274, 19686–19692 [DOI] [PubMed] [Google Scholar]
- 32. Lagace T. A., Ridgway N. D. (2005) Induction of apoptosis by lipophilic activators of CTP:phosphocholine cytidylyltransferase α (CCTα). Biochem. J. 392, 449–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lagace T. A., Miller J. R., Ridgway N. D. (2002) Caspase processing and nuclear export of CTP:phosphocholine cytidylyltransferase α during farnesol-induced apoptosis. Mol. Cell Biol. 22, 4851–4862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Geilen C. C., Wieder T., Boremski S., Wieprecht M., Orfanos C. E. (1996) c-Ha-ras oncogene expression increases choline uptake, CTP:phosphocholine cytidylyltransferase activity and phosphatidylcholine biosynthesis in the immortalized human keratinocyte cell line HaCaT. Biochim. Biophys. Acta 1299, 299–305 [DOI] [PubMed] [Google Scholar]
- 35. Momchilova A., Markovska T., Pankov R. (1999) Ha-ras-transformation alters the metabolism of phosphatidylethanolamine and phosphatidylcholine in NIH 3T3 fibroblasts. Cell Biol. Int. 23, 603–610 [DOI] [PubMed] [Google Scholar]
- 36. Bakovic M., Waite K., Vance D. E. (2003) Oncogenic Ha-Ras transformation modulates the transcription of the CTP:phosphocholine cytidylyltransferase α gene via p42/44MAPK and transcription factor Sp3. J. Biol. Chem. 278, 14753–14761 [DOI] [PubMed] [Google Scholar]
- 37. Khwaja A., Rodriguez-Viciana P., Wennström S., Warne P. H., Downward J. (1997) Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J. 16, 2783–2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Simpson C. D., Anyiwe K., Schimmer A. D. (2008) Anoikis resistance and tumor metastasis. Cancer Lett. 272, 177–185 [DOI] [PubMed] [Google Scholar]
- 39. Rak J., Mitsuhashi Y., Erdos V., Huang S. N., Filmus J., Kerbel R. S. (1995) Massive programmed cell death in intestinal epithelial cells induced by three-dimensional growth conditions. Suppression by mutant c-H-ras oncogene expression. J. Cell Biol. 131, 1587–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rosen K., Rak J., Jin J., Kerbel R. S., Newman M. J., Filmus J. (1998) Downregulation of the pro-apoptotic protein Bak is required for the ras-induced transformation of intestinal epithelial cells. Curr. Biol. 8, 1331–1334 [DOI] [PubMed] [Google Scholar]
- 41. Gehrig K., Morton C. C., Ridgway N. D. (2009) Nuclear export of the rate-limiting enzyme in phosphatidylcholine synthesis is mediated by its membrane binding domain. J. Lipid Res. 50, 966–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cornell R. (1989) Chemical cross-linking reveals a dimeric structure for CTP:phosphocholine cytidylyltransferase. J. Biol. Chem. 264, 9077–9082 [PubMed] [Google Scholar]
- 43. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 44. Rofstad E. K., Wahl A., Davies Cde L., Brustad T. (1986) Growth characteristics of human melanoma multicellular spheroids in liquid-overlay culture. Comparisons with the parent tumour xenografts. Cell Tissue Kinet. 19, 205–216 [DOI] [PubMed] [Google Scholar]
- 45. Tomayko M. M., Reynolds C. P. (1989) Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol. 24, 148–154 [DOI] [PubMed] [Google Scholar]
- 46. Janssen K. P., Abala M., El Marjou F., Louvard D., Robine S. (2005) Mouse models of K-ras-initiated carcinogenesis. Biochim. Biophys. Acta 1756, 145–154 [DOI] [PubMed] [Google Scholar]
- 47. Cornell R. B., Northwood I. C. (2000) Regulation of CTP:phosphocholine cytidylyltransferase by amphitropism and relocalization. Trends Biochem. Sci. 25, 441–447 [DOI] [PubMed] [Google Scholar]
- 48. Lagace T. A., Ridgway N. D. (2005) The rate-limiting enzyme in phosphatidylcholine synthesis regulates proliferation of the nucleoplasmic reticulum. Mol. Biol. Cell 16, 1120–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gehrig K., Cornell R. B., Ridgway N. D. (2008) Expansion of the nucleoplasmic reticulum requires the coordinated activity of lamins and CTP:phosphocholine cytidylyltransferase α. Mol. Biol. Cell 19, 237–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Karim M., Jackson P., Jackowski S. (2003) Gene structure, expression and identification of a new CTP:phosphocholine cytidylyltransferase β isoform. Biochim. Biophys. Acta 1633, 1–12 [DOI] [PubMed] [Google Scholar]
- 51. Wang Y., Sweitzer T. D., Weinhold P. A., Kent C. (1993) Nuclear localization of soluble CTP:phosphocholine cytidylyltransferase. J. Biol. Chem. 268, 5899–5904 [PubMed] [Google Scholar]
- 52. Liu Z., Li H., Derouet M., Filmus J., LaCasse E. C., Korneluk R. G., Kerbel R. S., Rosen K. V. (2005) ras oncogene triggers up-regulation of cIAP2 and XIAP in intestinal epithelial cells. epidermal growth factor receptor-dependent and -independent mechanisms of ras-induced transformation. J. Biol. Chem. 280, 37383–37392 [DOI] [PubMed] [Google Scholar]
- 53. Coll M. L., Rosen K., Ladeda V., Filmus J. (2002) Increased Bcl-xL expression mediates v-Src-induced resistance to anoikis in intestinal epithelial cells. Oncogene 21, 2908–2913 [DOI] [PubMed] [Google Scholar]
- 54. Yang W., Jackowski S. (1995) Lipid activation of CTP:phosphocholine cytidylyltransferase is regulated by the phosphorylated carboxyl-terminal domain. J. Biol. Chem. 270, 16503–16506 [DOI] [PubMed] [Google Scholar]
- 55. Kuhajda F. P. (2000) Fatty-acid synthase and human cancer. New perspectives on its role in tumor biology. Nutrition 16, 202–208 [DOI] [PubMed] [Google Scholar]
- 56. Ng M. N., Kitos T. E., Cornell R. B. (2004) Contribution of lipid second messengers to the regulation of phosphatidylcholine synthesis during cell cycle re-entry. Biochim. Biophys. Acta 1686, 85–99 [DOI] [PubMed] [Google Scholar]
- 57. Tessitore L., Dianzani I., Cui Z., Vance D. E. (1999) Diminished expression of phosphatidylethanolamine N-methyltransferase 2 during hepatocarcinogenesis. Biochem. J. 337, 23–27 [PMC free article] [PubMed] [Google Scholar]
- 58. Yu K., Ganesan K., Tan L. K., Laban M., Wu J., Zhao X. D., Li H., Leung C. H., Zhu Y., Wei C. L., Hooi S. C., Miller L., Tan P. (2008) A precisely regulated gene expression cassette potently modulates metastasis and survival in multiple solid cancers. PLoS Genet. 4, e1000129. [DOI] [PMC free article] [PubMed] [Google Scholar]